
Method Article
DNAzyme-dependent Analysis of rRNA 2’-O-Methylation
In This Article
Summary
Here we present a protocol for DNAzyme-dependent cleavage of RNA. This enables fast and site-dependent analysis of RNA 2’-O-methylation. This approach can be used for the preliminary or major assessment of snoRNA activity.
Abstract
Guide box C/D small nucleolar RNAs (snoRNAs) catalyze 2’-O-methylation of ribosomal and small nuclear RNA. However, a large number of snoRNA in higher eukaryotes may promiscuously recognize other RNA species and 2’-O-methylate multiple targets. Here, we provide step-by-step guide for the fast and non-expensive analysis of the site-specific 2’-O-methylation using a well-established method employing short DNA oligonucleotides called DNAzymes. These DNA fragments contain catalytic sequences which cleave RNA at specific consensus positions, as well as variable homology arms directing DNAzyme to its RNA targets. DNAzyme activity is inhibited by 2-’O-methylation of the nucleotide adjacent to the cleavage site in the RNA. Thus, DNAzymes, limited only by the consensus of the cleaved sequence, are perfect tools for the quick analysis of snoRNA-mediated RNA 2’-O-methylation. We analyzed snoRNA snR13- and snR47-guided 2’-O-methylation of 25S ribosomal RNA in Saccharomyces cerevisiae to demonstrate the simplicity of the technique and to provide a detailed protocol for the DNAzyme-dependent assay.
Introduction
RNA modifications play important roles in the regulation of gene expression. RNA 2’-O-methylation and pseudouridylation, which are guided by box C/D and box H/ACA small nucleolar RNAs (snoRNAs) respectively, protect RNA from degradation and stabilize their higher-order structures1,2,3. SnoRNA targets have been identified mainly in ribosomal RNAs (rRNA) and small nuclear RNAs (snRNAs). However, in higher eukaryotes, there are potentially hundreds of snoRNA with no assigned functions and some of them may recognize multiple RNAs1,4,5,6,7. Therefore, methods which allow for the identification and analysis of snoRNA-guided modifications are important tools in uncovering mechanisms governing cellular processes.
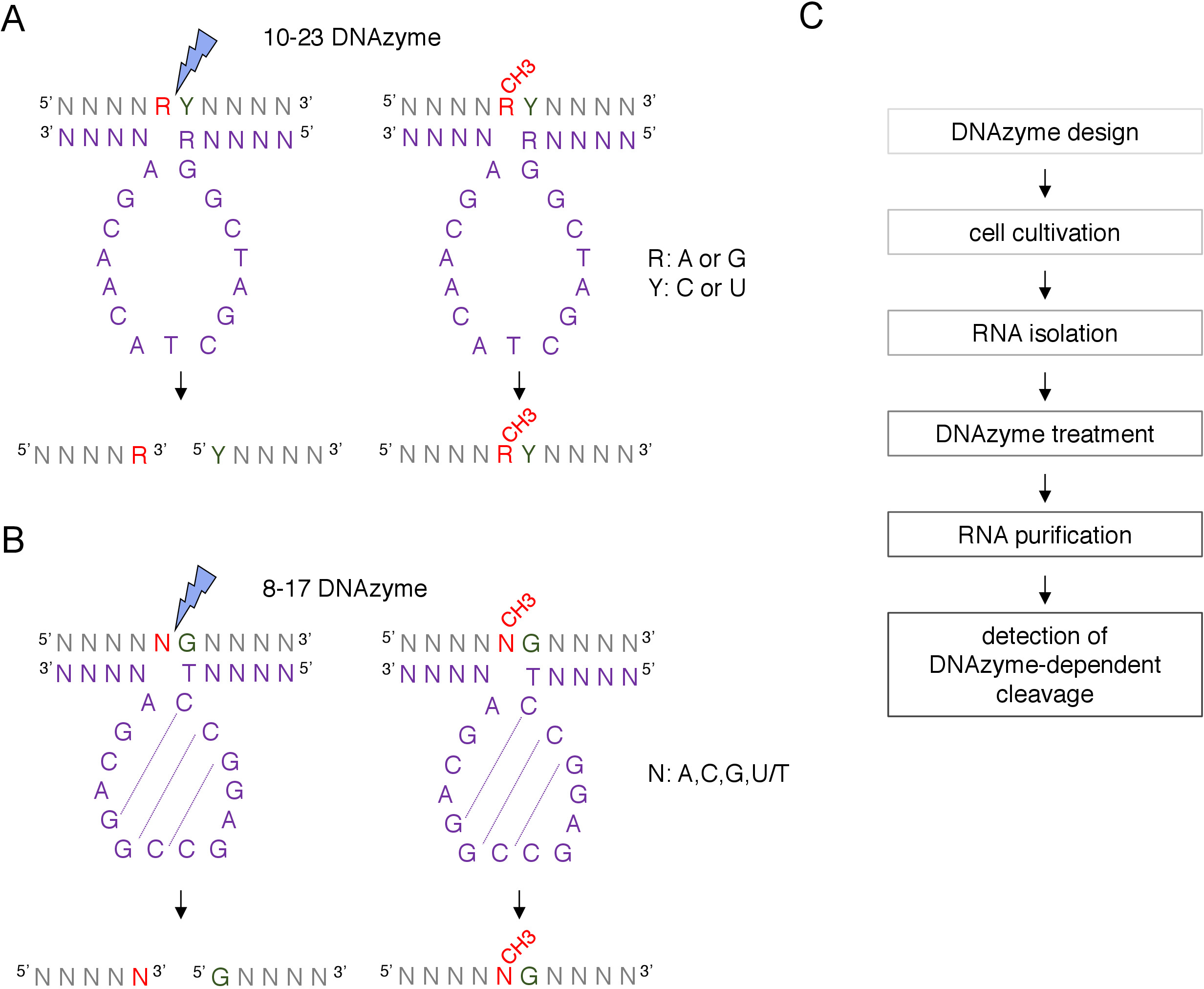
A box C/D snoRNA-guided putative 2’-O-methylation site can be identified bioinformatically and confirmed experimentally by many techniques, including RNase H-directed cleavage, or site-specific and genome-wide methods, which employ reverse transcription in low nucleotides (dNTPs) concentration approach8,9,10,11. These techniques are very sensitive but also laborious and expensive, therefore, may not be suitable for the initial or quick testing. One of the simplest and low-cost methods to identify 2’-O-methylation sites is DNAzyme-dependent RNA cleavage12. DNAzymes are short, single-stranded and catalytically active DNA molecules capable of endonucleolytic cleavage of RNA at specific positions. They consist of a conserved and catalytically active core sequence and 5’ and 3’ binding arms composed of variable sequences designed to hybridize by Watson-Crick base-pairing to the RNA target (Figure 1). Thus, the 5’ and 3’ arms deliver the catalytic sequence to the specific RNA site. DNAzyme-dependent cleavage is inhibited by 2’-O-methylation of the nucleotide positioned directly upstream of the cleavage site12,13. This makes DNAzymes very practical tools for the analysis of putative or known RNA 2’-O-methylation sites.
Two types of DNAzymes are used for RNA modifications analyses12. The active sequence of 10-23 DNAzyme (Figure 1A) consists of 15 nucleotides (5’RGGCTAGCTACAACGA3’) which form a loop around the targeted RNA purine-pyrimidine (RY) dinucleotide and catalyze the cleavage between these two nucleotides. The RNA purine (R) is not base-paired with the DNAzyme and the 2’-O-methylation presents on the DNAzyme inhibits the cleavage. The binding arms of 10-23 DNAzymes are usually 10-15 nucleotides long. The second DNAzyme class, 8-17 DNAzymes (Figure 1B) contain 14-nucleotide catalytic sequence (5’TCCGAGCCGGACGA3’). Nucleotides C2, C3 and G4 pair with C9 G10 and G11 forming a short stem-loop structure. 8-17 DNAzymes cleave RNA upstream of any guanine that is imperfectly paired with the first thymine from the DNAzyme active sequence. The RNA nucleotide upstream of the guanine is not base-paired with DNAzyme and its 2’-O-methylation impairs the cleavage. 8-17 DNAzymes require longer homology arms of around 20 nucleotides to direct DNAzyme to its specific sequence.
Here, we provide a step-by-step protocol for the analysis of 2’-O-methylation of rRNA in Saccharomyces cerevisiae using 10-23 and 8-17 DNAzyme-dependent approaches12,13 (Figure 1C). This protocol can be easily adapted for other organisms and RNA species and employed for the fast, preliminary or major analyses of site-specific RNA 2’-O-methylation.
Protocol
1. Strains, Media, and Buffer Recipes
- Prepare yeast (S. cerevisiae) media as detailed here: YP (1% w/v yeast extract, 2% w/v bacteriological peptone), and glucose and galactose stocks at 20% w/v.
- Prepare sodium acetate (NaAc)-EDTA (AE) buffer as detailed here: 50 mM NaAc pH 5.3 and 10 mM EDTA.
- Prepare 10-23 DNAzyme 4x Incubation Buffer as detailed here: 24 mM Tris pH 8.0, 60 mM NaCl and 10-23 DNAzyme 4x Reaction Buffer: 200 mM Tris pH 8.0 and 600 mM NaCl.
- Prepare 8-17 DNAzyme 2x Reaction Buffer as detailed here: 200 mM KCl, 800 mM NaCl, 100 mM HEPES pH 7.0, 15 mM MgCl2, and 15 mM MnCl2.
- Prepare 10x MOPS Buffer as detailed here: 200 mM MOPS, 50 mM NaAc, 1 mM EDTA; pH 7.0 and 1.5x Sample Denaturing Buffer: 50% v/v formamide, 20% v/v formaldehyde, 1.5x MOPS buffer.
- Obtain S. cerevisiae strains, BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0); GAL1::SNR13 (as BY4741 but GAL1::SNR13:KANmX); GAL1::SNR47 (as BY4741 but GAL1::SNR47:HIS3mX). Any other yeast strain can be used for this analysis.
2. DNAzyme Design
- Find RNA sequence of interest or putative methylation site using an appropriate database. For S. cerevisiae snoRNA targets, use yeast snoRNA database: http://people.biochem.umass.edu/fournierlab/snornadb/mastertable.php14
- To find the methylation site of interest, e.g., snR13-dependent site, select “snR13” and make a note of the position of the modified nucleotide (e.g., snR13-guided A2281 in 25S rRNA).
- Find sequences upstream and downstream of the modified nucleotide using appropriate database. For S. cerevisiae, use the Saccharomyces Genome Database: https://www.yeastgenome.org/
- Search for the target gene name e.g., RDN25 (coding 25S rRNA).
- From the “sequence” tab, select 10-15 nucleotides upstream (5’ arm) and downstream (3 ’arm) of the methylation site when using a 10-23 DNAzyme assay and 20 nucleotides upstream (5’ arm) and downstream (3’arm) of the methylation site for a 8-17 DNAzyme.
- Create complementary sequences of 5’ and 3’ arms.
- Flank 10-23 or 8-17 DNAzyme catalytic sequence with complementary sequences of 5’ and 3’ arms.
- Order DNAzyme as a normal DNA oligonucleotide from the supplier.
3. S. cerevisiae Growth Conditions
NOTE: S. cerevisiae BY4741 strain derivatives were used, in which the expression of either SNR13 or SNR47 snoRNA is driven from the inducible GAL1 promoter. In order to induce or inhibit their synthesis, grow cells on medium containing galactose (GAL1-dependent transcription on) or glucose (GAL1-dependent transcription off). As a control, use the wild type strain (BY4741) grown either on galactose or glucose.
- Grow yeast strains in an appropriate medium and conditions. To analyze GAL1::SNR13 and GAL1::SNR47 strains as well as the isogenic wild type strain, grow cells in 50 mL of YP media with either 2% glucose (YPD) or galactose (YPGal) at 30 °C to the middle exponential phase.
- Centrifuge cells at 1,000 x g, for 3 min at 4 °C.
- Discard the supernatant and keep the pellets.
- Freeze cell pellets in liquid nitrogen and store them at -80 °C.
CAUTION: Liquid nitrogen may cause severe cryogenic burns. Always wear protective clothing and exercise safety precautions.
NOTE: Cell pellets can be stored at -80 °C up to 1 month. The protocol can be paused here if needed.
4. RNA Isolation15
NOTE: Use the most appropriate method to isolate RNA. For yeast S. cerevisiae, hot-phenol RNA extraction can be used.
- Add 1 mL of ice-cold water, resuspend the pellets and transfer resuspended cells to 1.5 mL microtubes.
- Centrifuge at 20,000 x g for 10 s at 4 °C and remove the supernatant.
- Add 400 µL of AE buffer and resuspend the cells.
NOTE: Steps 4.4-4.15 are performed at room temperature unless stated otherwise. - Add 40 µL of 10% SDS and 400 µL of acid phenol (pH 4.5).
CAUTION: Phenol is toxic and should be handled under a fume hood. Always wear a lab coat, protective gloves, and glasses when working with phenol. Dispose of the waste according to the institutional regulations. - Mix well by vortexing for 20 s.
- Incubate at 65 °C for 10 min. Every 2 min, gently open and close the tube to release the pressure and flip the tube 2-3 times to mix the phases.
- Transfer the tubes to -80 °C and incubate for 10 min.
- Defrost the tubes on the bench and centrifuge at 20,000 x g, for 5 min at room temperature.
- Transfer the upper phase to a new tube containing 400 µL acid phenol:chloroform:isoamyl alcohol (25:24:1). Do not disrupt the interphase.
CAUTION: Chloroform is toxic and should be handled under a fume hood. Always wear a lab coat, protective gloves, and glasses when working with chloroform. Dispose of the waste according to institutional regulations. - Mix well by vortexing for 30 s and centrifuge at 20,000 x g for 10 min at room temperature.
- Transfer the upper phase (~400 µL) to a new tube containing 400 µL chloroform.
- Mix well by vortexing for 30 s and centrifuge at 20,000 x g for 5 min at room temperature.
- Transfer the upper phase (~300-350 µL) to a new tube containing 1 mL of EtOH and 40 µL of 7.5 M ammonium acetate (NH4AC). Mix by flipping the tube a few times.
- Incubate at -80 °C for 2 h or overnight at -20 °C.
NOTE: The procedure can be paused here. - Centrifuge at 20,000 x g, for 10 min at 4 °C. A small, white RNA pellet will become visible on the bottom of the tube.
- Remove EtOH by pipetting to avoid disturbing the pellet.
- Add 1 mL of 70% EtOH and centrifuge at 20,000 x g for 5 min at room temperature.
- Remove 70% EtOH by pipetting.
- Centrifuge at 20,000 x g for 15 s and remove the remaining EtOH with 2-20 µL pipette.
- Leave the tube open on the bench for 5 min to dry the RNA pellet.
NOTE: RNA pellet changes its color from white to transparent when dry. - Resuspend the RNA pellet in 30 µL of RNase/DNase-free H2O, transfer the tube immediately on the ice and measure RNA concentration on microspectrophotometer.
- Freeze the samples at -20 °C.
NOTE: RNA can be stored at -20 °C up to 1 month and at -80 °C up to 1 year. The procedure can be paused here or proceeded directly to the next step.
5. DNAzyme Digestion
- 10-23 DNAzyme digestion
- In 1.5 mL tubes prepare an incubation mix by combining 5 µg of RNA, 200 pmol of 10-23 DNAzyme (2 µL of 100 μM stock solution) and 2.5 µL of 4x 10-23 Incubation Buffer in a total volume of 10 µL. Keep the tubes on ice.
- Transfer the tubes to a dry heat block set at 95 °C and incubate for 3 min.
- Transfer the tubes immediately on ice and incubate for 5 min.
- Spin down briefly and put the tubes back on the ice.
- Add 20 U of RNase inhibitor (e.g., 0.5 µL RiboLock RNase inhibitor).
- Place the tubes in a dry heat block set for 25 °C and incubate for 10 min.
- In the meantime, prepare a reaction mixture in a 1.5 µL tube by combining 5 µL of 4x 10-23 Reaction Buffer with 4 µL of 300 mM MgCl2 and 1 µL H2O. Place the tube in a dry block set to 37 °C.
- Transfer the incubation mix to a dry heat block set for 37 °C and add 10 µL of pre-warmed reaction mix.
- Incubate the reaction at 37 °C for 1 h.
- Transfer the tubes on ice and proceed to step 5.3.1.
- 8-17 DNAzyme digestion
- Prepare a 1.5 mL microtube with 5 µg of RNA in a total volume of 6 µL. Keep the tube on ice.
- Prepare a 1.5 mL microtube with 400 pmol of 8-17 DNAzyme (4 µL out of 100 μM stock). Keep the tube on ice.
- Transfer the tubes to a dry heat block set for 95 °C and incubate for 2 min.
- Move the RNA sample on ice.
- Spin down the tube with DNAzyme for 5 s and incubate at 25 °C for 10 min.
- At the same time, prepare a 1.5 mL tube with 10 µL of 2x 8-17 Reaction Buffer and incubate at 25 °C.
- Prepare a reaction mixture by adding 10 µL of pre-warmed 2x Reaction Buffer to the tube with DNAzyme.
- Transfer 14 µL of the reaction mix to the tube with RNA and add 20 U of RNase inhibitor.
- Incubate the reaction at 25 °C for 2 h.
- Transfer the tube on ice and proceed to RNA purification (step 5.3.1).
- RNA purification
- Add 350 µL of water and 400 µL of chloroform to the reaction tube, mix well by vortexing for 30 s and centrifuge at 20,000 x g for 5 min at room temperature.
- Transfer the upper phase (~300-350 µL) to a new tube containing 1 mL of EtOH, 40 µL of 7.5 M NH4AC and 1 µL of glycogen (10 µg/µL). Mix by flipping the tube a few times.
- Incubate at -80 °C for 2 h or overnight at -20 °C.
NOTE: The procedure can be paused here. - Repeat steps from 4.15 to 4.21.
- Resuspend the RNA pellet in 10 µL of RNase/DNase-free H2O and transfer the tubes immediately on ice.
- Freeze the samples at -20 °C.
NOTE: RNA can be stored at -20 °C up to a month and at -80 °C up to 1 year. The procedure can be paused here or proceed to RNA electrophoresis.
6. RNA Electrophoresis
- Spray the electrophoresis equipment (tank, tray, comb) with 1% SDS, leave for 15 min and rinse with plenty of ddH2O.
- Dissolve 1.5 g of agarose in 127.5 mL of ddH2O by heating it in the microwave.
- Add 15 mL of 10x MOPS and 7.5 mL of 37% formaldehyde to the agarose solution (total volume is 150 mL).
CAUTION: Formaldehyde is toxic and should be handled under a fume hood. Always wear a lab coat, protective gloves, and glasses when working with formaldehyde. Dispose of the waste according to institutional regulations. - Add an appropriate amount of a gel stain of choice to the agarose solution (e.g., 15 µL of SYBR Safe DNA gel stain). Mix well and pour the agarose to the tray.
- Insert a comb in the gel immediately.
- Leave it for 45 min under the fume hood. Cover the tray with aluminum foil when using a light-sensitive gel stain.
- Prepare 600 mL of 1x MOPS buffer.
- RNA sample preparation
- In a 1.5 mL tube, combine 10 µL of the digested and purified RNA sample, 5 µL of Sample Denaturing Buffer and 0.5 µL of 6x Loading Dye.
CAUTION: Formamide is toxic and should be handled under a fume hood. Always wear a lab coat, protective gloves, and glasses when working with formamide. Dispose of the waste according to institutional regulations. - Incubate RNA samples at 70 °C for 5 min. Transfer the samples on ice. Incubate for 5 min.
- Spin down briefly before loading on the gel.
- In a 1.5 mL tube, combine 10 µL of the digested and purified RNA sample, 5 µL of Sample Denaturing Buffer and 0.5 µL of 6x Loading Dye.
- Put the gel in the electrophoresis tank and fill with 1x MOPS buffer. Load the entire volume of each sample (15 µL) on the gel. Run at 80 V until bromophenol blue reaches 2/3 of the gel length.
- Image the gel using an imager appropriate to detect the chosen gel stain (e.g., UV transilluminator).
Results
The utility of the DNAzyme-dependent cleavage in the analysis of rRNA modifications has been shown recently in the context of snoRNAs maturation13. The DNAzyme-dependent assay was used to show that lack of 5’-end pre-snoRNA processing affects 2’-O-methylation levels of 25S and 18S rRNA in S. cerevisiae13.
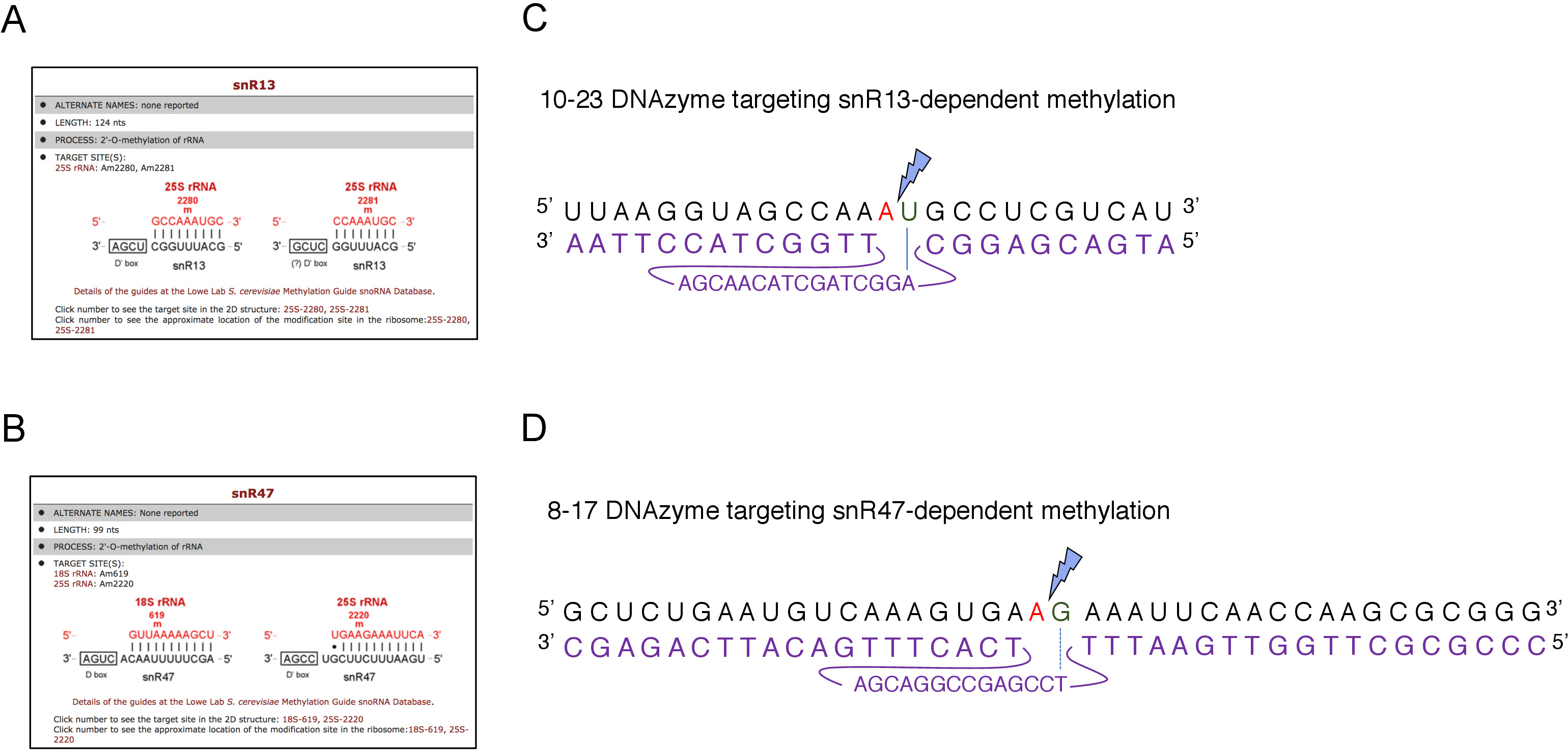
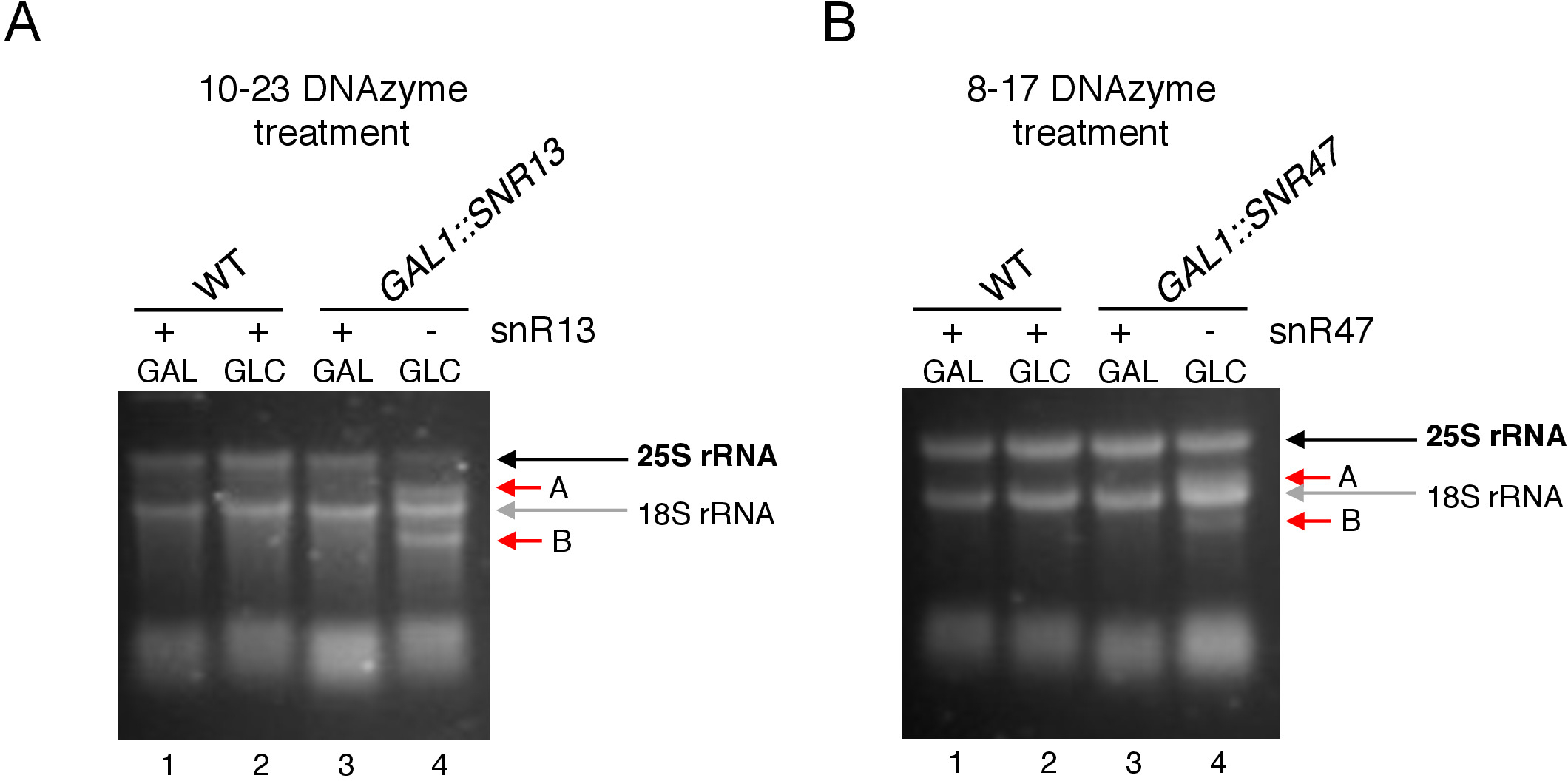
Here, we used an inducible snoRNA transcription system to demonstrate the effectiveness and simplicity of the technique. Box C/D snR13 guides methylation at two positions in 25S rRNA, including adenine 2281 (Figure 2A). This nucleotide is followed by uracil, which constitutes the consensus dinucleotide (RY) cleavable by a 10-23 DNAzyme. Box C/D snR47 also guides methylation of two nucleotides in 25S rRNA (Figure 2B). The adenine in position 2220 is followed by a guanine residue and this dinucleotide can be cleaved by an 8-17 DNAzyme. In order to induce or inhibit synthesis of either snR13 or snR47 snoRNA, we inserted the inducible GAL1 promoter upstream of either SNR13 or SNR47 genes and cultivated cells in medium containing galactose (GAL1-dependent transcription on) or glucose (GAL1-dependent transcription off). Next, RNA isolated from GAL1::SNR13 cells were incubated with 10-23 DNAzyme designed to cleave 25S rRNA at snR13-dependent site, in between nucleotides 2281 and 2282 (Figure 2C). RNA from GAL1::SNR47 strain was treated with 8-17 DNAzyme targeting snR47-dependent site in between nucleotides 2220 and 2221 (Figure 2D). As a control, RNA from the wild-type BY4741 strain growing on either galactose or glucose was incubated with both DNAzymes. Electrophoresis of the DNAzyme-treated RNA revealed that 25S rRNA extracted from GAL1::SNR13 and GAL1::SNR47 strains growing on galactose (GAL) remained intact (Figure 3A,B; lanes 3). In contrast, RNA isolated from GAL1::SNR13 and GAL1::SNR47 cells growing on glucose (GLC) was digested by respective DNAzymes (Figure 3A,B; lanes 4). In both cases, the 25S rRNA band decreased and 5’ and 3’ cut-off cleavage products (A and B) were observed. This indicates that in GAL1::SNR13 and GAL1::SNR47 strains, 25S rRNA was 2’-O-methylated at snR13- or snR47-guided sites when galactose was used as a carbon source and these snoRNA were expressed. The lack of 25S rRNA methylation when snR13 or snR47 expression was shut off on glucose allowed for DNAzyme-dependent cleavage. No RNA digestion was observed for wild-type samples (Figure 3A,B; lanes 1 and 2), as the expression of snR13 and snR47 is galactose/glucose-independent in this strain. Therefore, rRNA was normally methylated and so resistant to DNAzymes activity.
Overall, our experiment shows that the cleavage activity of 10-23 (Figure 3A) and 8-17 (Figure 3B) DNAzymes correlated with the absence of box C/D snR13 or snR47, clearly indicating that these snoRNA are responsible for 25S rRNA 2’-O-methylation at particular sites.

Figure 1: DNAzymes and their RNA substrates. (A) 10-23 DNAzymes cleave a purine-pyrimidine (RY) RNA dinucleotide. R in the RNA is not paired with DNAzyme, while Y is complementary to the R base in the DNAzyme. Methylation of the purine (R) in RNA suppresses DNAzyme-dependent cleavage. (B) 8-17 DNAzymes cleave RNA upstream of guanine that is imperfectly paired with the first thymine in the DNAzyme catalytic sequence. The nucleotide preceding guanine is not paired and its methylation protects from DNAzyme-dependent cleavage. RNA is shown in grey (apart from the methylation site), DNAzyme is shown in purple. N = any nucleotide, R = purine: adenine or guanine, Y = pyrimidine: cytosine or uracil; CH3- denotes RNA methylation. Base pairing within the DNAzyme active sequences is marked by dotted lines. A blue lightning bolt marks the cleavage site. (C) A flowchart showing the steps of a DNAzyme-dependent analysis. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: DNAzymes targeting snR13- and snR47-dependent methylation sites in 25S rRNA. (A, B) Browser screenshots showing snR13-dependent (A) and snR47-dependent (B) methylation sites in 25S rRNA (C) 25S rRNA sequence surrounding snR13-dependent methylation site (A2281) and 10-23 DNAzyme (shown in purple) designed to cleave RNA between A2281 and U2282. A2281 is not paired with the DNAzyme while U2282 forms a pair with the first nucleotide from the DNAzyme active sequence (marked by a blue line). A blue lightning bolt marks the cleavage. (D) 25S rRNA sequence surrounding snR47-dependent methylation site (A2220) and 8-17 DNAzyme (shown in purple) designed to cleave RNA between A2220 and G2221. A2220 is not hybridized with the DNAzyme while G2221 is imperfectly paired with thymine (denoted with a dashed line). A blue lightning bolt marks the cleavage site. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Analysis of site-specific 2’-O-methylation of 25S rRNA using 10-23 and 8-17 DNAzyme-dependent assay. (A) Analysis of snR13-dependent 25S rRNA methylation using 10-23 DNAzyme. (B) Analysis of snR47-dependent 25S rRNA methylation using 8-17 DNAzyme. RNA was visualized staining in a denaturing agarose gel. Cleavage products A and B are marked by red arrows. WT = wild-type strain; GAL = galactose, GLC = glucose. Please click here to view a larger version of this figure.
{kind=link}
Discussion
DNAzyme-dependent digestion can be used as a simple and quick method to analyze site-specific RNA 2’-O-methylation12,13. DNAzymes cleave RNA if the nucleotide upstream of the cleavage site is not methylated. In contrast to other approaches, including RNase H-directed digestion, alkaline degradation or reverse transcription in low nucleotides concentration followed by quantitative PCR or sequencing8,10,11,16, DNAzyme approach requires a simple DNA oligonucleotide and basic reagents that are present in any molecular biology laboratory. Moreover, DNAzymes may be used in a similar way to analyze RNA pseudouridylation mediated by box H/ACA snoRNA12, which makes them versatile tools in studying snoRNA targets.
DNAzyme-dependent approaches are limited only by cleavage site consensus sequences17. 10-23 DNAzymes can be used to analyze 2’-O-methylation only at position R of the RY dinucleotide, while 8-17 DNAzymes recognize the modification of the nucleotide located upstream of guanine. As a result, modifications like 2’-O-methylation of the first nucleotide in the dinucleotides guanine-adenine (GA), adenine-adenine (AA), pyrimidine-adenine (YA) and pyrimidine-pyrimidine (YY) cannot be analyzed. Moreover, the low efficiency of DNAzyme-dependent cleavage12 should be considered. Although some DNAzymes cleave RNA almost completely (Figure 3B), many DNAzymes only partially digest their targets (Figure 3B). The efficiency may depend on the sequence surrounding the cleavage site. For example, RNA regions with stretches of the same nucleotide may affect the correct positioning of the DNAzyme active sequence. Furthermore, RNA regions forming strong secondary structure may re-hybridize and suppress DNAzyme binding to the target sequence. To overcome these issues, cycles of heating and cooling of the 10-23 DNAzyme and its RNA substrate can be applied18.
We used the DNAzyme approach to investigate 2’-O-methylation of rRNA. One can also use this technique to analyze other RNA modifications, such as N6-methyladenosine19. Ribosomal RNA, due to its abundance, can be analyzed by electrophoresis and the cleavage products can be visualized under the UV light. However, this is not applicable for less abundant RNAs like RNA Polymerase II-generated coding RNAs (mRNA) and non-coding RNAs (ncRNA). These RNAs cannot usually be detected directly by RNA staining in agarose or polyacrylamide gels. In such cases, DNAzyme-dependent cleavage can be visualized by Northern blotting, indirectly detected by PCR/quantitative PCR or analyzed by quantitative PCR with polymerases (e.g., KlenTaq DNA Polymerase) capable of discriminating 2′-O-methylated RNA from unmethylated RNA20,21.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We thank Maya Wilson and Aneika Leney for the critical reading of the manuscript. This work was supported by a Sir Henry Dale Fellowship jointly funded by the Wellcome Trust and the Royal Society (200473/Z/16/Z).
Materials
| Name | Company | Catalog Number | Comments |
| Chemicals | |||
| Acid phenol | SIGMA | P4682 | |
| Agarose | VWR | A2114 | |
| Ammonium acetate | SIGMA | A1542 | |
| Chlorophorm | Fisher scientific | 10293850 | |
| DNase/RNase free water | Fischer Scientific | 10526945 | |
| DNAzyme | Integrated DNA Technology | Custom oligo DNA | |
| EDTA | SIGMA | E9884 | |
| Ethanol Absolute | Fisher scientific | 10437341 | |
| Formaldehyde | Sigma | F8775 | |
| Formamide | sigma | F9037 | |
| Galactose | SIGMA | G0750 | |
| Gel Loading Dye | Thermo Fisher Scientific | R0611 | |
| Glucose | SIGMA | G7021 | |
| Glycogen | Thermo Fisher Scientific | R0561 | |
| HEPES | SIGMA | H3375 | |
| Isoamyl | SIGMA | W205702 | |
| KCl | SIGMA | P9333 | |
| MgCl2 | SIGMA | M8266 | |
| MnCl2 | SIGMA | 244589 | |
| MOPS | SIGMA | M1254 | |
| NaCl | SIGMA | S7653 | |
| Oxoid Peptone Bacteriological | Thermo Fisher Scientific | LP0037 | |
| Oxoid Yeast Extract Powder | Thermo Fisher Scientific | LP0021 | |
| RiboLock RNase Inhibitor (40 U/µL) | Thermo Fisher Scientific | EO0382 | |
| SDS | SIGMA | 74255 | |
| Sodium acetate trihydrate | SIGMA | S8625 | |
| SYBR Safe DNA Gel Stain | Thermo Fisher Scientific | S33102 | |
| Tris base | SIGMA | TRIS-RO | |
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| 1.5 mL microtubes | Sarstedt | ||
| 152VR5C01M -80°C freezer | Thermo Fisher Scientific | ||
| 250 mL Erlenmeyer flasks | Cole-Parmer | ||
| 50 mL conical tubes | Sarstedt | ||
| Combicup VX200 vortex | Appleton Woods | ||
| DS-11 microspectrophotometer | Denovix | ||
| Electrophoresis chamber (20 cm tray) | SIGMA | ||
| FiveEasy F20 pH meter | Appleton Woods | ||
| Gel documentation system | Syngene | ||
| Heraeus Fresco 21 micro centrifuge | Fisher Scientific | ||
| Megafuge 8R centrifuge with rotator suitable for 50 mL conical tubes | Fisher Scientific | ||
| Mini Fuge Plus mini centrifuge | Starlab | ||
| Mixer HC thermal block | Starlab | ||
| OLS26 Shaking Water Bath | Grant | ||
| PowerPac power supplier | BioRad |
References
- Dieci, G., Preti, M., Montanini, B. Eukaryotic snoRNAs: a paradigm for gene expression flexibility. Genomics. 94 (2), 83-88 (2009).
- Watkins, N. J., Bohnsack, M. T. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdisciplinary Review RNA. 3 (3), 397-414 (2012).
- Kufel, J., Grzechnik, P. Small Nucleolar RNAs Tell a Different Tale. Trends in Genetics. , (2018).
- Li, T., Zhou, X., Wang, X., Zhu, D., Zhang, Y. Identification and characterization of human snoRNA core promoters. Genomics. 96 (1), 50-56 (2010).
- Jorjani, H., et al. An updated human snoRNAome. Nucleic Acids Research. 44 (11), 5068-5082 (2016).
- Hubbard, T. J., et al. Ensembl 2009. Nucleic Acids Research. 37 (Database issue), D690-D697 (2009).
- Makarova, J. A., Kramerov, D. A. SNOntology: Myriads of novel snoRNAs or just a mirage?. BMC Genomics. 12, 543 (2011).
- Yu, Y. T., Shu, M. D., Steitz, J. A. A new method for detecting sites of 2'-O-methylation in RNA molecules. RNA. 3 (3), 324-331 (1997).
- Decatur, W. A., Liang, X. H., Piekna-Przybylska, D., Fournier, M. J. Identifying effects of snoRNA-guided modifications on the synthesis and function of the yeast ribosome. Methods in Enzymology. 425, 283-316 (2007).
- Dong, Z. W., et al. RTL-P: a sensitive approach for detecting sites of 2'-O-methylation in RNA molecules. Nucleic Acids Research. 40 (20), e157 (2012).
- Birkedal, U., et al. Profiling of ribose methylations in RNA by high-throughput sequencing. Angewandte Chemie International Edition, England. 54 (2), 451-455 (2015).
- Buchhaupt, M., Peifer, C., Entian, K. D. Analysis of 2'-O-methylated nucleosides and pseudouridines in ribosomal RNAs using DNAzymes. Analytical Biochemistry. 361 (1), 102-108 (2007).
- Grzechnik, P., et al. Nuclear fate of yeast snoRNA is determined by co-transcriptional Rnt1 cleavage. Nature Communication. 9 (1), 1783 (2018).
- Piekna-Przybylska, D., Decatur, W. A., Fournier, M. J. New bioinformatic tools for analysis of nucleotide modifications in eukaryotic rRNA. RNA. 13 (3), 305-312 (2007).
- Schmitt, M. E., Brown, T. A., Trumpower, B. L. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Research. 18 (10), 3091-3092 (1990).
- Maden, B. E. Mapping 2'-O-methyl groups in ribosomal RNA. Methods. 25 (3), 374-382 (2001).
- Santoro, S. W., Joyce, G. F. A general purpose RNA-cleaving DNA enzyme. Proceedings of the National Academy of Science, U. S. A. 94 (9), 4262-4266 (1997).
- Hengesbach, M., Meusburger, M., Lyko, F., Helm, M. Use of DNAzymes for site-specific analysis of ribonucleotide modifications. RNA. 14 (1), 180-187 (2008).
- Sednev, M. V., et al. N(6) -Methyladenosine-Sensitive RNA-Cleaving Deoxyribozymes. Angewandte Chemie International Edition, England. 57 (6), 15117-15121 (2018).
- Aschenbrenner, J., Marx, A. Direct and site-specific quantification of RNA 2'-O-methylation by PCR with an engineered DNA polymerase. Nucleic Acids Research. 44 (8), 3495-3502 (2016).
- Lee, K. W., Bogenhagen, D. F. Assignment of 2'-O-methyltransferases to modification sites on the mammalian mitochondrial large subunit 16 S ribosomal RNA (rRNA). Journal of Biological Chemistry. 289 (36), 24936-24942 (2014).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved