Method Article
Expresión, purificación, cristalización y ensayos enzimáticos de proteínas que contienen el dominio de la hidrolasa fumarilacetoacetato
En este artículo
Resumen
La expresión y purificación de las proteínas que contienen el dominio de la hidrolasa fumarilacetoacetato se describe con ejemplos (expresión en E.coli, FPLC). Las proteínas purificadas se utilizan para la cristalización y la producción de anticuerpos y se emplean para ensayos enzimáticos. Los ensayos fotométricos seleccionados se presentan para mostrar la multifuncionalidad de FAHD1 como oxaloacetato decarboxilasa e hidrolasa de acilpuvato.
Resumen
Las proteínas que contienen el dominio de Fumarylacetoacetate hydrolase (FAH) (FAHD) son miembros identificados de la superfamilia FAH en eucariotas. Las enzimas de esta superfamilia generalmente muestran multifuncionalidad, involucrando principalmente mecanismos de hidrolasa y decarboxilasa. Este artículo presenta una serie de métodos consecutivos para la expresión y purificación de proteínas FAHD, principalmente ortologos de proteína FAHD 1 (FAHD1) entre especies (humanos, ratón, nematodos, plantas, etc.). Los métodos cubiertos son la expresión de proteínas en E.coli, cromatografía de afinidad, cromatografía de intercambio iónico, filtración de gel preparativo y analítico, cristalización, difracción de rayos X y ensayos fotométricos. Proteína concentrada de altos niveles de pureza (>98%) pueden emplearse para la cristalización o la producción de anticuerpos. Las proteínas de calidad similar o inferior pueden emplearse en ensayos enzimáticos o utilizarse como antígenos en sistemas de detección (Western-Blot, ELISA). En la discusión de este trabajo, los mecanismos enzimáticos identificados de FAHD1 se describen para describir su bifuncionalidad hidrolasa y decarboxilasa con más detalle.
Introducción
La superfamilia de enzimas fumarylacetoacetate hydrolase (FAH)1,2 describe un grupo de enzimas que comparten el dominio catalítico altamente conservadodeFAH 3,4,5,6 , 7 , 8 , 9 , 10. A pesar de su centro catalítico común, estas enzimas son multifuncionales, y la mayoría se encuentran en los procariotas, donde se utilizan para descomponer compuestos recuperados de fuentes complejas de carbono3. Sólo tres miembros de esta familia fueron identificados en eucariotas hasta el momento: el nombre que da FAH2, así como la proteína que contiene el dominio FAH 1 (FAHD1)11,12,13,14 ,15 y FAH proteína que contiene dominio 2 (FAHD2). El agotamiento de FAHD1 se ha asociado con insuficiencia de la respiración mitocondrial13,16 y con un tipo reversible de fenotipo14 de senescencia celular que está vinculado al potencial intermedio deficiencias en el sistema de transporte de electrones. FaHD1 humano y sus ortologos en sistemas modelo (ratón, nematodos, líneas celulares cancerosas, plantas, etc.), así como variantes de mutación puntual seleccionadas, se han convertido en objetivos farmacodrinables de interés potencial. Para esta investigación, la proteína recombinante a altos niveles de pureza, así como la información sobre los mecanismos catalíticos guiados por estructuras cristalinas y anticuerpos selectivos son vitales.
Este manuscrito describe los métodos para la expresión de proteína sorción de LA FAHD en E.coli, cromatografía de afinidad, cromatografía de intercambio iónico, precipitación de sulfato de amonio, filtración de gel preparativo y analítico, cristalización, difracción de rayos X y ensayos fotométricos. El propósito de los métodos y protocolos descritos aquí es proporcionar orientación para que los científicos que trabajan en diversos campos como la bacteriología, la biología vegetal, así como los estudios en animales y humanos, para caracterizar a los miembros de la superfamilia de la FAH, miembros de la superfamilia no caracterizados en caso de que sean relevantes en un campo en particular. Los protocolos descritos aquí pueden proporcionar un valioso apoyo para proyectos destinados a caracterizar a otros miembros de la superfamilia procariota o eucariota FAH.
La razón de ser detrás de los métodos descritos aquí es el hecho de que para la caracterización de proteínas mal descritas (en particular, enzimas metabólicas de relevancia fisiológica desconocida), el enfoque para comenzar con proteínas recombinantes purificadas permite desarrollo de herramientas de investigación invaluables y de alta calidad, como preparaciones enzimáticas activas in vitro, anticuerpos de alta calidad e inhibidores farmacológicos potentes y específicos para determinadas enzimas. Los métodos descritos requieren cromatografía líquida de proteínarápida rápida (FPLC) y cristalografía de rayos X. Los métodos alternativos (por ejemplo, para expresar proteínas sin inducción química, o para mostrar la purificación de proteínas por centrifugación después del tratamiento térmico seguido de la desalación y la cromatografía de exclusión de tamaño), se pueden encontrar en otros lugares17. Mientras que un espectro más amplio de métodos está disponible para la expresión y purificación de enzimas superfamilias FAH2,7,9,17,18, este trabajo se centra en la expresión y purificación de proteínas FAHD en particular.
En la sección de discusión de este manuscrito, los mecanismos catalíticos identificados para la proteína FAHD1 (hidrolasa, decarboxilasa)15 se describen con más detalle, con el fin de demostrar el carácter químico de las reacciones catalizadas. Los datos obtenidos sobre la base de trabajos anteriores7,15,18 (PDB: 6FOG, PDB:6FOH) implican una tercera actividad de la enzima como ceto-enol isomerasa.
Protocolo
1. Expresión de proteínas FAHD en E. coli competente
- Transformación de E. Coli con vectores para la expresión de proteína FAHD
NOTA: Los pasos descritos en la siguiente sección se resumen en el boceto de la Figura 1A,B. El mismo protocolo se aplica a cualquier proteína FAHD, incluidas las variantes de punto mutante. Estas variantes pueden obtenerse a través de la mutagénesis dirigida por el sitioy las técnicas de PCR19 (como la PCR 20 de SOE de dos lados) a partir de UN ADN de tipo salvaje.
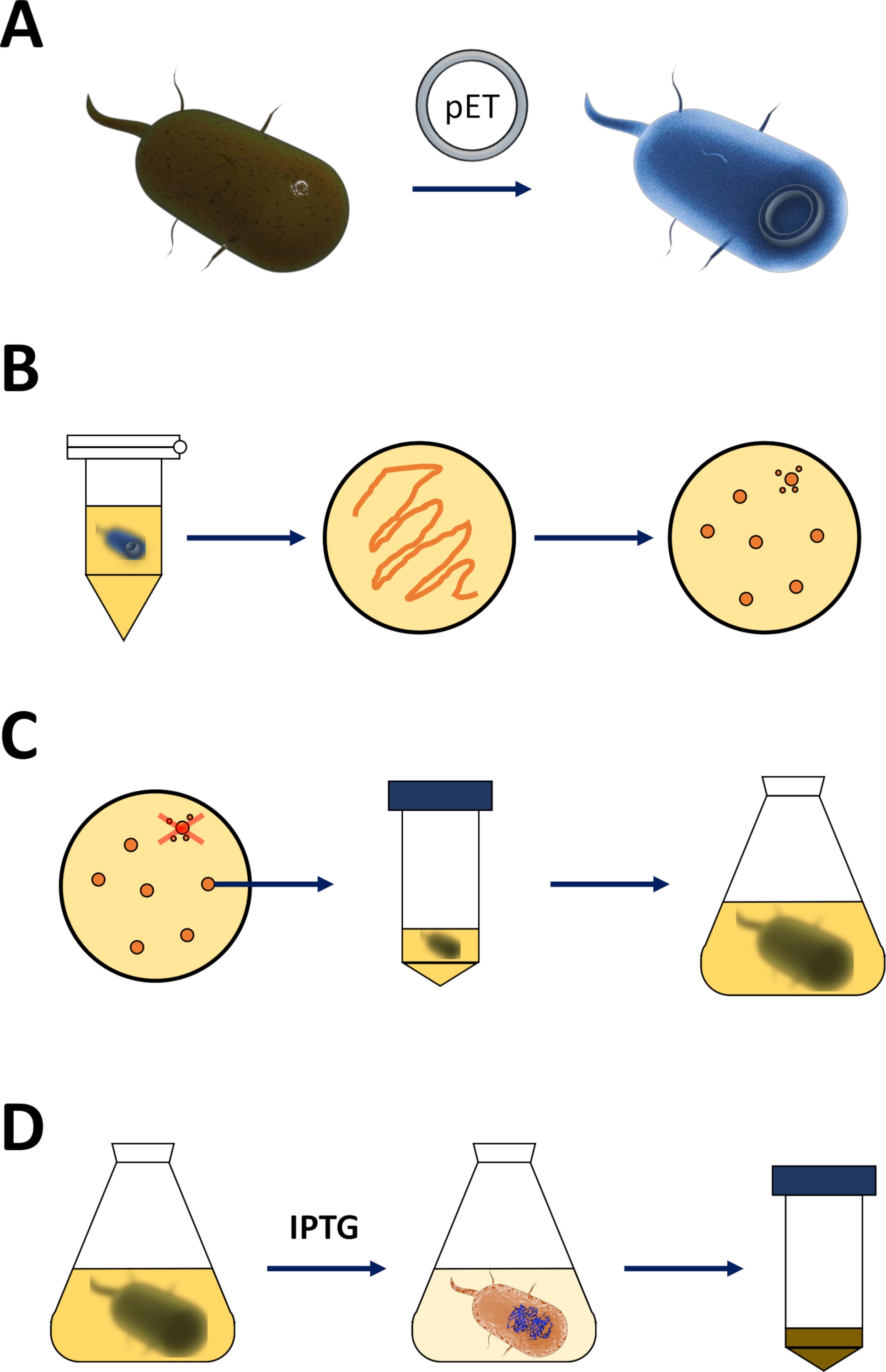
Figura 1 : Amplificación de E. coli competente e inducción de la expresión proteica.
(A) Inserción del vector pET en la bacteria BL21(DE3) pLysS E. coli competente, descrita en la sección 1. (B) Protocolo de choque térmico y chapado de la bacteria E. coli transformada con pET, descrito en el paso 1 del protocolo. Las bacterias transformadas se chapan en placas de agar LB con antibióticos para su selección. (C) Amplificación de la bacteria E. coli transformada con PET, descrita en la sección 1. Las colonias se recogen de una placa de agar LB y se amplifican en medio nutritivo (LB o NZCYM) hasta que la densidad bacteriana alcanzó el umbral empírico de 0.4. (D) Inducción de la expresión proteica a través del sistema DE3-IPTG-pET, descrito en la sección 1 y esbozado en la Figura 2. La producción de proteínas se inicia mediante la aplicación del IPTG químico. Al final de la sección 1, se cosecha el pellet bacteriano que contiene la proteína. Haga clic aquí para ver una versión más grande de esta figura.- Obtener bacterias competentes BL21(DE3) pLysS E. coli y un vector de expresión de pET (ver Tabla de Materiales). Preferiblemente elija un vector pET que también codifica una etiqueta Decaptura N-terminal o una etiqueta de captura relacionada para simplificar los siguientes pasos de purificación.
- Obtenga el ADNc de la proteína FAHD de elección e insértela en el sitio de clonación activo del vector de expresión pET, entre los sitios del promotor T7 y del terminador T7, respectivamente.
- Después de la amplificación y verificación exitosa del plásmido [a través de la secuenciación por un proveedor comercial (los imprimadores T7 se pueden utilizar con el sistema pET para mayor comodidad: promotor T7, imprimación directa: TAATACGACTCACTACTAGG; Terminador T7, imprimación inversa: GCTAGTTATTGCTCAGCGG)], inserte 5-10 ng de plásmido en 100 l de bacterias competentes BL21(DE3) pLysS E. coli sobre hielo. No aspirar hacia arriba y hacia abajo, pero toque ligeramente el tubo con para mezclar el contenido.
- Mantenga las bacterias en hielo durante 30 minutos, tocando suavemente el tubo cada pocos minutos.
- Calentar un dispositivo de calefacción o un baño de agua a 42 oC (exacto). Coloque el tubo que contiene las bacterias en el aparato y guárdelas durante 90 s (exacto). Póngalos en hielo inmediatamente (Figura1A).
- Después de 5-10 min sobre hielo, añadir 600 ol de medio NCZYM (ver Tabla de materiales) y poner el tubo en una incubadora de bacterias. Agitar el tubo a velocidad media orientada a lo largo de la dirección de agitación a 37 oC durante 1 h.
- Placa 200 l del cultivo bacteriano en una placa de agar LB de 10 cm (ver Tabla de materiales), que contiene antibióticos de selección de elección [por ejemplo, uno específico para la resistencia a la pLysS BL21(DE3) (cloranfenicol), y otro para la resistencia codificado en el vector pET (kanamicina o ampicilina, Figura 1B)].
- Cultivo de las bacterias en la placa de agar LB en una incubadora bacteriana a 37 oC durante la noche.
- Expresión de proteínas FAHD por inducción IPTG
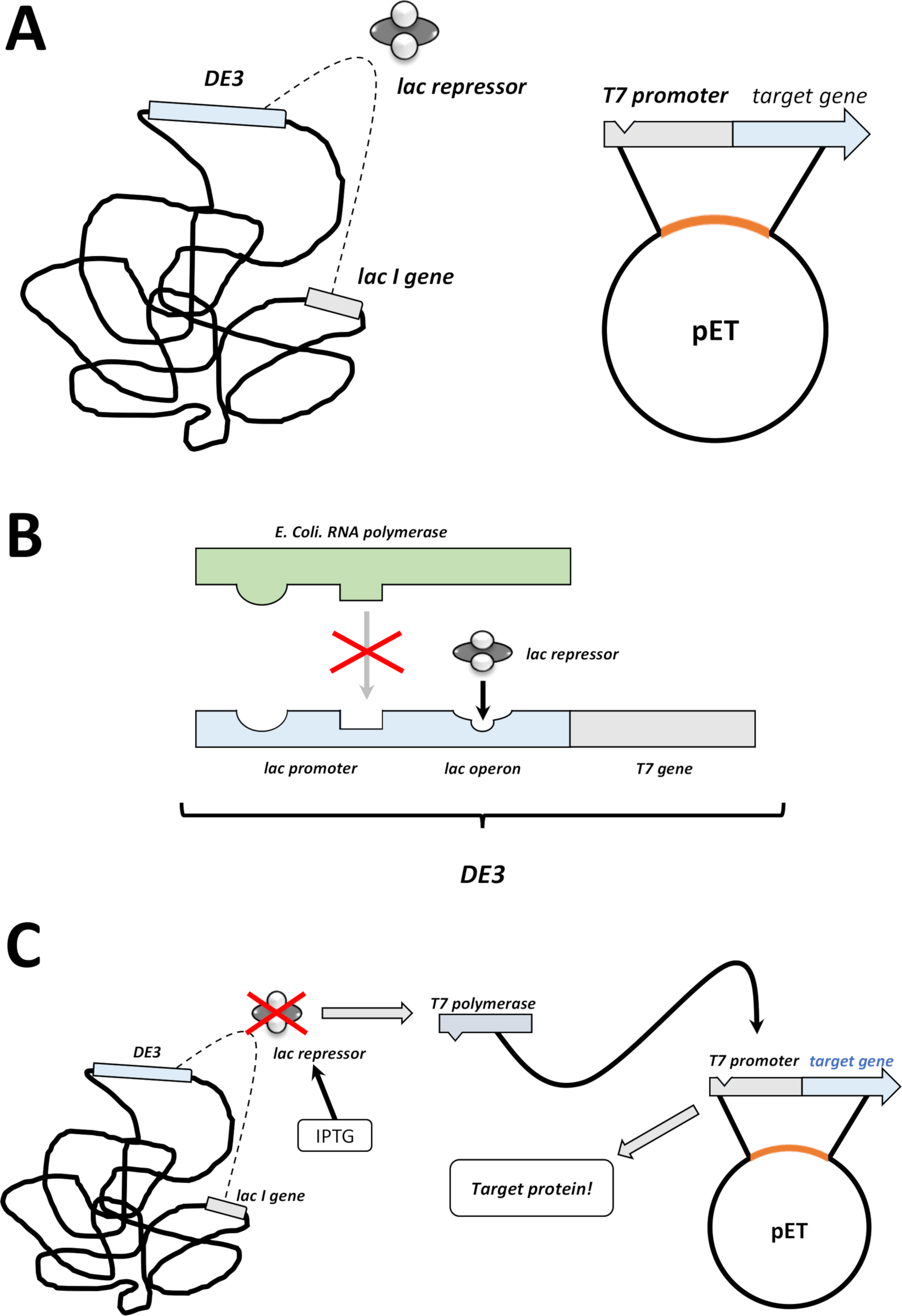
NOTA: Los primeros pasos que se describen en la siguiente sección se resumen como un boceto en la Figura 1C,D. El sistema de expresión T7 mediante la combinación del cassette bacteriano DE3 y el sistema vectorial pET se resumen en la Figura2.
Figura 2 : Se explicó el sistema dual vectorial DE3 cassette/pET.
(A) El genoma esbozado del vector pET transformó la bacteria BL21(DE3) pLysS E. coli. El genoma bacteriano nativo lleva un casete DE3 (ver panel B), así como un gen lac que expresa constantemente las unidades represoras de lac. El vector pET no nativo lleva el gen proteico insertado entre un promotor de la polimerasa T7 y una secuencia de terminador. Más detalles en el panel B. (B) El casete DE3 del genoma bacteriano nativo codifica la información para t7 polimerasa en términos de una e. coli ARN polimerasa operon. Esta proteína, sin embargo, no se expresa porque la unidad represora de lac impide que la proteína de la ARN polimerasa se adhiera. Por lo tanto, no se expresa ninguna polimerasa T7 y no se expresa ninguna proteína exógena. (C) La aplicación del IPTG químico (Tablade Materiales)distorsiona la estructura de las unidades represoras de lac y evita que se adhieran al casete DE3. Como resultado, la ARN polimerasa ahora puede unirse al casete, para el cual se expresa la polimerasa T7, al igual que la proteína exógena eventualmente. Haga clic aquí para ver una versión más grande de esta figura.- Después de una formación exitosa de colonias, escoge una sola colonia (sin colonias satelitales) y dispersa en 5 ml de nZCYM o medio LB con antibióticos, seleccionado como antes (paso 1.1.7). Cultivo en la incubadora bacteriana a 37oC durante la noche (Figura1C).
- Después de un crecimiento bacteriano exitoso, amplificar las bacterias en 250 ml, 500 ml, o 1 L lotes de medio, dependiendo de la demanda de cantidad de proteínas.
- Adecuado al volumen, aplicar antibióticos seleccionados como se hace en el paso 1.1.7 y añadir alrededor de 1%-2% de precultivo bacteriano denso (es decir, 2.5–5.0 mL a 250 ml de volumen de volumen medio, etc.). Tome una muestra que se utilizará en el paso 1.2.5 (1 ml o más) y compruebe la densidad óptica (OD) a 600 nm. Cultivar bacterias en la incubadora bacteriana a 37 oC durante 2–3 h (Figura1C).
- Después de 2–3 h, dibuje una muestra para el análisis fotométrico. Si el OD a 600 nm ha alcanzado 0.4, aplique 200 m hasta 1 mM isopropyl--D-thiogalactopyranosid (IPTG, véase Tabla de materiales).
NOTA: El valor real es empírico para cada proteína FAHD o variante de mutación puntual, donde 1 mM IPTG es el máximo que se debe aplicar. Esto induce la expresión de proteínas (Figura1D, Figura 2C). - Después de 3-5 horas más en la incubadora bacteriana a 37 oC, se agota la expresión de proteínas.
NOTA: Consulte la sección de discusión para ver los comentarios sobre el control de temperatura. No se recomienda más de 5 h de agitación después de la inducción. Tome una muestra para su uso en el paso 1.2.5 (1 ml o más) y compruebe la densidad óptica (OD) a 600 nm.- Cosechar el pellet bacteriano a través de centrifugación a 5.000 x g durante 5 min. Deseche el sobrenadante y congele el pellet a -80 oC para un almacenamiento más largo o -20 oC para un almacenamiento breve (Figura1D).
- Verifique la inducción a través de las dos muestras fotométricas recuperadas, que se etiquetan como "-I" (antes de la inducción) y "+I" (después de la inducción). Después de la centrifugación y la resuspensión del pellet bacteriano, analice las dos muestras por SDS-PAGE cargando la misma cantidad de proteína total.
NOTA: La muestra "+I" debe mostrar una banda fuerte asociada con el peso molecular de la proteína elegida, mientras que la muestra "-I" no debe contener esta banda. Un nivel de inducción bajo es un problema común para la producción de proteínas, sin embargo, el nivel de proteína expresada a menudo es suficiente para los siguientes pasos. Un nivel de inducción alto es una ventaja, pero no es obligatorio.
{kind=link}
{kind=link}
2. Lisis de pellets bacterianos y filtración de escombros
- Dependiendo de si la proteína elegida es Subúfer de ejecución -etiquetado o sin etiquetar, seleccione Ni-NTA running buffer (Su -etiquetado, consulte Tabla de materiales)o búfer de ejecución HIC helado (sin etiquetar).
- Por cada 250 ml de suspensión bacteriana original, aplicar 5 ml del tampón seleccionado al pellet bacteriano (5 ml para 250 ml, 10 ml para 500 ml, etc.). Añadir 10 sl de mercaptoetanol (-ME) por 5 ml de tampón aplicado. Utilice un pipeteo Pasteur de 10 ml para forzar mecánicamente el pellet en suspensión mediante arañazos y pipeteo (evitar la formación de burbujas de aire durante el pipeteo). Eventualmente transfiera toda la suspensión a un tubo de 50 ml.
- Preferiblemente sonicar (6x para 15 s a fuerza media) la suspensión.
- Centrífuga durante 30 min a alta velocidad (10.000 x g)a 4oC. Filtrar el sobrenadante consecutivamente con unidades de filtro (p. ej., 0,45 m, 0,22 m) sobre hielo.
NOTA: Dependiendo del paso de centrifugación anterior, la filtración directamente a través de un pequeño tamaño de poro de filtro puede ser tediosa y, por lo general, requiere prefiltración a través de un tamaño de poro más grande. DNAse puede agregarse para obtener mejores resultados. - Almacene la muestra en hielo y proceda inmediatamente con la sección 3 o 4, dependiendo de si la proteína es Su-etiquetada o sin etiquetar.
3. Purificación de susproteínas FAHD etiquetadas usando cromatografía de afinidad Ni-NTA
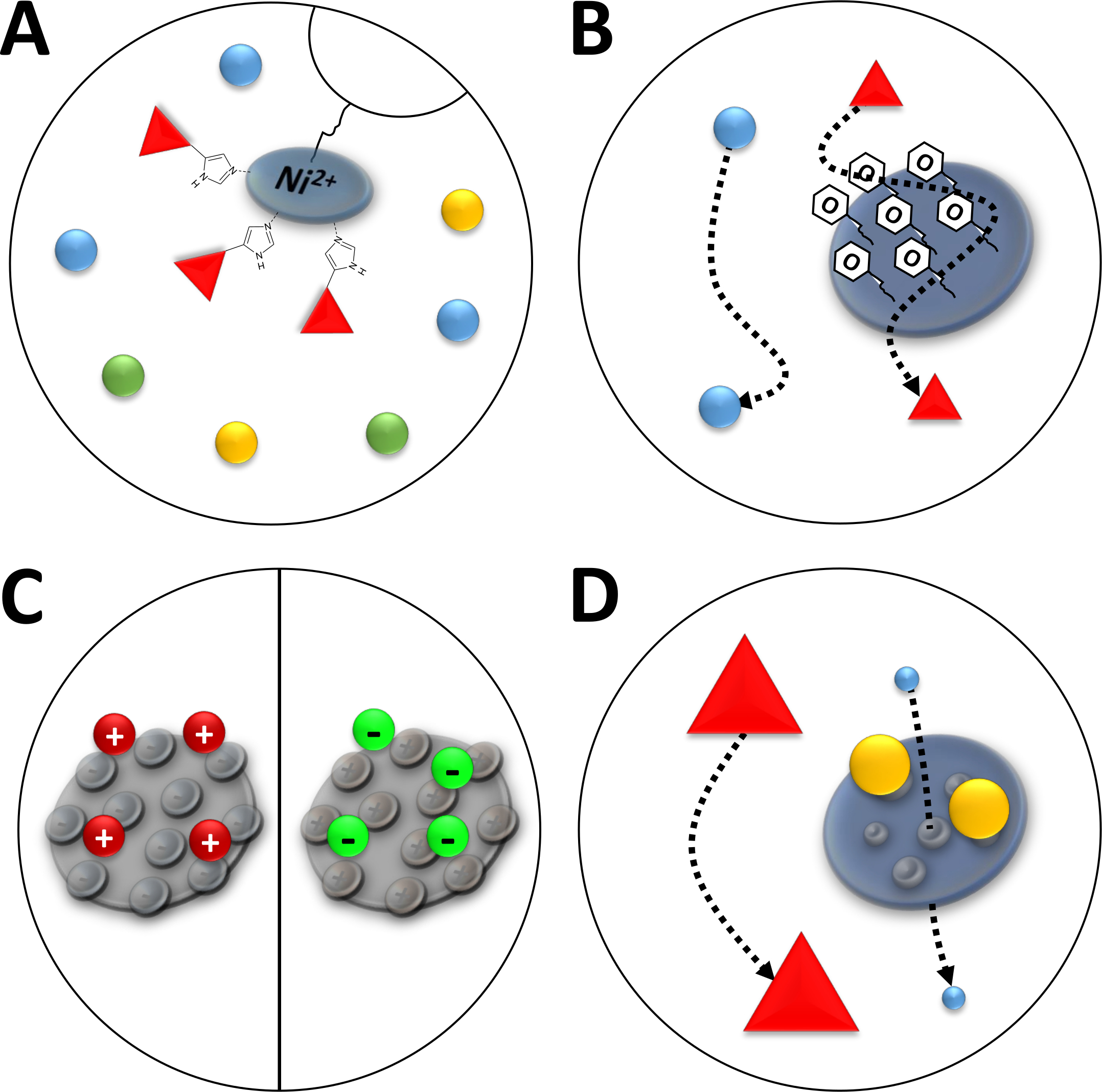
NOTA: Los iones Ni2+ se unen a través de ácido nitrilotriacético (NTA) a una resina de agarosa que se utiliza en la cromatografía de iones de afinidad (cromatografía de iones metálicos inmovilizados, IMAC, Figura 3A). Las etiquetas de aminoácidos de polihistidina se unen fuertemente a este Ni-quelato, y las proteínas his-tagged se pueden separar de la mayoría de las proteínas restantes. Una alternativa a la preparación descrita de columnas Ni-NTA es el uso de columnas Ni-NTA preempaquetadas y un sistema FPLC.

Figura 3 : Ilustraciones esbozadas de tipos comunes de cromatografía.
(A) La resina de una columna Ni-NTA. NTA posee iones de níquel bivalentes que se utilizan en términos de cromatografía de afinidad de iones metálicos inmovilizados (IMAC). Las etiquetas de polihistidina se unen preferentemente a este motivo y pueden ser eluted por imidazol. (B) El recubrimiento típico de partículas de sílice en una cromatografía de interacción hidrofóbica a base de fenilo (HIC-phenyl). Las proteínas hidrófobas interactúan con el material de recubrimiento y se retrasan en su migración, mientras que otras no. (C) El recubrimiento típico de partículas de sílice en cromatografía de interacción iónica. Las proteínas polarizadas y cargadas interactúan con el material de recubrimiento y se retrasan en su migración, mientras que otras no. (D) La resina de un gel de sílice en cromatografía de exclusión de tamaño (SEC). Sobre la base de los poros definidos en el material de sílice, las proteínas pueden separarse por su tamaño (en una primera aproximación correspondiente a su masa molecular). Las proteínas pequeñas impregnan el material de la columna porosa y son retardadas, mientras que las proteínas grandes migran más rápido alrededor de las partículas porosas. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Continúe con el paso 2.5 (es decir, la proteína está en el búfer de funcionamiento de Ni-NTA y filtrada por unidades de filtro de 0,22 m sobre hielo).
- Prepare una columna de plástico o vidrio vacía lavando la columna vacía y adjuntándola a un retenedor estable. Elija el tamaño de la columna dependiendo del volumen de la suspensión proteica.
- Por cada 10 ml de suspensión proteica, aplique 500 ml de lodo de agarosa Ni-NTA en la columna (agitar fuertemente antes de su uso). Aplique la suspensión lentamente y con gota en el filtro inferior de la columna utilizando una pipeta. Deje que la columna se asiente, lo que toma unos segundos.
- Llene la columna completamente con el búfer de funcionamiento Ni-NTA, asegurando no interrumpir la resina de agarosa. Deje que el búfer corra a través de la gravedad. El proceso puede acelerarse aplicando presión del pulgar sobre el líquido (usando una presión de tapa o guante y pulgar), pero tenga cuidado de no distorsionar la resina de agarosa.
- Aplicar la suspensión proteica. Como antes, deje que la muestra corra a través de la gravedad. No se recomienda acelerar este paso con la presión del pulgar, ya que la unión de proteínas a la columna se mejora si el caudal es bajo. Recoger el flujo en un tubo (Tabla de materiales).
- Una vez pasado el ejemplo, rellene toda la columna de nuevo con el búfer en ejecución de Ni-NTA. Tenga cuidado de no interrumpir la resina de agarosa. Deje que la muestra corra a través de la gravedad, pero a diferencia del paso anterior, se recomienda acelerar el proceso a través de la presión del pulgar, ya que las posibles contaminaciones debido a interacciones inespecíficas pueden interrumpirse de esta manera. Recoger la solución de lavado en un tubo. Repita este paso.
- Coloque una cubeta transparente UV debajo de la columna y aplique 1 ml de búfer de elución Ni-NTA. Recoja la muestra sin aplicar presión del pulgar a la resina.
- Compruebe la densidad óptica (OD) de la muestra a 280 nm frente a una muestra en blanco (es decir, búfer de elución Ni-NTA). De manera óptima, la muestra muestra una OD superior a 2,5. Una OD por debajo de 0,5 denota que no hay ninguna cantidad significativa de proteína en la muestra.
NOTA: Como se describe en la sección de discusión, las concentraciones de sal e imidazol del tampón de elución pueden tener que adaptarse para cada proteína FAHD individualmente. - Repita los pasos 3.1.7 y 3.1.8 hasta que el OD caiga por debajo de 0.5. Agrupa todas las muestras con un OD más alto en un tubo sobre hielo.
- Comience de nuevo con el paso 3.1.4, utilizando el flujo a través del paso 3.1.5 como nueva entrada para esta repetición del paso 3.1.5. Repita este proceso hasta que la primera muestra recopilada en el paso 3.1.6 muestre un OD por debajo de 0.5.
NOTA: Como se describe en la parte de solución de problemas de la sección de discusión, las proteínas etiquetadas pueden unirse insuficientemente a la resina Ni2+. En tales casos, se requiere la repetición de este paso o métodos alternativos (por ejemplo, cromatografía de intercambio iónico). - Tome muestras de todas las fracciones intermedias para el análisis SDS-PAGE.
- Las proteínas FAHD en el tampón de elución De Ni-NTA se precipitarán al congelarse y descongelarse. Por lo tanto, dializar la proteína contra un tampón diferente (durante la noche en hielo, utilizando 1 l de TDT por 100 ml de tampón de diálisis). Utilice tampón de sal baja en función del tipo de cromatografía de intercambio iónico que se debe realizar después de este paso. Utilice tubos de celulosa común con un corte típico de peso molecular de 14 kDa (Tablade materiales).
- Después de la diálisis durante la noche, concentre opcionalmente la proteína utilizando unidades de filtro de ultracentrifugación. Realizar análisis SDS-PAGE (12,5% gel de carrera, 4% gel de apilamiento) para comprobar la pérdida potencial de proteína, la elución insuficiente, y la pureza de la proteína en general. Si todo está bien, proceda a la sección 5.
4. Purificación de proteínas FAHD sin etiquetar mediante cromatografía de interacción hidrofóbica (HIC)
NOTA: Los grupos de fenilo en la superficie de recubrimiento de un gel de sílice en una columna HIC para FPLC (Figura3B)permiten la separación de proteínas según el carácter hidrófobo. Los pasos descritos deben realizarse con un sistema FPLC equipado con una columna HIC-phenyl de 5 ml. Las columnas se pueden lavar con 1 M NaOH para ser reutilizadas para diferentes proteínas. Sin embargo, las columnas que una vez se utilizaron para un tipo de proteína FAHD deben reutilizarse solo para este tipo de proteína.
-
Precipitación de sulfato de amonio (AS)
- Continúe con el paso 2.5. La proteína está en tampón de funcionamiento HIC helado (Tabla de materiales).
- Evaluar el volumen de la solución proteica preparada precisamente al microlitro (Vinicial). Adición lenta y gota de hic en cuanto a la solución AS de búfer en ejecución HIC, hasta que se alcance una saturación de AS de 35 volúmenes%: VAS agregado a Vinicial * 0.538. Revuelva suavemente la solución durante 30 minutos centrífuga durante 15 minutos a alta velocidad (10.000 x g) a 4 oC.
- Filtre el sobrenadante usando una unidad de filtro de 0,22 m sobre hielo. Opcionalmente, tome una muestra para el análisis SDS-PAGE: diluir 1:4 y calentar inmediatamente a 95 oC durante 5 min o de lo contrario la muestra se abultará. La muestra puede congelarse en este punto (-20 oC) para proceder otro día.
-
FPLC utilizando una columna HIC
- Configure el sistema FPLC y equilibre una columna HIC-phenyl de 5 ml con 5 volúmenes de columna (CV) de 20% EtOH (en H2O) seguido de 5 CV de H2O.
- Mezclar 260 ml de búfer de ejecución HIC (exacto) con 140 ml de búfer en ejecución HIC AS (exacto). Esto da como resultado una solución AS de 35 volúmenes. Compruebe el pH (7,0); este es el búfer A. El búfer B es 250 mL del búfer en ejecución. Agregue 1 mM DTT a ambos buffers A y B, después manténgalos en el hielo.
- Equilibrar la columna con 8 ml del buffer A, 8 mL del buffer B y 8 mL del buffer A en esta secuencia. Aplique la muestra preparada en el paso de protocolo 4.1. Lavar con tampón A, hasta que la absorción óptica basal a 280 nm alcance 1000–500 mAU.
- Aplique una mezcla de tampones A y B, de modo que la concentración de AS sea del 33 % (p/v). Lavar con 1 CV, lo que resulta en una meseta en el cromatograma. Configurar un gradiente de búfer B (hasta 100% tampón B en el tiempo): 1,5 ml de tampón B en 3,8 min (es decir, 5,7% buffer B con 1% B/mL de pendiente). Cuando la señal UV a 280 nm se eleva, comience a recoger la fracción y colóquela en hielo inmediatamente.
- Al final, lave la columna con el búfer B. Tome muestras de todas las fracciones para el análisis SDS-PAGE. Congele todas las muestras con nitrógeno líquido y guárdelas a -80 oC.
- Realizar análisis SDS-PAGE (y western blot), para detectar la proteína FAHD en las fracciones recolectadas. Las fracciones que contienen la proteína se agrupan y se aplican a una mayor purificación, como se describe en los siguientes pasos del protocolo. Lave la columna con H2O y 20% EtOH (en H2O).
5. Purificación de proteínas FAHD mediante cromatografía de intercambio iónico
NOTA: Las moléculas con grupos funcionales cargados están unidas a una columna de partículas de sílice para FPLC (Figura3C). Esto permite la diferenciación de proteínas según su carácter iónico, como la carga superficial. Los pasos descritos deben realizarse con una máquina FPLC y el know-how asociado, respectivamente. El método descrito es el mismo para la cromatografía de intercambio catiónico o aniónico, pero los tampón que se van a utilizar son ligeramente diferentes.
- Elija el sistema de cromatografía de intercambio catónico o aniónico. Esta opción es empírica y puede variar entre las proteínas FAHD. De manera óptima, ambos métodos se pueden utilizar consecutivamente.
- Configure el sistema FPLC y lave la columna con 5 CV de 20% EtOH (en H2O), seguido de 5 CV de H2O. Equilibrar la columna con 1 CV de búfer de sal baja, búfer de alta sal y de nuevo búfer de baja sal en esta secuencia.
- Aplique la muestra (dialyzed contra el búfer de sal baja correcto del paso 3.1.11) en la columna. Recoja el flujo a través. Lave la columna durante 1 CV con tampón de sal baja.
- Configurar una elución de gradiente: 100% de búfer de alta sal en 30 min a un caudal de 1 ml/min, o 60 min a un caudal de 0,5 ml/min. Esto puede ser re-seleccionado basado en un cromatograma FPLC ya conocido, con el fin de optimizar la purificación. Recoger todas las fracciones pico.
NOTA: Las condiciones de alta sal pueden variar entre las proteínas FAHD, como se describe en la sección de discusión. - Una vez finalizado el gradiente, ejecute con búfer de alta sal hasta que no se detecten más picos en el rango de 1 CV (recoja las fracciones).
- Tomar muestras de todas las fracciones recogidas y realizar análisis SDS-PAGE (12,5% gel de carrera, 4% gel de apilamiento). Congele las muestras individuales en nitrógeno líquido y guárdelas a -80 oC.
- Una vez completado el análisis SDS-PAGE, auna las muestras que contienen la proteína FAHD y desecha las demás. Opcionalmente, concentre la proteína utilizando unidades de filtro de ultracentrifugación.
- Aplique 1 ml de 25% de SDS en 0,5 M NaOH (u otros detergentes) para limpiar la columna. Lave la columna con H2O y 20% EtOH (en H2O).
- Opcionalmente, repita la sección 5 con la columna alternativa (cromatografía de intercambio catiónico o aniónico). La proteína obtenida de este método es lo suficientemente pura para realizar ensayos de actividad básica o se puede utilizar en ensayos de cribado para cristalografía. Para aplicaciones avanzadas, continúe con la sección 6.
6. Purificación de proteínas FAHD mediante cromatografía de exclusión de tamaño (SEC)
NOTA: Las partículas porosas en una columna de gel de sílice para FPLC permiten la diferenciación de proteínas según el tamaño molecular, como el radio hidrodinámico (Figura3D). Los pasos descritos deben realizarse con un sistema FPLC, utilizando columnas SEC.
- Elija una columna SEC, dependiendo de los pesos moleculares de las contaminaciones todavía presentes, como se detecta a través de SDS-PAGE y la tinción de plata. El método descrito es adecuado para ambas columnas. Lave la columna durante la noche con 400 ml de H2O y equilibre con el búfer de ejecución SEC. Se recomienda escribir un programa para que el sistema FPLC automatice este paso.
- Agregue 1 mM dTT a 300 mL de búfer de ejecución SEC y colóquelo en hielo. Este es el búfer en ejecución. Aplique 60 mL de este búfer a la columna.
- Centrifugar la muestra de proteína (10.000 x g durante 10 min) para eliminar cualquier microprecipitación. Aplique el sobrenadante a la columna. Por lo general, se recomienda filtrar el sobrenadante antes de FPLC.
- Aplique el búfer en ejecución a la columna hasta que se eluya toda la proteína. Recoger todos los picos en fracciones de volumen adecuado (por ejemplo, 2 ml). Tome muestras para SDS-PAGE y congele todas las fracciones usando nitrógeno líquido. Almacene las fracciones congeladas a -80 oC.
- Después del análisis SDS-PAGE (y western blot), recopile y acuse todas las fracciones que contienen la proteína FAHD. Se recomienda la tinción de plata para detectar contaminaciones menores que todavía pueden estar presentes.
- Utilice unidades de filtro de ultracentrifugación para concentrar la proteína. Aunque no es obligatorio para las proteínas FAHD, en general se recomienda un paso de desalación (por ejemplo, por diálisis) para ensayos enzimáticos y cristalización.
- Repita los pasos 6.3–6.6 varias veces con diferentes caudales y concentraciones de sal (empíricas) con el fin de mejorar la pureza de la proteína FAHD. Lave la columna durante la noche con H2O y 20% EtOH (en H2O).
7. Ensayos básicos de actividad FAHD con sustratos de oxaloacetato y acetilpiruvato
NOTA: FaHD proteína 1 (FAHD1) muestra la actividad de oxaloacetato decarboxilasa (ODx) y acipiruvato hidrolasa (ApH). Esto se describe con más detalle en la sección de discusión. Debido a la desestabilización por tautomerización de keto-enol en solución acuosa (es decir, enolización), el oxaloacetato se descompone por sí mismo con el tiempo (auto-descarboxilación) en función de la concentración de cofactores y el pH. A alrededor de un pH de 7 oC y una temperatura de 25 oC, este efecto no es dramático, pero los ensayos deben estar en blanco para tener en cuenta tanto la autodescarboxilación como la concentración enzimática. El esquema de pipeteo se describe en la Figura 4A. En general, se recomienda utilizar pipetas bien calibradas para este ensayo, ya que es bastante sensible a errores menores de pipeteo.

Figura 4 : Esquema de pipeteo esbozado para ensayos enzimáticos.
(A) Un esquema de pipeteo esbozado para ensayos básicos de enzimas de proteína Solución de sustrato. Sustrato en blanco: -S/-E; muestra de sustrato: +S/-E; enzima en blanco: -S/+E; enzima: +S/+E (S: sustrato, E: enzima). Consulte el paso 7 del protocolo para obtener más detalles. (B) Un esquema de pipeteo esbozado para evaluar la cinética de Michaelis-Menten de la proteína FAHD. Sustrato en blanco: -S/-E; muestra de sustrato: +S/-E; enzima en blanco: -S/+E; enzima: +S/+E (S: sustrato, E: enzima). Consulte la sección 8 del protocolo para obtener más detalles. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Poner en marcha un lector de microplacas y equilibrar durante 30 min a 25 oC. Configure un programa para leer 12 pozos (como se describe en la Figura 4A)a 255 nm. Se recomienda utilizar 25 lecturas múltiples con 5 ms de retardo de tiempo. Configure un ciclo para medir 15 veces cada 2 min (30 min total).
- Por defecto, prepare un tampón de ensayo enzimático (ver Tabla de Materiales)con 1 mM MgCl2 a pH 7.4. Las proteínas FAHD variantes pueden requerir diferentes cofactores o niveles de pH. Mg2+ y Mn2+ son cofactores conocidos para FAHD13,11,12,21.
- Crear una solución proteica de 1 g/L, diluyéndola con un tampón de ensayo enzimático (Tablade materiales).
- Configurar 1 ml de solución de 20 mM de un sustrato a probar (hasta ahora los sustratos identificados de proteínas FAHD se enumeran en otros lugares3) en el tampón de ensayo enzimático.
- De acuerdo con el esquema de pipeteo mostrado en la Figura 4A,prepare la enzima en blanco y los pozos de muestra: pipeta de 90 l de tampón de ensayo enzimático (Tablade materiales)en los pozos con 5 l (5 g) de solución enzimática.
- De acuerdo con el esquema de pipeteo mostrado en la Figura 4A,prepare el sustrato en blanco y los pozos de muestra: pipeta 95 l de tampón de ensayo enzimático en los pozos.
- Justo antes de medir, aplique 5 ml de tampón de ensayo enzimático en los seis pozos en blanco. Aplicar 5 ml de la solución de sustrato de 20 mM a los pozos de muestra. Se recomienda utilizar una pipeta multicanal.
- Utilice una pipeta multicanal a ajustes de 50 ml para mezclar suavemente todos los pozos. Comience con los espacios en blanco y continúe con los pozos de muestra. Tenga cuidado de no crear burbujas. Inserte la placa en un lector de microplacas y mida cada pocómetro a 255 nm (como se describe en el paso 7.1).
- Realice el análisis en una hoja de cálculo. Copie los datos sin procesar del fotómetro en una hoja de cálculo y escriba todos los ajustes (es decir, toda la documentación) en otra hoja. Promedio de los datos de los tres pozos de cada uno de los cuatro preparativos. Reste el espacio en blanco de la muestra. También calcular las desviaciones estándar y sumar las desviaciones de blanco y muestra.
- Trazar estos datos (y: densidad óptica, x: tiempo en min). Se debe mostrar una curva decreciente exponencial. Dependiendo del tipo de sustrato en uso, se puede observar un aumento inicial dentro de los primeros 10 minutos, después de lo cual la señal disminuye. Esto se atribuye a la tautomerización keto-enol del sustrato, como se describe con más detalle en la sección de discusión.
- Divida los datos de la señal óptica a lo largo del tiempo por el valor máximo de la gráfica, con el fin de reducir los datos al rango [0, 1] (se proporciona un ejemplo en la Figura 5A). Identifique el rango lineal de la curva, comenzando en la disminución inicial, y calcule la pendiente negativa (1/min).
- El curso de tiempo de la disminución de la DoS se asocia al sustrato a través de su concentración inicial: 100 nmol/pozo * pendiente. Utilizando la concentración de proteínas evaluada c0, se calcula la actividad específica: 100 nmol/pozo * pendiente * 1/c0. Expresando c0 en g/pozo, la actividad específica calculada de esta manera se expresa utilizando la unidad nmol/min/g, que es igual a émol/min/mg.
8. Evaluación de la cinética de Michaelis-Menten de proteínas FAHD
NOTA: La evaluación de la cinética Michaelis-Menten de las proteínas FAHD es tediosa, ya que la actividad proteica específica depende tanto de la concentración relativa de sustrato de proteína como del volumen físico en el que se está produciendo la reacción. La cinética en estado estacionario debe establecerse para obtener resultados fiables. Un protocolo probado en una placa transparente UV de 96 pocillos se describe en los siguientes pasos. Cada paso debe realizarse con mucho cuidado, ya que los errores menores generalmente estropean el experimento. Se recomienda dominar los ensayos descritos en la sección 7 antes de intentar el ensayo más complicado que se describe a continuación.

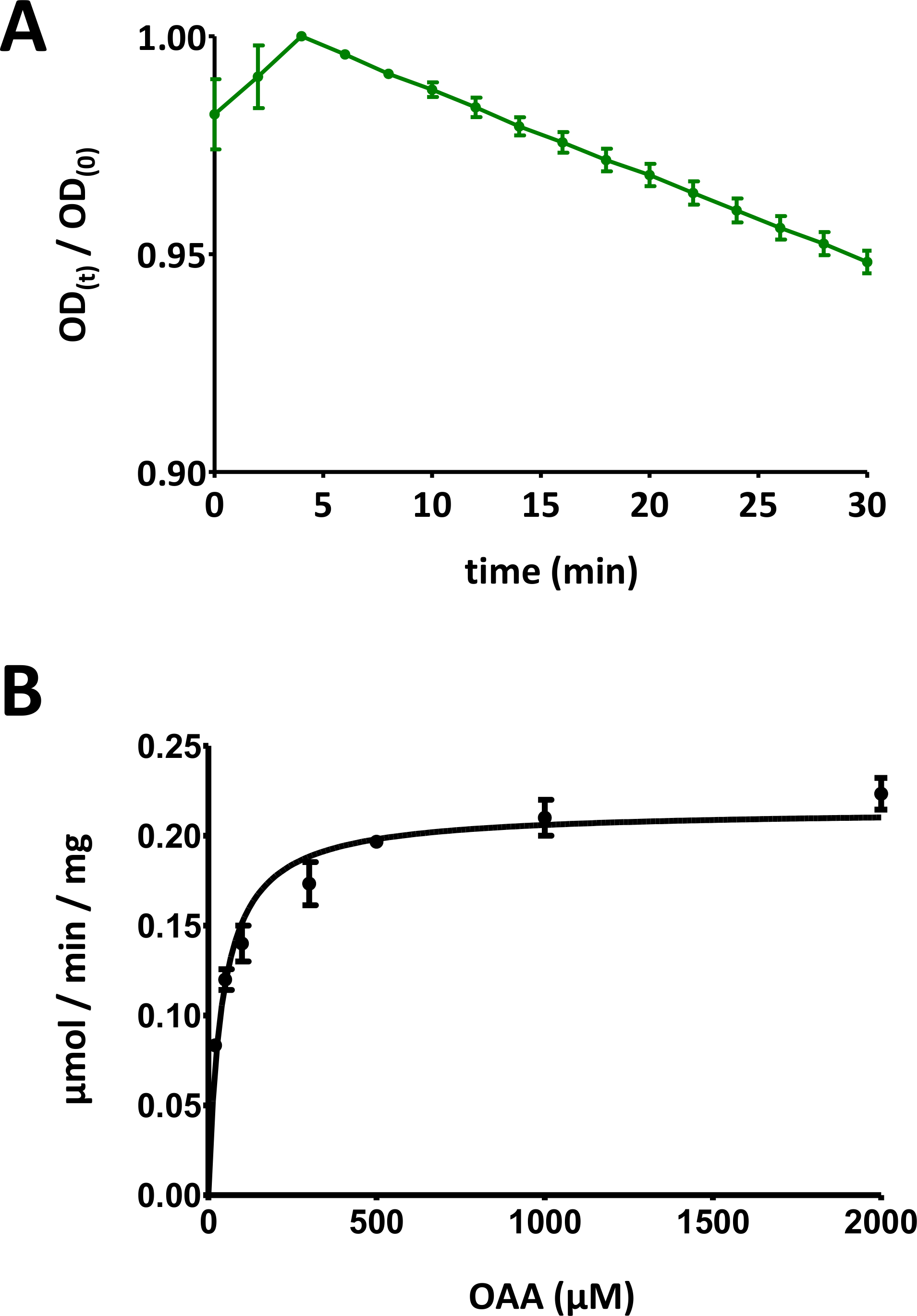
Figura 5 : Resultados ejemplares de ensayos enzimáticos.
(A) Una curva de absorción UV ejemplar obtenida para ensayos básicos de enzimas de proteína FAHD basados en sustratos (normalizados en el rango de 0 a 1) con desviación estándar. La relación de densidad óptica (OD) [OD(t)/OD(0)] en un momento dado t [OD(t)] se normaliza al OD inicial [t a 0; OD(0)]. Consulte la sección 7 del protocolo para obtener más detalles. (B) Cinética ejemplar Michaelis-Menten de la proteína humana FAHD1 con desviación estándar. Consulte la sección 8 del protocolo para obtener más detalles. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Inicie un lector de microplacas y equilibre durante 30 min a 25 oC. Configure un programa para leer 72 pozos (como se describe en la Figura 4B)a 255 nm. Se recomienda utilizar 25 lecturas múltiples con un retardo de tiempo de 5 ms. Configure un ciclo para medir 15 veces cada 2 min (30 min total).
- Realice los pasos 7.2 y 7.3. A continuación, configure 1 ml de solución de sustrato de 100 mM en tampón de ensayo enzimático.
- Preparar diluciones de la solución de sustrato en tampón de ensayo enzimático: 40 mM, 20 mM, 10 mM, 6 mM, 4 mM, 2 mM. El ensayo se realiza con concentraciones de enzimas/sustratos ("ajustadas") en pares. Para ello, prepare las siguientes diluciones de la solución enzimática en tampón de ensayo enzimático: 0,5 g/l, 0,4 g/l, 2,5 g/l, 2 g/l, 1,5 g/l, 1 g/l.
- En todos los pozos representados en la Figura 4B aplicar 180 l de tampón de ensayo enzimático. Aplicar 10 ml de tampón de ensayo enzimático en todos los pozos para el sustrato (en blanco y muestra). Aplicar 10 l de la serie de dilución proteica preparada en los pocillos para la enzima (en blanco y en la muestra). Aplicar 10 ml de tampón de ensayo enzimático en todos los pozos para los pozos para el sustrato en blanco y la enzima en blanco.
- Justo antes de la medición, aplique 10 ml de la serie de dilución de sustrato preparada en los pocillos para la muestra de sustrato y la muestra de enzimas.
- Utilice una pipeta multicanal a una configuración de 50 l para mezclar suavemente todos los pozos, comenzando con los espacios en blanco, procediendo a los pozos de muestra. Tenga cuidado de no crear burbujas.
- Inserte la placa en un lector de microplacas y mida cada pocómetro a 255 nm, como se describe en el paso 8.1. Realice el análisis en una hoja de cálculo. Copie los datos sin procesar del fotómetro en una hoja de cálculo, escriba todos los ajustes (es decir, toda la documentación) en otra hoja.
- Realice análisis de datos individuales por punto en la serie de dilución como se describe en los pasos 7.11. a las 7.14. Finalmente, obtener todas las actividades específicas y trazar contra la concentración inicial del sustrato: 2 mM, 1 mM, 0,5 mM, 0,3 mM, 0,2 mM, 0,1 mM.
- Visualice todos los puntos de datos con desviaciones estándar individuales. Computadora Michaelis-Menten cinética a través de ajuste de curva no lineal, o a través de análisis Lineweaver-Burk. Puede ser necesario volver a medir puntos individuales y adaptar las relaciones individuales de los pares proteína-concentración/sustrato-concentración en los pasos 8.5 y 8.6. El diagrama Michaelis-Menten para FAHD1 humano se proporciona en la Figura 5B.
9. Cristalización de proteínas FAHD
NOTA: La cristalización de las proteínas FAHD (FAHD1 humana descrita anteriormente15) puede lograrse mediante el método de difusión de vapor de gota colgante en un formato de 24 pozos (Figura6A). Un protocolo paso a paso sobre la cristalización de FAHD1 humano utilizando esta técnica se presenta a continuación15. En la sección de discusión se proporciona una descripción más detallada.

Figura 6 : Cristalización de proteínas FAHD.
(A) Placas de cristalización en 24 pocillos estándar o 96 pozos De la huella SBS. Consulte la sección 9 para obtener más detalles. (B) El proceso básico de configuración de la placa en la cristalización de proteínas FAHD. Esta cifra se vuelve a dibujar con el permiso23. Consulte la sección 9 para obtener más detalles. (C) Cristales FAHD1 humanos y patrones de difracción correspondientes (pequeñas inserciones). El espaciado de celosía más cercano se indica en las plaquitas como medida para la calidad de difracción de los cristales. Los números más bajos indican una resolución más alta y, por lo tanto, datos más informativos. Consulte la sección 9 del protocolo para obtener más detalles. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Asegúrese de que la proteína esté dializada contra el búfer de funcionamiento SEC. La proteína FAHD1 debe estar disponible a altas concentraciones (2-5 mg/ml). A concentraciones más bajas, la proteína puede no cristalizarse debido a la falta de nucleación espontánea.
- Preparar 20 ml de la solución del depósito para la cristalización. Realizar tres soluciones de stock, utilizando agua destilada o desionizada como disolvente: 1 M Na-HEPES (mínimo 25 ml, ajustado a pH 7,5), 50% (p/v) polietilenglicol 4000 (PEG4k) (mínimo 65 ml) y 1 M MgCl2 (10 mL).
- Configurar una rejilla de 4 x 6 (24 en total) diferentes tubos de 15 ml. Etiquetarlos de acuerdo con las posiciones correspondientes en la placa (por ejemplo, fila (A, B, C, D) contra columna (1–6) como "A1", "B5", "D6", etc.). Pipeta 1 mL de 1 M Na-HEPES en cada tubo.
- Pipeta 1 mL de 50% (p/v) PEG4k en la fila A de los tubos, 2 ml en la fila B, 3 ml en la fila C, y 4 ml en la fila D. Pipeta 100 l de 1 M MgCl2 en la columna 1 de los tubos, 250 ml en la columna 2 , 500 l en la columna 3, 1,0 ml en la columna 4, 1,5 ml en la columna 5 y 2,0 ml en la columna 6.
- Llene todos los tubos hasta un volumen de 10 ml con agua destilada o desionizada, donde la escala en los tubos es lo suficientemente precisa.
- Tomar la muestra de proteína FAHD1 humana (5 mg/ml) de la nevera (o del hielo) y girar hacia abajo a la velocidad máxima con una centrífuga de mesa a 4 oC durante al menos 10 minutos. Si se desea una cocristalización con oxalato, añadir oxalato de una solución en stock para que la muestra de proteína contenga una concentración final de oxalato de 2 mM. Aplicar 1 mM de TDT y almacenar en hielo.
- Mientras tanto, desempaquete una placa de cristalización de 24 pozos, idealmente dentro de una habitación con temperatura controlada a 18 oC. Distribuya una fina capa de aceite de parafina en el borde en la parte superior de cada pocil de la placa de 24 pozos con la ayuda de una varilla delgada de vidrio o plástico. Añadir 800 l de los cócteles de cristalización preparados (A1 a D6) en cada pocal correspondiente de la placa de cristalización.
- Coloque los labios frescos de 22 mm sobre una superficie limpia. Evite contaminar los resbalones de la cubierta con suciedad o polvo. Si es necesario, retire los restos del resbalón de la cubierta con aire comprimido o un spray de plumero.
- Una vez completada la centrifugación, evite agitar la muestra de proteína para que los agregados y desechos hilados en la parte inferior del tubo no vuelvan a flotar. En los siguientes pasos, pipeta de la muestra de proteína justo debajo de la superficie de la solución con el fin de evitar agitar agregados y depósitos desde la parte inferior.
- Para cada pocal (ver Figura 6B)pipeta 1 l de solución proteica en el centro de un resbalón de la cubierta y añadir 1 l del cóctel del depósito respectivo a la gota de proteína, evitando las burbujas. Gire el cubreobjetos hacia abajo y colóquelo en la parte superior del pozo para que el aceite selle el pozo con el cierre hermético. Repita hasta que se complete la placa de 24 pozos.
- Almacene la placa a 18 oC y observe las gotas en un horario progresivo con un microscopio adecuado. Los cristales FAHD1 humanos suelen aparecer durante la noche (ver Figura 6C).
Resultados
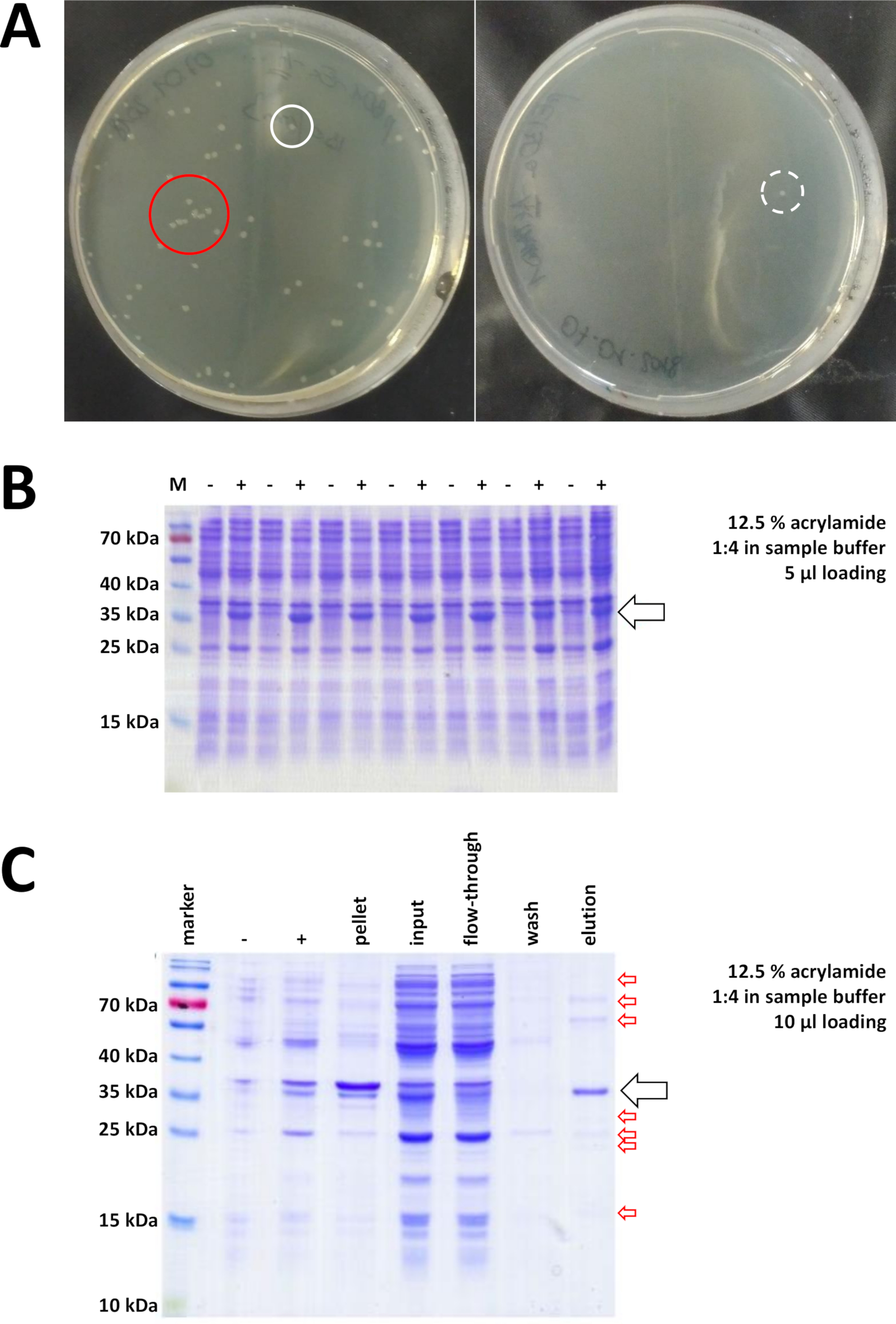
Comenzando con un vector de clonación preparado y comprado BL21(DE3) pLysS E. coli, el plásmido se inserta en la bacteria a través de choque térmico o cualquier método alternativo apropiado (Figura 1). Después de un corto período de amplificación, las bacterias transformadas se chapan en placas de agar LB, con el fin de crecer durante la noche. Placas en este punto pueden parecer diferentes, dependiendo de una variedad de fuentes de error potenciales. Las placas pueden estar vacías (es decir, sin colonias), completamente cubiertas de bacterias, o algo intermedio, respectivamente. En la Figura 7Ase describen dos ejemplos de placas de agar LB después de una transformación óptima y no óptima. Demasiadas colonias bacterianas indican que demasiadas bacterias fueron chapadas (probablemente) o que los antibióticos en uso pueden estar caducados (poco probable). Muy pocas colonias bacterianas pueden indicar que no se utilizó suficiente plásmido para la transformación (utilizar más la próxima vez) o que se utilizaron demasiados antibióticos para seleccionar las bacterias. En cualquier caso, si las colonias están presentes, deben estar bien, ya que el uso de dos antibióticos selectivos implica una probabilidad bastante insignificante de bacterias no transformadas para crecer. Ninguna colonia en absoluto, sin embargo, indica que o las bacterias perdieron su competencia de transformación (debido a almacenamiento o almacenamiento incorrectos durante períodos más largos, congelación y descongelación repetitiva, etc.), el choque de calor no tuvo éxito (sin absorción de plásmido o bacteriano muerte por demasiado calor), el vector de clonación está dañado, o por error se utilizó un conjunto incorrecto de antibióticos selectivos (verificar el gen de resistencia en el vector plásmido).

Figura 7 : Resultados representativos para la transformación de bacterias y el IMAC.
(A) Placas de agar LB representativas con transformada BL21(DE3) E. coli, obtenidas siguiendo el paso 1.1 del protocolo. Izquierda: Un plato con colonias bien distribuidas (ejemplo positivo). Derecha: Una placa con una sola colonia (ejemplo negativo). Los círculos blancos marcan buenas colonias. El círculo rojo marca las colonias que están creciendo demasiado cerca unas de otras y no deben ser recolectadas mientras las colonias aisladas estén disponibles. (B) Un análisis SDS-PAGE de acrilamida del 12,5% de una serie de controles de inducción ("-" indica antes de la inducción IPTG; "+" indica después de la inducción IPTG, antes de la cosecha de pellets, ajustado a cantidades iguales de proteína total. Esto se describe en el paso 1.2. (C) Un análisis ejemplar de 12,5% de acrilamida SDS-PAGE de la purificación De-NTA de suproteína FAHD1 etiquetada. Esto se describe en la sección 3 del protocolo. La cromatografía de afinidad produce proteínas de alta pureza (>70%, flecha negra), sin embargo, también se observan varias pequeñas contaminaciones (flechas rojas). Estas contaminaciones consisten en proteínas no FAHD que se unen a la columna, y de proteínas que se unen a la proteína FAHD. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Se seleccionan y recogen colonias validadas. Después de la amplificación en medio nutritivo, la expresión de proteína se desencadena mediante la aplicación del IPTG químico. El pellet bacteriano que contiene la proteína expresada en cantidades de miligramos se cosecha, y la expresión se verifica a través de SDS-PAGE (ver por ejemplo la figura 7B). Algunos problemas pueden ocurrir durante este proceso de otro modo simple. En primer lugar, algunas proteínas forman cuerpos de inclusión, porque aparentemente de alguna manera interfieren con el metabolismo natural de las bacterias huésped. Esto se observó para algunas mutaciones puntuales de FAHD1 humano y FAHD2. En tales casos, otros sistemas de expresión como las células de insectos pueden ser más apropiados y deben ser considerados. Después de cosechar un pellet de células de insectos, por ejemplo, la purificación de las proteínas sigue los mismos pasos que se describen en este protocolo. En segundo lugar, a veces se encuentra que el sistema DE3-pET es "leaky" (es decir, la proteína ya se expresa hasta cierto punto antes de la inducción de IPTG). La razón potencial de esto no es bien entendida, pero puede ayudar a expresar la proteína lentamente durante la noche en una incubadora de sala fría. En tercer lugar, no se expresa ninguna proteína. Este es probablemente el peor escenario, ya que probablemente indica un vector plásmido dañado y por lo tanto aconsejable secuenciar el plásmido.
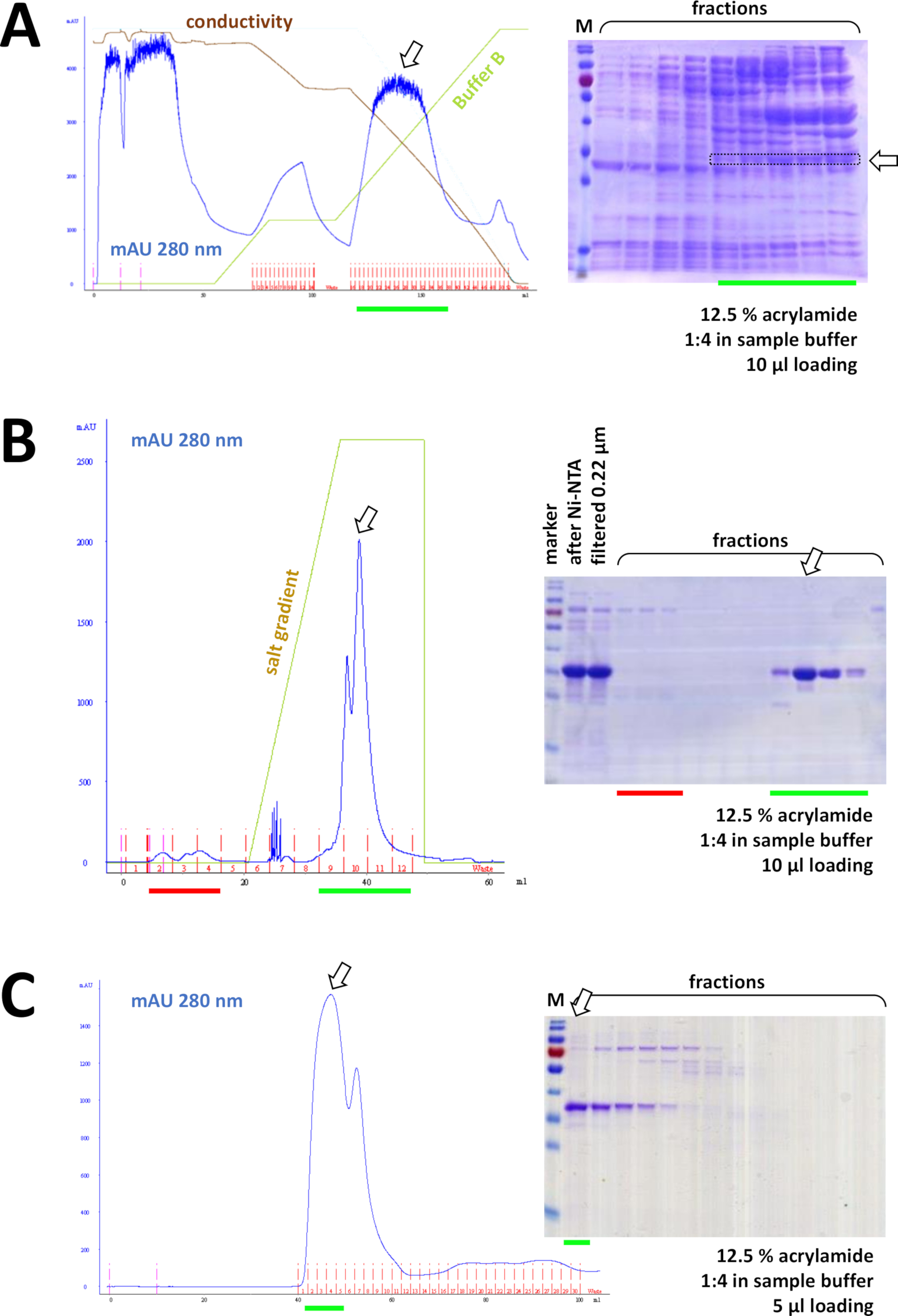
Si se utilizó una etiqueta Suyapara etiquetar la proteína, la cromatografía de afinidad con la agarosa de Ni-NTA es un método de captura fácil y barato que elimina la mayoría de las contaminaciones (Figura7C). Existen métodos similares para otros sistemas de etiquetas (por ejemplo, STREP-II). Si no se utilizó ninguna etiqueta, una combinación de precipitación de sulfato de amonio y cromatografía de intercambio hidrófoba consecutiva también puede separar la proteína de la mayoría de otras proteínas (Figura8A). Sin embargo, comparando los dos métodos (Figura7C frente a la Figura 8A),el análisis DeDS-PAGE puede demostrar la superioridad de los métodos Ni-NTA. Por lo tanto, se recomienda el uso de suproteína etiquetada.

Figura 8 : Resultados representativos de los experimentos FPLC (HIC, intercambio iónico, SEC).
(A) Un cromatógrafo típico y un análisis Delagrilamida SDS-PAGE del 12,5% de cromatografía HIC-fenil después de la precipitación de sulfato de amonio (AS) de la proteína FAHD1 sin etiquetar, como se describe en la sección 4 del protocolo. La línea verde refleja el degradado del búfer B que no contiene AS. Durante el proceso AS se lava gradualmente fuera del sistema. La comparación de este panel con la Figura 7C muestra el poder de la cromatografía de afinidad Ni-NTA en comparación con el método HIC-phenyl, y la ventaja de usar un sistema de etiquetas Supara la purificación de proteínas. (B) Un cromatograma ejemplar y un análisis SDS-PAGE de acrilamida del 12,5% de cromatografía de intercambio catiónico de Su-etiquetada FAHD tras la purificación de Ni-NTA. Usando un gradiente de sal, la muestra aplicada se separa en proteínas individuales. (C) Un cromatografía ejemplar y un análisis SDS-PAGE de acrilamida del 12,5% de cromatografía de exclusión de tamaño G75 de Su-etiquetada FAHD tras cromatografía de intercambio catiónico. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Consecutivamente, la proteína se separa aún más de las contaminaciones sobrantes por cromatografía de intercambio catión/anión (por ejemplo, ver Figura 8B), seguida de cromatografía de exclusión de tamaño (por ejemplo, ver Figura 8C). Se aconseja establecer una estrategia de purificación inicial en este orden; sin embargo, estas columnas deben utilizarse en combinación, posteriormente y en variación, hasta que la proteína sea lo suficientemente pura.
Los ensayos de actividad simples, con el fin de probar las decisiones del "sí o no" en sustratos activos y/o cofactores, se pueden realizar con susproteínas etiquetadas después de la purificación de Ni-NTA, o proteínas sin etiquetar después de la columna de intercambio iónico. Las actividades específicas y las constantes cinéticas deben determinarse con proteínas de mayor pureza. La cristalización se puede intentar con proteínas después de la columna de intercambio iónico, pero la calidad de los cristales casi siempre se correlaciona con la pureza de las proteínas. Los anticuerpos policlonales pueden criarse contra proteínas en cualquier etapa del protocolo de purificación; sin embargo, aquí la calidad también se correlaciona con la pureza de la proteína.
Discusión
Pasos críticos
Las proteínas FAHD son muy sensibles a las concentraciones de sal. A bajas concentraciones de NaCl, las proteínas pueden precipitarse al descongelarse, pero por lo general se pueden reconstituir completamente a concentraciones de sal más altas. Es decir, si una proteína FAHD se precipita por alguna razón, puede recuperarse o volver a plegarse con concentraciones de sal más altas (>300 m). Sin embargo, es posible que no se recuperen algunas proteínas más hidrófobas (por ejemplo, FAHD2 humana), pero detergentes como CHAPS (máximo 1%) o glicerol (10%) puede utilizarse para mantenerlos en solución estable. En cualquier caso, se recomienda la congelación de choque utilizando nitrógeno líquido y el almacenamiento a -80 oC, ya que es un proceso suave y lento de descongelación.
Algunos problemas inesperados pueden ocurrir durante la purificación de Ni-NTA en el paso 3.1.10. Cabe destacar que una Mayor Do en la segunda muestra recolectada que en la primera muestra indica un volumen demasiado alto de la resina de agarosa (tome nota y use menos resina en el siguiente experimento). Además, la resina de agarosa en sí conduce a una señal DeA a 280 nm (es decir, la interrupción del lecho de resina de agarosa dará señales artificiales). En caso de duda, se recomienda utilizar otros métodos como un ensayo de Bradford o BSA para determinar las concentraciones de proteínas.
En los ensayos enzimáticos, hay tres aspectos críticos a tener en cuenta. En primer lugar, evaluar la concentración de proteínas es fundamental para obtener las actividades específicas correctas. El nivel de pureza de la proteína está influyendo en el resultado y debe estimarse. En el caso de la proteína etiquetada, la masa de la parte de la etiqueta tiene que ser calculada, y la actividad específica tiene que ser corregida correspondientemente. Para ensayos simples descritos en la sección 7 del protocolo, la pureza de Ni-NTA es suficiente para distinguir entre sustratos activos e inactivos, cofactores, etc. En el caso de la cinética más compleja de Michaelis-Menten, todas las concentraciones de reactivos y sustratos deben determinarse correctamente. Especialmente cuando se utiliza oxaloacetato (que auto-descarboxila con el tiempo) la parte enzimática de la reacción debe corregirse para la auto-descarboxilación (bajo el supuesto de que ambas reacciones se producen simultáneamente). Deberán tenerse en cuenta los cambios iniciales en la señal de densidad óptica dirigida a la tautomerización keto-enol del sustrato. En tercer lugar, deben ajustarse las concentraciones y los volúmenes. Una reacción con concentraciones definidas de enzima y sustrato puede dar resultados diferentes dependiendo del volumen del ensayo. Si hay demasiada enzima por pozo, la adhesión del líquido puede, de hecho, sesgar el resultado.
Para evaluar la cinética de Michaelis-Menten se recomienda realizar experimentos iniciales en lotes de 100 , 200 l y 300 l para encontrar la combinación óptima. Aspectos similares se aplican a la relación entre las concentraciones de enzimas y sustratos para ensayos cinéticos. Demasiada enzima por sustrato o demasiado sustrato por enzima coloca el sistema fuera del rango lineal de estado estacionario Michaelis. Se requieren experimentos iniciales para optimizar estas condiciones. El ajuste ejemplar para la proteína FAHD1 humana (tipo salvaje) se proporciona en la sección 8, lo que resulta en diagramas cinéticos (como se presenta en la Figura 5B,por ejemplo).
Para la cristalización, una gota de solución proteica se entuba en el centro de un encubridor y se mezcla con una gota de cóctel de cristalización, que generalmente se compone de un tampón (por ejemplo, Tris-HCl, HEPES) y un precipitante (por ejemplo, polietilenglicol, amonio sulfato). Opcionalmente, se puede aplicar una gota de solución inhibidora para la cocristalización (como el oxalato en este protocolo). El cubreobjetos se coloca al revés por encima de un pozo de depósito que contiene cóctel de cristalización, sellando el pozo hermético con la ayuda de aceite sellante (Figura6B). Idealmente, no se produce precipitación dentro de la caída al principio del experimento, lo que significa que la proteína permanece en solución. Dado que la concentración precipitada en el embalse es mayor que en la gota, la gota comienza a perder agua por evaporación en la atmósfera del pozo hasta que se alcanza el equilibrio con el depósito. La difusión de agua en el depósito provoca una disminución lenta del volumen de la caída que a su vez provoca un aumento de la concentración de proteínas y precipitantes en la gota. Si la solución proteica alcanza el estado requerido de supersaturación y, por lo tanto, la metaestabilidad, puede producirse una nucleación espontánea seguida de un crecimiento cristalino. Alcanzar el estado sobresaturado es una condición necesaria pero no suficiente para la cristalización. La cristalización de proteínas necesita ambas condiciones termodinámicas y cinéticas favorables, y depende en gran medida de las propiedades impredecibles de la proteína a cristalizar22.
Modificaciones y solución de problemas
La expresión de proteína en E. coli puede ser ineficiente. Es posible que sea necesario analizar las concentraciones de IPTG, la temperatura de expresión y el tiempo de amplificación, como la temperatura ambiente durante varias horas o en una sala fría durante la noche, para detectar cada nueva proteína para encontrar condiciones óptimas. A veces se observa precipitación de proteínas en los cuerpos de inclusión para proteínas FAHD más hidrófobas. En tales casos, se recomienda la expresión de proteínas en otros sistemas modelo como las células de insectos, ya que los cuerpos de inclusión son menos propensos a formar26.
Como las proteínas FAHD son sensibles a las concentraciones de sal y cofactores, así como al pH, las estrategias de purificación para diferentes homólogos, ortologos y variantes de mutación puntual pueden diferir en entornos individuales. Los métodos de purificación descritos se desarrollan para la proteína FAHD1 humana y de ratón de tipo salvaje. Las concentraciones de productos químicos, como El NaCl y el imidazol, así como el pH, pueden tener que adaptarse para proteínas individuales con un punto isoeléctrico (pI) diferente. También es de destacar, no todas susproteínas etiquetadas pueden unirse bien a una resina Ni-NTA. Si la unión a proteínas a la columna Ni-NTA es ineficiente, las concentraciones adaptadas de NaCl e imidazol, así como las diferentes condiciones de pH en el tampón de funcionamiento de Ni-NTA pueden ayudar a mejorar la calidad del resultado. Si no es así, saltarse el paso Ni-NTA y proceder al paso de la cromatografía de intercambio iónico también puede conducir a una estrategia de purificación exitosa. Si una proteína se une a la columna Ni-NTA pero no se puede eluir de la columna, la adición de un poco de mM EDTA puede ayudar a interrumpir el complejo Ni2+.
En cuanto al proceso de cristalización, es necesario entender que la autoorganización de moléculas de proteínas grandes y complejas en una celosía periódica regular es un proceso intrínsecamente improbable que depende en gran medida de parámetros cinéticos difíciles de controlar. Incluso pequeños cambios en la configuración utilizada para la cristalización pueden alterar dramáticamente el resultado y no se formarán cristales. La pureza de las proteínas es generalmente de suma importancia. Como regla general, un gel SDS-PAGE muy sobrecargado no debe mostrar otras bandas. Además, la secuencia en la que se realizan los pasos puede afectar al resultado. Como ejemplo, para garantizar la reproducibilidad, a menudo es necesario mantener la secuencia de pipeteo igual, luego primero agregar la proteína, y finalmente agregar precipitant a la gota de cristalización (o viceversa). Cualquiera que sea el método utilizado, debe mantenerse igual al intentar reproducir o escalar experimentos. Si no se observan cristales siguiendo este protocolo, la composición del precipitante químico, el pH, el tamaño de la gota y la relación proteína-precipitada pueden variar en pequeños incrementos. La paciencia y las observaciones consistentes de las gotas son de virtud.
Observaciones a los Mecanismos Catalíticos de FAHD1
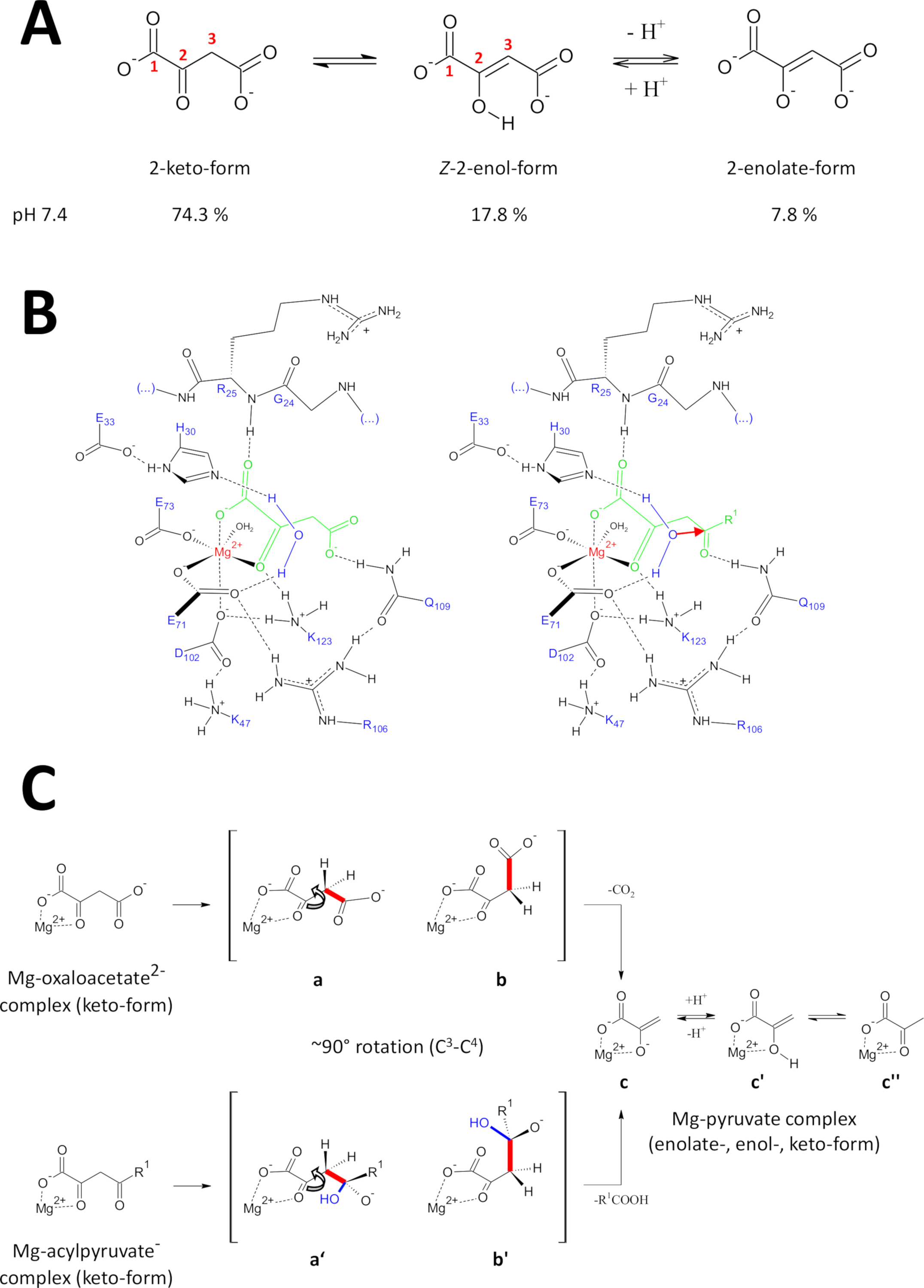
Los métodos presentados han sido desarrollados específicamente para obtener proteínas FAHD1 de alta calidad. Esto permitió el crecimiento de cristales FAHD1, así como la ingeniería de cristales que contienen FAHD1 complejo a un inhibidor (oxalato, PDB:6FOG). Las estructuras de rayos X proporcionan una arquitectura 3D de la cavidad catalítica de la enzima. Estos resultados establecen una descripción completa de los residuos potencialmente importantes para los mecanismos catalíticos de esta enzima intrigante. FAHD1 fue descrito por primera vez para ser capaz de celpiriruvates (acetilpiruvato, fumarylpyruvate)11. Más tarde, se encontró que FAHD1 funciona también como una decarboxilasa de oxaloacetato12. Aunque los sustratos de acipiruvato y oxaloacetato son diferentes mitades químicas, las transformaciones químicas comparten mecanicidad el escote estratégico de un enlace común de C3-C4, cedida energéticamente si el C3 -C4 orbitales de unión permanecen ortogonales a los orbitales de la C2-carbonyl15. Tal conformación permite la estabilizaciónpor resonancia del C 3-carbanión formada transitoriamente durante el proceso de escisión. Los sustratos de FAHD1 (oxaloacetato y acipiruvatos) son moléculas flexibles y pueden existir enformas tautoméricas (keto-enol) así como en formas termoréticas (Figura9A). Los equilibrios entre las diferentes especies están determinados principalmente por la naturaleza de la composición tampón utilizada, el pH y la presencia de iones metálicos. A continuación discutimos escenarios hipóticos mecanicistas inspirados en el análisis de estructuras cristalinas de rayos X que revelaron el centro catalítico de FAHD1.

Figura 9 : Detalles sobre el mecanismo catalítico propuesto de FAHD1 humano.
(A) Oxaloacetato existe en estado cristalino, así como en solución neutra principalmente en la forma Z-enol24. Sin embargo, en condiciones fisiológicas de pH, la forma 2-keto es la representación predominante25. (B) Boceto químico de la cavidad hFAHD115 con oxaloacetato ligado a Mg (izquierda) y acipiruvato (derecha, con R1 como resto orgánico; la flecha roja denota un ataque nucleófilo de la molécula de agua estabilizada adyacente) (ver discusión). (C) Comparación de las conformaciones favorecidas para la escisión C3-C4 en la descarboxilasa (b a c) y el mecanismo de hidrolasa (b' a c) de FAHD1: ambos procesos dan como resultado piruvato-enolado complejo con Mg (véase el debate). Se espera que los intermedios b y b' sean estabilizados por Q109, tal como se esboza en el panel B (véase la discusión). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
La actividad de descarboxilasa de FAHD1
Oxaloacetato existe en estado cristalino, así como en solución neutra principalmente en la Z-enol forma24. Pero se demostró que en condiciones fisiológicas de pH (condiciones de pH a pH 7.4) la forma de 2-keto es la representación predominante de oxaloacetato25 (Figura9A),y que la enolización no es un requisito previo para la descarboxilación27 . Cabe destacar que los iones Mg2+ no influyen en la proporción de las especies de oxaloacetato a un pH de 7,4 o inferior a28. La transposición del keto de oxaloacetato se forma en el centro catalítico de FAHD1 (guiado por el oxalato encuadernado en la enzima compleja (PDB: 6FOG15)) reveló el residuo Q109 como regulador de conformación del oxaloacetato enlazado15. Como se describe en otro artículo15,la unión de hidrógeno al grupo de carbamoyl de Q109 estabiliza una conformación de oxaloacetato resultante de la rotación alrededor del enlace C2-C3 (Figura9B,panel izquierdo). Como consecuencia de esta rotación, el enlace C3-C4 (a cleaved) adopta una disposición ortogonal cercana a la disposición ortogonal en relación con los orbitales de la C2-carbonyl (Figura9C). El dióxido de carbono se puede liberar. El producto principal de este proceso sería la resonancia estabilizada Mg-enolate de piruvato. Se sabe a partir de investigaciones de complejos de oxaloacetato-Mg que el enolado forma el complejo más estable28,29. Suponiendo una estabilidad comparable para un complejo de enolato de Mg-piruvato el cofactor de FAHD1 podría ser bloqueado, pero el residuo de lisina K123 puede protonizar el piruvato-enolato en un equilibrio para prohibir la pérdida del cofactor15.
La interpretación dada sugiere piruvato enol como un intermedio distinto en la función catalítica ODx de FAHD1. En este paso del modelo hipotética, los datos experimentales no proporcionan ninguna indicación adicional de por qué la tapa cerrada debe abrirse para liberar el producto. Se puede deducir, sin embargo, que el mecanismo propuesto parece una inhibición enzimática por el producto: La estructura cristalina revela una molécula de agua conservada mantenida en orientación direccional hacia el centro catalítico FAHD1 por los residuos H30 y E33 presentados en un hélice corta15,que se induce sobre la unión de ligando y el cierre de la tapa. Si el enol primario se mantuviera en equilibrio con el enolado, la enolación estabilizada por resonancia podría ser saciada para piruvate por la molécula de agua. El hidroxilo resultante sería capaz de desplazar el piruvato del Mg-cofactor sobre el cual se abriría la tapa. Finalmente, el centro catalítico sería restaurado en el entorno mitocondrial. En este escenario hipotético, la molécula de agua de la cavidad funcionaría como un ácido, respectivamente.
Actividad hidrolasa de FAHD1
La actividad hidrolasa de una enzima requiere implícitamente la formación intermedia de un nucleófilo hidroxilo. Este mecanismo se encuentra generalmente en combinación con la actividad catalítica de base ácida. El estado de transición de la reacción tiene que ser preparado a través del control conformación por cadenas laterales de aminoácidos críticos en la cavidad. En analogía con la discusión de la función de la decarboxilasa, la acilpiruvato ligada a enzimas en forma de 2-ceto se pondrá bajo control de la conformación mediante la unión al hidrógeno del oxígeno 4-carbonilo a Q109 (Figura9B,panel derecho). La estructura cristalina de FAHD1 ligada a oxalato (PDB:6FOG) revela una molécula de agua conservada en orientación direccional hacia el centro catalítico FAHD1 por los residuos H30 y E33 presentados en una hélice corta15. El diadido E33-H30 es competente para desprotonar el agua posicionada direccional y el hidroxilo resultante está en disposición ideal para atacar el 4-carbonilo de acilpiruvato presentado bajo control conforme por Q10915.
Cabe destacar que se ha propuesto un mecanismo similar para FAH18. Se espera que el ataque por el nucleófilo hidroxilo dé lugar a una especie de oxianion, que se estabiliza sobre el escote de enlace C3-C4 controlado orbital (Figura9C). En este modelo, la rotación de la unión C3-C4 (Figura9C)ocurre después del ataque nucleófilo por el hidroxilo formado indicado en la Figura 9B (es decir, prepara el acylpyruvate para el escote de la unión). Los productos principales serían ácido acético y enolato de Mg-piruvato. En este escenario hipotético, el ácido acético podría saciar el enol a piruvato y posteriormente ayudar al desplazamiento del producto. Por encima de un pH de 7,5 y en presencia de iones Mg, los aciroluvados existen en un equilibrio entre las formas de keto- y enol, este último en ligera preferencia30. Lo más probable es que ambas formas sean capaces de unirse al cofactor de FAHD1 bajo el cierre posterior de la tapa. El procesamiento de sustratos de acirolpiruvato enólico por la enzima se ve obstaculizado debido a la estructura plana de la forma enol. El escote C3-C4 resultaría en un carbanión vinánica sin estabilización por resonancia.
Por lo tanto, proponemos un paso de cetonización catalítica para prepararel para el ataque del nucleófilo hidroxilo en el carbonilo de acilo. Este proceso de ketonización, sin embargo, requeriría el control sobre las transposiciones de protones por los residuos de FAHD1, lo que atribuiría una actividad isomerasa inherente a FAHD1. Se informa que la acidez del hidrógeno enol mg-bound revela un aumento de diez mil veces en comparación con la forma no compleja28. Una deprotonación de la forma de enol encuadernado Mg sería factible por K123 no protensado. La desprotonación de K123 puede ser asistida por el carboxilato de D102. Una red de enlace de hidrógeno formada por residuos D102-K47-K123 podría funcionar como el relé de protones necesario en el centro catalítico de FAHD115. Un enolato intermedio tal formado podría ser aplacado por una tríada E33-H30-H20 bajo la ketonización del sustrato15. La forma de 2-ceto entraría bajo control conformacional de Q109, y el hidroxilo formado concomitantemente atacaría el carbonilo de acilo. La discusión resumida implica un control de FAHD1 sobre una molécula de agua para cambiar entre ácido y base a través de la interacción de residuos formados de cavidades.
Aplicaciones futuras o instrucciones del método
Las aplicaciones futuras de los métodos descritos aquí son numerosas. Una plétora de miembros procariotas de la superfamilia FAH todavía espera la caracterización funcional. Incluso la información disponible sobre las actividades catalíticas de los miembros conocidos de la superfamilia FAH es escasa y, en la mayoría de los casos, se basa en suposiciones teóricas en lugar de datos experimentales. La aplicación de los métodos descritos aquí para los miembros de la superfamilia prokaryotic FAH depende de los intereses específicos de investigación en bacteriología. Por otro lado, la reciente demostración de que los miembros de la superfamilia eucariota FAH desempeñan un papel esencial en varios compartimentos celulares (por ejemplo, citosol frente a mitocondrias) pone de relieve la necesidad de caracterizar mejor estas proteínas (tres de las cuales han sido identificado hasta ahora), en particular porque los datos actuales sugieren que algunas proteínas no caracterizadas pueden llevar a cabo diferentes funciones en el contexto de la biología mitocondrial, la investigación del envejecimiento y la investigación del cáncer. Se propone que la caracterización molecular y fisiológica completa de estos miembros de la superfamilia eucariota FAH pueda proporcionar una visión importante de los principales campos de la investigación contemporánea en el sector biomédico. Se necesitan más investigaciones sobre los mecanismos de FAHD1 (y enzimas relacionadas) para comprender mejor los mecanismos subyacentes a la bifuncionalidad de FAHD1, que todavía no está completamente aclarado. Estudios adicionales con mutantes FAHD1, investigaciones de RMN y estudios estructurales sobre complejos inhibidores pueden ayudar a resolver los verdaderos escenarios mecánicos para los que FAHD1 parece ser competente. Además, el diseño asistido por ordenador de imitaciones de enol capaces de unirse al mg-cofactor eventualmente conducirá a inhibidores potentes de FAHD1.
Divulgaciones
Los autores no tienen nada que revelar y declarar que no hay intereses financieros en competencia. H. G. es CEOCSO en MoleculeCrafting.HuGs e.U. y proporcionó acylpyruvates para este estudio a través de la síntesis personalizada. El trabajo en el laboratorio de P. J. D. contó con el apoyo del Fondo Austriaco de Ciencia (FWF): número de proyecto P 31582-B26. Las tasas de publicación de este manuscrito han sido cubiertas en parte por el Fondo De Ciencia de Austria (FWF) bajo el número de proyecto P 31582-B26. A. N. y B. R. cuentan con el apoyo del Fondo De Ciencia de Austria (FWF) en el marco del proyecto P28395-B26.
Agradecimientos
Los autores están muy agradecidos por la asistencia técnica experta de Annabella Pittl y el desarrollo del método piloto por Haymo Pircher.
Materiales
| Name | Company | Catalog Number | Comments |
| BL21(DE3) pLysS competent E. coli | Promega | L1195 | High-efficiency protein expression from gene with T7 promoter and ribosome binding site |
| pET E. coli T7 Expression Vectors | MERCK | - | http://www.merckmillipore.com/AT/de/life-science-research/genomic-analysis/dna-preparation-cloning/pet-expression-vectors/qFSb.qB.mLQAAAFA6.VkiQ0G,nav |
| 0.45 µm filter units | MERCK | SLHP033NS | Millex-HP, 0.45 µm, PES 33 mm, not steril |
| 0.22 µm filter units | MERCK | SLGP033RS | Millex-HP, 0.22 µm, PES 33 mm, not steril |
| Eppendof tubes 1.5 mL | VWR | 525-1042 | microcentrifugal tubes; autoclaved |
| 15 mL Falcon | VWR | 734-0451 | centrifugal tubes |
| 50 mL Falcon | VWR | 734-0448 | centrifugal tubes |
| PS Cuvettes Spectrophotometer Semi-Micro | VWR | 30622-758 | VIS transparent cuvettes |
| UV Cuvettes Spectrophotometer Semi-Micro | VWR | 47727-024 | UV/VIS transparent cuvettes |
| isopropyl-β-D-thiogalactopyranosid (IPTG) | ROTH | 2316 | chemical used for induction of protein expression with the DE3/pET system |
| imidazole | ROTH | X998 | chemical used for elution of polyhistidine (6xHis) sequences from a nickel-charged affinity resin |
| Glass Econo-Column Columns | Bio-Rad | - | http://www.bio-rad.com/de-at/product/glass-econo-column-columns?ID=2cfb1c6e-32e8-4c72-b532-dd39013d707d&pcp_loc=catprod |
| chloramphenicol | Sigma-Aldrich | C0378 | antibiotic for bacterial growth selection; resistance endióded in pLysS plasmid of BL21(DE3) E. coli; 25 µg/mL final concentration |
| kanamycin | Sigma-Aldrich | 60615 | antibiotic for bacterial growth selection; to be used if this resistance is encoded in the employed pET vector; 50 µg/mL final concentration |
| ampicillin | Sigma-Aldrich | A1593 | antibiotic for bacterial growth selection; to be used if this resistance is encoded in the employed pET vector; 100 µg/mL final concentration |
| Ultra-15, MWCO 10 kDa | Sigma-Aldrich | Z706345 | centrifigal filters for protein enrichment; https://www.sigmaaldrich.com/catalog/product/sigma/z706345?lang=de®ion=AT |
| Ultra-0.5 Centrifugal Filter Units | Sigma-Aldrich | Z677108 | centrifigal filters for protein enrichment; https://www.sigmaaldrich.com/catalog/product/ALDRICH/Z677108?lang=de®ion=AT&cm_sp=Insite-_-prodRecCold_xviews-_-prodRecCold5-2 |
| oxaloacetic acid | Sigma-Aldrich | O4126 | TCA metabolite |
| sodium oxlalate | Sigma-Aldrich | 71800 | a competitive inhibitor of FAH superfamily enzymes |
| Dialysis tubing cellulose membrane | Sigma-Aldrich | D9277 | https://www.sigmaaldrich.com/catalog/product/sigma/d9277; or comparable |
| Ni-NTA agarose | Thermo-Fischer | R90101 | a nickel-charged affinity resin that can be used to purify recombinant proteins containing a polyhistidine (6xHis) sequence |
| 96-Well UV Microplate | Thermo-Fischer | 8404 | UV/VIS transparent flat-bottom 96 well plates |
| PageRuler Prestained Protein Ladder, 10 to 180 kDa | Thermo-Fischer | 26616 | https://www.thermofisher.com/order/catalog/product/26616?SID=srch-hj-26616 |
| ÄKTA FPLC system | GE Healthcare Life Sciences | - | using the FPLC system by GE Healthcare; different custom versions exist; this work used the "ÄKTA pure" system |
| HiTrap Phenyl HP column | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/it/shop/chromatography/prepacked-columns/hydrophobic-interaction/hitrap-phenyl-hp-p-05630 |
| Mono S 10/100 GL | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/ch/shop/chromatography/prepacked-columns/ion-exchange/mono-s-cation-exchange-chromatography-column-p-00723 |
| Mono Q 10/100 GL | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/ch/shop/chromatography/prepacked-columns/ion-exchange/mono-q-anion-exchange-chromatography-column-p-00608 |
| HiLoad Superdex column 75 pg (G75) | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/ch/shop/chromatography/prepacked-columns/size-exclusion/hiload-superdex-75-pg-preparative-size-exclusion-chromatography-columns-p-05800 |
| HiLoad Superdex column 200 pg (G200) | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/ch/shop/chromatography/prepacked-columns/size-exclusion/hiload-superdex-200-pg-preparative-size-exclusion-chromatography-columns-p-06283 |
| TECAN microplate reader | TECAN Life Sciences | - | https://lifesciences.tecan.com/microplate-readers |
| acetylpyruvate | MoleculeCrafting.HuGs e.U. | - | custom synthesis |
| benzoylpyruvate | MoleculeCrafting.HuGs e.U. | - | custom synthesis |
| VDX™ plate (24 wells) | Hampton | HR3-142 | 24 well plates used for crystallization via Hanging Drop Vapor Diffusion |
| paraffin oil | Hampton | HR3-411 | used for crystallization via Hanging Drop Vapor Diffusion |
| coverslips (22 mm) | Karl Hecht KG | 14043 | coverslips used for crystallization via Hanging Drop Vapor Diffusion |
| Luria broth (LB) medium | self-prepared | - | a general growth medium for E. coli: 5 g/L yeast extract; 10 g/L peptone from casein; 10 g/L sodium chloride; 12 g/L agar-agar |
| NZCYM medium | self-prepared | - | a better growth medium for E. coli, used for amplification: 10 g/L NZ amine; 5 g/L NaCl; 5 g/L yeast extract; 1 g/L casamino acids; 2 g/L MgSO4; adjust pH to 7.4 |
| Luria broth (LB) agarose plates | self-prepared | - | autoclaved agarose plates containing LB-medium and antibiotics for bacterial groth selection; https://www.addgene.org/protocols/pouring-lb-agar-plates/ |
| Ni-NTA running buffer | self-prepared | - | 20 mM Tris-HCl pH 7,4; 50-300 mM NaCl; 10-200 mM imidazole; ranges: optimal value varies among FAHD proteins |
| Ni-NTA elution buffer | self-prepared | - | 20 mM Tris-HCl pH 7,4; 50-300 mM NaCl; 200-500 mM imidazole; ranges: optimal value varies among FAHD proteins |
| HIC running buffer | self-prepared | - | 44 mM NaH2PO4; 6 mM Na2HPO4; 100 mM NaCl; 20 mM DTT; adjust to pH 7 |
| HIC running buffer AS | self-prepared | - | HIC running buffer saturated with ammonium sulfate (AS); adjust to pH 7: 70 g ammonium sulfate + 90 mL buffer, stirred overnight in the cold room; adjust to pH 7.0 |
| Mono S low salt buffer | self-prepared | - | 44 mM NaH2PO4; 6 mM Na2HPO4; 10-300 mM NaCl; ranges: optimal value varies among FAHD proteins |
| Mono S high salt buffer | self-prepared | - | 44 mM NaH2PO4; 6 mM Na2HPO4; 1-2 M NaCl; ranges: optimal value varies among FAHD proteins |
| Mono Q low salt buffer | self-prepared | - | 20 mM Tris-HCl; 15 mM NaCl; adjust to pH 8.0 |
| Mono Q high salt buffer | self-prepared | - | 20 mM Tris-HCl; 1 M NaCl; 10 % glycerol; adjust to pH 8.0 |
| G75 / G200 running buffer | self-prepared | - | 15 mM Tris-HCl; 300 mM NaCl; adjust to pH 7.4 |
| enzyme assay buffer | self-prepared | - | 50 mM Tris-HCl pH7.4; 100 mM KCl; 1 mM MgCl2 |
| protein crystallization buffer | self-prepared | - | G75 / G200 running buffer with 1 mM DTT |
| reservoir solution for crystallization | self-prepared | - | 100 mM Na-HEPES pH 7.5; 5-20 % (w/v) PEG4k; 10 mM-200 mM MgCl2 |
Referencias
- Brouns, S. J. J., et al. Structural Insight into Substrate Binding and Catalysis of a Novel 2-Keto-3-deoxy-d-arabinonate Dehydratase Illustrates Common Mechanistic Features of the FAH Superfamily. Journal of Molecular Biology. 379, 357-371 (2008).
- Timm, D. E., Mueller, H. A., Bhanumoorthy, P., Harp, J. M., Bunick, G. J. Crystal structure and mechanism of a carbon-carbon bond hydrolase. Structure (London, England: 1993). 7, 1023-1033 (1999).
- Weiss, A. K. H., Loeffler, J. R., Liedl, K. R., Gstach, H., Jansen-Dürr, P. The fumarylacetoacetate hydrolase (FAH) superfamily of enzymes: multifunctional enzymes from microbes to mitochondria. Biochemical Society Transactions. 46, 295 (2018).
- Guimarães, S. L., et al. Crystal Structures of Apo and Liganded 4-Oxalocrotonate Decarboxylase Uncover a Structural Basis for the Metal-Assisted Decarboxylation of a Vinylogous β-Keto Acid. Biochemistry. 55, 2632 (2016).
- Zhou, N. Y., Fuenmayor, S. L., Williams, P. A. nag genes of Ralstonia (formerly Pseudomonas) sp. strain U2 encoding enzymes for gentisate catabolism. Journal of Bacteriology. 183, 700 (2001).
- Izumi, A., et al. Structure and Mechanism of HpcG, a Hydratase in the Homoprotocatechuate Degradation Pathway of Escherichia coli. Journal of Molecular Biology. 370, 899-911 (2007).
- Manjasetty, B. A., et al. X-ray structure of fumarylacetoacetate hydrolase family member Homo sapiens FLJ36880. Biological Chemistry. 385, 935-942 (2004).
- Tame, J. R. H., Namba, K., Dodson, E. J., Roper, D. I. The crystal structure of HpcE, a bifunctional decarboxylase/isomerase with a multifunctional fold. Biochemistry. 41, 2982-2989 (2002).
- Ran, T., et al. Crystal structures of Cg1458 reveal a catalytic lid domain and a common catalytic mechanism for the FAH family. The Biochemical Journal. 449, 51-60 (2013).
- Ran, T., Wang, Y., Xu, D., Wang, W. Expression, purification, crystallization and preliminary crystallographic analysis of Cg1458: A novel oxaloacetate decarboxylase from Corynebacterium glutamicum. Acta Crystallographica Section F: Structural Biology and Crystallization Communications. 67, 968-970 (2011).
- Pircher, H., et al. Identification of human Fumarylacetoacetate Hydrolase Domain-containing Protein 1 (FAHD1) as a novel mitochondrial acylpyruvase. Journal of Biological Chemistry. 286, 36500-36508 (2011).
- Pircher, H., et al. Identification of FAH domain-containing protein 1 (FAHD1) as oxaloacetate decarboxylase. Journal of Biological Chemistry. 290, 6755-6762 (2015).
- Petit, M., Koziel, R., Etemad, S., Pircher, H., Jansen-Dürr, P. Depletion of oxaloacetate decarboxylase FAHD1 inhibits mitochondrial electron transport and induces cellular senescence in human endothelial cells. Experimental Gerontology. 92, 7-12 (2017).
- Etemad, S., et al. Oxaloacetate decarboxylase FAHD1 – a new regulator of mitochondrial function and senescence. Mechanisms of Ageing and Development. 177, 22-29 (2019).
- Weiss, A. K. H., et al. Structural basis for the bi-functionality of human oxaloacetate decarboxylase FAHD1. Biochemical Journal. 475, 3561-3576 (2018).
- Taferner, A., et al. FAH domain-containing protein 1 (FAHD-1) Is required for mitochondrial function and locomotion activity in C. elegans. PLoS ONE. 10, 1-15 (2015).
- Mizutani, H., Kunishima, N. Purification, crystallization and preliminary X-ray analysis of the fumarylacetoacetase family member TTHA0809 from Thermus thermophilus HB8. Acta Crystallographica Section F Structural Biology and Crystallization Communications. 63, 792-794 (2007).
- Bateman, R. L., Bhanumoorthy, P., Witte, J. F., McClard, R. W., Grompe, M., Timm, D. E., et al. Mechanistic Inferences from the Crystal Structure of Fumarylacetoacetate Hydrolase with a Bound Phosphorus-based Inhibitor. Journal of Biological Chemistry. 276, 15284-15291 (2001).
- Zeng, F., et al. Efficient strategy for introducing large and multiple changes in plasmid DNA. Scientific Reports. 8, 1714 (2018).
- Higuchi, R., Krummel, B., Saiki, R. K. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Research. 16, 7351-7367 (1988).
- Jansen-Duerr, P., Pircher, H., Weiss, A. K. H. The FAH Fold Meets the Krebs Cycle. Molecular Enzymology and Drug Targets. 2, 1-5 (2016).
- Rupp, B. Origin and use of crystallization phase diagrams. Acta Crystallographica Section F Structural Biology Communications. 71, 247-260 (2015).
- Rupp, B. . Biomolecular Crystallography: Principles, Practice, and Application to Structural Biology. , (2010).
- Flint, D. H., Nudelman, A., Calabrese, J. C., Gottlieb, H. E. Enol oxalacetic acid exists in the Z form in the crystalline state and in solution. The Journal of Organic Chemistry. 57, 7270-7274 (1992).
- Pogson, C. I. I., Wolfe, R. G. G. Oxaloacetic acid tautomeric and hydrated forms in solution. Biochemical and Biophysical Research Communications. 46, 1048-1054 (1972).
- Kost, T. A., Condreay, J. P., Jarvis, D. L., Kost, A. T. Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nature Biotechnology. 23, 567-575 (2005).
- Steinberger, R., Westheimer, F. H. Metal Ion-catalyzed Decarboxylation: A Model for an Enzyme System 1. Journal of the American Chemical Society. 73, 429-435 (1951).
- Tate, S. S., Grzybowski, A. K., Datta, S. P. The stability constants of the magnesium complexes of the keto and enol isomers of oxaloacetic acid at 25. Journal of Chemical Society. , 1381-1389 (1964).
- Tate, S. S., Grzybowski, A. K., Datta, S. P. The acid dissociations of the keto and enol isomers of oxaloacetic acid at 25. Journal of Chemical Society. 1372, 1380 (1964).
- Brecker, L., et al. Synthesis of 2,4-diketoacids and their aqueous solution structures. New Journal of Chemistry. 23, 437-446 (1999).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados