
Method Article
Expression, Reinigung, Kristallisation und Enzym-Assays von Fumarylacetoacetat-Hydrolase-Domänen-haltigen Proteinen
In diesem Artikel
Zusammenfassung
Die Expression und Reinigung von Fumarylacetoacetat-Hydrolase-Domänen-haltigen Proteinen wird anhand von Beispielen beschrieben (Expression in E. coli, FPLC). Gereinigte Proteine werden zur Kristallisation und Antikörperproduktion verwendet und für Enzym-Assays eingesetzt. Ausgewählte fotometrische Assays werden vorgestellt, um die Multifunktionalität von FAHD1 als Oxaloacetat-Decarboxylase und Acylpyruvathydrolase darzustellen.
Zusammenfassung
Fumarylacetoacetathydrolase (FAH) domänenhaltige Proteine (FAHD) sind identifizierte Mitglieder der FAH-Überfamilie in Eukaryoten. Enzyme dieser Superfamilie zeigen in der Regel Multifunktionalität, die hauptsächlich Hydrolase- und Decarboxylase-Mechanismen beinhalten. Dieser Artikel stellt eine Reihe von aufeinanderfolgenden Methoden zur Expression und Reinigung von FAHD-Proteinen dar, hauptsächlich FAHD-Protein 1 (FAHD1) Orthologen unter Arten (Mensch, Maus, Nematoden, Pflanzen, etc.). Abgedeckte Methoden sind Proteinexpression in E. coli, Affinitätschromatographie, Ionenaustauschchromatographie, präparative und analytische Gelfiltration, Kristallisation, Röntgenbeugung und photometrische Assays. Konzentriertes Protein mit hohem Reinheitsgrad (>98%) kann für die Kristallisation oder Antikörperproduktion verwendet werden. Proteine ähnlicher oder niedrigerer Qualität können in Enzymtests oder als Antigene in Detektionssystemen (Western-Blot, ELISA) verwendet werden. In der Diskussion dieser Arbeit werden die identifizierten enzymatischen Mechanismen von FAHD1 skizziert, um seine Hydrolase und Decarboxylase-Bifunktionalität genauer zu beschreiben.
Einleitung
Die Fumarylacetoacetathydrolase (FAH)1,2 Überfamilie von Enzymen beschreibt eine Gruppe von Enzymen, die die hochkonservierte katalytische FAH-Domäne3,4,5,6 teilen , 7 , 8 , 9 , 10. Trotz ihrer gemeinsamen katalytischen Zentrum, Diese Enzyme sind multifunktional, und die meisten sind in Prokaryotengefunden, wo sie verwendet werden, um Verbindungen aus komplexen Kohlenstoffquellen abgerufen 3 . Nur drei Mitglieder dieser Familie wurden bisher in Eukaryoten identifiziert: der Name, der FAH2gibt, sowie DAS FAH-Domänenhaltige Protein 1 (FAHD1)11,12,13,14 ,15 und FAH-Domänen-haltiges Protein 2 (FAHD2). Die Erschöpfung von FAHD1 wurde mit einer beeinträchtigten mitochondrialen Atmung13,16 und mit einer reversiblen Art von zellulärem Seneszenzpnotyp14 in Verbindung gebracht, der mit dem Zwischenpotential Mängel im Elektronentransportsystem. Human FAHD1 und seine Orthologe in Modellsystemen (Maus, Nematode, Krebszelllinien, Pflanzen, etc.) sowie ausgewählte Punktmutationsvarianten sind zu medikamentenfähigen Zielen von potenziellem Interesse geworden. Für diese Forschung sind rekombinantes Protein bei hoher Reinheit sowie Informationen über katalytische Mechanismen, die von Kristallstrukturen und selektiven Antikörpern geleitet werden, von entscheidender Bedeutung.
Dieses Manuskript beschreibt Methoden zur FAHD-Proteinexpression in E. coli, Affinitätschromatographie, Ionenaustauschchromatographie, Ammoniumsulfatfällung, präparative und analytische Gelfiltration, Kristallisation, Röntgenbeugung und photometrische Assays. Der Zweck der hier beschriebenen Methoden und Protokolle besteht darin, Wissenschaftlern, die in verschiedenen Bereichen wie Bakteriologie, Pflanzenbiologie sowie Tier- und Humanstudien arbeiten, Orientierungshilfen zu geben, um Mitglieder der FAH-Überfamilie zu charakterisieren, uncharakterisierte Überfamilienmitglieder, wenn sie in einem bestimmten Bereich relevant werden. Die hier beschriebenen Protokolle können wertvolle Unterstützung für Projekte bieten, die darauf abzielen, andere prokaryotische oder eukaryotische FA-Überfamilienmitgliedn zu charakterisieren.
Der Grund gedanke hinter den hier beschriebenen Methoden ist die Tatsache, dass für die Charakterisierung schlecht beschriebener Proteine (insbesondere metabolische Enzyme von unbekannter physiologischer Relevanz) der Ansatz, mit gereinigten rekombinanten Proteinen zu beginnen, die Entwicklung von unschätzbaren, qualitativ hochwertigen Forschungsinstrumenten wie in vitro aktiven Enzympräparaten, hochwertigen Antikörpern und potenten und spezifischen pharmakologischen Inhibitoren für ausgewählte Enzyme. Die beschriebenen Methoden erfordern eine schnelle Proteinflüssigkeitschromatographie (FPLC) und Röntgenkristallographie. Alternative Methoden (z.B. zur Expression von Protein ohne chemische Induktion oder zur Anzeige der Proteinreinigung durch Zentrifugation nach Wärmebehandlung, gefolgt von Entsalzung und Größenausschlusschromatographie), können an anderer Stelle gefunden werden17. Während ein breiteres Spektrum von Methoden für die Expression und Reinigung von FAH-Superfamilienenzymen2,7,9,17,18zur Verfügung steht, konzentriert sich diese Arbeit auf die Expression und insbesondere die Reinigung von FAHD-Proteinen.
Im Diskussionsteil dieses Manuskripts werden die für das FAHD1-Protein (Hydrolase, Decarboxylase)15 identifizierten katalytischen Mechanismen ausführlicher beschrieben, um den chemischen Charakter der katalysierten Reaktionen zu demonstrieren. Die auf früheren Arbeiten7,15,18 (PDB: 6FOG, PDB:6FOH) gewonnenen Daten implizieren eine dritte Aktivität des Enzyms als Keto-Enol-Isomerase.
Protokoll
1. Expression von FAHD-Proteinen in kompetentem E. coli
- Transformation von E. Coli mit Vektoren zur Expression von FAHD-Protein
ANMERKUNG: Die im folgenden Abschnitt beschriebenen Schritte sind in der Skizze in Abbildung 1A,Bzusammengefasst. Das gleiche Protokoll gilt für jedes FAHD-Protein, einschließlich punktmutierter Varianten. Solche Varianten können über standortgesteuerte Mutagenese- und PCR-Techniken19 (z. B. beidseitige SOE PCR20) aus Wildtyp cDNA gewonnen werden.
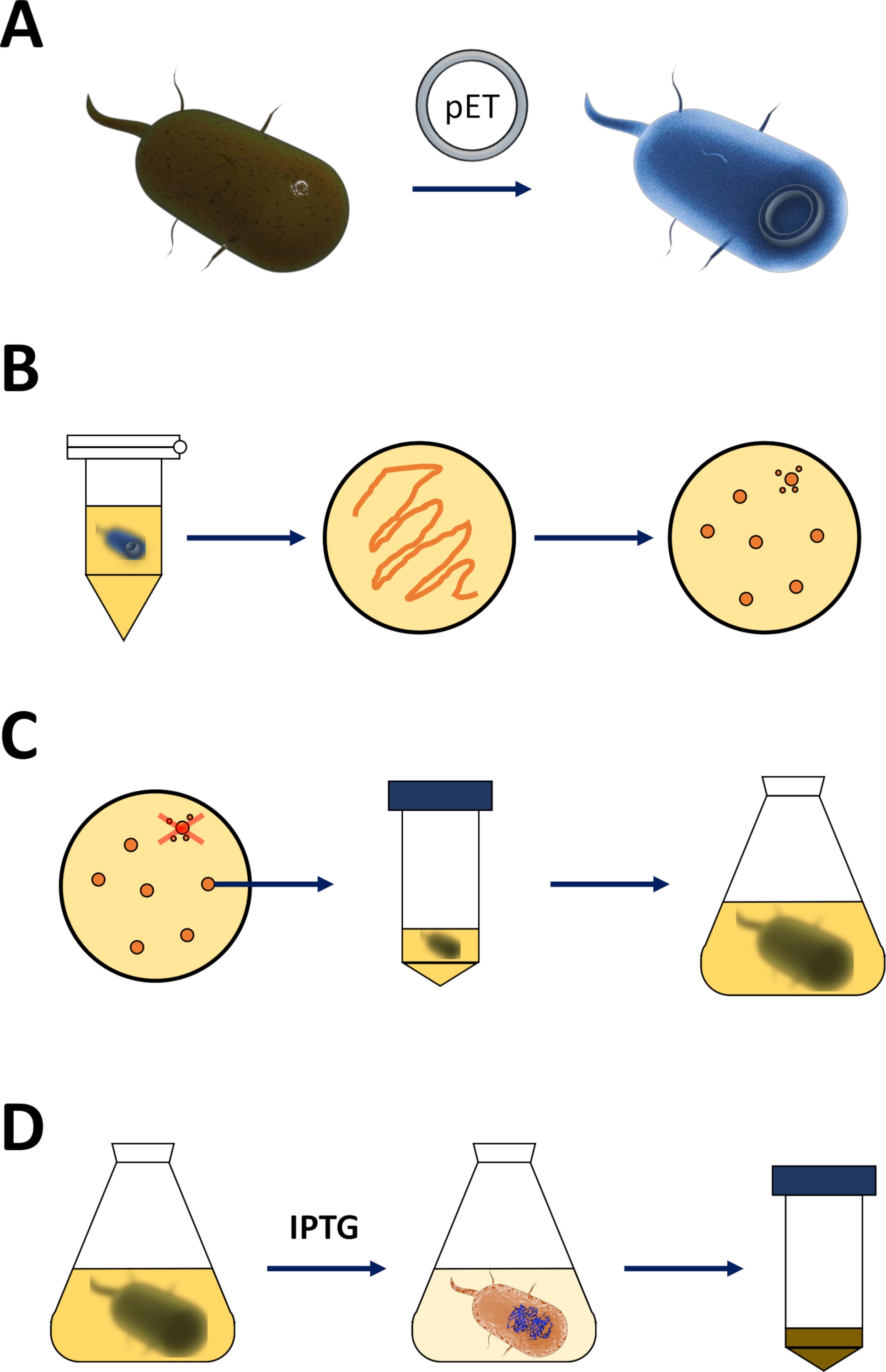
Abbildung 1 : Verstärkung der kompetenten E. coli und Induktion der Proteinexpression.
(A) Einfügung des pET-Vektors in kompetente BL21(DE3) pLysS E. coli-Bakterien, beschrieben in Abschnitt 1. (B) Hitzeschockprotokoll und Beschichtung der pET-transformierten E. coli-Bakterien, beschrieben in Schritt 1 des Protokolls. Transformierte Bakterien werden auf LB-Agarplatten mit Antibiotika zur Selektion plattiert. (C) Amplifikation von pET-transformierten E. coli-Bakterien, beschrieben in Abschnitt 1. Kolonien werden aus einer LB-Agarplatte entnommen und im nährenden Medium (LB oder NZCYM) verstärkt, bis die Bakteriendichte die empirische Schwelle von 0,4 erreicht hat. (D) Induktion der Proteinexpression über das DE3-IPTG- pET-System, beschrieben in Abschnitt 1 und skizziert in Abbildung 2. Die Proteinproduktion wird durch die Anwendung der Chemikalie IPTG gestartet. Am Ende von Abschnitt 1 wird das bakterielle Pellet, das das Protein enthält, geerntet. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.- Erhalten Sie kompetente BL21(DE3) pLysS E. coli-Bakterien und einen pET-Expressionsvektor (siehe Materialtabelle). Wählen Sie vorzugsweise einen pET-Vektor, der auch ein N-terminal His-Tag oder ein zugehöriges Capture-Tag kodiert, um die folgenden Reinigungsschritte zu vereinfachen.
- Beziehen Sie cDNA des FAHD-Proteins Ihrer Wahl und fügen Sie es in die aktive Klonstelle des pET-Expressionsvektors zwischen den T7-Promotor- bzw. T7-Terminator-Sites ein.
- Nach erfolgreicher Plasmidverstärkung und -verifizierung [durch Sequenzierung durch einen kommerziellen Lieferanten (T7-Primer können mit dem pET-System zur Bequemlichkeit verwendet werden: T7-Promoter, Vorwärtsprimer: TAATACGACTCACTATAGGG; T7-Terminator, Reverse primer: GCTAGTTATTGCTCAGCGG)], 5–10 ng Plasmid in 100 l kompetente BL21(DE3) pLysS E. coli-Bakterien auf Eis einsetzen. Nicht nach oben und unten ansaugen, sondern leicht auf die Röhre tippen, um den Inhalt zu mischen.
- Halten Sie die Bakterien 30 min auf Eis und tippen Sie alle paar Minuten sanft auf die Röhre.
- Erhitzen Sie ein Heizgerät oder Wasserbad auf 42 °C (genau). Legen Sie das Rohr, das die Bakterien enthält, in das Gerät und halten Sie sie 90 s (genau). Legen Sie sie sofort auf Eis (Abbildung 1A).
- Nach 5–10 min auf Eis 600 l NCZYM-Medium hinzufügen (siehe Materialtabelle) und die Röhre in einen Bakterien-Inkubator geben. Schütteln Sie das Rohr bei mittlerer Geschwindigkeit entlang der Schüttelrichtung bei 37 °C für 1 h.
- Platte 200 l der Bakterienkultur auf einer 10 cm LB-Agar-Platte (siehe Materialtabelle),die Selektionsantibiotika der Wahl enthält [z. B. eine spezifische für die BL21(DE3) PLysS-Resistenz (Chloramphenicol) und eine für die Resistenz auf dem pET-Vektor (Kanamycin oder Ampicillin, Abbildung 1B)] kodiert.
- Die Bakterien auf der LB-Agar-Platte in einem Bakterien-Inkubator über Nacht bei 37 °C ansleben.
- Expression von FAHD-Proteinen durch IPTG-Induktion
HINWEIS: Die ersten Schritte im folgenden Abschnitt werden als Skizze in Abbildung 1C,Dzusammengefasst. Das T7-Expressionssystem über die Kombination der bakteriellen DE3-Kassette und des pET-Vektorsystems sind in Abbildung 2zusammengefasst.
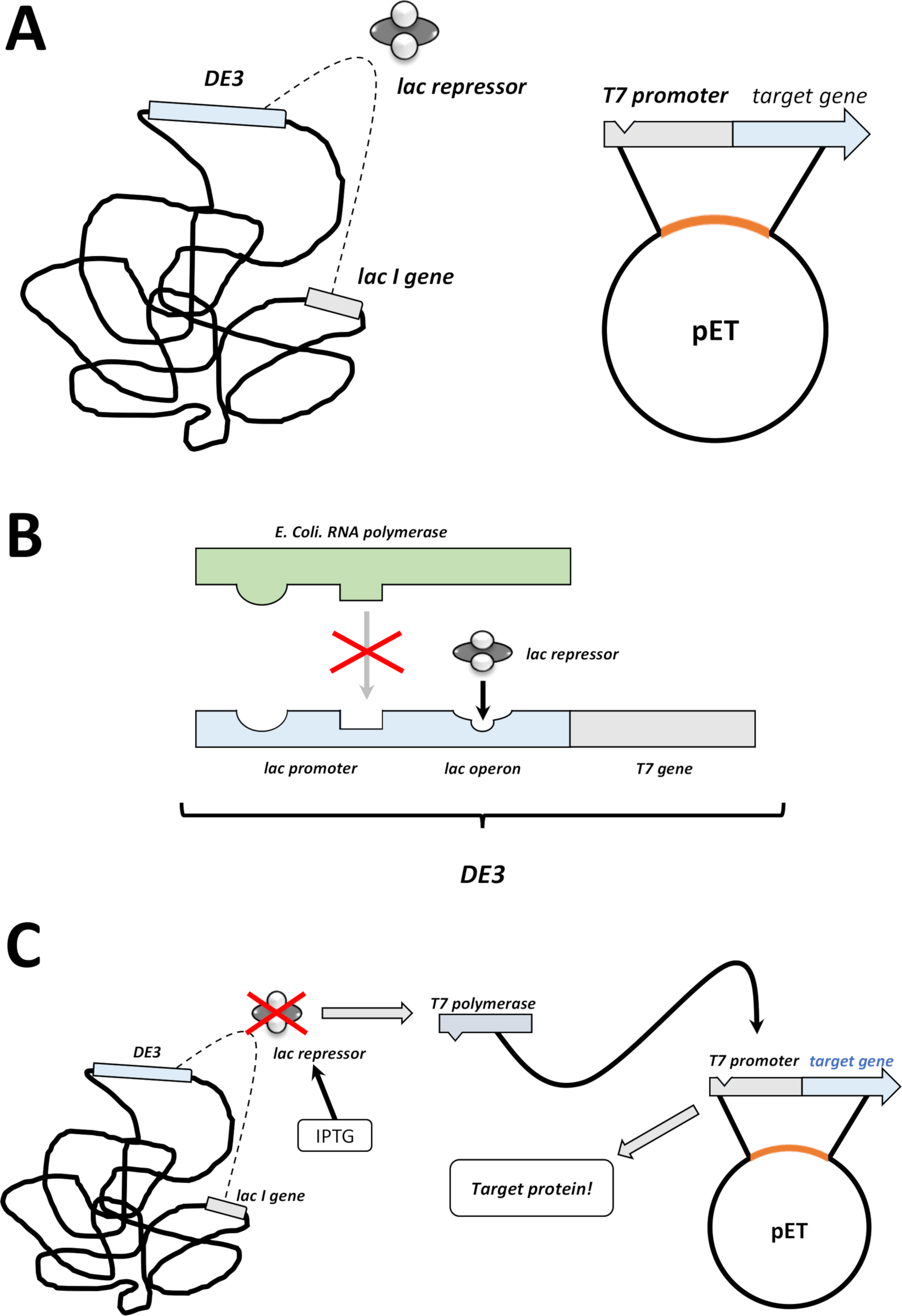
Abbildung 2 : Die DE3-Kassette/pET-Vektor-Dual-System erklärt.
(A) Das skizzierte Genom von pET-Vektor transformiert BL21(DE3) pLysS E. coli Bakterien. Das native bakterielle Genom trägt eine DE3-Kassette (siehe Panel B) sowie ein Lac-Gen, das ständig Lac-Repressor-Einheiten ausdrückt. Der nicht native pET-Vektor trägt das Proteingen, das zwischen einem T7-Polymerase-Promotor und einer Terminatorsequenz eingefügt wird. Weitere Details in Panel B. (B) Die DE3-Kassette des nativen bakteriellen Genoms kodiert die Informationen für T7-Polymerase in Bezug auf einen E. coli-RNA-Polymerase-Operon. Dieses Protein wird jedoch nicht exprimiert, da die Lac-Repressor-Einheit verhindert, dass das RNA-Polymerase-Protein bindet. Daher wird keine T7-Polymerase und kein exogenes Protein exprimiert. (C) Die Anwendung der Chemikalie IPTG (Materialtabelle) verzerrt die Struktur von Lac-Repressor-Einheiten und verhindert, dass sie an die DE3-Kassette gebunden werden. Dadurch kann die RNA-Polymerase nun an die Kassette binden, für die T7-Polymerase exprimiert wird, ebenso wie schließlich exogenes Protein. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.- Nach erfolgreicher Koloniebildung eine einzelne Kolonie (ohne Satellitenkolonien) pflücken und in 5 ml NZCYM oder LB-Medium mit Antibiotika dispergieren, die wie zuvor ausgewählt wurden (Schritt 1.1.7). Kultur im Bakterien-Inkubator bei 37 °C über Nacht (Abbildung 1C).
- Nach erfolgreichem Bakterienwachstum verstärken Sie die Bakterien in 250 ml, 500 ml oder 1 L Chargen Medium, abhängig von der Nachfrage nach Proteinmenge.
- Entsprechend dem Volumen Antibiotika anwenden, die wie in Schritt 1.1.7 ausgewählt wurden, und fügen Sie etwa 1%–2% der dichten bakteriellen Vorkultur hinzu (d. h. 2,5–5,0 ml bis 250 ml Volumen des Mediums usw.). Nehmen Sie eine Probe, die in Schritt 1.2.5 (1 ml oder mehr) verwendet werden soll, und überprüfen Sie die optische Dichte (OD) bei 600 nm. Kulturbakterien im Bakterien-Inkubator bei 37 °C für 2–3 h (Abbildung1C).
- Zeichnen Sie nach 2:3 h eine Probe für die photometrische Analyse. Wenn die OD bei 600 nm 0,4 erreicht hat, wenden Sie 200 m bis 1 mM Isopropyl-D-Thiogalactopyranosid (IPTG, siehe Tabelle der Materialien) an.
HINWEIS: Der tatsächliche Wert ist empirisch für jede FAHD-Protein- oder Punktmutationsvariante, wobei 1 mM IPTG das Maximum ist, das angewendet werden sollte. Dies induziert eine Proteinexpression (Abbildung 1D, Abbildung 2C). - Nach 3–5 weiteren Stunden im Bakterien-Inkubator bei 37 °C ist die Proteinexpression erschöpft.
HINWEIS: Im Diskussionsabschnitt finden Sie Kommentare zur Temperaturregelung. Länger als 5 h Schütteln nach Induktion wird nicht empfohlen. Nehmen Sie eine Probe für die Verwendung in Schritt 1.2.5 (1 ml oder mehr) und überprüfen Sie die optische Dichte (OD) bei 600 nm.- Das bakterielle Pellet über Zentrifugation bei 5.000 x g 5 min ernten. Den Überstand entsorgen und bei -80 °C für längere Lagerung oder -20 °C für kurze Lagerung einfrieren (Abbildung 1D).
- Überprüfen Sie die Induktion über die beiden abgerufenen photometrischen Proben, die mit "-I" (vor der Induktion) und "+I" (nach Induktion) beschriftet sind. Nach Zentrifugation und Resuspension des bakteriellen Pellets analysieren Sie die beiden Proben per SDS-PAGE, indem Sie die gleiche Menge an Gesamtprotein laden.
HINWEIS: Die "+I"-Probe sollte ein starkes Band aufweisen, das mit dem Molekulargewicht des gewählten Proteins verbunden ist, während die "-I"-Probe dieses Band nicht enthalten sollte. Ein niedriger Induktionsgrad ist ein häufiges Problem für die Produktion von Proteinen, aber der Gehalt an exprimiertem Protein ist oft ausreichend für die folgenden Schritte. Ein hohes Induktionsniveau ist ein Vorteil, ist aber nicht obligatorisch.
{kind=link}
{kind=link}
2. Lyse von bakteriellen Pellets und Filtration von Schmutz
- Je nach, ob das gewählte Protein Sein-tagged oder untaggt ist, wählen Sie Ni-NTA-Laufpuffer (Sein-tagged, siehe Tabelle der Materialien) oder eiskalten HIC-Laufpuffer (nicht markiert).
- Für jede 250 ml originalbakterielle Suspension 5 ml des ausgewählten Puffers auf das bakterielle Pellet auftragen (5 ml für 250 ml, 10 ml für 500 ml usw.). Fügen Sie 10 l -L-Mercaptoethanol (-ME) pro 5 ml des aufgebrachten Puffers hinzu. Verwenden Sie eine 10 ml Pasteur Pipette, um das Pellet durch Kratzen und Pipettieren mechanisch in die Suspension zu zwingen (Luftblasenbildung beim Pipetieren vermeiden). Übertragen Sie schließlich die gesamte Aufhängung in ein 50 ml Rohr.
- Vorzugsweise beschallen (6x für 15 s bei mittlerer Kraft) die Suspension.
- Zentrifuge für 30 min bei hoher Geschwindigkeit (10.000 x g) bei 4 °C. Filtern Sie den Überstand nacheinander mit Filtereinheiten (z.B. 0,45 m, 0,22 m) auf Eis.
HINWEIS: Abhängig vom vorherigen Zentrifugationsschritt kann die Filtration direkt durch eine kleine Filterporengröße mühsam sein und erfordert in der Regel eine Vorfiltration durch eine größere Porengröße. DNAse können hinzugefügt werden, um bessere Ergebnisse zu erzielen. - Bewahren Sie die Probe auf Eis auf und fahren Sie sofort mit Abschnitt 3 oder 4 fort, je nachdem, ob das Protein mit seinem-tagagged oder ohne Tags versehen ist.
3. Reinigung seiner-markierten FAHD-Proteine mit Ni-NTA-Affinitätschromatographie
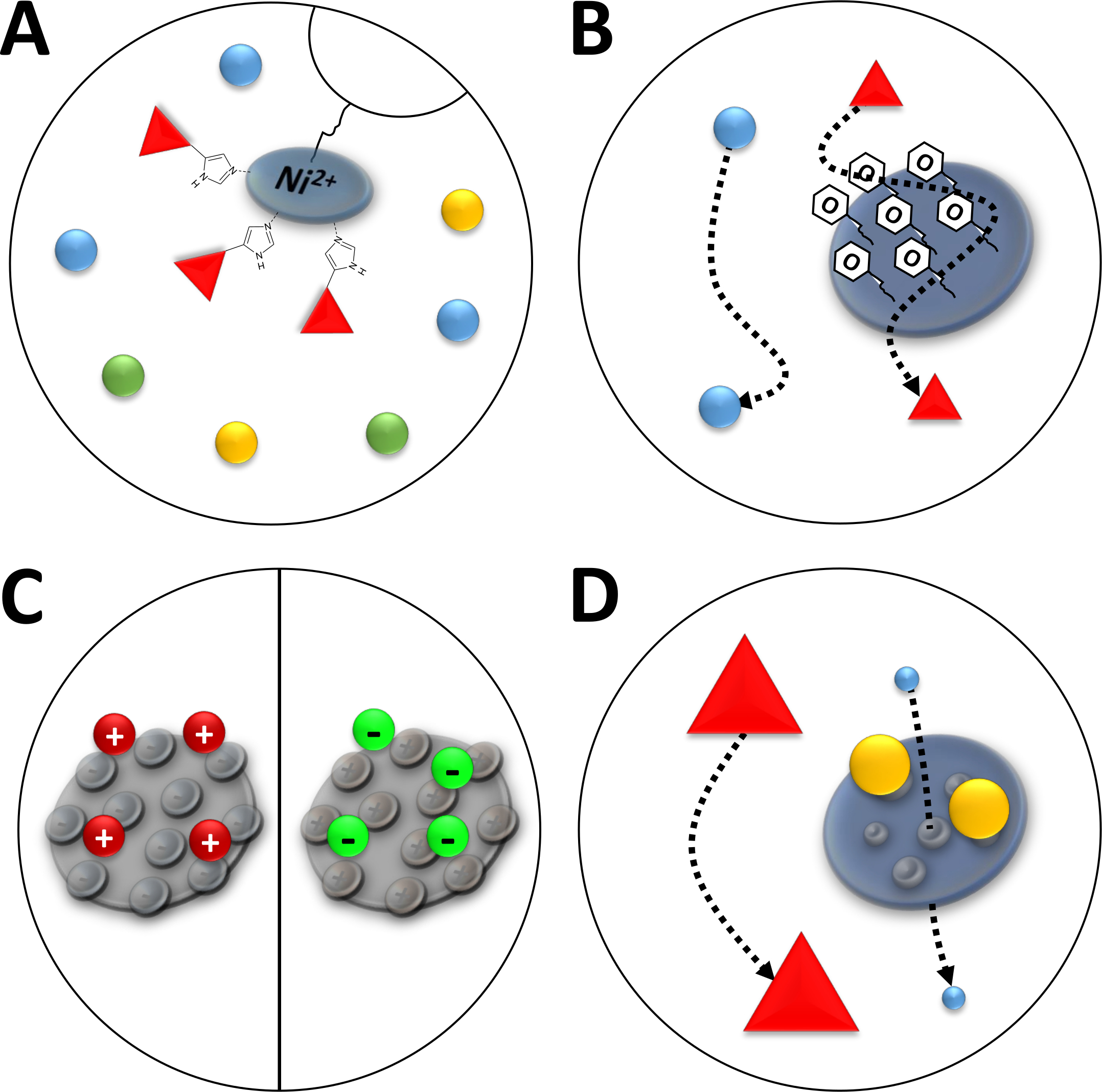
HINWEIS: Ni 2+-Ionen werden über Nittrilotriaticsäure (NTA) an ein Agaroseharz gebunden, das in der Affinitätschromatographie verwendet wird (immobilisierte Metallionenchromatographie, IMAC, Abbildung 3A). Polyhistidin-Aminosäure-Tags binden stark an dieses Ni-Chelat, und Seine-getaggten Proteine können von der Mehrheit der verbleibenden Proteine getrennt werden. Eine Alternative zur beschriebenen Herstellung von Ni-NTA-Säulen ist die Verwendung vorverpackter Ni-NTA-Säulen und eines FPLC-Systems.

Abbildung 3 : Skizzierte Abbildungen gängiger Chromatographie.
(A) Das Harz einer Ni-NTA-Säule. NTA enthält bivalente Nickelionen, die in Bezug auf die immobilisierte Metallionenaffinitätschromatographie (IMAC) verwendet werden. Polyhistidin-Tags binden vorzugsweise an dieses Motiv und können durch Imidazol eluiert werden. (B) Die typische Beschichtung von Kieselsäurepartikeln in einer phenylbasierten hydrophoben Wechselwirkungschromatographie (HIC-Phenyl). Hydrophobe Proteine interagieren mit dem Beschichtungsmaterial und verzögern sich bei ihrer Migration, andere nicht. (C) Die typische Beschichtung von Kieselsäurepartikeln in der ionenkotonischen Chromatographie. Polarisierte und geladene Proteine interagieren mit dem Beschichtungsmaterial und verzögern sich bei ihrer Migration, andere nicht. (D) Das Harz eines Kieselgels in der Größenausschlusschromatographie (SEC). Basierend auf definierten Poren im Kieselsäurematerial können Proteine durch ihre Größe getrennt werden (in einer ersten Annäherung, die ihrer Molekularmasse entspricht). Kleine Proteine durchdringen das poröse Säulenmaterial und werden verzögert, während große Proteine schneller um die porösen Partikel wandern. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Fahren Sie mit Schritt 2.5 fort (d. h. das Protein befindet sich im Ni-NTA-Laufpuffer und wird von 0,22 m Filtereinheiten auf Eis gefiltert).
- Bereiten Sie eine leere Kunststoff- oder Glassäule vor, indem Sie die leere Säule waschen und an einem stabilen Halter befestigen. Wählen Sie die Größe der Säule in Abhängigkeit vom Volumen der Proteinsuspension.
- Für jede 10 ml Proteinsuspension 500 l Ni-NTA-Agarose-Schlämme in die Säule auftragen (vor gebrauchenschwer schütteln). Tragen Sie die Gülle langsam auf und lassen Sie sie mit einer Pipette auf den unteren Filter der Säule fallen. Lassen Sie die Spalte absetzen, was einige Sekunden dauert.
- Füllen Sie die Säule vollständig mit Ni-NTA Laufpuffer, um sicherzustellen, dass das Agaroseharz nicht gestört wird. Lassen Sie den Puffer durch die Schwerkraft laufen. Der Prozess kann beschleunigt werden, indem Daumendruck auf die Flüssigkeit ausgeübt wird (mit einem Deckel oder Handschuh und Daumendruck), aber achten Sie darauf, das Agaroseharz nicht zu verzerren.
- Tragen Sie die Proteinsuspension auf. Lassen Sie die Probe wie bisher durch die Schwerkraft laufen. Es wird nicht empfohlen, diesen Schritt mit Dementol druck zu beschleunigen, da die Bindung von Proteinen an die Säule verbessert wird, wenn die Durchflussrate niedrig ist. Sammeln Sie den Durchfluss in einem Rohr (Tabelle der Materialien).
- Nachdem das Beispiel durchlaufen wurde, füllen Sie die gesamte Spalte erneut mit dem Ni-NTA-Laufpuffer. Achten Sie darauf, das Agaroseharz nicht zu stören. Lassen Sie die Probe durch die Schwerkraft laufen, aber im Gegensatz zum vorherigen Schritt wird empfohlen, den Prozess über den Daumendruck zu beschleunigen, da potenzielle Verunreinigungen aufgrund unspezifischer Wechselwirkungen auf diese Weise gestört werden können. Sammeln Sie die Waschlösung in einem Rohr. Wiederholen Sie diesen Schritt.
- Legen Sie eine UV-transparente Küvette unter die Säule und wenden Sie 1 ml Ni-NTA-Elutionspuffer auf. Sammeln Sie die Probe, ohne einen Daumendruck auf das Harz auszuüben.
- Überprüfen Sie die optische Dichte (OD) der Probe bei 280 nm gegenüber einer leeren Probe (d. h. Ni-NTA-Elutionspuffer). Optimalerweise zeigt das Beispiel einen OD von mehr als 2,5 an. Ein OD unter 0,5 bedeutet, dass sich keine signifikante Menge Protein in der Probe befindet.
HINWEIS: Wie im Diskussionsbereich beschrieben, müssen die Salz- und Imidazolkonzentrationen des Elutionspuffers möglicherweise individuell für jedes FAHD-Protein angepasst werden. - Wiederholen Sie die Schritte 3.1.7 und 3.1.8, bis der OD unter 0,5 fällt. Alle Proben mit höherem OD in einer Röhre auf Eis bündeln.
- Beginnen Sie erneut mit Schritt 3.1.4, wobei Sie den Durchfluss von Schritt 3.1.5 als neuen Eingang für diese Wiederholung von Schritt 3.1.5 verwenden. Wiederholen Sie diesen Vorgang, bis in der ersten in Schritt 3.1.6 gesammelten Probe ein OD unter 0,5 angezeigt wird.
HINWEIS: Wie im Abschnitt zur Fehlerbehebung beschrieben, können seine mit Tagged-Proteinen nicht ausreichend an das Ni2+-Harzbinden. In solchen Fällen ist eine Wiederholung dieses Schritts oder alternativer Methoden (z. B. Ionenaustauschchromatographie) erforderlich. - Nehmen Sie Proben aller Zwischenfraktionen für die SDS-PAGE-Analyse.
- FAHD-Proteine im Ni-NTA-Elutionspuffer fallen beim Einfrieren und Auftauen aus. Dialyse das Protein gegen einen anderen Puffer (über Nacht auf Eis, mit 1 l DTT pro 100 ml Dialysepuffer). Verwenden Sie einen Salzpuffer, je nachdem, welche Art von Ionenaustauschchromatographie nach diesem Schritt durchgeführt werden soll. Verwenden Sie gemeinsame Zelluloseschläuche mit einem typischen Molekulargewichtsabgrenzung von 14 kDa (Materialtabelle).
- Nach der nächtlichen Dialyse konzentrieren Sie das Protein optional mit Ultrazentrifugationsfiltereinheiten. Führen Sie eine SDS-PAGE-Analyse (12,5 % Laufgel, 4 % Stapelgel) durch, um einen potenziellen Proteinverlust, unzureichende Elution und Proteinreinheit im Allgemeinen zu überprüfen. Wenn alles in Ordnung ist, fahren Sie mit Abschnitt 5 fort.
4. Reinigung von nicht markierten FAHD-Proteinen durch hydrophobe Wechselwirkungschromatographie (HIC)
HINWEIS: Phenylgruppen auf der Beschichtungsoberfläche eines Kieselgels in einer HIC-Säule für FPLC (Abbildung 3B) ermöglichen die Trennung von Proteinen nach hydrophoben Charakter. Die beschriebenen Schritte sollten mit einem FPLC-System durchgeführt werden, das mit einer 5 ml HIC-Phenylsäule ausgestattet ist. Säulen können mit 1 M NaOH gewaschen werden, um für verschiedene Proteine wiederverwendet zu werden. Allerdings sollten Spalten, die einst für eine Art von FAHD-Protein verwendet wurden, nur für diese Art von Protein wiederverwendet werden.
-
Ammoniumsulfat (AS) Niederschlag
- Fahren Sie mit Schritt 2.5 fort. Das Protein befindet sich im eiskalten HIC-Laufpuffer (Tabelle der Materialien).
- Bewerten Sie das Volumen der vorbereiteten Proteinlösung genau auf den Mikroliter (Vinitial). Langsam und tropfenweise vorgekühlte HIC-Laufpuffer-AS-Lösung hinzufügen, bis eine 35 Volume-% AS-Sättigung erreicht ist: VAS added = Vinitial * 0.538. Die Lösung für 30 min. Zentrifuge 15 min bei hoher Geschwindigkeit (ca. 10.000 x g) bei 4 °C vorsichtig rühren.
- Filtern Sie den Überstand mit einer 0,22 m Filtereinheit auf Eis. Optional nehmen Sie eine Probe für die SDS-PAGE-Analyse: 1:4 verdünnen und sofort bei 95 °C für 5 min erhitzen, sonst wird die Probe verklumpt. Die Probe kann an dieser Stelle (-20 °C) eingefroren werden, um an einem anderen Tag fortzufahren.
-
FPLC mit einer HIC-Spalte
- Richten Sie das FPLC-System ein und gleichnden Sie eine 5ml HIC-Phenyl-Säule mit 5 Spaltenvolumen (CV) von 20% EtOH (in H2O) gefolgt von 5 CV von H2O.
- Mischen Sie 260 ml HIC-Laufpuffer (genau) mit 140 ml HIC-Laufpuffer AS (genau). Dies führt zu einer 35-Volumen-% AS-Lösung. Überprüfen Sie den pH-Wert (7,0); Dies ist Puffer A. Puffer B ist 250 ml laufender Puffer. Fügen Sie 1 mM DTT zu den beiden Puffern A und B hinzu, und halten Sie sie dann auf Eis.
- Equilibrate die Spalte mit 8 ml Puffer A, 8 ml Puffer B und 8 ml Puffer A in dieser Reihenfolge. Wenden Sie die in Protokollschritt 4.1 vorbereitete Probe an. Mit Puffer A waschen, bis die optische Grundabsorption bei 280 nm 1000–500 mAU erreicht.
- Wenden Sie eine Mischung der Puffer A und B an, so dass die Konzentration von AS 33 % (w/v) beträgt. Waschen Sie mit 1 CV, was zu einem Plateau im Chromatogramm führt. Einrichten eines Gradienten von Puffer B (bis zu 100 % Puffer B im Zeitverlauf): 1,5 ml Puffer B in 3,8 min (d. h. 5,7 % Puffer B mit 1% B/ml Steigung). Wenn das UV-Signal bei 280 nm steigt, beginnen Sie, den Bruch zu sammeln und legen Sie ihn sofort auf Eis.
- Waschen Sie die Säule am Ende mit Puffer B. Nehmen Sie Proben aller Fraktionen für die SDS-PAGE-Analyse. Alle Proben mit flüssigem Stickstoff einfrieren und bei -80 °C lagern.
- Führen Sie eine SDS-PAGE-Analyse (und einen westlichen Blot) durch, um das FAHD-Protein in den gesammelten Fraktionen zu erkennen. Brüche, die das Protein enthalten, werden gepoolt und auf die weitere Reinigung angewendet, wie in den folgenden Protokollschritten beschrieben. Waschen Sie die Säule mit H2O und 20% EtOH (in H2O).
5. Reinigung von FAHD-Proteinen durch Ionenaustauschchromatographie
HINWEIS: Moleküle mit geladenen funktionellen Gruppen sind an eine Kieselsäure-Partikelsäule für FPLC gebunden (Abbildung 3C). Dies ermöglicht die Differenzierung von Proteinen nach ihrem ionischen Charakter, wie z. B. Oberflächenladung. Die beschriebenen Schritte sollten mit einer FPLC-Maschine bzw. dem dazugehörigen Know-how durchgeführt werden. Die beschriebene Methode ist die gleiche für die kationische oder anionische Austauschchromatographie, aber die zu verwendenden Puffer sind etwas unterschiedlich.
- Wählen Sie das kationische oder anionische Austauschchromatographiesystem. Diese Wahl ist empirisch und kann zwischen FAHD-Proteinen variieren. Optimalerweise können beide Methoden nacheinander eingesetzt werden.
- Richten Sie das FPLC-System ein und waschen Sie die Säule mit 5 CV von 20% EtOH (in H2O), gefolgt von 5 CV von H2O. Equilibrate die Säule mit 1 CV Salzpuffer, Hochsalzpuffer und wieder Salzpuffer in dieser Reihenfolge.
- Tragen Sie die Probe (dialyziert gegen den richtigen niedrigen Salzpuffer ab Schritt 3.1.11) auf die Spalte auf. Sammeln Sie den Durchfluss. Die Säule für 1 CV mit niedrigem Salzpuffer waschen.
- Einrichten einer Gradientenelution: 100% Hochsalzpuffer in 30 min bei einer Durchflussrate von 1 ml/min oder 60 min bei einer Durchflussrate von 0,5 mL/min. Dies kann auf DerGrundlage eines bereits bekannten FPLC-Chromatogramms erneut ausgewählt werden, um die Reinigung zu optimieren. Sammeln Sie alle Spitzenfraktionen.
HINWEIS: Die Hochsalzbedingungen können zwischen FAHD-Proteinen variieren, wie im Diskussionsabschnitt beschrieben. - Nachdem der Gradient beendet ist, führen Sie mit hohem Salzpuffer aus, bis keine weiteren Spitzen über den Bereich von 1 CV erkannt werden (sammeln Sie die Brüche).
- Nehmen Sie Proben aller gesammelten Fraktionen und führen Sie eine SDS-PAGE-Analyse durch (12,5 % Laufgel, 4 % Stapelgel). Die einzelnen Proben in flüssigem Stickstoff einfrieren und bei -80 °C lagern.
- Nachdem die SDS-PAGE-Analyse abgeschlossen ist, bündeln Sie die Proben, die das FAHD-Protein enthalten, und entsorgen Sie die anderen. Konzentrieren Sie das Protein optional mit Ultrazentrifugationsfiltereinheiten.
- 1 ml 25% SDS in 0,5 M NaOH (oder anderen Reinigungsmitteln) auftragen, um die Säule zu reinigen. Waschen Sie die Säule mit H2O und 20% EtOH (in H2O).
- Optional Abschnitt 5 mit der alternativen Säule (kationische oder anionische Austauschchromatographie) wiederholen. Das mit dieser Methode gewonnene Protein ist ausreichend rein, um grundlegende Aktivitätstests durchzuführen, oder kann in Screening-Assays für die Kristallographie verwendet werden. Für fortgeschrittene Anwendungen fahren Sie mit Abschnitt 6 fort.
6. Reinigung von FAHD-Proteinen durch Größenausschlusschromatographie (SEC)
HINWEIS: Poröse Partikel in einer Kieselgelsäule für FPLC ermöglichen die Differenzierung von Proteinen nach molekularer Größe, wie z. B. hydrodynamischen Radius (Abbildung 3D). Die beschriebenen Schritte sind mit einem FPLC-System unter Verwendung von SEC-Spalten durchzuführen.
- Wählen Sie eine SEC-Säule, abhängig von den Molekulargewichten der noch vorhandenen Kontaminationen, wie sie über SDS-PAGE und Silberfärbung nachgewiesen werden. Die skizzierte Methode ist für beide Spalten geeignet. Waschen Sie die Säule über Nachtmit 400 ml H2 O und gleichnden Sie mit dem SEC-Laufpuffer. Es wird empfohlen, ein Programm für das FPLC-System zu schreiben, um diesen Schritt zu automatisieren.
- Fügen Sie 1 mM DTT zu 300 ml SEC-Laufpuffer hinzu und legen Sie ihn auf Eis. Dies ist der laufende Puffer. Wenden Sie 60 ml dieses Puffers auf die Spalte an.
- Zentrifugieren Sie die Proteinprobe (10.000 x g für 10 min), um eventuelle Mikrofällungen zu entfernen. Wenden Sie den Überstand auf die Spalte an. Es wird allgemein empfohlen, den Überstand vor FPLC zu filtern.
- Wenden Sie den laufenden Puffer auf die Spalte an, bis das gesamte Protein eluiert ist. Sammeln Sie alle Spitzen in Bruchteilen des geeigneten Volumens (z. B. 2 ml). Nehmen Sie Proben für SDS-PAGE und frieren Sie alle Fraktionen mit flüssigem Stickstoff ein. Die gefrorenen Fraktionen bei -80 °C lagern.
- Sammeln und bündeln Sie nach der SDS-PAGE-Analyse (und Western Blot) alle Fraktionen, die das FAHD-Protein enthalten. Silberfärbung wird empfohlen, um kleinere Verunreinigungen zu erkennen, die noch vorhanden sein können.
- Verwenden Sie Ultrazentrifugationsfiltereinheiten, um das Protein zu konzentrieren. Obwohl für FAHD-Proteine nicht obligatorisch, wird im Allgemeinen ein Entsalzungsschritt (z.B. durch Dialyse) für Enzymassays und Kristallisation empfohlen.
- Wiederholen Sie die Schritte 6.3–6.6 mehrmals mit unterschiedlichen Durchflussraten und Salzkonzentrationen (empirisch), um die Reinheit des FAHD-Proteins zu erhöhen. Waschen Sie die Säule über Nacht mit H2O und 20% EtOH (in H2O).
7. Grundlegende FAHD-Aktivitätstests mit Substraten Oxaloacetat und Acetylpyruvat
HINWEIS: FAHD-Protein 1 (FAHD1) zeigt Oxaloacetat-Decarboxylase (ODx) und Acylpyruvathydrolase (ApH) Aktivität an. Dies wird im Diskussionsabschnitt ausführlicher beschrieben. Aufgrund der Destabilisierung durch Keto-Enol-Tautomerisierung in wässriger Lösung (d.h. Enolisierung) zerfällt Oxaloacetat im Laufe der Zeit (Auto-Decarboxylierung) in Abhängigkeit von Cofaktorkonzentration und pH-Wert von selbst.. Bei etwa einem pH-Wert von 7 und einer Temperatur von 25 °C ist dieser Effekt nicht dramatisch, aber Assays müssen ausgeblendet werden, um sowohl die Auto-Decarboxylation als auch die Enzymkonzentration zu berücksichtigen. Das Pipettierschema ist in Abbildung 4Abeschrieben. Im Allgemeinen wird empfohlen, gut kalibrierte Pipetten für diesen Test zu verwenden, da er sehr empfindlich auf kleinere Pipettierfehler reagiert.

Abbildung 4 : Skizziertes Pipettierschema für Enzym-Assays.
(A) Ein skizziertes Pipettierschema für grundlegende auf Substraten basierende FAHD-Proteinenzym-Assays. Substratrohling: -S/-E; Substratprobe: +S/-E; Enzym roh: -S/+E; Enzymprobe: +S/+E (S: Substrat, E: Enzym). Weitere Informationen finden Sie unter Protokollschritt 7. (B) Ein skizziertes Pipettierschema zur Beurteilung der Michaelis-Menten-Kinetik des FAHD-Proteins. Substratrohling: -S/-E; Substratprobe: +S/-E; Enzym roh: -S/+E; Enzymprobe: +S/+E (S: Substrat, E: Enzym). Weitere Informationen finden Sie in Abschnitt 8 des Protokolls. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Starten Sie einen Mikroplattenleser und equilibrate für 30 min bei 25 °C. Richten Sie ein Programm zum Lesen von 12 Brunnen (wie in Abbildung 4Abeschrieben) bei 255 nm ein. Es wird empfohlen, 25 Mehrfachauslesungen mit 5 ms Zeitverzögerung zu verwenden. Richten Sie einen Zyklus ein, der 15x alle 2 min misst (30 min insgesamt).
- Bereiten Sie standardmäßig einen Enzym-Assay-Puffer (siehe Materialtabelle)mit 1 mM MgCl2 bei pH 7,4 vor. Variante FAHD-Proteine können unterschiedliche Kofaktoren oder pH-Werte erfordern. Mg2+ und Mn2+ sind bekannte Kofaktoren für FAHD13,11,12,21.
- Erstellen Sie eine Proteinlösung von 1 g/l, die mit einem Enzym-Assay-Puffer verdünnt wird (Tabelle der Materialien).
- Richten Sie 1 ml 20 mM Lösung eines zu testenden Substrats ein (bisher identifizierte Substrate von FAHD-Proteinen sind an anderer Stelle aufgeführt3) im Enzym-Assay-Puffer.
- Nach dem in Abbildung 4Adargestellten Pipettierschema bereiten Sie das Enzym Leer- und Probenbrunnen vor: Pipetten 90 l Enzym-Assay-Puffer (Tabelle der Materialien) in die Brunnen mit 5 l (5 g) Enzymlösung.
- Entsprechend dem in Abbildung 4Adargestellten Pipettierschema bereiten Sie das Substratrohling vor und proben Brunnen: Pipetten 95 L Enzym-Assay-Puffer in die Brunnen.
- Unmittelbar vor der Messung 5 l Enzym-Assay-Puffer in die sechs leeren Brunnen auftragen. Tragen Sie 5 l der 20 mM Substratlösung auf die Probenbrunnen auf. Es wird empfohlen, eine Mehrkanalpipette zu verwenden.
- Verwenden Sie eine Mehrkanalpipette mit 50 L-Einstellungen, um alle Bohrungen sanft zu mischen. Beginnen Sie mit den Rohlingen und fahren Sie mit den Probenbrunnen fort. Achten Sie darauf, keine Blasen zu erstellen. Setzen Sie die Platte in einen Mikroplattenleser ein und messen Sie jeden Brunnen bei 255 nm (wie in Schritt 7.1 beschrieben).
- Führen Sie die Analyse in einer Kalkulationstabelle durch. Kopieren Sie die Rohdaten aus dem Photometer in eine Kalkulationstabelle, und schreiben Sie alle Einstellungen (d. h. die gesamte Dokumentation) in ein anderes Blatt. Durchschnitt der Daten der drei Brunnen jeder der vier Präparate. Subtrahieren Sie das Leerzeichen von der Probe. Berechnen Sie auch Standardabweichungen und summiert die Abweichungen von Leerling und Probe.
- Zeichnen Sie diese Daten (y: optische Dichte, x: Zeit in min). Es sollte eine exponentiell abnehmende Kurve angezeigt werden. Abhängig von der Art des substrats kann eine anfängliche Erhöhung innerhalb der ersten 10 min beobachtet werden, nach der das Signal abnimmt. Dies wird der Keto-Enol-Tautomerisierung des Substrats zugeschrieben, wie im Diskussionsteil ausführlicher beschrieben.
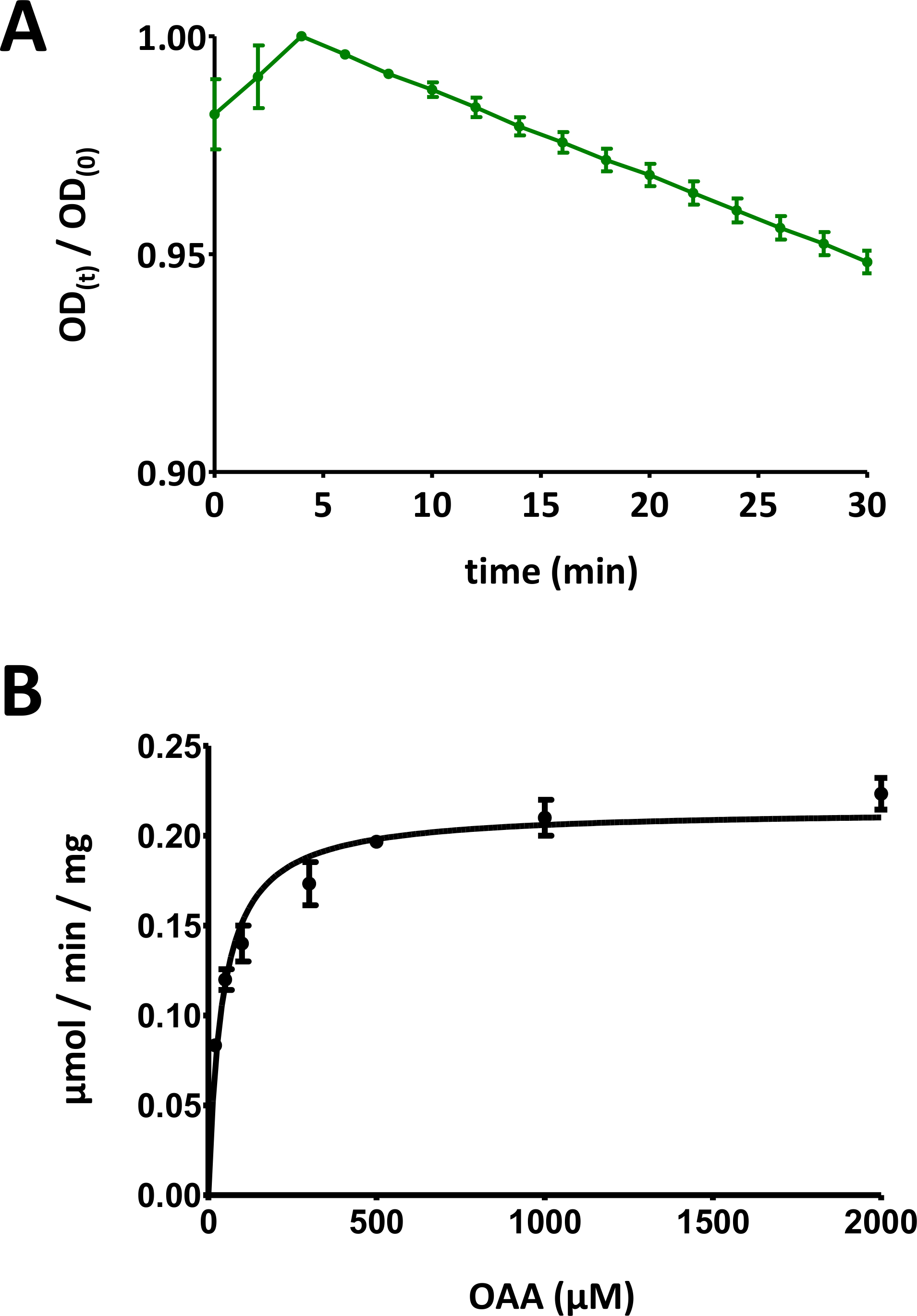
- Teilen Sie die optischen Signaldaten im Zeitverlauf durch den maximalen Wert des Diagramms, um die Daten in den Bereich [0, 1] zu skalieren (ein Beispiel ist in Abbildung 5Aangegeben). Identifizieren Sie den linearen Bereich der Kurve, beginnend mit der anfänglichen Abnahme, und berechnen Sie die negative Steigung (1/min).
- Der Zeitverlauf der Abnahme der OD wird dem Substrat durch seine Anfangskonzentration zugeordnet: 100 nmol/well * Steigung. Unter Verwendung der bewerteten Proteinkonzentration c0 wird die spezifische Aktivität berechnet: 100 nmol/well * Steigung * 1/c0. Die auf diese Weise berechnete spezifische Aktivität wird mit der Einheit nmol/min/g ausgedrückt, die gleich nmol/min/mg ist.
8. Beurteilung der Michaelis-Menten-Kinetik von FAHD-Proteinen
HINWEIS: Die Beurteilung der Michaelis-Menten-Kinetik von FAHD-Proteinen ist mühsam, da die spezifische Proteinaktivität sowohl von der relativen Protein-Substrat-Konzentration als auch von dem physikalischen Volumen abhängt, in dem die Reaktion stattfindet. Die Festzustandskinetik muss hergestellt werden, um zuverlässige Ergebnisse zu erhalten. In den folgenden Schritten wird ein getestetes Protokoll auf einer 96 gut UV-transparenten Platte beschrieben. Jeder Schritt muss mit großer Sorgfalt durchgeführt werden, da kleinere Fehler in der Regel das Experiment verderben. Es wird empfohlen, die in Abschnitt 7 beschriebenen Assays zu meistern, bevor Sie den komplizierteren Test versuchen, der unten beschrieben wird.

Abbildung 5 : Beispielhafte Ergebnisse von Enzym-Assays.
(A) Eine beispielhafte UV-Absorptionskurve, die für grundlegende auf Substraten basierende FAHD-Proteinenzym-Assays (normalisiert im Bereich von 0 bis 1) mit Standardabweichung erhalten wurde. Das optische Dichteverhältnis [OD(t)/OD(0)] zu einem bestimmten Zeitpunkt t [OD(t)] wird auf die ursprüngliche OD [t = 0; OD(0)]. Weitere Informationen finden Sie in Abschnitt 7 des Protokolls. (B) Beispielhafte Michaelis-Menten-Kinetik des humanen FAHD1-Proteins mit Standardabweichung. Weitere Informationen finden Sie in Abschnitt 8 des Protokolls. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Starten Sie einen Mikroplattenleser und equilibrate für 30 min bei 25 °C. Richten Sie ein Programm zum Lesen von 72 Brunnen (wie in Abbildung 4Bbeschrieben) bei 255 nm ein. Es wird empfohlen, 25 Mehrfachauslesungen mit einer Zeitverzögerung von 5 ms zu verwenden. Richten Sie einen Zyklus ein, der 15x pro 2 min (30 min insgesamt) misst.
- Führen Sie die Schritte 7.2 und 7.3 aus. Dann richten Sie 1 ml 100 mM Substratlösung im Enzym-Assay-Puffer ein.
- Vorbereiten von Verdünnungen der Substratlösung im Enzym-Assay-Puffer: 40 mM, 20 mM, 10 mM, 6 mM, 4 mM, 2 mM. Der Test wird mit paarweisen ("angepassten") Enzym-/Substratkonzentrationen durchgeführt. Bereiten Sie dazu die folgenden Verdünnungen der Enzymlösung im Enzym-Assay-Puffer vor: 0,5 g/l, 0,4 g/l, 2,5 g/l, 2 g/l, 1,5 g/l, 1,5 g/l, 1 g/l.
- In alle in Abbildung 4B dargestellten Brunnen werden 180 L Enzym-Assay-Puffer aufgebracht. Tragen Sie 10 L Enzym-Assay-Puffer in alle Brunnen für das Substrat (Leer und Probe) auf. Tragen Sie 10 l der vorbereiteten Proteinverdünnungsreihe in die Brunnen für das Enzym (Leer und Probe) auf. Tragen Sie 10 L Enzym-Assay-Puffer in alle Brunnen für Brunnen für das Substrat-Rohling und das Enzym leer auf.
- Unmittelbar vor der Messung 10 l der vorbereiteten Substratverdünnungsreihe in die Brunnen für die Substratprobe und die Enzymprobe auftragen.
- Verwenden Sie eine Mehrkanalpipette bei 50 L-Einstellungen, um alle Bohrungen sanft zu mischen, beginnend mit den Rohlingen, und gehen Sie zu den Probenbrunnen. Achten Sie darauf, keine Blasen zu erstellen.
- Setzen Sie die Platte in einen Mikroplattenleser ein und messen Sie jeden Brunnen bei 255 nm, wie in Schritt 8.1 beschrieben. Führen Sie die Analyse in einer Kalkulationstabelle durch. Kopieren Sie die Rohdaten aus dem Photometer in eine Kalkulationstabelle, schreiben Sie alle Einstellungen (d. h. alle Dokumentation) in ein anderes Blatt.
- Führen Sie individuelle Datenanalysen pro Punkt in der Verdünnungsreihe durch, wie in den Schritten 7.11 beschrieben. bis 7.14. Schließlich erhalten Sie alle spezifischen Aktivitäten und Parzellen gegen die anfängliche Substratkonzentration: 2 mM, 1 mM, 0,5 mM, 0,3 mM, 0,2 mM, 0,1 mM.
- Zeigt alle Datenpunkte mit individuellen Standardabweichungen an. Computer Michaelis-Menten Kinetik über nichtlineare Kurvenanpassung oder lineweaver-Burk-Analyse. Es kann erforderlich sein, einzelne Punkte neu zu messen und einzelne Protein-Konzentrations-/Substrat-Konzentrationspaarverhältnisse in den Schritten 8.5 und 8.6 anzupassen. Das Michaelis-Menten-Diagramm für menschliches FAHD1 ist in Abbildung 5Bdargestellt.
9. Kristallisation von FAHD-Proteinen
HINWEIS: Die Kristallisation von FAHD-Proteinen (menschliches FAHD1, das zuvor15beschrieben wurde) kann durch die Hängende Tropfendampfdiffusionsmethode in einem 24-Well-Format erreicht werden (Abbildung 6A). Ein Schritt-für-Schritt-Protokoll zur Kristallisation von menschlichem FAHD1 mit dieser Technik wird unter15dargestellt. Eine ausführlichere Beschreibung finden Sie im Diskussionsbereich.

Abbildung 6 : Kristallisation von FAHD-Proteinen.
(A) Kristallisationsplatten in Standard 24 gut oder 96 gut SBS Fußabdruck. Weitere Informationen finden Sie in Abschnitt 9. (B) Der grundlegende Plattenaufbauprozess bei der Kristallisation von FAHD-Proteinen. Diese Zahl wird mit Genehmigung23neu gezeichnet. Weitere Informationen finden Sie in Abschnitt 9. (C) Menschliche FAHD1-Kristallen und entsprechende Beugungsmuster (kleine Einsätze). Der engste Gitterabstand wird in den Einsätzen als Maß für die Beugungsqualität der Kristalle angegeben. Niedrigere Zahlen weisen auf eine höhere Auflösung und damit informativere Daten hin. Weitere Informationen finden Sie in Abschnitt 9 des Protokolls. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Stellen Sie sicher, dass das Protein gegen den SEC-Laufpuffer dialyziert wird. Das FAHD1-Protein sollte in hohen Konzentrationen (2–5 mg/ml) erhältlich sein. Bei niedrigeren Konzentrationen kann das Protein aufgrund mangelnder spontaner Keimbildung nicht kristallisieren.
- Bereiten Sie 20 ml der Reservoirlösung für die Kristallisation vor. Herstellung von drei Lagerlösungen unter Verwendung von destilliertem oder entionisiertem Wasser als Lösungsmittel: 1 M Na-HEPES (mindestens 25 ml, angepasst auf pH 7,5), 50% (w/v) Polyethylenglykol 4000 (PEG4k) (mindestens 65 ml) und 1 M MgCl2 (10 ml).
- Richten Sie ein Raster von 4 x 6 (24 insgesamt) verschiedenen 15 ml Rohren ein. Beschriften Sie sie entsprechend den entsprechenden Positionen auf der Platte (z.B. Reihe (A, B, C, D) vs. Spalte (1–6) wie "A1", "B5", "D6", etc.). Pipette 1 ml von 1 M Na-HEPES in jedes Rohr.
- Pipette 1 ml von 50% (w/v) PEG4k in Reihe A der Rohre, 2 ml in Reihe B, 3 ml in Reihe C und 4 ml in Reihe D. Pipette 100 'L von 1 M MgCl2 in Spalte 1 der Rohre, 250 l in Spalte 2 , 500 l in Spalte 3, 1,0 ml in Spalte 4, 1,5 ml in Spalte 5 und 2,0 ml in Spalte 6.
- Füllen Sie alle Rohre bis zu einem Volumen von 10 ml mit destilliertem oder entionisiertem Wasser, wobei die Skala auf den Rohren ausreichend genau ist.
- Nehmen Sie die menschliche FAHD1-Proteinprobe (ca. 5 mg/ml) aus dem Kühlschrank (oder aus Eis) und drehen Sie sie mit maximaler Geschwindigkeit mit einer Tischzentrifuge bei 4 °C für mindestens 10 min. Wenn eine Kokristallisation mit Oxalat gewünscht wird, Oxalat aus einer Stammlösung hinzufügen, so dass die Proteinprobe eine endgültige Oxalatkonzentration von 2 mM enthält. 1 mM DTT auftragen und auf Eis lagern.
- In der Zwischenzeit eine 24 Brunnenkristallisationsplatte auspacken, idealerweise in einem temperaturgeregelten Raum bei 18 °C. Mit Hilfe einer dünnen Glas- oder Kunststoffstange eine dünne Schicht Paraffinöl auf die Felge auf jeden Brunnen der 24 Brunnenplatte verteilen. Fügen Sie 800 l der vorbereiteten Kristallisationscocktails (A1 bis D6) in jeden entsprechenden Brunnen der Kristallisationsplatte ein.
- Frische 22 mm Abdeckungen auf eine saubere Oberfläche legen. Vermeiden Sie die Verunreinigtder Abdeckung mit Schmutz oder Staub. Entfernen Sie bei Bedarf Schmutz aus dem Abdeckschlupf mit Druckluft oder einem Staubspray.
- Nachdem die Zentrifugation abgeschlossen ist, vermeiden Sie das Schütteln der Proteinprobe, damit die aufgesponnenen Aggregate und Ablagerungen am Boden des Rohres nicht wieder nach oben schweben. In den folgenden Schritten Pipette aus der Proteinprobe direkt unter der Oberfläche der Lösung, um das Aufrühren von Aggregaten und Ablagerungen von unten zu vermeiden.
- Für jeden Brunnen (siehe Abbildung 6B) Pipette 1 L Proteinlösung auf der Mitte eines Deckelschlupfes und fügen Sie 1 l des jeweiligen Reservoir-Cocktails in das Proteintröpfchen, um Blasen zu vermeiden. Drehen Sie den Deckelrutsch auf den Kopf und legen Sie ihn auf die Oberseite des Brunnens, so dass das Öl den Brunnen mit dem Deckelrutsch luftdicht abdichtet. Wiederholen Sie dies, bis die 24 Wellplatte fertig ist.
- Bewahren Sie die Platte bei 18 °C auf und beobachten Sie die Tropfen nach einem progressiven Zeitplan mit einem geeigneten Mikroskop. Menschliche FAHD1-Kristallen erscheinen in der Regel über Nacht (siehe Abbildung 6C).
Ergebnisse
Beginnend mit einem vorbereiteten Klonvektor und gekauftem BL21(DE3) pLysS E. coliwird das Plasmid über Einen Hitzeschock oder eine geeignete alternative Methode in die Bakterien eingeführt (Abbildung 1). Nach einer kurzen Phase der Verstärkung werden die transformierten Bakterien auf LB-Agarplatten plattiert, um über Nacht zu wachsen. Platten an dieser Stelle können unterschiedlich aussehen, abhängig von einer Vielzahl von potenziellen Fehlerquellen. Platten können leer sein (d.h. keine Kolonien), völlig von Bakterien überwuchert, oder etwas dazwischen, bzw. etwas dazwischen. Zwei Beispiele von LB-Agarplatten nach optimaler und nicht optimaler Transformation sind in Abbildung 7Adargestellt. Zu viele Bakterienkolonien deuten darauf hin, dass entweder zu viele Bakterien plattiert wurden (wahrscheinlich) oder dass die verwendeten Antibiotika abgelaufen sein können (unwahrscheinlich). Zu wenige Bakterienkolonien können darauf hindeuten, dass entweder nicht genügend Plasmid für die Transformation verwendet wurde (verwenden Sie beim nächsten Mal mehr) oder dass zu viele Antibiotika verwendet wurden, um die Bakterien auszuwählen. In jedem Fall, wenn Kolonien vorhanden sind, sollten sie in Ordnung sein, da die Verwendung von zwei selektiven Antibiotika eine ziemlich unbedeutende Chance von untransformierten Bakterien impliziert, zu wachsen. Keine Kolonien deutet jedoch darauf hin, dass entweder die Bakterien ihre Transformationskompetenz verloren haben (wegen falscher Lagerung oder Lagerung über längere Zeiträume, wiederholtes Einfrieren und Tauwetter usw.), der Hitzeschock war nicht erfolgreich (keine Plasmidaufnahme oder bakterielle Tod durch zu viel Hitze), ist der Klonvektor korrumpiert, oder versehentlich wurde ein falscher Satz selektiver Antibiotika verwendet (verifizieren Sie das Resistenzgen auf dem Plasmidvektor).

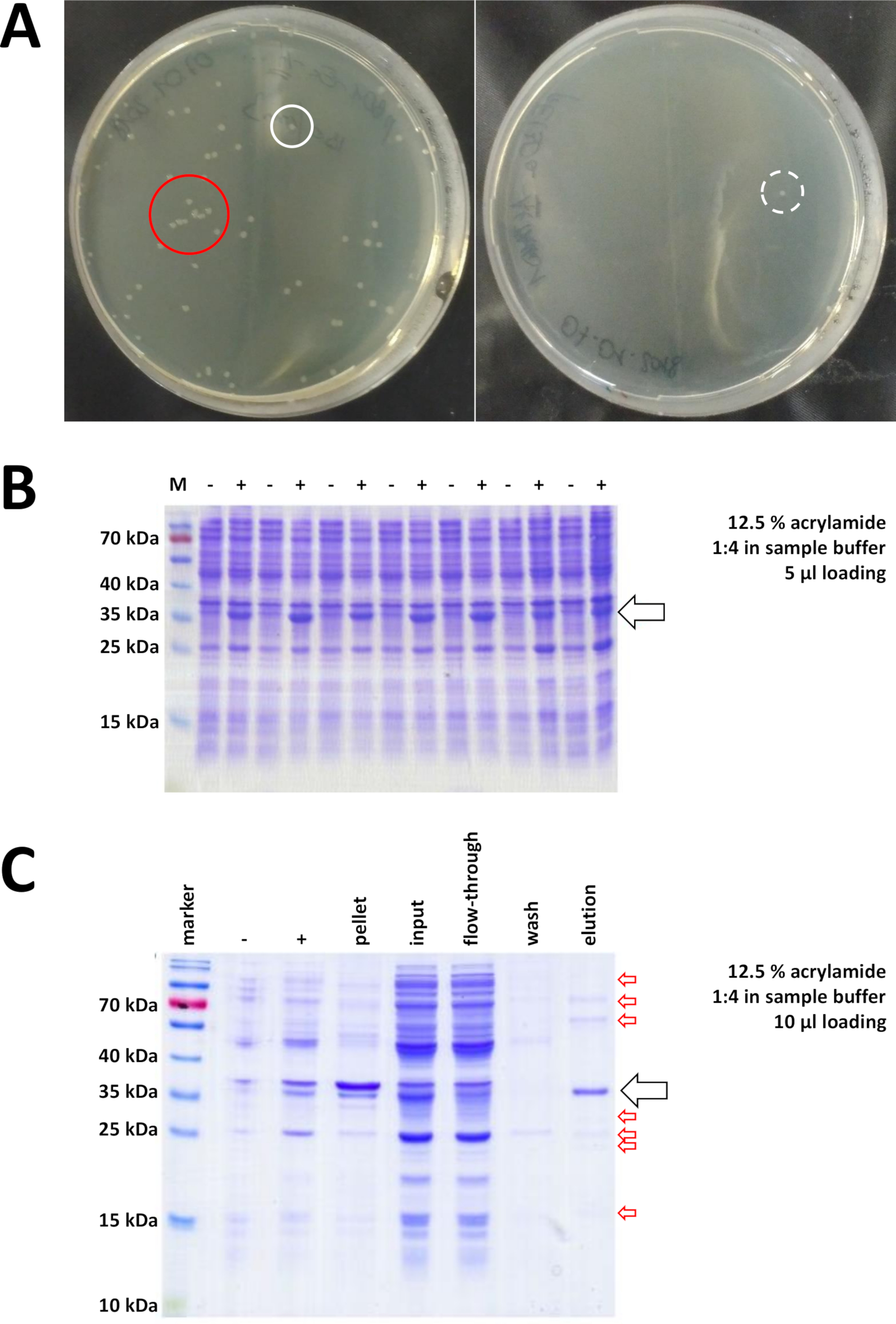
Abbildung 7 : Repräsentative Ergebnisse für die Bakterientransformation und IMAC.
(A) Repräsentative LB-Agarplatten mit transformiertem BL21(DE3) E. coli, erhalten durch Protokollschritt 1.1. Links: Eine Platte mit gut verteilten Kolonien (positives Beispiel). Rechts: Eine Platte mit nur einer einzigen Kolonie (negatives Beispiel). Weiße Kreise markieren gute Kolonien. Der rote Kreis markiert Kolonien, die zu nah beieinander wachsen und nicht gepflückt werden sollten, solange isolierte Kolonien verfügbar sind. (B) Eine 12,5%-Acrylamid-SDS-PAGE-Analyse einer Reihe von Induktionskontrollen ("-" zeigt vor IPTG-Induktion; "+" zeigt nach der IPTG-Induktion vor der Pelleternte an, angepasst an die gleichen Mengen des Gesamtproteins. Dies wird in Schritt 1.2 beschrieben. (C) Eine beispielhafte 12,5% Acrylamid SDS-PAGE Analyse der Ni-NTA-Reinigung seines-tagged FAHD1 Proteins. Dies wird in Abschnitt 3 des Protokolls beschrieben. Die Affinitätschromatographie liefert Protein von hoher Reinheit (>70%, schwarzer Pfeil), aber auch einige kleine Verunreinigungen werden beobachtet (rote Pfeile). Diese Kontaminationen bestehen aus Nicht-FAHD-Proteinen, die an die Säule binden, und aus Proteinen, die an das FAHD-Protein binden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Validierte Kolonien werden ausgewählt und ausgewählt. Nach der Amplifikation im nährenden Medium wird die Proteinexpression durch Anwendung der Chemikalie IPTG ausgelöst. Das bakterielle Pellet, das das exprimierte Protein in Milligrammmengen enthält, wird geerntet und die Expression wird über SDS-PAGE überprüft (siehe z.B. Abbildung 7B). Einige Probleme können während dieses ansonsten einfachen Prozesses auftreten. Erstens bilden einige Proteine Inklusionskörper, weil sie offenbar irgendwie den natürlichen Stoffwechsel der Wirtsbakterien stören. Dies wurde bei einigen Punktmutationen von humanen FAHD1 und FAHD2 beobachtet. In solchen Fällen können andere Expressionssysteme wie Insektenzellen geeigneter sein und sollten in Betracht gezogen werden. Nach der Ernte eines Pellets aus Insektenzellen beispielsweise folgt die Reinigung der Proteine den gleichen Schritten wie in diesem Protokoll beschrieben. Zweitens wird das DE3-pET-System manchmal als "leaky" (d. h. Protein wird bereits in gewissem Umfang vor der IPTG-Induktion exprimiert) gefunden. Der mögliche Grund dafür ist nicht gut verstanden, aber es kann helfen, das Protein langsam über Nacht in einem KaltenRaum-Inkubator auszudrücken. Drittens wird kein Protein exprimiert. Dies ist wahrscheinlich das Worst-Case-Szenario, da es wahrscheinlich auf einen beschädigten Plasmidvektor hindeutet und daher ratsam ist, das Plasmid zu sequenzieren.
Wenn ein His-Tag verwendet wurde, um das Protein zu markieren, ist die Affinitätschromatographie mit Ni-NTA-Agarose eine einfache und kostengünstige Erfassungsmethode, die die Mehrheit der Kontaminationen eliminiert (Abbildung 7C). Ähnliche Methoden gibt es auch für andere Tag-Systeme (z.B. STREP-II). Wenn kein Tag verwendet wurde, kann eine Kombination aus Ammoniumsulfat-Ausfällung und aufeinanderfolgender hydrophober Austauschchromatographie auch das Protein von der Mehrheit der anderen Proteine trennen (Abbildung 8A). Ein Vergleich der beiden Methoden (Abbildung 7C vs. Abbildung 8A) kann jedoch die Überlegenheit der Ni-NTA-Methoden durch SDS-PAGE-Analysen demonstriert werden. Die Verwendung seines-getaggten Proteins wird daher empfohlen.

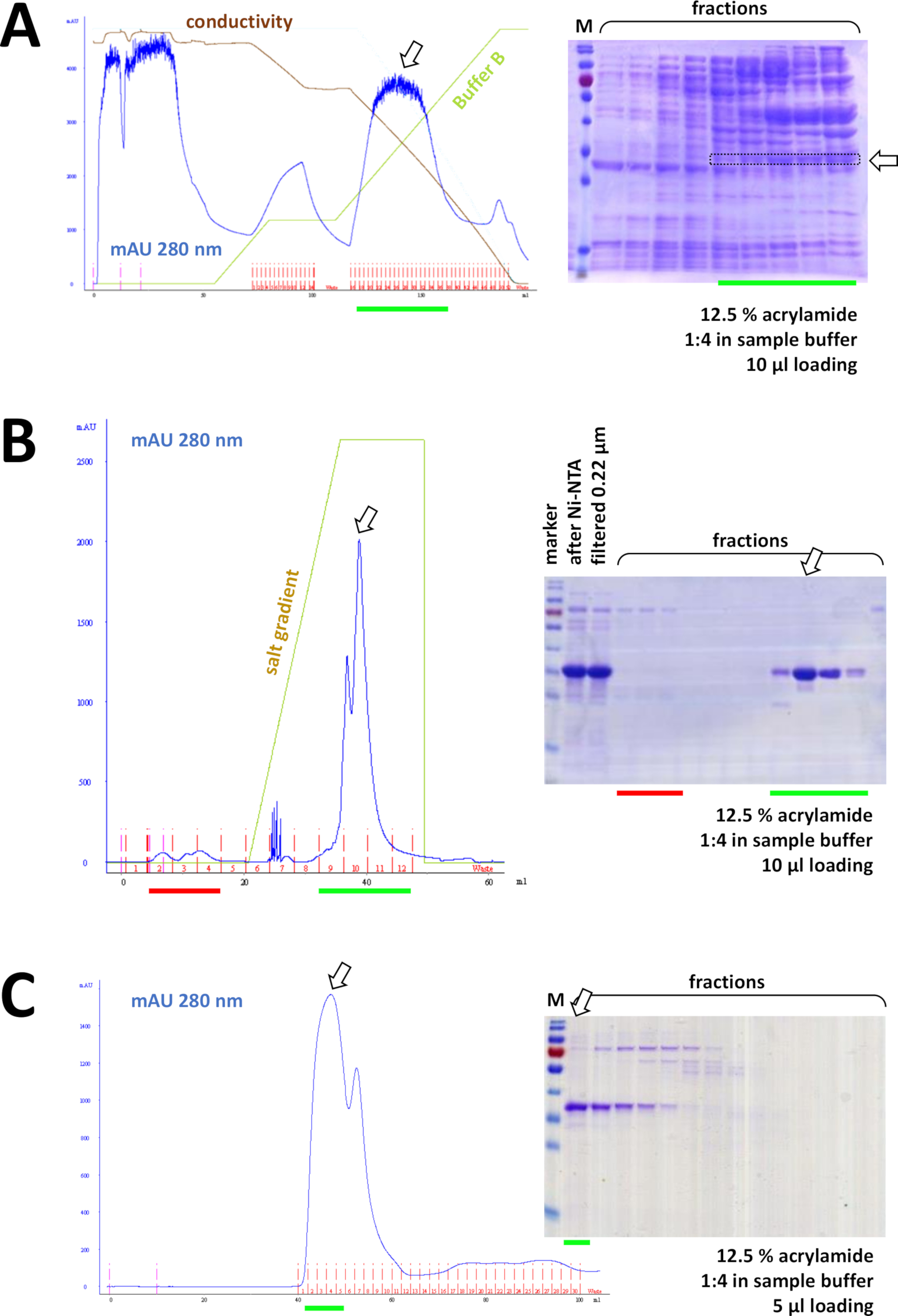
Abbildung 8 : Repräsentative Ergebnisse für FPLC-Experimente (HIC, Ionenaustausch, SEC).
(A) Eine typische Chromatogramm- und 12,5%-Acrylamid-SDS-PAGE-Analyse der HIC-Phenylchromatographie nach Ammoniumsulfat (AS) Ausfällung des nicht markierten FAHD1-Proteins, wie in Abschnitt 4 des Protokolls beschrieben. Die grüne Linie spiegelt den Gradienten von Puffer B wider, der as nicht enthält. Während des Prozesses wird AS nach und nach aus dem System ausgewaschen. Der Vergleich dieses Panels mit Abbildung 7C zeigt die Leistungsfähigkeit der Ni-NTA-Affinitätschromatographie im Vergleich zur HIC-Phenyl-Methode und den Vorteil der Verwendung eines His-tag-Systems zur Proteinreinigung. (B) Ein beispielhaftes Chromatogramm und 12,5% Acrylamid SDS-PAGE Analyse der kationischen Austauschchromatographie seiner-tagged FAHD nach Ni-NTA-Reinigung. Mit Hilfe eines Salzverlaufs wird die aufgetragene Probe in einzelne Proteine getrennt. (C) Ein beispielhaftes Chromatogramm und 12,5% Acrylamid SDS-PAGE Analyse der G75 Größe Ausschlusschromatographie seiner-tagged FAHD nach kationischer Austauschchromatographie. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Nacheinander wird das Protein durch Kation/Anionenaustauschchromatographie weiter von Restkontaminationen getrennt (siehe Abbildung 8B),gefolgt von größenausschlusschromatographie (siehe Abbildung 8C). Es wird empfohlen, in dieser Reihenfolge eine erste Reinigungsstrategie zu entwickeln; diese Säulen sollten jedoch in Kombination, anschließend und in Variation verwendet werden, bis das Protein ausreichend rein ist.
Einfache Aktivitätstests, um "Ja oder Nein"-Entscheidungen auf aktive Substrate und/oder Kofaktoren zu testen, können mit seinen-markierten Proteinen nach der Ni-NTA-Reinigung oder nicht markierten Proteinen nach der Ionenaustauschsäule durchgeführt werden. Spezifische Aktivitäten und kinetische Konstanten müssen mit Protein von höchster Reinheit bestimmt werden. Die Kristallisation kann mit Proteinen nach der ionenaustauschenden Säule versucht werden, aber die Qualität der Kristalle korreliert fast immer mit der Proteinreinheit. Polyklonale Antikörper können in jeder Phase des Reinigungsprotokolls gegen Proteine aufgebracht werden; Hier korreliert die Qualität aber auch mit der Proteinreinheit.
Diskussion
Kritische Schritte
FAHD-Proteine sind sehr empfindlich auf Salzkonzentrationen. Bei niedrigen NaCl-Konzentrationen können die Proteine beim Auftauen ausbrechen, aber sie können in der Regel bei höheren Salzkonzentrationen vollständig rekonstituiert werden. Das heißt, wenn ein FAHD-Protein aus irgendeinem Grund ausfällt, kann es mit höheren Salzkonzentrationen (>300 m) zurückgewonnen oder wieder gefaltet werden. Einige weitere hydrophobe Proteine können jedoch nicht zurückgewonnen werden (z. B. menschliches FAHD2), aber Waschmittel wie CHAPS (maximal 1%) oder Glycerin (10%) kann verwendet werden, um sie in einer stabilen Lösung zu halten. In jedem Fall wird das Einfrieren von Stößen mit flüssigem Stickstoff und die Lagerung bei -80 °C empfohlen, da es sich um einen sanften und langsamen Auftauprozess handelt.
Einige unerwartete Probleme können während der Ni-NTA-Reinigung in Schritt 3.1.10 auftreten. Bemerkenswert ist, dass eine höhere OD in der zweiten entnommenen Probe als in der ersten Probe auf ein zu hohes Volumen des Agaroseharzes hindeutet (beachten Sie dies und verwenden Sie im nächsten Experiment weniger Harz). Auch das Agaroseharz selbst führt zu einem OD-Signal bei 280 nm (d.h. störung des Agaroseharzbettes gibt künstliche Signale). Im Zweifelsfall wird empfohlen, andere Methoden wie einen Bradford- oder BSA-Assay zu verwenden, um Proteinkonzentrationen zu bestimmen.
In enzymatischen Assays gibt es drei kritische Aspekte, die berücksichtigt werden müssen. Erstens ist die Bewertung der Proteinkonzentration entscheidend, um die richtigen spezifischen Aktivitäten zu erhalten. Der Reinheitsgrad des Proteins beeinflusst das Ergebnis und muss geschätzt werden. Bei getaggten Proteinen muss die Masse des Tag-Teils berechnet und die spezifische Aktivität entsprechend korrigiert werden. Für einfache Assays, die in Abschnitt 7 des Protokolls beschrieben werden, reicht die Ni-NTA-Reinheit aus, um zwischen aktiven und inaktiven Substraten, Kofaktoren usw. zu unterscheiden. Bei komplexerer Michaelis-Menten-Kinetik müssen alle Reaktanten- und Substratkonzentrationen korrekt bestimmt werden. Insbesondere bei der Verwendung von Oxaloacetat (das sich im Laufe der Zeit automatisch decarboxyliert) muss der enzymatische Teil der Reaktion für die automatische Decarboxylierung korrigiert werden (unter der Annahme, dass beide Reaktionen gleichzeitig auftreten). Erste Änderungen des optischen Dichtesignals, das an die Keto-Enol-Tautomerisierung des Substrats adressiert ist, müssen berücksichtigt werden. Drittens müssen Konzentrationen und Mengen angepasst werden. Eine Reaktion mit definierten Konzentrationen von Enzym und Substrat kann je nach Assayvolumen unterschiedliche Ergebnisse liefern. Wenn es zu viel Enzym pro Brunnen gibt, kann die Haftung der Flüssigkeit in der Tat das Ergebnis verzerrt.
Für die Beurteilung der Michaelis-Menten-Kinetik wird empfohlen, erste Experimente in 100 L-, 200-L- und 300-L-Chargen durchzuführen, um die optimale Kombination zu finden. Ähnliche Aspekte gelten für das Verhältnis von Enzym-Substrat-Konzentrationen für kinetische Assays. Zu viel Enzym pro Substrat oder zu viel Substrat pro Enzym brachte das System außerhalb des linearen stabilen Michaelis-Bereichs. Erste Experimente sind erforderlich, um diese Bedingungen zu optimieren. Beispielhafte Anpassungen für menschliches FAHD1-Protein (Wildtyp) sind in Abschnitt 8 vorgesehen, was zu kinetischen Diagrammen führt (z. B. in Abbildung 5B).
Zur Kristallisation wird ein Tröpfchen Proteinlösung in der Mitte eines Deckelrutsches pipettiert und mit einem Tröpfchen Kristallisationscocktail vermischt, der in der Regel aus einem Puffer (z.B. Tris-HCl, HEPES) und einem Niederschlagsmittel (z.B. Polyethylenglykol, Ammonium Sulfat). Optional kann ein Tröpfchen der Inhibitorlösung zur Kokristallisation (wie Oxalat in diesem Protokoll) angewendet werden. Der Deckelschlupf wird dann kopfüber über einem Reservoir mit Kristallisationscocktail platziert, das den Brunnen mit Hilfe von Dichtöl dicht versiegelt (Abbildung 6B). Im Idealfall findet zu Beginn des Experiments kein Niederschlag innerhalb des Tropfens statt, was bedeutet, dass das Protein in Lösung bleibt. Da die Niederschlagskonzentration im Reservoir höher ist als im Tropfen, beginnt der Tropfen wasserdurchdampfend in die Atmosphäre des Brunnens zu verlieren, bis das Gleichgewicht mit dem Reservoir erreicht ist. Die Diffusion von Wasser in das Reservoir verursacht eine langsame Volumenabnahme des Tropfens, was wiederum zu einer Erhöhung von protein- und niederschlagsmitteler Konzentration im Tropfen führt. Erreicht die Proteinlösung den erforderlichen Zustand der Supersättigung und damit der Metastabilität, kann eine spontane Keimbildung mit anschließendem Kristallwachstum auftreten. Das Erreichen des übersättigten Zustands ist eine notwendige, aber nicht ausreichende Bedingung für die Kristallisation. Die Kristallisation von Proteinen benötigt sowohl günstige thermodynamische als auch kinetische Bedingungen und hängt stark von den unvorhersehbaren Eigenschaften des zu kristallisierenden Proteins ab22.
Änderungen und Fehlerbehebung
Die Expression von Protein in E. coli kann ineffizient sein. Unterschiedliche IPTG-Konzentrationen, Expressionstemperatur und Amplifikationszeit, wie Raumtemperatur für mehrere Stunden oder in Kühlräumen über Nacht, müssen möglicherweise für jedes neue Protein getestet werden, um optimale Bedingungen zu finden. Die Ausfällung von Proteinen in Inklusionskörpern wird manchmal bei hydrophoben FAHD-Proteinen beobachtet. In solchen Fällen wird die Proteinexpression in anderen Modellsystemen wie Insektenzellen empfohlen, da Einschlusskörper weniger wahrscheinlich26bilden.
Da FAHD-Proteine empfindlich auf Salz- und Kofaktorkonzentrationen sowie pH-Wert reagieren, können sich Reinigungsstrategien für verschiedene Homologe, Orthologe und Punktmutationsvarianten in den einzelnen Einstellungen unterscheiden. Die beschriebenen Reinigungsmethoden werden für das Wildtyp-Human- und Maus-FAHD1-Protein entwickelt. Chemikalienkonzentrationen wie NaCl und Imidazol sowie pH-Wert müssen möglicherweise für einzelne Proteine mit einem anderen isoelektrischen Punkt (pI) angepasst werden. Bemerkenswert ist auch, dass nicht jedes seine-getaggten Proteine gut an ein Ni-NTA-Harz binden kann. Wenn die Proteinbindung an die Ni-NTA-Säule ineffizient ist, können angepasste Konzentrationen von NaCl und Imidazol sowie unterschiedliche pH-Bedingungen im Ni-NTA-Laufpuffer dazu beitragen, die Qualität des Ergebnisses zu verbessern. Andernfalls kann das Überspringen des Ni-NTA-Schritts und das Fortfahren zum Schritt der ionenaustauscheren Chromatographie auch zu einer erfolgreichen Reinigungsstrategie führen. Wenn ein Protein an die Ni-NTA-Säule bindet, aber nicht aus der Spalte eluiert werden kann, kann die Zugabe von einigen mM EDTA dazu beitragen, den Ni2+-Komplex zu stören.
Was den Kristallisationsprozess betrifft, so ist zu verstehen, dass die Selbstorganisation großer und komplexer Proteinmoleküle in ein regelmäßiges periodisches Gitter ein von Natur aus unwahrscheinlicher Prozess ist, der stark von schwer zu kontrollierenden kinetischen Parametern abhängt. Selbst kleine Änderungen im Aufbau, die für die Kristallisation verwendet werden, können das Ergebnis dramatisch verändern und es bilden sich keine Kristalle. Proteinreinheit ist im Allgemeinen von größter Bedeutung. Als Faustregel gilt, dass ein stark überlastetes SDS-PAGE-Gel keine anderen Bands zeigen sollte. Außerdem kann sich die Reihenfolge, in der Schritte ausgeführt werden, auf das Ergebnis auswirken. Um die Reproduzierbarkeit zu gewährleisten, ist es oft notwendig, die Pipettiersequenz gleich zu halten, dann zuerst das Protein hinzuzufügen und schließlich das Kristallisationströpfchen (oder umgekehrt) ausgefällt zu fügen. Unabhängig davon, welche Methode verwendet wird, sollte es gleich gehalten werden, wenn versucht wird, Experimente zu reproduzieren oder zu skalieren. Wenn nach diesem Protokoll keine Kristalle beobachtet werden, können die chemische Niederschlagszusammensetzung, der pH-Wert, die Tropfengröße und das Protein-zu-Gefällt-um-Gefällt-um-Gefällt-um-Gefällt-um-Gefällt-um-Gefällt-um-Gefällt-um-Gefällt-um-Verhältnis in kleinen Schritten variiert werden. Geduld und konsequente Beobachtungen der Tropfen sind von Tugend.
Bemerkungen zu katalytischen Mechanismen von FAHD1
Die vorgestellten Methoden wurden speziell zur Gewinnung von FAHD1-Proteinen von hoher Qualität entwickelt. Dies ermöglichte das Wachstum von FAHD1-Kristallen sowie die Entwicklung von Kristallen, die FAHD1 enthalten, die zu einem Inhibitor komplexiert sind (Oxalat, PDB:6FOG). Die Röntgenstrukturen bieten eine 3D-Architektur der katalytischen Kavität des Enzyms. Diese Ergebnisse stellen eine umfassende Beschreibung von Rückständen fest, die für die katalytischen Mechanismen dieses faszinierenden Enzyms potenziell wichtig sind. FAHD1 wurde zuerst beschrieben, um Acylpyruvate (Acetylpyruvat, Fumarylpyruvat) spalten zu können11. Später wurde festgestellt, dass FAHD1 auch als Decarboxylase von Oxaloacetat12arbeitet. Obwohl die Substrate Acylpyruvat und Oxaloacetat unterschiedliche chemische Moieties sind, teilen die chemischen Transformationen mechanistisch die strategische Spaltung einer gemeinsamen einzigen C3-C4-Bindung, die energetisch erleichtert wird, wenn dieC3 -C4-Bindungsorbitale bleiben orthogonal zu den A-Orbitalen des C2-Carbonyl15. Eine solche Konformation ermöglicht dieResonanzstabilisierung des während des Spaltungsprozesses vorübergehend gebildeten C3-Carbanion. Die FAHD1-Substrate (Oxaloacetat und Acylpyruvate) sind flexible Moleküle und können in tautomer (Keto-Enol) sowie C2-hydratisierten Formen existieren (Abbildung 9A). Die Gleichgewichte zwischen den verschiedenen Arten werden hauptsächlich durch die Art der verwendeten Pufferzusammensetzung, den pH-Wert und das Vorhandensein von Metallionen bestimmt. Im Folgenden diskutieren wir hypothetische mechanistische Szenarien, inspiriert von der Analyse von Röntgenkristallstrukturen, die das katalytische Zentrum von FAHD1 offenbarten.

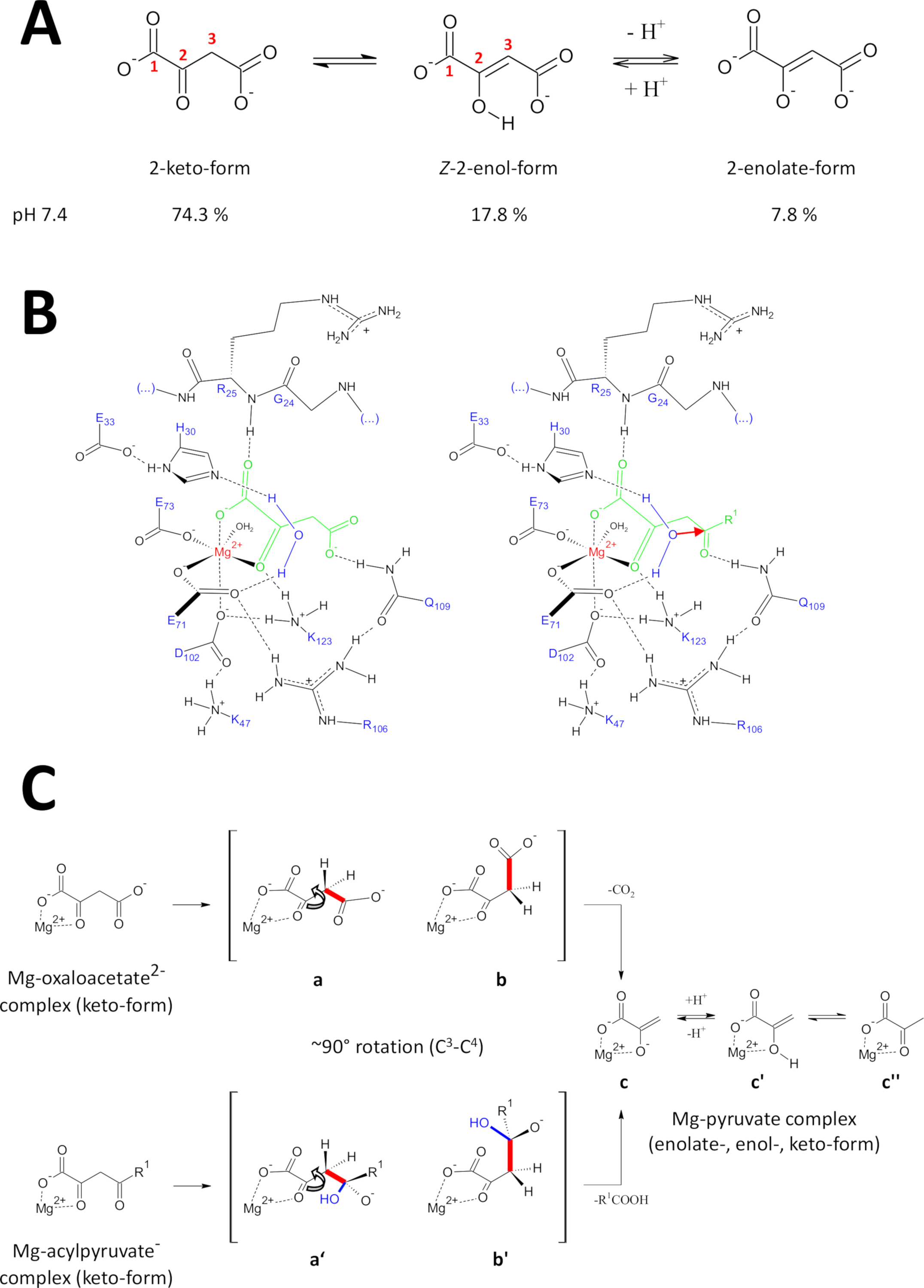
Abbildung 9 : Einzelheiten zum vorgeschlagenen katalysatorischen Mechanismus des menschlichen FAHD1.
(A) Oxaloacetat existiert im kristallinen Zustand sowie in neutraler Lösung hauptsächlich in der Z-Enolform24. Unter physiologischen pH-Bedingungen ist jedoch die 2-Keto-Form die vorherrschende Darstellung25. (B) Chemische Skizze des hFAHD1 Hohlraums15 mit Mg-gebundenem Oxaloacetat (links) und Acylpyruvat (rechts, mit R1 als organischer Rest; der rote Pfeil bezeichnet einen nukleophilen Angriff des angrenzenden stabilisierten Wassermoleküls) (siehe Diskussion). (C) Vergleich der bevorzugtenKonkonformationen für C3-C4-Spaltung in Decarboxylase (b bis c) und Hydrolase (b' bis c) Mechanismus von FAHD1: Beide Prozesse führen zu Mg-komplexiertem Pyruvat-Enolate (siehe Diskussion). Die Zwischenprodukte b und b' sollen bis Q109 stabilisiert werden, wie in Panel B skizziert (siehe Diskussion). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Die Decarboxylase-Aktivität von FAHD1
Oxaloacetat existiert im kristallinen Zustand sowie in neutraler Lösung hauptsächlich in der Z-Enolform24. Es zeigte sich jedoch, dass unter physiologischen pH-Bedingungen (Pufferbedingungen bei pH 7,4) die 2-Keto-Form die vorherrschende Darstellung von Oxaloacetat25 (Abbildung 9A) ist und dass die Enolisierung keine Voraussetzung für die Decarboxylierung ist27 . Bemerkenswert ist, dass Mg 2+-Ionen keinen Einfluss auf das Verhältnis der Oxaloacetat-Arten bei einem pH-Wert von 7,4 oder unter28haben. Die Transposition der Oxaloacetat-Ketoform in das katalytische Zentrum von FAHD1 (geführt durch das gebundene Oxalat im komplexen Enzym (PDB: 6FOG15)) zeigte Rückstand Q109 als Konformationsregulator des gebundenen Oxaloacetat15. Wie in einem anderen Artikel15beschrieben, stabilisiert die Wasserstoffbindung mit der Carbamoyl-Gruppe von Q109eine Oxaloacetat-Konformation, die durch Rotation um die C2-C3-Bindung entsteht (Abbildung 9B, linkes Panel). Als Folge dieser Rotation nimmtdie C3-C4-Bindung (zu spalten) eine nahezu orthogonale Disposition relativ zu den Orbitalen des C2-Carbonylaners an (Abbildung 9C). Kohlendioxid kann freigesetzt werden. Das Primäreprodukt dieses Prozesses wäre Resonanz stabilisiertMg-Enoat von Pyruvat. Aus Untersuchungen von Oxaloacetat-Mg-Komplexen ist bekannt, dass das Enolate den stabilsten Komplex bildet28,29. Unter der Annahme einer vergleichbaren Stabilität für einen Mg-Pyruvat-Enolate-Komplex könnte der Kofaktor von FAHD1 blockiert werden, aber Lysinrückstand K123 kann das Pyruvat-Enolate in einem Gleichgewicht protonieren, um den Verlust des Cofaktors15zu verhindern.
Die gegebene Interpretation schlägt Pyruvat-Enol als eigenständiges Zwischenprodukt in der katalytischen ODx-Funktion von FAHD1 vor. In diesem Schritt des hypothetischen Modells liefern experimentelle Daten keinen weiteren Hinweis darauf, warum sich der geschlossene Deckel öffnen sollte, um das Produkt freizusetzen. Daraus lässt sich jedoch ableiten, dass der vorgeschlagene Mechanismus wie eine Enzymhemmung durch das Produkt aussieht: Die Kristallstruktur zeigt ein konserviertes Wassermolekül, das in Richtungsausrichtung zum KATAlysezentrum FAHD1 durch die in einem kurzheis15, die bei Ligandenbindung und Deckelverschluss induziert wird. Wenn das primäre Enol in einem Gleichgewicht mit dem Enolate bleiben würde, könnte die Resonanz stabilisiert Enolate durch das Wassermolekül zu Pyruvat gelöscht werden. Das resultierende Hydroxyl wäre in der Lage, das Pyruvat aus dem Mg-Kofaktor zu verdrängen, auf dem sich der Deckel öffnen würde. Schließlich würde das Katalysezentrum in der mitochondrialen Umgebung wiederhergestellt werden. In diesem hypothetischen Szenario würde das Hohlraumwassermolekül als Säure bzw. als Säure wirken.
Hydrolase Aktivität von FAHD1
Die Hydrolase-Aktivität eines Enzyms erfordert implizit die Zwischenbildung eines Hydroxylnucleophilen. Dieser Mechanismus findet sich in der Regel in Kombination mit säure-basenkatalytischer Aktivität. Der Übergangszustand der Reaktion muss durch konforme Kontrolle durch kritische Aminosäureseitenketten in der Kavität hergestellt werden. Analog zur Diskussion der Decarboxylase-Funktion wird enzymgebundenes Acylpyruvat in 2-Keto-Form durch Wasserstoffbindung des 4-Carbonyl-Sauerstoffs mit Q109 (Abbildung9B,rechtes Panel) unter Konformationskontrolle gestellt. Die Kristallstruktur des oxalatgebundenen FAHD1 (PDB:6FOG) zeigt ein konserviertes Wassermolekül, das in Richtungsausrichtung zum KATAlysezentrum FAHD1 durch die Ineinere H30 und E33 in einer kurzen Helix15gehalten wird. Die Dyade E33-H30 ist für die Deprotonation des gerichteten Wassers zuständig und das resultierende Hydroxyl ist in idealer Disposition, um das 4-Carbonyl von Acylpyruvat anzugreifen, das unter Konformationskontrolle durch Q10915präsentiert wird.
Bemerkenswert ist, dass ein ähnlicher Mechanismus für FAH18vorgeschlagen wurde. Ein Angriff des Hydroxylnucleophilen wird voraussichtlich zu einer Oxyanionsart führen,die bei orbitalgesteuerter C3-C4-Bindungsspaltung stabilisiert wird (Abbildung 9C). In diesem Modellerfolgt die C3-C4-Bindungsrotation (Abbildung 9C) nach dem nucleophilen Angriff durch das in Abbildung 9B angegebene gebildete Hydroxyl (d. h. es bereitet das Acylpyruvat für die Bindungsspaltung). Die Hauptprodukte wären Essigsäure und Mg-Pyruvat-Enoat. In diesem hypothetischen Szenario könnte die Essigsäure das Enol zu Pyruvat abschrecken und anschließend die Verschiebung des Produkts unterstützen. Oberhalb eines pH-Werts von 7,5 und in Gegenwart von Mg-Ionen existieren Acylpyruvate in einem Gleichgewicht zwischen Keto- und Enolformen, letztere in leichter Präferenz30. Wahrscheinlich sind beide Formen in der Lage, sich unter anschließendem Deckelverschluss an den Cofaktor von FAHD1 zu binden. Die Verarbeitung von enolic acylpyruvat Substraten durch das Enzym wird durch die flache Struktur der Enolform behindert. Die Spaltung C3-C4 würde zu einem Vinyl-Carbanion ohne Resonanzstabilisierung führen.
Daher schlagen wir einen katalytischen Ketonisierungsschritt vor, um sich auf den Angriff des Hydroxylnucleophilen auf das Acylcarbonyl vorzubereiten. Dieser Ketonisierungsprozess würde jedoch die Kontrolle über die Protonentranspositionen durch FAHD1-Rückstände erfordern, was FAHD1 eine inhärente Isomerase-Aktivität zuschreiben würde. Es wird berichtet, dass der Säuregehalt von Mg-gebundenem Enolwasserstoff einen Zehntausendfachen Anstieg im Vergleich zur unkomplexen Form28zeigt. Eine Deprotonation der Mg-gebundenen Enolform wäre durch unprotoniertes K123 möglich. Die Deprotonation von K123 kann durch das Carboxylat von D102 unterstützt werden. Ein aus Rückständen d102-K47-K123 gebildetes Wasserstoff-Bindungsnetz könnte als notwendiges Protonenrelais im Katalysatorzentrum von FAHD115betrieben werden. Ein so geformtes Zwischenenoat könnte dann durch eine E33-H30-H20 Triade unter Ketonisierung des Substrats15abgeschreckt werden. Die 2-Keto-Form würde unter konforme Kontrolle von Q109 kommen, und das gleichzeitig gebildete Hydroxyl würde das Acylcarbonyl angreifen. Die zusammengefasste Diskussion impliziert eine Kontrolle von FAHD1 über ein Wassermolekül zum Umschalten zwischen Säure und Base durch Zusammenspiel von hohlraumbildenden Rückständen.
Zukünftige Anwendungen oder Anweisungen der Methode
Zukünftige Anwendungen der hier beschriebenen Methoden sind zahlreich. Eine Fülle von prokaryotischen Mitgliedern der FAH-Überfamilie wartet noch immer auf eine funktionale Charakterisierung. Selbst die verfügbaren Informationen über die katalytischen Aktivitäten bekannter FA-Überfamilienmitglieder sind rar und basieren in den meisten Fällen eher auf theoretischen Annahmen als auf experimentellen Daten. Die Anwendung der hier beschriebenen Methoden für prokaryotische FA-Überfamilienmitgliedn hängt von den spezifischen Forschungsinteressen in der Bakteriologie ab. Auf der anderen Seite zeigt die jüngste Demonstration, dass eukaryotische FA-Überfamilienmitgliedn wesentliche Rollen in verschiedenen Zellkompartimenten spielen (z. B. Zytosol vs. Mitochondrien), die Notwendigkeit, diese Proteine besser zu charakterisieren (drei davon wurden wurden bisher identifiziert), insbesondere weil aktuelle Daten darauf hindeuten, dass einige nicht charakterisierte Proteine verschiedene Funktionen im Kontext der mitochondrialen Biologie, der Alterungsforschung und der Krebsforschung erfüllen können. Es wird vorgeschlagen, dass die vollständige molekulare und physiologische Charakterisierung dieser eukaryotischen FA-Überfamilienmitgliedn wichtige Einblicke in wichtige Bereiche der zeitgenössischen Forschung im biomedizinischen Sektor liefern kann. Mehr Forschung über die Mechanismen von FAHD1 (und verwandten Enzymen) sind notwendig, um Mechanismen besser zu verstehen, die der Bi-Funktionalität von FAHD1 zugrunde liegen, die immer noch nicht vollständig geklärt ist. Zusätzliche Studien mit FAHD1-Mutanten, NMR-Untersuchungen und Strukturstudien zu Inhibitorkomplexen können helfen, die wahren mechanistischen Szenarien zu lösen, für die FAHD1 zu kompetent zu sein scheint. Darüber hinaus wird die computergestützte Entwicklung von Enol-Imitiden, die in der Lage sind, an den Mg-Kofaktor zu binden, schließlich zu potenten Inhibitoren von FAHD1 führen.
Offenlegungen
Die Autoren haben nichts zu verraten und erklären keine konkurrierenden finanziellen Interessen. H. G. ist CEOCSO bei MoleculeCrafting.HuGs e.U. und stellte Acylpyruvate für diese Studie über die kundenspezifische Synthese zur Verfügung. Die Arbeit im Labor von P. J. D. wurde vom Wissenschaftsfonds (FWF) unterstützt: Projektnummer P 31582-B26. Die Veröffentlichungsgebühren für dieses Manuskript wurden teilweise vom Wissenschaftsfonds (FWF) unter der Projektnummer P 31582-B26 übernommen. A. N. und B. R. werden vom Wissenschaftsfonds (FWF) im Rahmen des Projekts P28395-B26 unterstützt.
Danksagungen
Die Autoren sind sehr dankbar für die fachkundige technische Unterstützung durch Annabella Pittl und die Pilotmethodenentwicklung von Haymo Pircher.
Materialien
| Name | Company | Catalog Number | Comments |
| BL21(DE3) pLysS competent E. coli | Promega | L1195 | High-efficiency protein expression from gene with T7 promoter and ribosome binding site |
| pET E. coli T7 Expression Vectors | MERCK | - | http://www.merckmillipore.com/AT/de/life-science-research/genomic-analysis/dna-preparation-cloning/pet-expression-vectors/qFSb.qB.mLQAAAFA6.VkiQ0G,nav |
| 0.45 µm filter units | MERCK | SLHP033NS | Millex-HP, 0.45 µm, PES 33 mm, not steril |
| 0.22 µm filter units | MERCK | SLGP033RS | Millex-HP, 0.22 µm, PES 33 mm, not steril |
| Eppendof tubes 1.5 mL | VWR | 525-1042 | microcentrifugal tubes; autoclaved |
| 15 mL Falcon | VWR | 734-0451 | centrifugal tubes |
| 50 mL Falcon | VWR | 734-0448 | centrifugal tubes |
| PS Cuvettes Spectrophotometer Semi-Micro | VWR | 30622-758 | VIS transparent cuvettes |
| UV Cuvettes Spectrophotometer Semi-Micro | VWR | 47727-024 | UV/VIS transparent cuvettes |
| isopropyl-β-D-thiogalactopyranosid (IPTG) | ROTH | 2316 | chemical used for induction of protein expression with the DE3/pET system |
| imidazole | ROTH | X998 | chemical used for elution of polyhistidine (6xHis) sequences from a nickel-charged affinity resin |
| Glass Econo-Column Columns | Bio-Rad | - | http://www.bio-rad.com/de-at/product/glass-econo-column-columns?ID=2cfb1c6e-32e8-4c72-b532-dd39013d707d&pcp_loc=catprod |
| chloramphenicol | Sigma-Aldrich | C0378 | antibiotic for bacterial growth selection; resistance endióded in pLysS plasmid of BL21(DE3) E. coli; 25 µg/mL final concentration |
| kanamycin | Sigma-Aldrich | 60615 | antibiotic for bacterial growth selection; to be used if this resistance is encoded in the employed pET vector; 50 µg/mL final concentration |
| ampicillin | Sigma-Aldrich | A1593 | antibiotic for bacterial growth selection; to be used if this resistance is encoded in the employed pET vector; 100 µg/mL final concentration |
| Ultra-15, MWCO 10 kDa | Sigma-Aldrich | Z706345 | centrifigal filters for protein enrichment; https://www.sigmaaldrich.com/catalog/product/sigma/z706345?lang=de®ion=AT |
| Ultra-0.5 Centrifugal Filter Units | Sigma-Aldrich | Z677108 | centrifigal filters for protein enrichment; https://www.sigmaaldrich.com/catalog/product/ALDRICH/Z677108?lang=de®ion=AT&cm_sp=Insite-_-prodRecCold_xviews-_-prodRecCold5-2 |
| oxaloacetic acid | Sigma-Aldrich | O4126 | TCA metabolite |
| sodium oxlalate | Sigma-Aldrich | 71800 | a competitive inhibitor of FAH superfamily enzymes |
| Dialysis tubing cellulose membrane | Sigma-Aldrich | D9277 | https://www.sigmaaldrich.com/catalog/product/sigma/d9277; or comparable |
| Ni-NTA agarose | Thermo-Fischer | R90101 | a nickel-charged affinity resin that can be used to purify recombinant proteins containing a polyhistidine (6xHis) sequence |
| 96-Well UV Microplate | Thermo-Fischer | 8404 | UV/VIS transparent flat-bottom 96 well plates |
| PageRuler Prestained Protein Ladder, 10 to 180 kDa | Thermo-Fischer | 26616 | https://www.thermofisher.com/order/catalog/product/26616?SID=srch-hj-26616 |
| ÄKTA FPLC system | GE Healthcare Life Sciences | - | using the FPLC system by GE Healthcare; different custom versions exist; this work used the "ÄKTA pure" system |
| HiTrap Phenyl HP column | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/it/shop/chromatography/prepacked-columns/hydrophobic-interaction/hitrap-phenyl-hp-p-05630 |
| Mono S 10/100 GL | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/ch/shop/chromatography/prepacked-columns/ion-exchange/mono-s-cation-exchange-chromatography-column-p-00723 |
| Mono Q 10/100 GL | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/ch/shop/chromatography/prepacked-columns/ion-exchange/mono-q-anion-exchange-chromatography-column-p-00608 |
| HiLoad Superdex column 75 pg (G75) | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/ch/shop/chromatography/prepacked-columns/size-exclusion/hiload-superdex-75-pg-preparative-size-exclusion-chromatography-columns-p-05800 |
| HiLoad Superdex column 200 pg (G200) | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/ch/shop/chromatography/prepacked-columns/size-exclusion/hiload-superdex-200-pg-preparative-size-exclusion-chromatography-columns-p-06283 |
| TECAN microplate reader | TECAN Life Sciences | - | https://lifesciences.tecan.com/microplate-readers |
| acetylpyruvate | MoleculeCrafting.HuGs e.U. | - | custom synthesis |
| benzoylpyruvate | MoleculeCrafting.HuGs e.U. | - | custom synthesis |
| VDX™ plate (24 wells) | Hampton | HR3-142 | 24 well plates used for crystallization via Hanging Drop Vapor Diffusion |
| paraffin oil | Hampton | HR3-411 | used for crystallization via Hanging Drop Vapor Diffusion |
| coverslips (22 mm) | Karl Hecht KG | 14043 | coverslips used for crystallization via Hanging Drop Vapor Diffusion |
| Luria broth (LB) medium | self-prepared | - | a general growth medium for E. coli: 5 g/L yeast extract; 10 g/L peptone from casein; 10 g/L sodium chloride; 12 g/L agar-agar |
| NZCYM medium | self-prepared | - | a better growth medium for E. coli, used for amplification: 10 g/L NZ amine; 5 g/L NaCl; 5 g/L yeast extract; 1 g/L casamino acids; 2 g/L MgSO4; adjust pH to 7.4 |
| Luria broth (LB) agarose plates | self-prepared | - | autoclaved agarose plates containing LB-medium and antibiotics for bacterial groth selection; https://www.addgene.org/protocols/pouring-lb-agar-plates/ |
| Ni-NTA running buffer | self-prepared | - | 20 mM Tris-HCl pH 7,4; 50-300 mM NaCl; 10-200 mM imidazole; ranges: optimal value varies among FAHD proteins |
| Ni-NTA elution buffer | self-prepared | - | 20 mM Tris-HCl pH 7,4; 50-300 mM NaCl; 200-500 mM imidazole; ranges: optimal value varies among FAHD proteins |
| HIC running buffer | self-prepared | - | 44 mM NaH2PO4; 6 mM Na2HPO4; 100 mM NaCl; 20 mM DTT; adjust to pH 7 |
| HIC running buffer AS | self-prepared | - | HIC running buffer saturated with ammonium sulfate (AS); adjust to pH 7: 70 g ammonium sulfate + 90 mL buffer, stirred overnight in the cold room; adjust to pH 7.0 |
| Mono S low salt buffer | self-prepared | - | 44 mM NaH2PO4; 6 mM Na2HPO4; 10-300 mM NaCl; ranges: optimal value varies among FAHD proteins |
| Mono S high salt buffer | self-prepared | - | 44 mM NaH2PO4; 6 mM Na2HPO4; 1-2 M NaCl; ranges: optimal value varies among FAHD proteins |
| Mono Q low salt buffer | self-prepared | - | 20 mM Tris-HCl; 15 mM NaCl; adjust to pH 8.0 |
| Mono Q high salt buffer | self-prepared | - | 20 mM Tris-HCl; 1 M NaCl; 10 % glycerol; adjust to pH 8.0 |
| G75 / G200 running buffer | self-prepared | - | 15 mM Tris-HCl; 300 mM NaCl; adjust to pH 7.4 |
| enzyme assay buffer | self-prepared | - | 50 mM Tris-HCl pH7.4; 100 mM KCl; 1 mM MgCl2 |
| protein crystallization buffer | self-prepared | - | G75 / G200 running buffer with 1 mM DTT |
| reservoir solution for crystallization | self-prepared | - | 100 mM Na-HEPES pH 7.5; 5-20 % (w/v) PEG4k; 10 mM-200 mM MgCl2 |
Referenzen
- Brouns, S. J. J., et al. Structural Insight into Substrate Binding and Catalysis of a Novel 2-Keto-3-deoxy-d-arabinonate Dehydratase Illustrates Common Mechanistic Features of the FAH Superfamily. Journal of Molecular Biology. 379, 357-371 (2008).
- Timm, D. E., Mueller, H. A., Bhanumoorthy, P., Harp, J. M., Bunick, G. J. Crystal structure and mechanism of a carbon-carbon bond hydrolase. Structure (London, England: 1993). 7, 1023-1033 (1999).
- Weiss, A. K. H., Loeffler, J. R., Liedl, K. R., Gstach, H., Jansen-Dürr, P. The fumarylacetoacetate hydrolase (FAH) superfamily of enzymes: multifunctional enzymes from microbes to mitochondria. Biochemical Society Transactions. 46, 295 (2018).
- Guimarães, S. L., et al. Crystal Structures of Apo and Liganded 4-Oxalocrotonate Decarboxylase Uncover a Structural Basis for the Metal-Assisted Decarboxylation of a Vinylogous β-Keto Acid. Biochemistry. 55, 2632 (2016).
- Zhou, N. Y., Fuenmayor, S. L., Williams, P. A. nag genes of Ralstonia (formerly Pseudomonas) sp. strain U2 encoding enzymes for gentisate catabolism. Journal of Bacteriology. 183, 700 (2001).
- Izumi, A., et al. Structure and Mechanism of HpcG, a Hydratase in the Homoprotocatechuate Degradation Pathway of Escherichia coli. Journal of Molecular Biology. 370, 899-911 (2007).
- Manjasetty, B. A., et al. X-ray structure of fumarylacetoacetate hydrolase family member Homo sapiens FLJ36880. Biological Chemistry. 385, 935-942 (2004).
- Tame, J. R. H., Namba, K., Dodson, E. J., Roper, D. I. The crystal structure of HpcE, a bifunctional decarboxylase/isomerase with a multifunctional fold. Biochemistry. 41, 2982-2989 (2002).
- Ran, T., et al. Crystal structures of Cg1458 reveal a catalytic lid domain and a common catalytic mechanism for the FAH family. The Biochemical Journal. 449, 51-60 (2013).
- Ran, T., Wang, Y., Xu, D., Wang, W. Expression, purification, crystallization and preliminary crystallographic analysis of Cg1458: A novel oxaloacetate decarboxylase from Corynebacterium glutamicum. Acta Crystallographica Section F: Structural Biology and Crystallization Communications. 67, 968-970 (2011).
- Pircher, H., et al. Identification of human Fumarylacetoacetate Hydrolase Domain-containing Protein 1 (FAHD1) as a novel mitochondrial acylpyruvase. Journal of Biological Chemistry. 286, 36500-36508 (2011).
- Pircher, H., et al. Identification of FAH domain-containing protein 1 (FAHD1) as oxaloacetate decarboxylase. Journal of Biological Chemistry. 290, 6755-6762 (2015).
- Petit, M., Koziel, R., Etemad, S., Pircher, H., Jansen-Dürr, P. Depletion of oxaloacetate decarboxylase FAHD1 inhibits mitochondrial electron transport and induces cellular senescence in human endothelial cells. Experimental Gerontology. 92, 7-12 (2017).
- Etemad, S., et al. Oxaloacetate decarboxylase FAHD1 – a new regulator of mitochondrial function and senescence. Mechanisms of Ageing and Development. 177, 22-29 (2019).
- Weiss, A. K. H., et al. Structural basis for the bi-functionality of human oxaloacetate decarboxylase FAHD1. Biochemical Journal. 475, 3561-3576 (2018).
- Taferner, A., et al. FAH domain-containing protein 1 (FAHD-1) Is required for mitochondrial function and locomotion activity in C. elegans. PLoS ONE. 10, 1-15 (2015).
- Mizutani, H., Kunishima, N. Purification, crystallization and preliminary X-ray analysis of the fumarylacetoacetase family member TTHA0809 from Thermus thermophilus HB8. Acta Crystallographica Section F Structural Biology and Crystallization Communications. 63, 792-794 (2007).
- Bateman, R. L., Bhanumoorthy, P., Witte, J. F., McClard, R. W., Grompe, M., Timm, D. E., et al. Mechanistic Inferences from the Crystal Structure of Fumarylacetoacetate Hydrolase with a Bound Phosphorus-based Inhibitor. Journal of Biological Chemistry. 276, 15284-15291 (2001).
- Zeng, F., et al. Efficient strategy for introducing large and multiple changes in plasmid DNA. Scientific Reports. 8, 1714 (2018).
- Higuchi, R., Krummel, B., Saiki, R. K. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Research. 16, 7351-7367 (1988).
- Jansen-Duerr, P., Pircher, H., Weiss, A. K. H. The FAH Fold Meets the Krebs Cycle. Molecular Enzymology and Drug Targets. 2, 1-5 (2016).
- Rupp, B. Origin and use of crystallization phase diagrams. Acta Crystallographica Section F Structural Biology Communications. 71, 247-260 (2015).
- Rupp, B. . Biomolecular Crystallography: Principles, Practice, and Application to Structural Biology. , (2010).
- Flint, D. H., Nudelman, A., Calabrese, J. C., Gottlieb, H. E. Enol oxalacetic acid exists in the Z form in the crystalline state and in solution. The Journal of Organic Chemistry. 57, 7270-7274 (1992).
- Pogson, C. I. I., Wolfe, R. G. G. Oxaloacetic acid tautomeric and hydrated forms in solution. Biochemical and Biophysical Research Communications. 46, 1048-1054 (1972).
- Kost, T. A., Condreay, J. P., Jarvis, D. L., Kost, A. T. Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nature Biotechnology. 23, 567-575 (2005).
- Steinberger, R., Westheimer, F. H. Metal Ion-catalyzed Decarboxylation: A Model for an Enzyme System 1. Journal of the American Chemical Society. 73, 429-435 (1951).
- Tate, S. S., Grzybowski, A. K., Datta, S. P. The stability constants of the magnesium complexes of the keto and enol isomers of oxaloacetic acid at 25. Journal of Chemical Society. , 1381-1389 (1964).
- Tate, S. S., Grzybowski, A. K., Datta, S. P. The acid dissociations of the keto and enol isomers of oxaloacetic acid at 25. Journal of Chemical Society. 1372, 1380 (1964).
- Brecker, L., et al. Synthesis of 2,4-diketoacids and their aqueous solution structures. New Journal of Chemistry. 23, 437-446 (1999).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten