
Method Article
Espressione, purificazione, cristallizzazione e saggi enzimatici di Fumarylacetoacetate Hydrolase Domain-Containing Proteins
In questo articolo
Riepilogo
L'espressione e la purificazione delle proteine contenenti idrolasi fumarylacetoacetate sono descritte con esempi (espressione in E. coli, FPLC). Le proteine purificate sono utilizzate per la cristallizzazione e la produzione di anticorpi e impiegate per i saggi enzimatici. Vengono presentati alcuni saggi fotometrici per mostrare la multifunzionalità di FAHD1 come oxaloacetate decarboxylase e idrolasi acylpyruvate.
Abstract
Le proteine che contengono idrolasi (FAH) di Fumarylacetoacetate (FAH) sono membri identificati della superfamiglia FAH negli eucarioti. Gli enzimi di questa superfamiglia mostrano generalmente la multi-funzionalità, che coinvolge principalmente meccanismi idrolasi e decarboxylase. Questo articolo presenta una serie di metodi consecutivi per l'espressione e la purificazione delle proteine FAHD, principalmente proteina FAHD 1 (FAHD1) ortologhi tra le specie (umane, topi, nematodi, piante, ecc.). I metodi coperti sono espressione proteica in E. coli, cromatografia di affinità, cromatografia dello scambio iostico, filtrazione del gel preparativo e analitico, cristallizzazione, diffrazione a raggi X e saggi fotometrici. Proteine concentrate con alti livelli di purezza (>98%) possono essere impiegati per la cristallizzazione o la produzione di anticorpi. Proteine di qualità simile o inferiore possono essere impiegate nei saggi enzimatici o utilizzate come antigeni nei sistemi di rilevamento (Western-Blot, ELISA). Nella discussione di questo lavoro, i meccanismi enzimatici identificati di FAHD1 sono delineati per descrivere la sua bifunzionalità idrolasi e decarboxylase in modo più dettagliato.
Introduzione
La cogiuria fumarylacetoacetate (FAH)1,2 superfamiglia di enzimi descrive un gruppo di enzimi che condividono il dominio FAH catalitico altamente conservato3,4,5,6 , 7 (in questo stato , 8 (IN vio , 9 (in vie , 10. Nonostante il loro comune centro catalitico, questi enzimi sono multifunzionali, e la maggior parte si trovano nei procarioti, dove vengono utilizzati per abbattere i composti recuperati da complesse fonti di carbonio3. Solo tre membri di questa famiglia sono stati identificati negli eucarioti finora: il nome che dà FAH2, così come la proteina che contiene il dominio FAH 1 (FAHD1)11,12,13,14 ,15 e proteina che contiene il dominio FAH 2 (FAHD2). L'esaurimento di FAHD1 è stato associato a una respirazione mitocondriale alterata13,16 e associata a un tipo reversibile di fenotipo14 della senescenza cellulare che è collegato al potenziale intermedio carenze nel sistema di trasporto degli elettroni. FaHD1 umano e i suoi ortologhi nei sistemi modello (topo, nematode, linee cellulari tumorali, piante, ecc.), così come varianti di mutazione punto selezionati, sono diventati bersagli drogabili di potenziale interesse. Per questa ricerca, le proteine ricombinanti ad alti livelli di purezza, nonché le informazioni sui meccanismi catalitici guidati da strutture cristalline e anticorpi selettivi sono vitali.
Questo manoscritto descrive i metodi per l'espressione della proteina FAHD in E. coli, la cromatografia di affinità, la cromatografia allo scambio di ioni, la precipitazione del solfato di ammonio, la filtrazione del gel preparativo e analitico, la cristallizzazione, la diffrazione a raggi X e saggi fotometrici. Lo scopo dei metodi e dei protocolli qui descritti è quello di fornire indicazioni per gli scienziati che lavorano in diversi campi come la batteriologia, la biologia vegetale, nonché gli studi sugli animali e sull'uomo, per caratterizzare i membri della superfamiglia FAH, tra cui membri di superfamiglia non caratterizzati se diventano rilevanti in un particolare campo. I protocolli qui descritti possono fornire un valido supporto per i progetti che mirano a caratterizzare altri membri della superfamiglia faH procariotica o eucatermica o eucatermica.
La logica alla base dei metodi qui descritti è il fatto che per la caratterizzazione di proteine mal descritte (in particolare, enzimi metabolici di rilevanza fisiologica sconosciuta), l'approccio per iniziare con proteine ricombinanti purificate consente sviluppo di strumenti di ricerca inestimabili e di alta qualità, come preparati in vitro attivi, anticorpi di alta qualità e inibitori farmacologici potenti e specifici per enzimi selezionati. I metodi descritti richiedono la cromatografia liquida a proteine veloce (FPLC) e la cristallografia a raggi X. I metodi alternativi (ad esempio, per esprimere proteine senza induzione chimica, o per mostrare la purificazione delle proteine per centrifugazione dopo il trattamento termico seguita da dealare e la cromatografia di esclusione delle dimensioni), possono essere trovati altrove17. Mentre è disponibile una gamma più ampia di metodi per l'espressione e la purificazione degli enzimi della superfamiglia FAH2,7,9,17,18, questo lavoro si concentra sull'espressione e purificazione delle proteine FAHD in particolare.
Nella sezione di discussione di questo manoscritto, i meccanismi catalitici identificati per la proteina FAHD1 (hydrolase, decarboxylase)15 sono descritti in modo più dettagliato, al fine di dimostrare il carattere chimico delle reazioni catalizzate. I dati ottenuti sulla base del lavoro precedente7,15,18 (PDB: 6FOG, PDB:6FOH) implicano una terza attività dell'enzima come cheto-enol isomerase.
Protocollo
1. Espressione di proteine FAHD in competente E. coli
- Trasformazione di E. Coli con vettori per l'espressione della proteina FAHD
NOTA: i passaggi descritti nella sezione seguente sono riepilogati nello schizzo nella figura 1A,B. Lo stesso protocollo si applica a qualsiasi proteina FAHD, comprese le varianti point-mutant. Tali varianti possono essere ottenute tramite la mutagenesi site-directed e le tecniche PCR19 (come SOE PCR20a due lati) da cDNA wild-type.
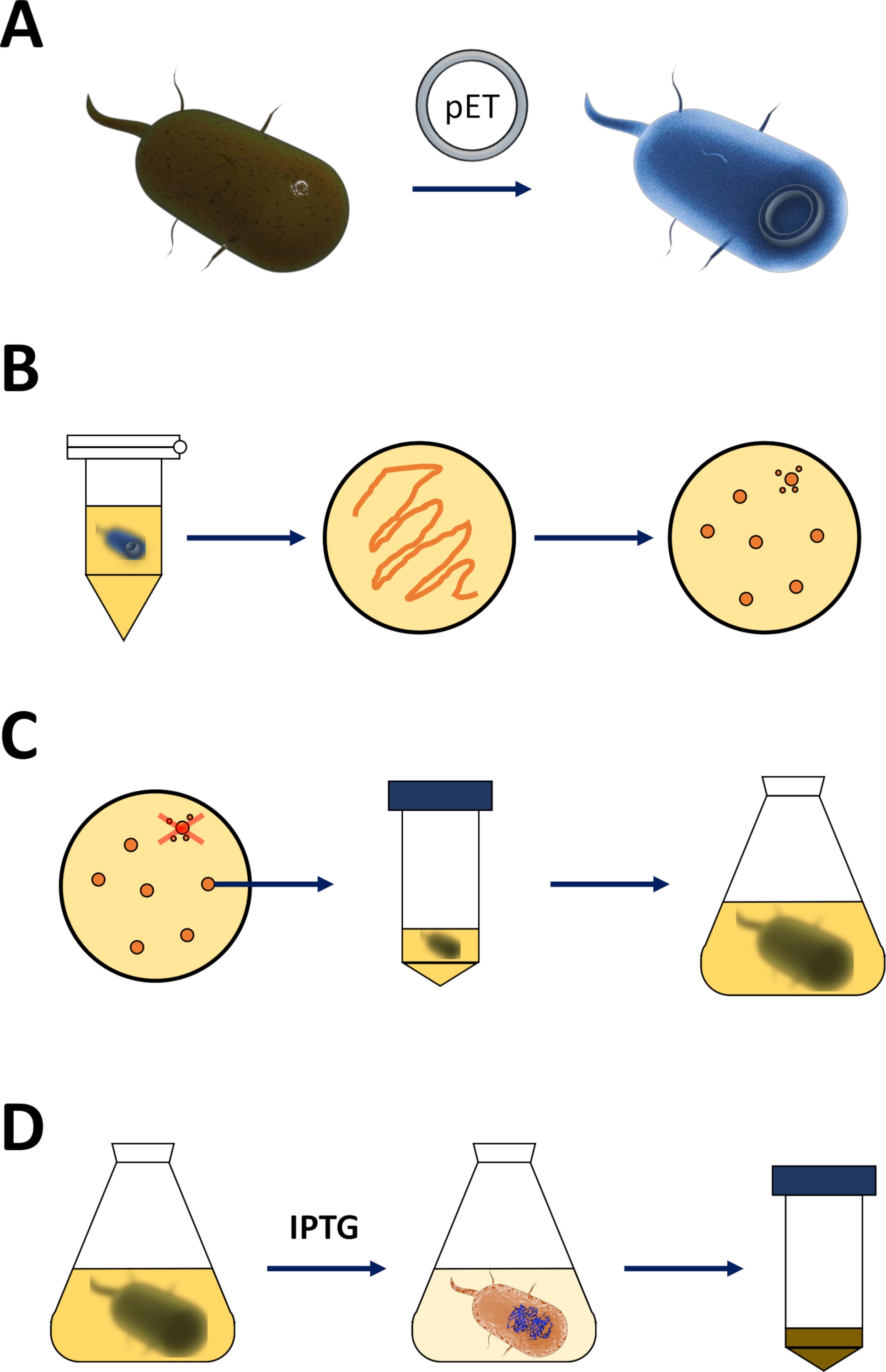
Figura 1 : Amplificazione dell'e. coli competente e induzione dell'espressione proteica.
(A) Inserimento del vettore pET nei batteri competenti BL21(DE3) pLysS E. coli, descritto nella sezione 1. (B) Protocollo di shock termico e placcatura dei batteri E. coli trasformati, descritti nel passaggio 1 del protocollo. I batteri trasformati sono placcati su piastre di agar LB con antibiotici per la selezione. (C) Amplificazione dei batteri E. coli trasformati in pET, descritti nella sezione 1. Le colonie vengono prelevate da una placca di agar LB e amplificate in mezzi nutritivi (LB o N'CYM) fino a quando la densità batterica non ha raggiunto la soglia empirica di 0,4. (D) Induzione dell'espressione proteica tramite il sistema DE3-IPTG-pET, descritto nella sezione 1 e delineato nella figura 2. La produzione di proteine è iniziata dall'applicazione della sostanza chimica IPTG. Alla fine della sezione 1 viene raccolto il pellet batterico contenente la proteina. Fare clic qui per visualizzare una versione più grande di questa figura.- Ottenere i batteri competenti BL21(DE3) pLysS E. coli e un vettore di espressione pET (vedere Tabella dei materiali). Preferibilmente scegliere un vettore pET che codifica anche un N-terminal His-tag o tag di cattura correlati per comodità per semplificare i seguenti passaggi di purificazione.
- Ottenere cDNA della proteina FAHD di scelta e inserirlo nel sito di clonazione attivo del vettore di espressione pET, tra i siti promotori T7 e T7 terminator, rispettivamente.
- Dopo il successo dell'amplificazione e della verifica plasmide [tramite sequenziamento da parte di un fornitore commerciale (i primer T7 possono essere utilizzati con il sistema pET per comodità: Promotore T7, primer in avanti: TAATACGACTCACTATAGGG; T7 terminatore, primer inverso: GCTAGTTATTTGCTGCGG)], inserire 5-10 ng di plasmide in 100 -L di competenti batteri BL21(DE3) pLysS E. coli sul ghiaccio. Non aspirare su e giù, ma toccare leggermente il tubo con al fine di mescolare il contenuto.
- Tenere i batteri sul ghiaccio per 30 min, toccando delicatamente il tubo ogni pochi min.
- Riscaldare un dispositivo di riscaldamento o un bagno d'acqua a 42 gradi (esatto). Mettere il tubo contenente i batteri nell'apparecchio e tenerli per 90 s (esatto). Metterli immediatamente in ghiaccio (Figura 1A).
- Dopo 5-10 min sul ghiaccio, aggiungere 600 -L di supporto NC-YM (vedi Tabella dei materiali) e mettere il tubo in un'incubatrice di batteri. Agitare il tubo a velocità media orientata lungo la direzione di agitazione a 37 gradi centigradi per 1 h.
- Piastra 200 - L della coltura batterica su una piastra LB-agar di 10 cm (vedi Tabella deimateriali), contenente antibiotici di selezione prescelta [ad esempio, uno specifico per la resistenza BL21(DE3) pLysS (cloramfenicolo) e uno per la resistenza codificato sul vettore pET (kanamycin o ampicillin, Figura 1B)].
- Coltura i batteri sulla piastra LB-agar in un'incubatrice batterica a 37 gradi durante la notte.
- Espressione delle proteine FAHD per induzione IPTG
NOTA: i primi passaggi illustrati nella sezione seguente sono riepilogati come schizzo nella figura 1C,D. Il sistema di espressione T7 tramite la combinazione della cassetta batterica DE3 e del sistema vettoriale pET è riassunto nella Figura 2.
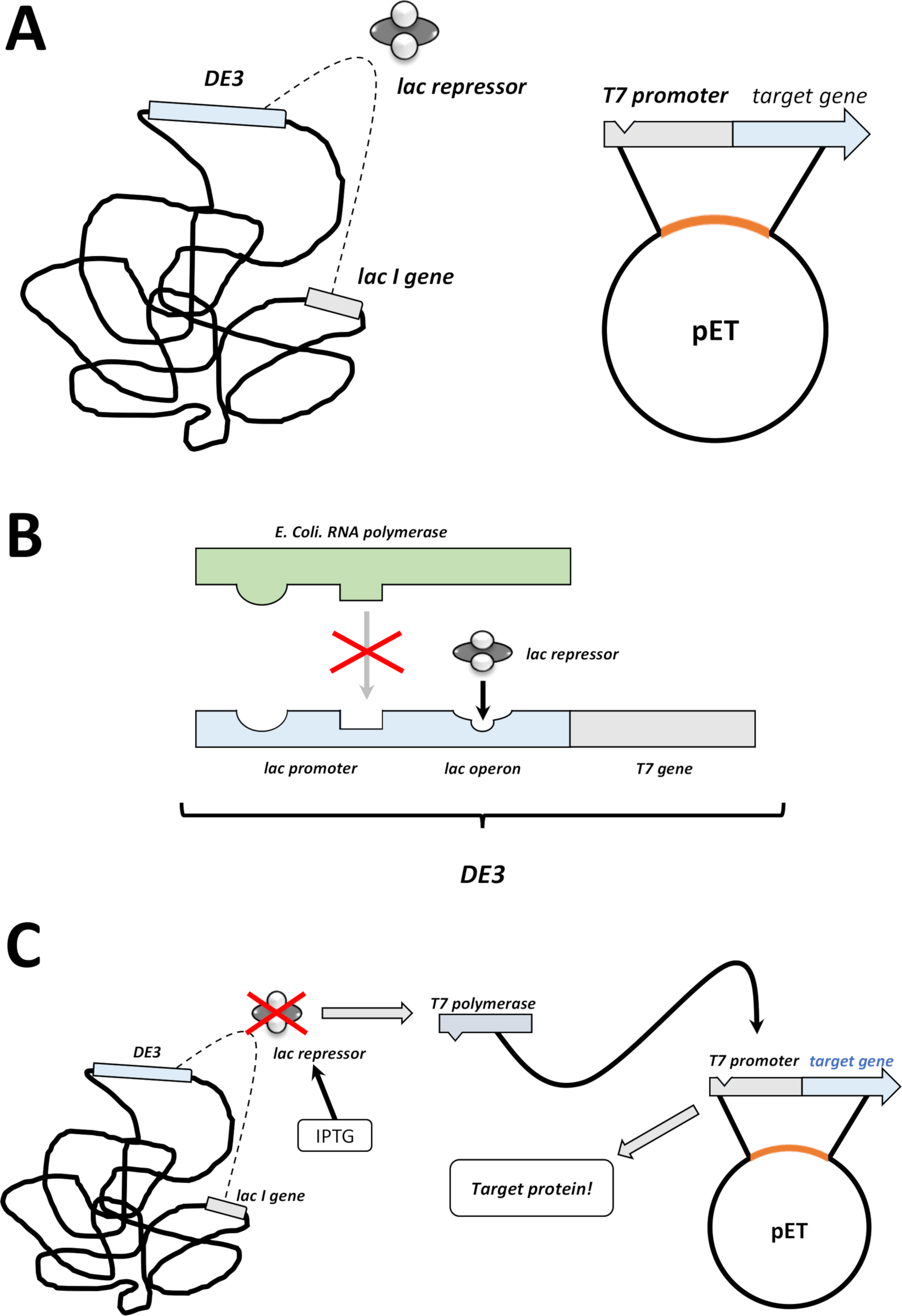
Figura 2 : Spiegazione del sistema duale vettoriale DE3 cassetta/pET.
(A) Il genoma disegnato del vettore pET ha trasformato i batteri BL21(DE3) pLysS E. coli. Il genoma batterico nativo trasporta una cassetta DE3 (vedi pannello B), così come un gene lac che esprime costantemente unità reprimere lac. Il vettore pET non nativo trasporta il gene proteico inserito tra un promotore di polimerasi T7 e una sequenza di terminatore. Maggiori dettagli nel pannello B. (B) La cassetta DE3 del genoma batterico nativo codifica le informazioni per la polimerasi T7 in termini di un operone polimerasi E. coli RNA. Questa proteina, tuttavia, non è espressa perché l'unità repressore lac impedisce alla proteina polimerasi dell'RNA di legarsi. Quindi non si esprime nessun aipo polimerasi T7 e non viene espressa alcuna proteina esogena. (C) L'applicazione della sostanza chimica IPTG (Tabella dei materiali) distorce la struttura delle unità repressive lac e impedisce loro di legarsi alla cassetta DE3. Di conseguenza, la polimerasi dell'RNA può ora legarsi alla cassetta, per la quale viene espressa la polimerasi T7, così come la proteina esogena alla fine. Fare clic qui per visualizzare una versione più grande di questa figura.- Dopo aver avuto successo nella formazione delle colonie, scegli una singola colonia (senza colonie satellitari) e disperdela in 5 mL di n.CYM o LB media con antibiotici, selezionata come prima (passaggio 1.1.7). Coltura nell'incubatrice batterica a 37 gradi durante la notte (Figura 1C).
- Dopo una crescita batterica di successo, amplificare i batteri in 250 mL, 500 mL, o 1 L lotti di media, a seconda della domanda di quantità di proteine.
- Adeguato al volume, applicare gli antibiotici selezionati nel passaggio 1.1.7 e aggiungere circa l'1-2% della pre-coltura batterica densa (ad esempio, da 2,5 a 5,0 mL a 250 mL di volume medio, ecc.). Prendere un campione da utilizzare nel passaggio 1.2.5 (1 mL o più) e controllare la densità ottica (OD) a 600 nm. Batteri di coltura nell'incubatrice batterica a 37 gradi centigradi per 2-3 h (Figura 1C).
- Dopo 2-3 h, disegnare un campione per l'analisi fotometrica. Se l'OD a 600 nm ha raggiunto 0,4, applicare 200 m fino a 1 mM di isopropile-z-D-thiogalactopyranosid (IPTG, vedere Tabella dei materiali).
NOTA: Il valore effettivo è empirico per ogni proteina FAHD o variante di mutazione puntiforme, dove 1 mM IPTG è il massimo da applicare. Ciò induce l'espressione della proteina (Figura 1D, Figura 2C). - Dopo 3-5 ore in più nell'incubatrice batterica a 37 gradi centigradi, l'espressione proteica si esaurisce.
NOTA: vedere la sezione Discussione per i commenti sul controllo della temperatura. Più di 5 h di agitazione dopo l'induzione non è raccomandato. Prendere un campione da utilizzare nel passaggio 1.2.5 (1 mL o più) e controllare la densità ottica (OD) a 600 nm.- Raccogliere il pellet batterico via centrifugazione a 5.000 x g per 5 min. Scartare il supernatante e congelare il pellet a -80 gradi centigradi per un deposito più lungo o -20 gradi centigradi per una breve conservazione (Figura 1D).
- Verificare l'induzione tramite i due campioni fotometrici recuperati, che sono etichettati "-I" (prima dell'induzione) e "-I" (dopo l'induzione). Dopo la centrifugazione e la sospensione del pellet batterico, analizzare i due campioni da SDS-PAGE caricando la stessa quantità di proteine totali.
NOTA: Il campione "I" dovrebbe mostrare una forte banda associata al peso molecolare della proteina prescelta, mentre il campione "-I" non dovrebbe contenere questa banda. Un basso livello di induzione è un problema comune per la produzione di proteine, ma il livello di proteine espresse è spesso sufficiente per i passaggi successivi. Un alto livello di induzione è un vantaggio, ma non è obbligatorio.
{kind=link}
{kind=link}
2. Lisidi di pellet batterici e filtrazione dei detriti
- A seconda che la proteina scelta sia contag O senza tag, selezionare buffer di corsa Ni-NTA (His-tagged, vedere Table of Materials) o buffer di running HIC ghiacciato (senza tag).
- Per ogni 250 mL di sospensione batterica originale, applicare 5 mL del buffer selezionato al pellet batterico (5 mL per 250 mL, 10 mL per 500 mL, ecc.). Aggiungere 10 l'oratoetanolo (z-ME) per 5 mL di buffer applicato. Utilizzare un tubo Pasteur da 10 ml per forzare meccanicamente il pellet in sospensione graffiando e pipettando (evitare la formazione di bolle d'aria durante la pipa). Infine trasferire tutte le sospensioni in un tubo da 50 mL.
- Preferibilmente sonicare (6x per 15 s a media forza) la sospensione.
- Centrifuga per 30 min ad alta velocità (10.000 x g)a 4 gradi centigradi. Filtrare il supernatante consecutivamente con unità filtranti (ad es., 0,45 m, 0,22 m) sul ghiaccio.
NOTA: A seconda della precedente fase di centrifugazione, la filtrazione direttamente attraverso una piccola dimensione del poro del filtro può essere noiosa e di solito richiede la pre-filtrazione attraverso una dimensione dei pori più grande. Il DNAse può essere aggiunto per ottenere risultati migliori. - Conservare il campione sul ghiaccio e procedere immediatamente con la sezione 3 o 4, a seconda che la proteina sia marcata o senza tag.
3. Purificazione delle sueproteine FAHD taggate utilizzando la cromatografia di affinità Ni-NTA
NOTA: gli ioni Ni2 sono legati tramite acido nitrilotriacetic (NTA) a una resina agarose che viene utilizzata nella cromatografia di affinità (cromatografia degli ioni metallici immobilizzati, IMAC, Figura 3A). I tag degli amminoacidi poli-istidini si legano fortemente a questo Ni-chelato, e le proteine his-tagged possono essere separate dalla maggior parte delle proteine rimanenti. Un'alternativa alla preparazione descritta delle colonne Ni-NTA consiste nell'utilizzare colonne Ni-NTA preconfezionate e un sistema FPLC.

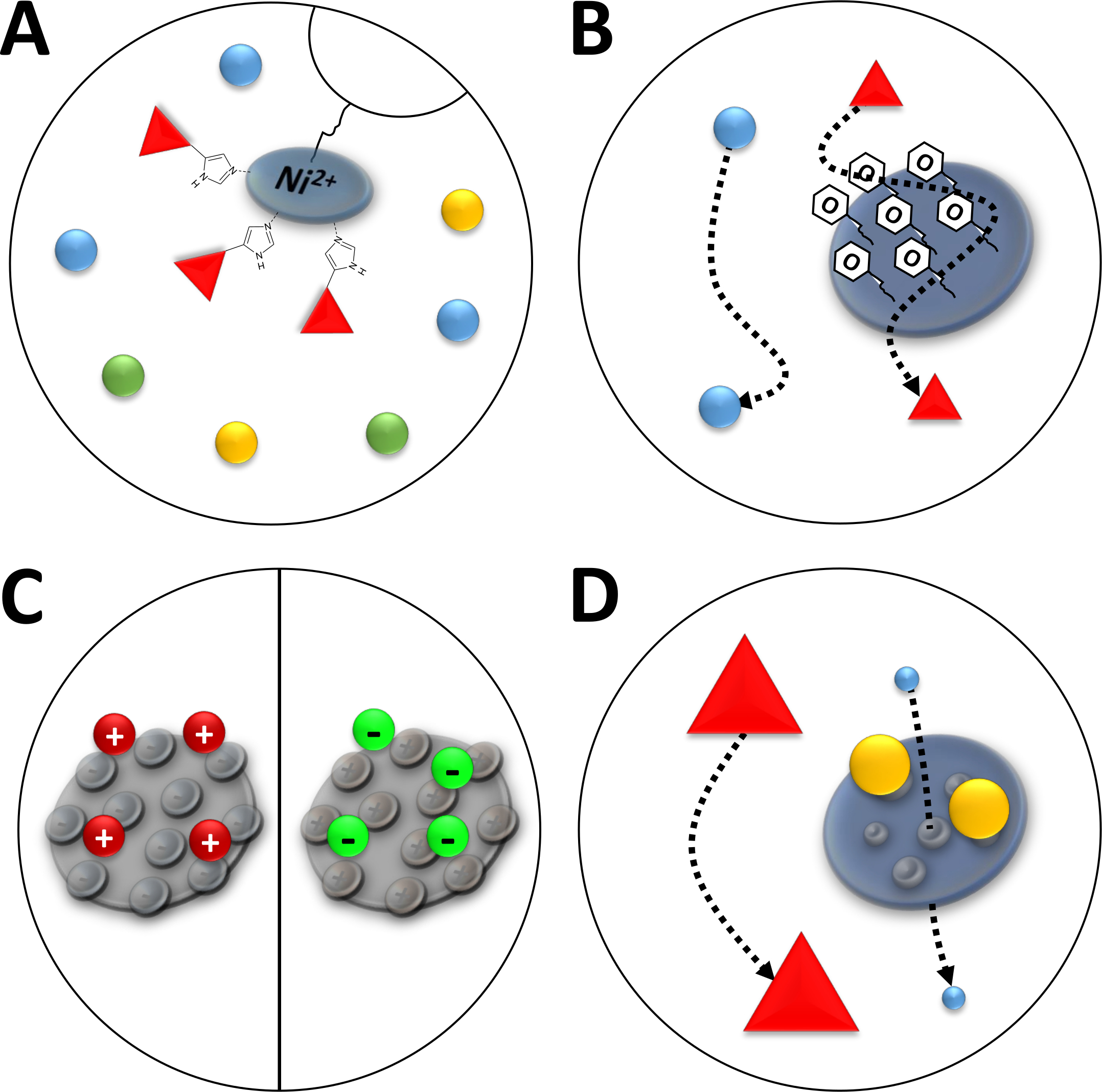
Figura 3 : Illustrazioni di schizzi di tipi comuni di cromatografia.
(A) La resina di una colonna Ni-NTA. NTA contiene ioni di nichel bivalenti che vengono utilizzati in termini di cromatografia di affinità degli ioni metallici immobilizzati (IMAC). I tag poli-istadina si legano preferibilmente a questo motivo e possono essere eluiti da imidazole. (B) Il rivestimento tipico delle particelle di silice in una cromatografia di interazione idrofobica a base di fenil (HIC-phenyl). Le proteine idrofobiche interagiscono con il materiale di rivestimento e vengono ritardate nella loro migrazione, mentre altre non lo sono. (C) Il rivestimento tipico delle particelle di silice nella cromatografia dell'interazione ionica. Le proteine polarizzate e cariche interagiscono con il materiale di rivestimento e vengono ritardate nella loro migrazione, mentre altre non lo sono. (D) La resina di un gel di silice in cromatografia di dimensioni-esclusione (SEC). Sulla base di pori definiti nel materiale di silice, le proteine possono essere separate per la loro dimensione (in una prima approssimazione corrispondente alla loro massa molecolare). Piccole proteine permeano il materiale colonna poroso e sono ritardate, mentre le grandi proteine migrano più velocemente intorno alle particelle porose. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Procedere dal punto 2.5 (cioè, la proteina si trova nel tampone di corsa Ni-NTA e filtrata da 0,22 unità di filtro su ghiaccio).
- Preparare una colonna di plastica o vetro vuota lavando la colonna vuota e attaccandola a un fermo stabile. Scegliere la dimensione della colonna a seconda del volume della sospensione proteica.
- Per ogni 10 mL di sospensione proteica, applicare 500 l of Ni-NTA agarose liquame nella colonna (agitare pesantemente prima dell'uso). Applicare il liquame lentamente e dropwise sul filtro inferiore della colonna utilizzando una pipetta. Lasciare che la colonna si stabilizzi, il che richiede alcuni secondi.
- Riempire completamente la colonna con buffer in esecuzione Ni-NTA, assicurandosi di non interrompere la resina agarose. Lasciate che il buffer attraversa per gravità. Il processo può essere accelerato applicando la pressione del pollice sul liquido (utilizzando un coperchio o guanti e la pressione del pollice), ma fare attenzione a non distorcere la resina agarose.
- Applicare la sospensione proteica. Come prima, lasciate che il campione attraversa per gravità. Non è consigliabile accelerare questo passaggio utilizzando la pressione del pollice, poiché il legame delle proteine alla colonna è migliorato se la portata è bassa. Raccogliere il flow-through in un tubo (Tabella dei materiali).
- Dopo che l'esempio è passato, riempire nuovamente l'intera colonna con il buffer in esecuzione Ni-NTA. Fare attenzione a non interrompere la resina di agarose. Lasciate che il campione attraversa per gravità, ma a differenza della fase precedente, si consiglia di accelerare il processo attraverso la pressione del pollice, poiché potenziali contaminazioni a causa di interazioni non specifiche possono essere interrotte in questo modo. Raccogliere la soluzione di lavaggio in un tubo. Ripetere questo passaggio.
- Posizionare una cuvette UV-trasparente sotto la colonna e applicare 1 mL di buffer di eluzione Ni-NTA. Raccogliere il campione senza applicare alcuna pressione del pollice alla resina.
- Controllare la densità ottica (OD) dell'esempio a 280 nm rispetto a un campione vuoto (ad esempio, buffer di eluzione Ni-NTA). In modo ottimale, nel campione viene visualizzato un OD maggiore di 2,5. Un OD inferiore a 0,5 indica che nel campione non è presente una quantità significativa di proteine.
NOTA: Come descritto nella sezione di discussione, le concentrazioni di sale e imidazole del tampone di eluizione potrebbero dover essere adattate individualmente per ogni proteina FAHD. - Ripetere i passaggi 3.1.7 e 3.1.8 finché l'OD non scende al di sotto di 0,5. Mettere in comune tutti i campioni con OD più alto in un tubo sul ghiaccio.
- Ricominciare dal passaggio 3.1.4, usando il flow-through del passaggio 3.1.5 come nuovo input per questa ripetizione del passaggio 3.1.5. Ripetere questo processo finché il primo campione raccolto nel passaggio 3.1.6 visualizza un OD inferiore a 0,5.
NOTA: Come descritto nella parte relativa alla risoluzione dei problemi della sezionedi discussione, le proteine con etichettatura His potrebbero legarsi in modo insufficiente alla resina Ni 2. In questi casi, è necessaria la ripetizione di questo passaggio o di metodi alternativi (ad esempio, cromatografia dello scambio ionomi) . - Preleva campioni di tutte le frazioni intermedie per l'analisi SDS-PAGE.
- Le proteine FAHD nel buffer di eluizione Ni-NTA precipiteranno al congelamento e allo scongelamento. Pertanto, dialze la proteina contro un buffer diverso (durante la notte sul ghiaccio, utilizzando 1 L di DTT per 100 mL di tampone di dialisi). Utilizzare buffer a basso contenuto di sale in base al tipo di cromatografia dello scambio ioleomatico da eseguire dopo questo passaggio. Utilizzare tubi di cellulosa comuni con un tipico peso molecolare cut-off di 14 kDa (Tabella dei materiali).
- Dopo una notte di dialisi, facoltativamente concentrare la proteina utilizzando unità di filtro ultra-centrifugation. Eseguire l'analisi SDS-PAGE (12,5% gel da corsa, 4% gel impilamento) per verificare la potenziale perdita di proteine, eluizione insufficiente, e la purezza delle proteine in generale. Se tutto va bene, procedere alla sezione 5.
4. Purificazione delle proteine FAHD senza tag tramite cromatografia di interazione idrofobica (HIC)
NOTA: I gruppi fenili sulla superficie di rivestimento di un gel di silice in una colonna HIC per FPLC (Figura 3B) consentono la separazione delle proteine in base al carattere idrofobico. I passaggi descritti devono essere eseguiti con un sistema FPLC dotato di una colonna di 5 mL di HIC-phenyl. Le colonne possono essere lavate con 1 M NaOH per essere riutilizzate per diverse proteine. Tuttavia, le colonne un tempo utilizzate per un tipo di proteina FAHD devono essere riutilizzate solo per questo tipo di proteine.
-
Precipitazioni a zolfo di ammonio (AS)
- Procedere dal passaggio 2.5. La proteina è nel tampone di corsa HIC ghiacciato (Tabella dei materiali).
- Valutare il volume della soluzione proteica preparata con precisione al microlitro (Viniziale). Lentamente e drop-saggio aggiungere pre-raffreddato HIC in esecuzione soluzione di buffer AS, fino a quando non viene raggiunta una saturazione 35 volume-% AS: VAS aggiunto - Viniziale - 0.538. Mescolare delicatamente la soluzione per 30 min. Centrifughe per 15 min ad alta velocità (10.000 x g) a 4 gradi centigradi.
- Filtrare il supernatante utilizzando un'unità filtro 0,22 m sul ghiaccio. Se lo si desidera, prendere un campione per l'analisi SDS-PAGE: diluire 1:4 e riscaldare immediatamente a 95 gradi centigradi per 5 min, altrimenti il campione sarà grumo. Il campione può essere congelato a questo punto (-20 gradi centigradi) per procedere con un altro giorno.
-
FPLC utilizzando una colonna HIC
- Impostare il sistema FPLC ed equilibrare una colonna HIC-phenyyl 5mL con 5 volumi di colonna (CV) del 20% EtOH (in H2O) seguiti da 5 CV di H2O.
- Mescolare 260 mL di BUFFER in esecuzione HIC (esatto) con 140 mL di HIC in esecuzione buffer AS (esatto). Ciò si traduce in una soluzione AS 35 volume-%. Controllare il pH (7.0); questo è il buffer A. Buffer B è 250 mL di buffer in esecuzione. Aggiungere 1 mM DTT a entrambi i buffer A e B, quindi tenerli sul ghiaccio.
- E la colonna con 8 mL di buffer A, 8 mL del buffer B e 8 mL del buffer A in questa sequenza. Applicare l'esempio preparato nel passaggio 4.1 del protocollo. Lavare con buffer A, fino a quando l'assorbimento ottico di base a 280 nm raggiunge 1000-500 mAU.
- Applicare una miscela di buffer A e B, in modo che la concentrazione di AS sia 33 % (w/v). Lavare con 1 CV, risultando in un altopiano nel cromatogramma. Impostare una sfumatura di buffer B (fino al 100% tampone B nel tempo): 1,5 mL di buffer B in 3,8 min (ad esempio, 5,7% buffer B con pendenza B/mL dell'1%). Quando il segnale UV a 280 nm si alza, inizia a raccogliere la frazione e posizionala immediatamente sul ghiaccio.
- Alla fine, lavare la colonna con il tamponi B. Preleva campioni di tutte le frazioni per l'analisi SDS-PAGE. Congelare tutti i campioni utilizzando l'azoto liquido e conservarli a -80 gradi centigradi.
- Eseguire l'analisi SDS-PAGE (e western blot), per rilevare la proteina FAHD nelle frazioni raccolte. Le frazioni che contengono la proteina vengono raggruppate e applicate per un'ulteriore purificazione, come descritto nei seguenti passaggi di protocollo. Lavare la colonna con H2O e 20% EtOH (in H2O).
5. Purificazione delle proteine FAHD tramite cromatografia di scambio iosto
NOTA: Le molecole con gruppi funzionali caricati sono legate a una colonna di particelle di silice per FPLC (Figura 3C). Ciò consente la differenziazione delle proteine in base al loro carattere ionico, come la carica superficiale. I passaggi descritti devono essere eseguiti rispettivamente con un computer FPLC e il know-how associato. Il metodo descritto è lo stesso per la cromatografia di scambio cationico o anionico, ma i buffer da utilizzare sono leggermente diversi.
- Ha scelto il sistema di cromatografia di scambio cationico o anionico. Questa scelta è empirica e può variare tra le proteine FAHD. In modo ottimale, entrambi i metodi possono essere utilizzati consecutivamente.
- Impostare il sistema FPLC e lavare la colonna con 5 CV del 20% EtOH (in H2O), seguito da 5 CV di H2O. Equilibrate la colonna con 1 CV di buffer a basso contenuto di sale, tampone ad alto sale e di nuovo buffer a basso contenuto di sale in questa sequenza.
- Applicare il campione (dialyzed rispetto al buffer di sale basso corretto dal passaggio 3.1.11) nella colonna. Raccogliere il flow-through. Lavare la colonna per 1 CV con basso buffer di sale.
- Impostazione di un'elusione graduale: 100% buffer di sale alto in 30 min ad una portata di 1 mL/min, o 60 min ad una velocità di flusso di 0,5 mL/min. Questo può essere riselezionato in base a un cromatogramma FPLC già noto, al fine di ottimizzare la purificazione. Raccogli tutte le frazioni di picco.
NOTA: Le condizioni di sale elevato possono variare tra le proteine FAHD, come descritto nella sezione di discussione. - Al termine del gradiente, eseguire con buffer ad alto sale fino a quando non vengono rilevati più picchi nell'intervallo di 1 CV (raccogliere le frazioni).
- Prendete campioni di tutte le frazioni raccolte ed eseguite l'analisi SDS-PAGE (12,5% gel in esecuzione, 4% gel impilamento). Congelare i singoli campioni in azoto liquido e conservarli a -80 gradi centigradi.
- Al termine dell'analisi SDS-PAGE, raggruppare i campioni contenenti la proteina FAHD ed eliminare gli altri. Facoltativamente, concentrare la proteina utilizzando unità filtrante ultra-centrifugazione.
- Applicare 1 mL del 25% di SDS in 0,5 M NaOH (o altri detergenti) per pulire la colonna. Lavare la colonna con H2O e 20% EtOH (in H2O).
- Facoltativamente, ripetere la sezione 5 con la colonna alternativa (cromatografia di scambio cationico o anionico). La proteina ottenuta da questo metodo è sufficientemente pura per eseguire saggi di attività di base o può essere utilizzata nei saggi di screening per la cristallografia. Per applicazioni avanzate, procedere con la sezione 6.
6. Purificazione delle proteine FAHD tramite cromatografia taglia-esclusione (SEC)
NOTA: Le particelle di popolate in una colonna di gel di silice per FPLC consentono la differenziazione delle proteine in base alle dimensioni molecolari, come il raggio idrodinamico (Figura 3D). I passaggi descritti devono essere eseguiti con un sistema FPLC, utilizzando le colonne SEC.
- Scegliere una colonna SEC, a seconda dei pesi molecolari delle contaminazioni ancora presenti, come rilevato tramite SDS-PAGE e colorazione d'argento. Il metodo strutturato è adatto per entrambe le colonne. Lavare la colonna durante la notte con 400 mL di H2O ed equilibrate con tampone di funzionamento SEC. Si consiglia di scrivere un programma per il sistema FPLC per automatizzare questo passaggio.
- Aggiungere 1 mM DTT a 300 mL di SEC tampone di esecuzione e metterlo sul ghiaccio. Questo è il buffer in esecuzione. Applicare 60 mL di questo buffer alla colonna.
- Centrifugare il campione proteico (10.000 x g per 10 min) per rimuovere eventuali micro-precipitazioni. Applicare il super-atuante alla colonna. Generalmente si raccomanda di filtrare il super-natante prima di FPLC.
- Applicare il buffer di corsa alla colonna fino a quando tutte le proteine non vengono eluite. Raccogliere tutti i picchi in frazioni di volume adatto (ad esempio, 2 mL). Prelevare campioni per SDS-PAGE e congelare tutte le frazioni utilizzando azoto liquido. Conservare le frazioni congelate a -80 gradi centigradi.
- Dopo l'analisi SDS-PAGE (e western blot), raccogliere e mettere in comune tutte le frazioni contenenti la proteina FAHD. La colorazione dell'argento è raccomandata per rilevare piccole contaminazioni che potrebbero essere ancora presenti.
- Utilizzare unità di filtro ultra-centrifugazione per concentrare la proteina. Anche se non è obbligatorio per le proteine FAHD, in generale una fase di dealificazione (ad esempio, per dialisi) è raccomandata per i saggi enzimatici e la cristallizzazione.
- Ripetere i passaggi da 6,3 a 6,6 più volte con diverse portate e concentrazioni di sale (empiriche) al fine di migliorare la purezza della proteina FAHD. Lavare la colonna durante la notte con H2O e 20% EtOH (in H2O).
7. Saggi di attività FAHD di base con substrati oxaloacetate e acetilpyruvate
NOTA: la proteina FAHD 1 (FAHD1) mostra l'attività di oxaloacetate decarboxylase (ODx) e idrolasi acylpyruvate (ApH). Questo è delineato in modo più dettagliato nella sezione di discussione. A causa della destabilizzazione mediante la tautomerizzazione acquosa in soluzione acquosa (ad esempio, l'enolizzazione), l'oxaloacetate decade da solo nel tempo (auto-decarboxylation) in funzione della concentrazione di co-factor e del pH. A circa un pH di 7 e una temperatura di 25 gradi centigradi, questo effetto non è drammatico, ma i saggi devono essere svuotati per tenere conto sia dell'auto-decarboxylazione che della concentrazione degli enzimi. Lo schema di pipettaggio è delineato in Figura 4A. In generale, si consiglia di utilizzare pipette ben calibrate per questo saggio, in quanto è abbastanza sensibile agli errori di pipettatura minori.

Figura 4 : Schema di pipettaggio schizzato per saggi enzimatici.
(A) Uno schema di pipettaggio abbozzato per saggi di base sulle proteine FAHD a base di substrato. Substrato vuoto: -S/-E; campione di substrato: S/-E; enzima vuoto: -S/ enzima: S / E (S: substrato, E: enzima). Vedere il passaggio 7 del protocollo per ulteriori dettagli. (B) Uno schema di pipettaggio abbozzato per valutare la cinetica Michaelis-Menten della proteina FAHD. Substrato vuoto: -S/-E; campione di substrato: S/-E; enzima vuoto: -S/ enzima: S / E (S: substrato, E: enzima). Vedere la sezione 8 del protocollo per maggiori dettagli. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Avviare un lettore di microplacie ed equilimare per 30 min a 25 gradi centigradi. Impostare un programma per la lettura di 12 pozzi (come descritto nella Figura 4A) a 255 nm. Si consiglia di utilizzare 25 letture multiple con ritardo di 5 ms. Impostare un ciclo per misurare 15x ogni 2 min (30 min totale).
- Per impostazione predefinita, preparare un buffer di analisi enzimatica (vedere Tabella dei materiali) con 1 mM MgCl2 a pH 7.4. Le proteine FAHD varianti possono richiedere diversi cofattori o livelli di pH. Mg2 e Mn2 sono noti cofactorper per FAHD13,11,12,21.
- Creare una soluzione proteica da 1 g/l, diluindo con il buffer di asve (Tabella dei Materiali).
- Impostare 1 mL di soluzione da 20 mM di un substrato da testare (finora i substrati identificati delle proteine FAHD sono elencati altrove3) nel buffer di analisi enzimatica.
- Secondo lo schema di pipettaggio mostrato nella Figura 4A, preparare l'enzima vuoto e pozzi campione: pipet 90 - L di assaggio enzimatico buffer (Tabella dei materiali) nei pozzi con 5 .L (5 g) di soluzione enzimatica.
- Secondo lo schema di pipettaggio mostrato in Figura 4A, preparare il substrato vuoto e pozzi campione: pipet 95 -L di enzima assaggio buffer nei pozzi.
- Subito prima della misurazione, applicare 5 l di assaggio di enzima buffer nei sei pozzi vuoti. Applicare 5 l della soluzione di substrato da 20 mM ai pozze di campionamento. Si consiglia di utilizzare una pipetta multicanale.
- Utilizzare una pipetta multicanale a impostazioni 50 per mescolare delicatamente tutti i pozze. Iniziare con gli spazi vuoti e procedere con i pozzi campione. Fare attenzione a non creare bolle. Inserire la piastra in un lettore di micropla e misurare ogni bene a 255 nm (come descritto nel passaggio 7.1).
- Eseguire l'analisi in un foglio di calcolo. Copia i dati grezzi dal fotometro in un foglio di calcolo e scrivi tutte le impostazioni (ad esempio, tutta la documentazione) in un altro foglio. Calcolare la media dei dati dei tre pozze di ciascuno dei quattro preparativi. Sottrarre lo spazio vuoto dal campione. Calcolare anche le deviazioni standard e sommare le deviazioni di vuoto e campione.
- Traccia questi dati (y: densità ottica, x: tempo in min). Dovrebbe essere visualizzata una curva esponenzialmente decrescente. A seconda del tipo di substrato in uso, si può osservare un aumento iniziale entro i primi 10 min, dopo di che il segnale diminuisce. Questo è attribuito alla tautomerizzazione keto-enol del substrato, come descritto più in dettaglio nella sezione di discussione.
- Dividere i dati del segnale ottico nel tempo per il valore massimo del grafico, al fine di scalare i dati verso il basso nell'intervallo [0, 1] (un esempio è fornito nella Figura 5A). Identificare l'intervallo lineare della curva, a partire dalla diminuzione iniziale, e calcolare la pendenza negativa (1/min).
- Il corso di tempo della diminuzione della OD è associato al substrato attraverso la sua concentrazione iniziale: pendenza 100 nmol/well. Utilizzando la concentrazione di proteine valutata c0, viene calcolata l'attività specifica: 100 nmol/well : pendenza 1/c0. Esprimendo c0 nel pozzo, l'attività specifica calcolata in questo modo viene espressa utilizzando l'unità nmol/min/g, che equivale a .mol/min/mg.
8. Valutazione della cinetica Michaelis-Menten delle proteine FAHD
NOTA: La valutazione della cinetica Michaelis-Menten delle proteine FAHD è noiosa, poiché l'attività specifica delle proteine dipende sia dalla relativa concentrazione di proteine-substrato che dal volume fisico in cui si sta verificando la reazione. Per ottenere risultati affidabili, è necessario stabilire una cinetica stabile. Un protocollo testato su una piastra trasparente UV ben 96 è delineato nei seguenti passaggi. Ogni passo deve essere eseguito con grande cura, come errori minori di solito rovinare l'esperimento. Si consiglia di padroneggiare i saggi descritti nella sezione 7 prima di tentare l'esempio più complicato descritto di seguito.

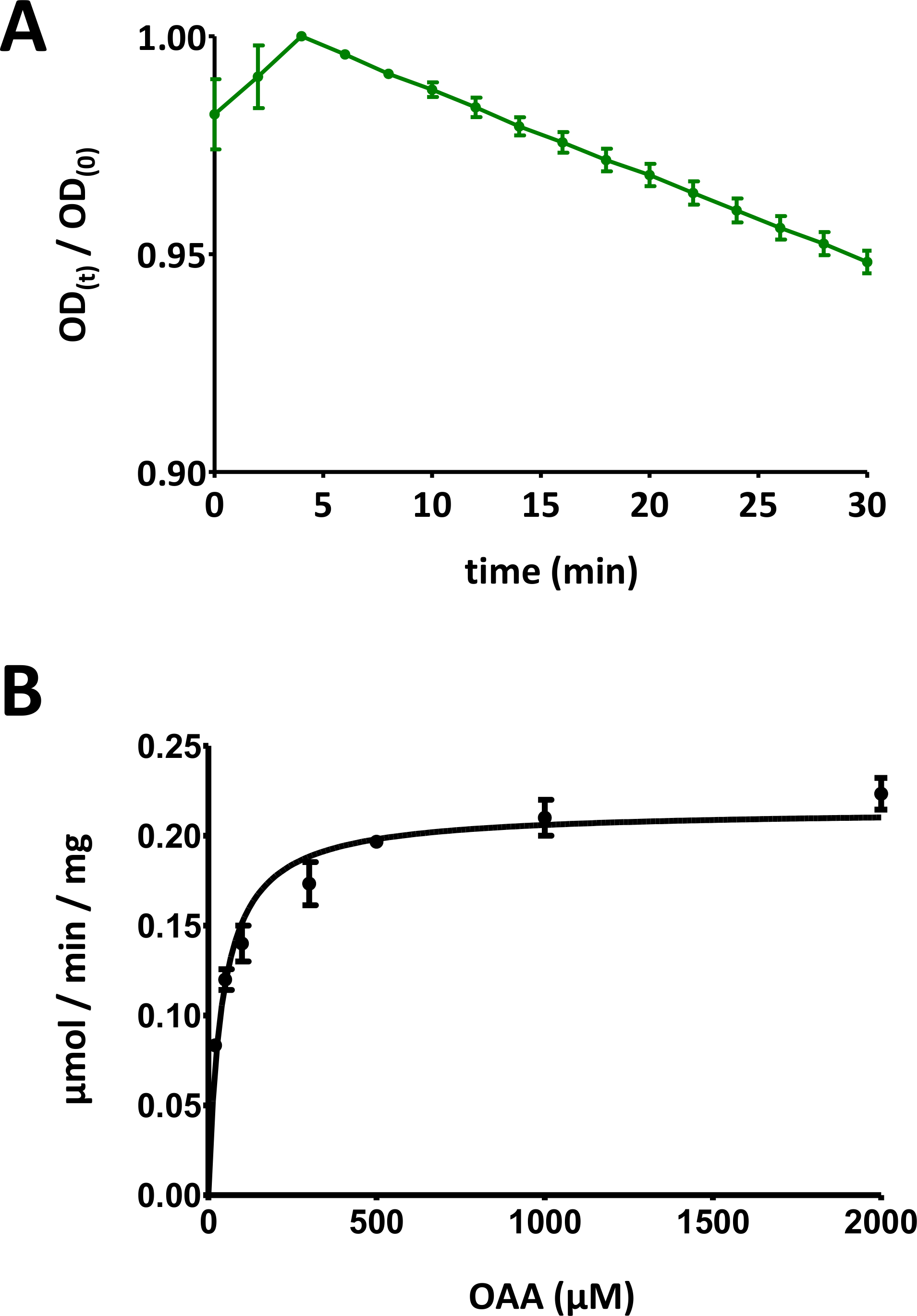
Figura 5 : Risultati esemplari dei saggi enzimatici.
(A) Una curva esemplare di assorbimento UV ottenuta per i saggi di base delle proteine FAHD a base di substrato (normalizzati nell'intervallo da 0 a 1) con deviazione standard. Il rapporto densità ottica (OD) [OD(t)/OD(0)] in un dato momento t [OD(t)] viene normalizzato all'OD iniziale [t - 0; OD(0)]. Vedere la sezione 7 del protocollo per maggiori dettagli. (B) Cinetica Michaelis-Menten esemplare della proteina umana FAHD1 con deviazione standard. Vedere la sezione 8 del protocollo per maggiori dettagli. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Avviare un lettore di micropille ed equilipare per 30 min a 25 gradi centigradi. Impostare un programma per la lettura di 72 pozzi (come descritto nella Figura 4B) a 255 nm. Si consiglia di utilizzare 25 letture multiple con un ritardo di 5 ms. Impostare un ciclo per misurare 15x ogni 2 min (30 min totale).
- Eseguire i passaggi 7.2 e 7.3. Quindi, impostare 1 mL di 100 mM di soluzione di substrato in buffer di aspo enzima.
- Preparare le diluizioni della soluzione di substrato nel buffer di analisi enzimatica: 40 mM, 20 mM, 10 mM, 6 mM, 4 mM, 2 mM. Il saggio viene eseguito con concentrazioni di enzimi/substrati a coppie ("regolate"). A tale scopo, preparare le seguenti diluizioni della soluzione enzimatica nel buffer di asve': 0,5 g/l, 0,4 g/l, 2,5 g/l, 1,5 g/l, 1 g/l.
- In tutti i pozzi raffigurati nella Figura 4B applicare 180 l di buffer di analisi enzimatica. Applicare 10 l di assaggio di enzima buffer in tutti i pozzi per il substrato (vuoto e campione). Applicare 10 l della serie di diluizione di proteine preparate nei pozze per l'enzima (vuoto e campione). Applicare 10 l di assaggio di enzima buffer in tutti i pozzi per pozzi per il substrato vuoto e l'enzima vuoto.
- Subito prima della misurazione, applicare 10 ll della serie di diluizione del substrato preparata nei pozze per il campione di substrato e il campione di enzima.
- Utilizzare una pipetta multicanale a impostazioni 50 per mescolare delicatamente tutti i pozze, a partire dagli spazi vuoti, procedendo ai pozze campione. Fare attenzione a non creare bolle.
- Inserire la piastra in un lettore di micropla e misurare ogni bene a 255 nm, come descritto nel passaggio 8.1. Eseguire l'analisi in un foglio di calcolo. Copia i dati grezzi dal fotometro in un foglio di calcolo, scrivi tutte le impostazioni (cioè tutta la documentazione) in un altro foglio.
- Eseguire l'analisi dei singoli dati per punto della serie di diluizione, come descritto nei passaggi 7.11. a 7,14. Alla fine, ottenere tutte le attività specifiche e la trama contro la concentrazione iniziale del substrato: 2 mM, 1 mM, 0,5 mM, 0,3 mM, 0,2 mM, 0,1 mM.
- Visualizzare tutti i punti dati con singole deviazioni standard. Computer Michaelis-Menten cinetica tramite raccordo curva non lineare, o tramite l'analisi Lineweaver-Burk. Può essere necessario ri-misurare i singoli punti e adattare i singoli rapporti di coppia proteina-concentrazione/concentrazione di substrato nei passaggi 8.5 e 8.6. Il diagramma Michaelis-Menten per faHD1 umano è disponibile in Figura 5B.
9. Cristallizzazione delle proteine FAHD
NOTA: la cristallizzazione delle proteine FAHD (FAHD1 umano descritto in precedenza15) può essere ottenuta con il metodo di diffusione del vapore a goccia appesa in un formato a 24 pozzi (Figura 6A). Un protocollo passo-passo sulla cristallizzazione del FAHD1 umano utilizzando questa tecnica è presentato sotto15. Una descrizione più dettagliata è fornita nella sezione discussione.

Figura 6 : Cristallizzazione delle proteine FAHD.
(A) Piastre di cristallizzazione in standard 24 pozzo o 96 pozzo SBS impronta. Vedere la sezione 9 per maggiori dettagli. (B) Il processo di configurazione della piastra di base nella cristallizzazione delle proteine FAHD. Questa cifra viene ridisegnata con il permesso23. Vedere la sezione 9 per maggiori dettagli. (C) Cristalli faHD1 umani e corrispondenti modelli di diffrazione (piccoli inserti). La spaziatura reticolare più vicina è indicata negli inserti come misura per la qualità della diffrazione dei cristalli. Numeri più bassi indicano una risoluzione più alta e quindi dati più informativi. Vedere la sezione 9 del protocollo per maggiori dettagli. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Assicurarsi che la proteina sia diallyzed contro SEC tampone di corsa. La proteina FAHD1 deve essere disponibile ad alte concentrazioni (2-5 mg/mL). A concentrazioni più basse, la proteina potrebbe non cristallizzarsi a causa della mancanza di nucleazione spontanea.
- Preparare 20 mL della soluzione del serbatoio per la cristallizzazione. Effettuare tre soluzioni stock, utilizzando l'acqua distillata o deionizzata come solvente: 1 M Na-HEPES (minimo 25 mL, regolato al pH 7,5), 50% (w/v) glietilene 4000 (PEG4k) (minimo 65 mL) e 1 M MgCl2 (10 mL).
- Impostare una griglia di 4 x 6 (24 totale) diversi tubi da 15 mL. Etichettarli in base alle posizioni corrispondenti sulla piastra (ad esempio, riga (A, B, C, D) rispetto alla colonna (1-6) come "A1", "B5", "D6" e così via. Pipetta 1 mL di 1 M Na-HEPES in ogni tubo.
- Pipette 1 mL del 50% (w/v) PEG4k nella riga A dei tubi, 2 mL nella riga B, 3 mL nella riga C e 4 mL nella fila D. Pipette 100 L di 1 M MgCl2 nella colonna 1 dei tubi, 250 - L nella colonna 2 , 500 l nella colonna 3, 1,0 mL nella colonna 4, 1,5 mL nella colonna 5 e 2,0 mL nella colonna 6.
- Riempire tutti i tubi fino a un volume di 10 mL con acqua distillata o divinizzata, dove la scala sui tubi è sufficientemente accurata.
- Prendi il campione umano di proteine FAHD1 (5 mg/mL) dal frigorifero (o dal ghiaccio) e gira verso il basso alla massima velocità con una centrifuga superiore a 4 gradi centigradi per almeno 10 min. Se si desidera la cocristallizzazione con l'ossalato, aggiungere l'ossialato da una soluzione di stock in modo che il campione proteico contenga una concentrazione finale di ossalato di 2 mM. Applicare 1 mM DTT e conservare sul ghiaccio.
- Nel frattempo, disfare una piastra di cristallizzazione 24 bene, idealmente all'interno di una stanza a temperatura controllata a 18 gradi centigradi. Distribuire un sottile strato di olio di paraffina sul bordo sopra ogni pozzo del pozzo 24 con l'aiuto di un sottile vetro o asta di plastica. Aggiungere 800 l dei cocktail di cristallizzazione preparati (da A1 a D6) in ogni pozzetto corrispondente della piastra di cristallizzazione.
- Posizionare i freschi copricopre2piedi su una superficie pulita. Evitare di contaminare gli scivolamenti del coperchio con sporcizia o polvere. Se necessario, rimuovere eventuali detriti dallo slittamento del coperchio utilizzando aria compressa o uno spray spolverino.
- Una volta completata la centrifugazione, evitare di scuotere il campione proteico in modo che gli aggregati filati verso il basso e i detriti nella parte inferiore del tubo non fluttuino di nuovo. Nei passaggi seguenti, pipetta dal campione di proteine appena sotto la superficie della soluzione, al fine di evitare di agitare aggregati e depositi dal basso.
- Per ogni pozzo (vedere la figura 6B) pipetta 1 - L di soluzione proteica al centro di uno slittamento di copertura e aggiungere 1 -L del rispettivo cocktail serbatoio alla gocciolina proteica, evitando bolle. Capovolgere il coperchio a testa in giù e posizionarlo sulla parte superiore del pozzo in modo che l'olio sigilli il bene con la coverslip a tenuta d'aria. Ripetere fino a quando la piastra 24 pozzo è completata.
- Conservare la piastra a 18 gradi centigradi e osservare le gocce in base a un programma progressivo con un microscopio adeguato. Cristalli FAHD1 umani di solito appaiono durante la notte (vedere Figura 6C).
Risultati
A partire da un vettore di clonazione preparato e acquistato BL21(DE3) pLysS E. coli, il plasmide viene inserito nei batteri tramite scossa di calore o qualsiasi metodo alternativo appropriato (Figura 1). Dopo un breve periodo di amplificazione, i batteri trasformati vengono placcati su piastre di agar LB, al fine di crescere durante la notte. Piastre a questo punto possono apparire diverse, a seconda di una varietà di potenziali fonti di errore. Le piastre possono essere vuote (cioè senza colonie), completamente invase da batteri, o qualcosa in mezzo, rispettivamente. Due esempi di piastre di agar LB dopo la trasformazione ottimale e non ottimale sono illustrati nella Figura 7A. Troppe colonie batteriche indicano o che troppi batteri sono stati placcati (probabilmente) o che gli antibiotici in uso possono essere scaduti (improbabile). Troppo poche colonie batteriche possono indicare che non abbastanza plasmide è stato utilizzato per la trasformazione (utilizzare più la prossima volta) o che troppi antibiotici sono stati utilizzati per selezionare i batteri. In ogni caso, se le colonie sono presenti, dovrebbero andare bene, poiché l'uso di due antibiotici selettivi implica una possibilità piuttosto insignificante di crescita dei batteri non trasformati. Nessuna colonia, tuttavia, indica che i batteri hanno perso la loro competenza di trasformazione (a causa di conservazione o stoccaggio sbagliato per periodi più lunghi, congelamento e disgelo ripetitivi, ecc.), lo shock termico non ha avuto successo (nessun assorbimento di plasmide o batteri morte per troppo calore), il vettore di clonazione è danneggiato, o per errore è stato utilizzato un insieme sbagliato di antibiotici selettivi (verificare il gene di resistenza sul vettore plasmide).

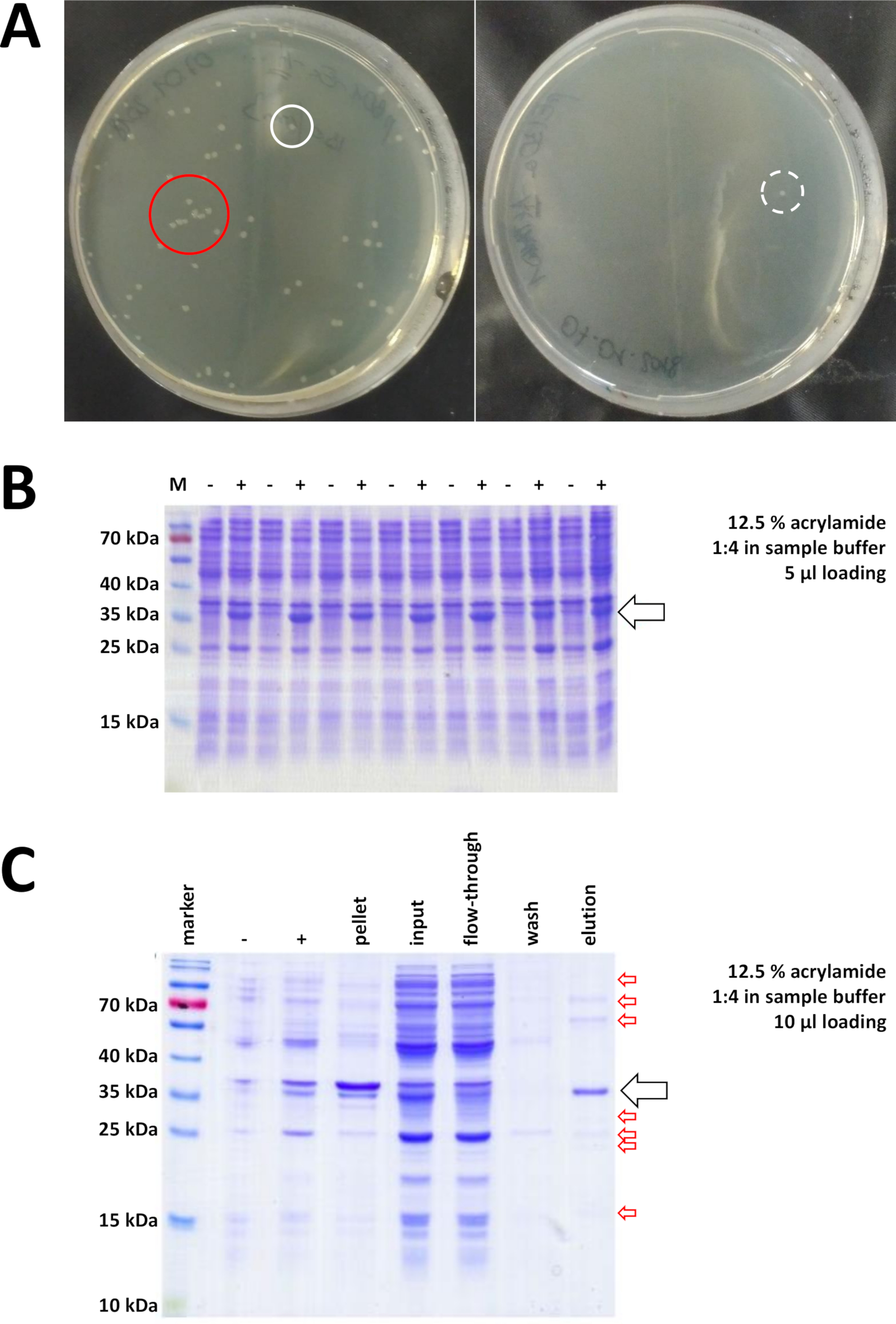
Figura 7 : Risultati rappresentativi per la trasformazione dei batteri e IMAC.
(A) Piastre di agar LB rappresentative con trasformazione BL21(DE3) E. coli, ottenute seguendo il passo 1.1 del protocollo. Sinistra: Piastra con colonie ben distribuite (esempio positivo). Destra: una piastra con una sola colonia (esempio negativo). I cerchi bianchi segnano buone colonie. Il cerchio rosso segna colonie che crescono troppo vicine l'una all'altra e non devono essere raccolte finché sono disponibili colonie isolate. (B) Un'analisi SDS-PAGE acrilammide del 12,5% di una serie di controlli di induzione ("-" indica prima dell'induzione di IPTG; Il " s" indica dopo l'induzione IPTG, prima del raccolto di pellet), regolato a pari quantità di proteine totali. Ciò è descritto nel passaggio 1.2. (C) Un'analisi esemplare del 12,5% di acrilammide SDS-PAGE della purificazione Ni-NTA della proteina FAHD1 con etichettatura del12,5%. Questo è descritto nella sezione 3 del protocollo. La cromatografia di affinità produce proteine ad alta purezza (>70%, freccia nera), tuttavia, si osservano anche diverse piccole contaminazioni (frecce rosse). Queste contaminazioni consistono di proteine non-FAHD che si legano alla colonna, e da proteine che si legano alla proteina FAHD. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Le colonie convalidate vengono selezionate e selezionate. Dopo l'amplificazione nel mezzo nutritivo, l'espressione proteica viene innescata dall'applicazione della sostanza chimica IPTG. Il pellet batterico contenente la proteina espressa in quantità di milligrammo viene raccolto e l'espressione viene verificata tramite SDS-PAGE (vedere ad esempio la figura 7B). Alcuni problemi possono verificarsi durante questo processo altrimenti semplice. In primo luogo, alcune proteine formano corpi di inclusione, perché a quanto pare in qualche modo interferiscono con il metabolismo naturale dei batteri ospiti. Questo è stato osservato per alcune mutazioni punto di FAHD1 umano e FAHD2. In questi casi, altri sistemi di espressione come le cellule degli insetti possono essere più appropriati e dovrebbero essere considerati. Dopo aver raccolto un pellet dalle cellule degli insetti, ad esempio, la purificazione delle proteine segue gli stessi passaggi descritti in questo protocollo. In secondo luogo, il sistema DE3-pET è talvolta trovato per essere "perdente" (cioè, la proteina è già espressa in una certa misura prima dell'induzione IPTG). La ragione potenziale di questo non è ben compresa, ma può aiutare ad esprimere la proteina lentamente durante la notte in un'incubatrice di celle frigorifere. In terzo luogo, non viene espressa alcuna proteina. Questo è probabilmente lo scenario peggiore, in quanto probabilmente indica un vettore di plasmide danneggiato e quindi consigliabile sequenziare il plasmide.
Se un suotag è stato utilizzato per etichettare la proteina, la cromatografia di affinità con agarose Ni-NTA è un metodo di cattura facile ed economico eliminando la maggior parte delle contaminazioni (Figura 7C). Metodi simili esistono per altri sistemi di tag (ad esempio, STREP-II). Se non è stato utilizzato alcun tag, una combinazione di precipitazione solfato di ammonio e cromatografia di scambio idrofobico consecutivo può anche separare la proteina dalla maggior parte delle altre proteine (Figura 8A). Tuttavia, confrontando i due metodi (Figura 7C rispetto figura 8A), la superiorità dei metodi Ni-NTA può essere dimostrata dall'analisi SDS-PAGE. Si consiglia quindi di utilizzare la suaproteina marcata.

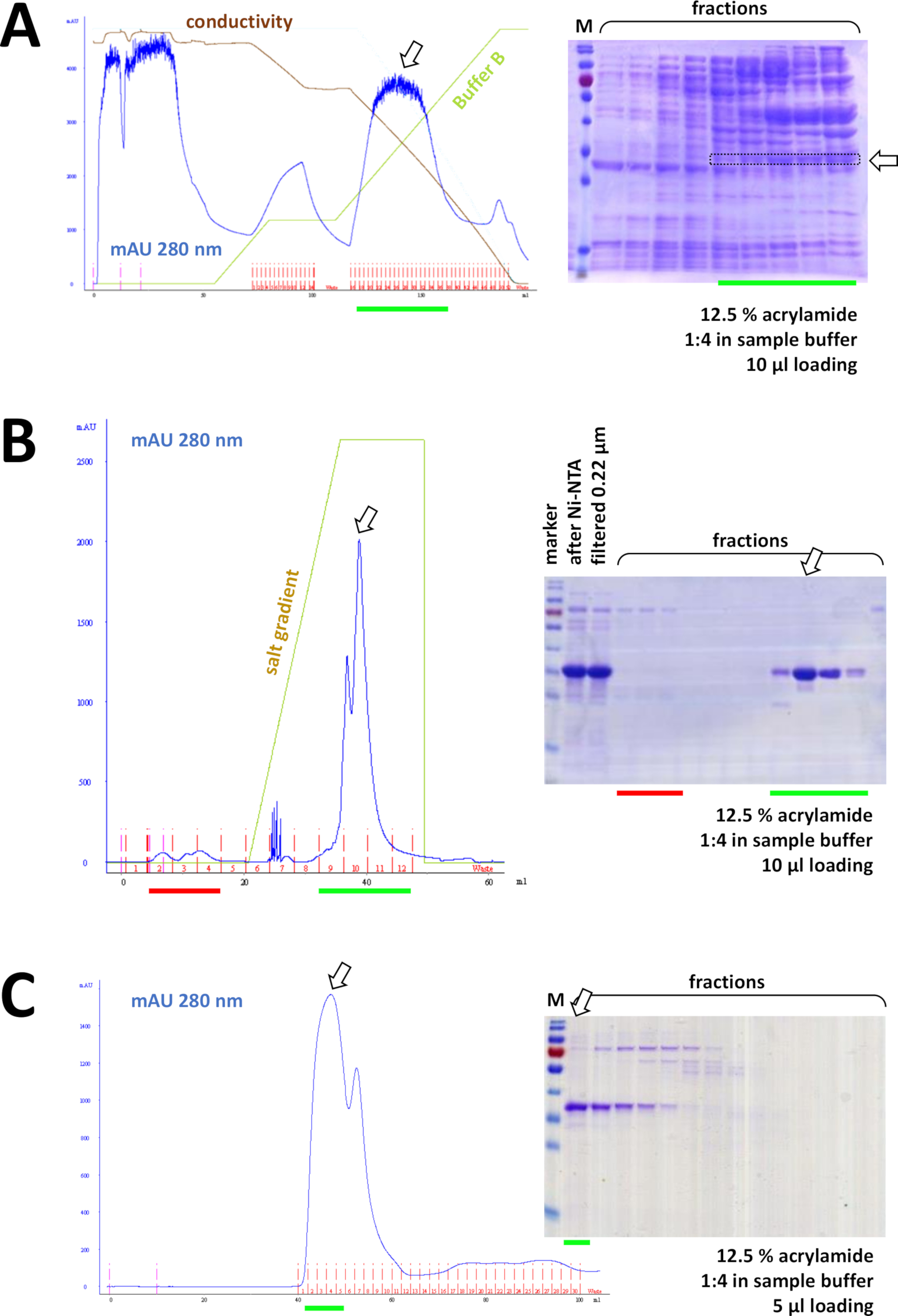
Figura 8 : risultati rappresentativi per gli esperimenti FPLC (HIC, scambio iole, SEC).
(A) Un tipico cromatogramma e 12,5% di acrilammide SDS-PAGE analisi della cromatografia HIC-fenyl dopo la precipitazione del solfato di ammonio (AS) della proteina FAHD1 senza tag, come descritto nella sezione 4 del protocollo. La linea verde riflette la sfumatura del buffer B che non contiene AS. Durante il processo AS viene gradualmente lavato fuori dal sistema. Confrontando questo pannello con la figura 7C si visualizza la potenza della cromatografia di affinità Ni-NTA rispetto al metodo HIC-fenil e il vantaggio di utilizzare un sistema His-tag per la purificazione delle proteine. (B) Un cromatogramma esemplare e 12,5% di acillamina SDS-PAGE analisi della cromatografia di scambio cationico della suaFAHD con tag dopo la purificazione Ni-NTA. Utilizzando un gradiente di sale, il campione applicato viene separato in singole proteine. (C) Un cromatogramma esemplare e un'analisi SDS-PAGE acrilamide del 12,5% della cromatografia di esclusione delle dimensioni G75 della suaFAHD con tag FAHD in seguito alla cromatografia di scambio cationico. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Consecutivamente, la proteina è ulteriormente separata dalle contaminazioni resimittuate dalla cromatografia di scambio di cation/anione (ad esempio, figura 8B), seguita dalla cromatografia di esclusione delle dimensioni (ad esempio, vedere la figura 8C). Si consiglia di istituire una strategia di purificazione iniziale in questo ordine; tuttavia, queste colonne devono essere utilizzate in combinazione, successivamente e in variazione, fino a quando la proteina non è sufficientemente pura.
Semplici analisi di attività, al fine di testare le decisioni "sì o no" su substrati attivi e/o cofattori, possono essere eseguiti con le sueproteine con tag dopo la purificazione Ni-NTA, o proteine senza tag dopo la colonna di scambio ionico. Le attività specifiche e le costanti cinetiche devono essere determinate con proteine di massima purezza. La cristallizzazione può essere tentata con le proteine dopo la colonna di scambio ionico, ma la qualità dei cristalli è quasi sempre correlata alla purezza delle proteine. Gli anticorpi policlonali possono essere sollevati contro le proteine in qualsiasi fase del protocollo di purificazione; tuttavia, qui la qualità è anche correlata con la purezza delle proteine.
Discussione
Passaggi critici
Le proteine FAHD sono molto sensibili alle concentrazioni di sale. A basse concentrazioni di NaCl, le proteine possono precipitare allo scongelamento, ma di solito possono essere completamente ricostituite a concentrazioni di sale più elevate. Cioè, se una proteina FAHD precipita per qualche motivo, può essere recuperata o ripiegata con concentrazioni di sale più elevate (>300 m). Alcune altre proteine idrofobiche, tuttavia, non possono essere recuperate (ad esempio, FAHD2 umano), ma detergenti come CHAPS (massimo 1%) o glicerolo (10%) possono essere utilizzati per mantenerli in soluzione stabile. In ogni caso, si raccomanda il congelamento degli urti con azoto liquido e di stoccaggio a -80 gradi centigradi, in quanto si tratta di un processo delicato e lento di scongelamento.
Alcuni problemi imprevisti possono verificarsi durante la purificazione Ni-NTA nel passaggio 3.1.10. Da notare che un OD più alto nel secondo campione raccolto rispetto al primo campione indica un volume troppo elevato della resina agarosa (prendere una nota e utilizzare meno resina nell'esperimento successivo). Inoltre, la resina di agarose stessa porta ad un segnale OD a 280 nm (cioè, l'interruzione del letto di resina di agarose darà segnali artificiali). In caso di dubbio, si consiglia di utilizzare altri metodi come un saggio Bradford o BSA per determinare le concentrazioni di proteine.
Nei saggi enzimatici, ci sono tre aspetti critici da considerare. In primo luogo, valutare la concentrazione proteica è fondamentale per ottenere le corrette attività specifiche. Il livello di purezza della proteina sta influenzando il risultato e deve essere stimato. Nel caso di proteine etichettate, la massa della parte tag deve essere calcolata e l'attività specifica deve essere corretta di conseguenza. Per i semplici saggi descritti nella sezione 7 del protocollo, la purezza Ni-NTA è sufficiente a distinguere tra substrati attivi e inattivi, cofattori, ecc. Nel caso di cinetica Michaelis-Menten più complessa, tutte le concentrazioni di reattori e substrati devono essere determinate correttamente. Soprattutto quando si utilizza l'oxaloacetate (che auto-decarboxylates on-time) la parte enzimatica della reazione deve essere corretta per l'auto-decarboxylation (presupponendo che entrambe le reazioni si verificano contemporaneamente). Devono essere prese in considerazione le modifiche iniziali nel segnale di densità ottica indirizzato alla tautomerizzazione del substrato. In terzo luogo, le concentrazioni e i volumi devono essere rettificati. Una reazione con concentrazioni definite di enzimi e substrati può dare risultati diversi a seconda del volume del saggio. Se c'è troppo enzima per bene, l'adesione del liquido può infatti bias il risultato.
Per la valutazione della cinetica Michaelis-Menten si consiglia di eseguire esperimenti iniziali in 100 lotti ll, 200 e 300 per trovare la combinazione ottimale. Aspetti simili si applicano al rapporto delle concentrazioni enzimatico-substrato per i saggi cinetici. Troppo enzima per substrato o troppo substrato per enzima mettono il sistema al di fuori della gamma Michaelis lineare a stato costante. Sono necessari esperimenti iniziali per ottimizzare queste condizioni. Aggiustamento esemplare per la proteina umana FAHD1 (wild-type) sono forniti nella sezione 8, con conseguente diagrammi cinetici (come presentato nella Figura 5B, per esempio).
Per la cristallizzazione una goccia di soluzione proteica viene filtrata al centro di una coverslip e mescolata con una goccia di cocktail di cristallizzazione, che di solito è composta da un buffer (ad esempio, Tris-HCl, HEPES) e un precipitante (ad esempio, polietilene glicole ammon, ammonie solfato). Una goccia di soluzione inibitore per la co-cristallizzazione (come l'ossilato in questo protocollo) può essere facoltativamente applicata. Il coperchio viene quindi posizionato a testa in giù sopra un pozzo di serbatoio contenente cocktail di cristallizzazione, sigillando il pozzo a tenuta d'aria con l'aiuto di olio sigillante (Figura 6B). Idealmente, nessuna precipitazione si verifica all'interno della goccia all'inizio dell'esperimento, il che significa che la proteina rimane in soluzione. Poiché la concentrazione precipitosa nel serbatoio è maggiore rispetto alla goccia, la goccia inizia a perdere acqua per evaporazione nell'atmosfera del pozzo fino a raggiungere l'equilibrio con il serbatoio. La diffusione dell'acqua nel serbatoio provoca una lenta diminuzione del volume della caduta che a sua volta provoca un aumento di entrambe, proteine e concentrazione precipitante nella caduta. Se la soluzione proteica raggiunge lo stato richiesto di super-saturazione e quindi di meta-stabilità, può verificarsi una nucleazione spontanea seguita dalla crescita cristallina. Raggiungere lo stato supersaturi è una condizione necessaria ma non sufficiente per la cristallizzazione. La cristallizzazione delle proteine ha bisogno sia, condizioni termodinamiche e cinetiche favorevoli, e dipende fortemente dalle proprietà imprevedibili della proteina da cristallizzare22.
Modifiche e risoluzione dei problemi
L'espressione della proteina in E. coli può essere inefficiente. Variare le concentrazioni IPTG, la temperatura di espressione e il tempo di amplificazione, come la temperatura ambiente per diverse ore o nella stanza fredda durante la notte, potrebbe essere necessario testare per ogni nuova proteina per trovare condizioni ottimali. Precipitazioni di proteine nei corpi di inclusione è talvolta osservato per le proteine FAHD più idrofobiche. In questi casi, si raccomanda l'espressione proteica in altri sistemi modello come le cellule degli insetti, poiché i corpi di inclusione hanno meno probabilità di formare26.
Poiché le proteine FAHD sono sensibili alle concentrazioni di sale e cofattore, così come al pH, le strategie di purificazione per diversi omologhi, gli ortologhi e le varianti di mutazione puntiforme possono differire in singole impostazioni. I metodi di purificazione descritti sono sviluppati per la proteina faHD1 umana e topo di tipo selvatico. Le concentrazioni di sostanze chimiche, come NaCl e imidazolo, così come il pH, potrebbero dover essere adattate per singole proteine con un punto isoelettrico diverso (pI). Da notare anche che non tutte le proteine conetichettatura possono legarsi bene a una resina Ni-NTA. Se il legame proteico alla colonna Ni-NTA è inefficiente, le concentrazioni adattate di NaCl e imidazole, nonché le diverse condizioni di pH nel buffer di corsa Ni-NTA possono contribuire a migliorare la qualità del risultato. In caso contrario, saltare il passo Ni-NTA e procedere alla fase della cromatografia dello scambio ionico può anche portare a una strategia di purificazione di successo. Se una proteina si lega alla colonna Ni-NTA ma non può essere elatata dalla colonna, l'aggiunta di alcuni mM EDTA può aiutare a disturbare il complesso di Ni 2.
Per quanto riguarda il processo di cristallizzazione, occorre capire che l'auto-organizzazione di molecole proteiche grandi e complesse in un reticolo periodico regolare è un processo intrinsecamente improbabile che dipende fortemente dai parametri cinetici difficili da controllare. Anche piccoli cambiamenti nel set-up utilizzato per la cristallizzazione possono alterare drasticamente il risultato e non si formeranno cristalli. La purezza delle proteine è generalmente di fondamentale importanza. Come regola generale, un gel SDS-PAGE pesantemente sovraccarico non dovrebbe mostrare altre bande. Inoltre, la sequenza in cui vengono eseguiti i passaggi può influire sul risultato. Ad esempio, per garantire la riproducibilità, è spesso necessario mantenere la sequenza di pipettaggio la stessa, quindi aggiungere prima la proteina e infine aggiungere precipitante alla goccia di cristallizzazione (o viceversa). Qualunque sia il metodo utilizzato, dovrebbe essere mantenuto lo stesso quando si tenta di riprodurre o scale-up esperimenti. Se non si osservano cristalli in base a questo protocollo, la composizione chimica precipitante, il pH, la dimensione della goccia e il rapporto proteina-precipitato possono essere variati in piccoli incrementi. La pazienza e le osservazioni coerenti delle gocce sono di virtù.
Osservazioni ai meccanismi catalitici della FAHD1
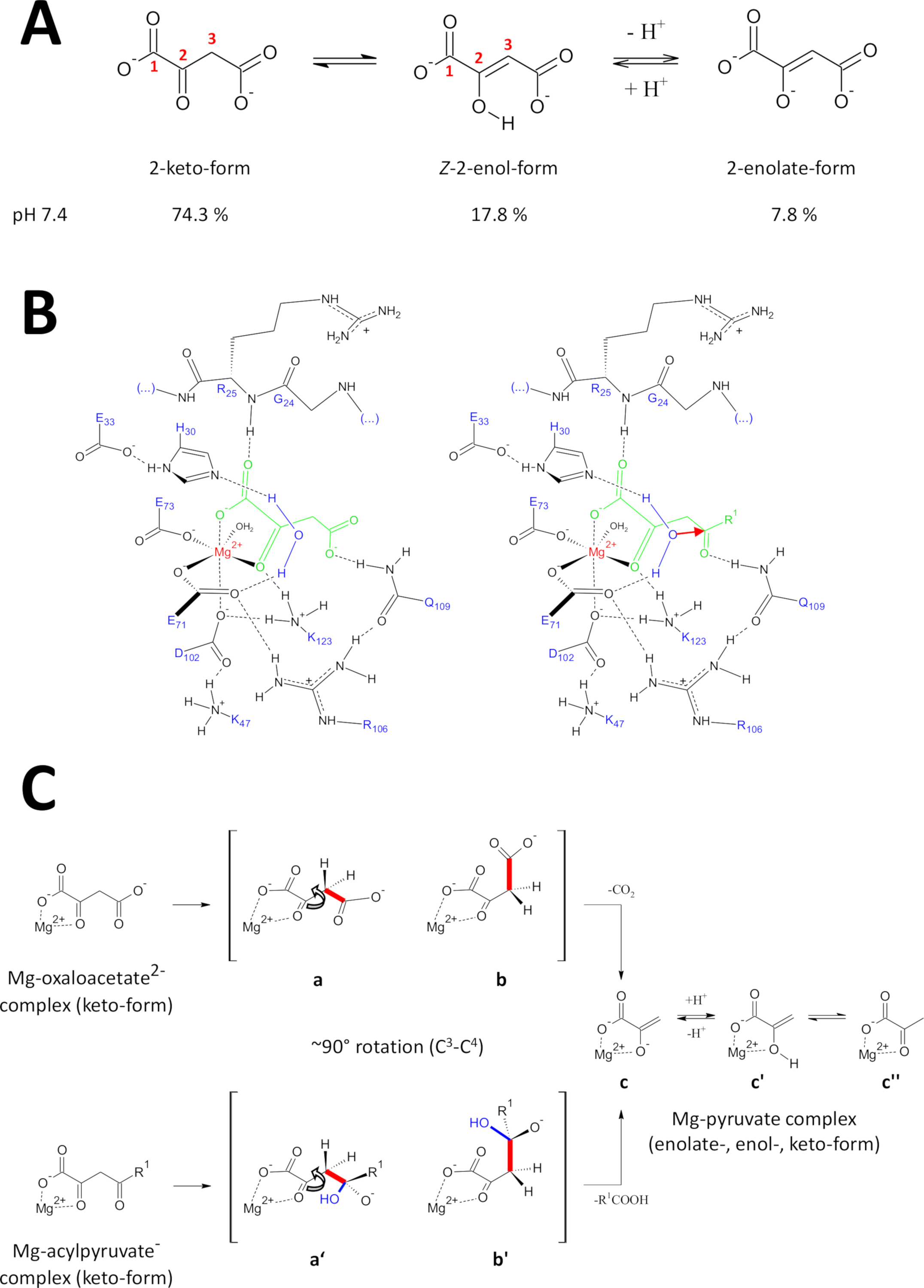
I metodi presentati sono stati sviluppati specificamente per ottenere proteine FAHD1 di alta qualità. Questo ha permesso la crescita di cristalli FAHD1 così come l'ingegneria di cristalli contenenti FAHD1 complesso ad un inibitore (ossialato, PDB:6FOG). Le strutture a raggi X forniscono un'architettura 3D della cavità catalitica dell'enzima. Questi risultati stabiliscono una descrizione completa dei residui potenzialmente importanti per i meccanismi catalitici di questo enzima intrigante. FAHD1 è stato descritto per la prima volta per essere in grado di fendere acylpyruvates (acetilpyruvate, fumarylpyruvate)11. Più tardi, si è scoperto che FAHD1 opera anche come un decarboxylase di oxaloacetate12. Sebbene i substrati acylpyruvate e oxaloacetate siano diverse moieties chimiche, le trasformazioni chimiche condividono meccanicamente la scissione strategica di un singolo legame C3-C4 comune, agevocato energicamente se il C3 -C4 legami orbitali rimangono ortogonali agli orbitali z del C2-carbonil1. Tale conformazione consente la stabilizzazione della risonanza del C3-carbanion formatosi transitoriamente durante il processo di scissione. I substrati FAHD1 (oxaloacetate e acylpyruvates) sono molecole flessibili e possono esistere in tautomierico (keto-enol) così come nelle forme idratate C2(Figura 9A). Gli equilibri tra le diverse specie sono determinati principalmente dalla natura della composizione tampone utilizzata, dal pH e dalla presenza di ioni metallici. Di seguito discutiamo ipotetici scenari meccanicistici ispirati all'analisi delle strutture a cristalli a raggi X che hanno rivelato il centro catalitico di FAHD1.

Figura 9 : Dettagli sul meccanismo catalitico proposto della FAHD1 umana.
(A) L'oxaloaceto esiste in stato cristallino e in soluzione neutra principalmente nella forma -enol24. Tuttavia, in condizioni fisiologiche di pH la forma a 2 keto è la rappresentazione predominante25. (B) Schizzo chimico della cavità hFAHD115 con oxaloaceto legato a Mg (a sinistra) e acylpyruvate (a destra, con R1 come riposo organico; la freccia rossa indica un attacco nucleofilo della molecola d'acqua stabilizzata adiacente) (vedi discussione). (C) Confronto delle conformazioni preferite per la scissione C3-C4 in decarboxylase (da b a c) e idrolasi (b' a c) meccanismo di FAHD1: entrambi i processi provocano la funzione pyruvate-enolate (cfr. discussione). Gli intermedi a dire i mari b e b' dovrebbero essere stabilizzati entro il Q109, come indicato nella giuria B (vedi discussione). Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
L'attività Decarboxylase di FAHD1
L'oxaloacetate esiste in stato cristallino e in soluzione neutra principalmente nella forma -enol24. Ma è stato dimostrato che in condizioni fisiologiche di pH (condizioni di buffer a pH 7.4) la forma a 2 keto è la rappresentazione predominante di oxaloacetate25 (Figura9A) e che l'enolizzazione non è un prerequisito per la decarboxylation27 . Da notare che gli ioni di Mg2 non hanno alcuna influenza sul rapporto tra le specie di oxaloaceto a un pH di 7,4 o inferiore a28. Trasposizione della forma di osaloacetate keto nel centro catalitico di FAHD1 (guidato dall'ossito tecnico legato nell'enzima complesso (PDB: 6FOG15)) ha rivelato residuo Q109 come regolatore conformazionale dell'ossaacelottato legato15. Come descritto in un altro articolo15, il legame con l'idrogeno al gruppo di carboidrati del Q109 stabilizza una conformazione di oxaloacetate risultante dalla rotazione intorno al legame C2-C3 (Figura9B, pannello sinistro). Come conseguenza di questa rotazione, il legame C3-C4 (da scissionare) adotta una disposizione quasi ortogonale rispetto agli orbitali z del C2-carbonyl (Figura9C). L'anidride carbonica può essere rilasciata. Il prodotto primario di questo processo sarebbe risonanza stabilizzata Mg-enolato di piravato. È noto dalle indagini sui complessi oxaloacetate-Mg che l'enolato forma il complesso più stabile28,29. Supponendo una stabilità comparabile per un enolato-complesso Mg-pyruvate il cofattore di FAHD1 potrebbe essere bloccato, ma il residuo di lisina K123 può protonare l'enotopo di piravato in un equilibrio per vietare la perdita del cofattore15.
L'interpretazione data suggerisce pyruvate enol come un intermedio distinto nella funzione catalitica ODx di FAHD1. In questa fase del modello ipotizzato, i dati sperimentali non forniscono alcuna ulteriore indicazione sul motivo per cui il coperchio chiuso dovrebbe aprirsi per rilasciare il prodotto. Si può dedurre, tuttavia, che il meccanismo proposto si presenta come un'inibizione enzimatica dal prodotto: La struttura cristallina rivela una molecola d'acqua conservata tenuta in orientamento direzionale verso il centro catalitico FAHD1 dai residui H30 ed E33 presentati in un elica corta15, che viene indotta al legame del ligando e alla chiusura del coperchio. Se l'enol primario rimanesse in equilibrio con l'enolato, l'enolato stabilizzato della risonanza potrebbe essere spento per pirovare dalla molecola d'acqua. L'idrossile risultante sarebbe in grado di sposto il pirone dal cofattore Mg su cui si aprirebbe il coperchio. Infine, il centro catalitico sarebbe stato restaurato nell'ambiente mitocondriale. In questo ipotetico scenario, la molecola dell'acqua della cavità opererebbe rispettivamente come acido.
Attività idrolasi di FAHD1
L'attività idrolasi di un enzima richiede implicitamente la formazione intermedia di un nucleofilo idrossile. Questo meccanismo si trova di solito in combinazione con l'attività catalitica acido-base. Lo stato transitorio della reazione deve essere preparato tramite il controllo conformazionale da parte di catene laterali critiche di amminoacidi nella cavità. In analogia con la discussione della funzione decarboxylase, l'acylpyruvate legato agli enzimi in forma di 2-keto sarà messo sotto controllo conformazionale mediante l'incollaggio dell'idrogeno dell'ossigeno 4-carbonilico al Q109 (Figura9B, pannello destro). La struttura cristallina di FAHD1 (PDB:6FOG) legata all'ossalato rivela una molecola d'acqua conservata tenuta in orientamento direzionale verso il centro catalitico FAHD1 dai residui H30 ed E33 presentati in un breve elica15. Il diadee E33-H30 è competente a deprotonare l'acqua posizionata direzionale e l'idrossile risultante è in disposizione ideale per attaccare il 4-carbonyl di acylpyruvate presentato sotto controllo conformativo da Q10915.
Da notare che è stato proposto un meccanismo analogo per il FAH18. L'attacco da parte del nucleofilo idrossile dovrebbe portare a una specie di ossianion, che è stabilizzata sulla scissione orbitale c3-C4 (Figura 9C). In questo modello, la rotazione del legame C3-C4 (Figura 9C) avviene dopo l'attacco nucleofilo da parte dell'idrossile formato indicato nella figura 9B (cioè, prepara l'acylpyruvate per la scissione del legame). I prodotti primari sarebbero l'acido acetico e l'enolato di Mg-pyruvate. In questo ipotetico scenario, l'acido acetico potrebbe placare l'enol a pyruvate e successivamente assistere lo spostamento del prodotto. Sopra un pH di 7,5 e in presenza di ioni Mg, gli acylpyruvates esistono in un equilibrio tra keto- e enol-forms, quest'ultimo in leggera preferenza30. Molto probabilmente entrambe le forme sono in grado di legarsi al cofattore di FAHD1 sotto la successiva chiusura del coperchio. L'elaborazione dei substrati acylpyruvate enolici da parte dell'enzima è ostacolata dalla struttura piatta della forma enol. La scissione C3-C4 si tradurrà in una carbanion in vinile senza stabilizzazione di risonanza.
Pertanto, proponiamo un passo catalitico di ketonizzazione per prepararsi all'attacco del nucleofilo idrossile sul carbonio acilo. Questo processo di ketonizzazione, tuttavia, richiederebbe il controllo sulle trasposizioni dei protoni da parte dei residui di FAHD1, che attribuirebbero un'attività intrinseca di isomerase a FAHD1. Si dice che l'acidità dell'idrogeno enol legato a Mg riveli un aumento di diecimila volte rispetto alla forma non complessa28. Una deprotonazione della forma enolina legata mg sarebbe fattibile da K123 non protonato. La deprotonazione di K123 può essere assistita dal carboxylato di D102. Una rete di legame di idrogeno formata da residui D102-K47-K123 potrebbe funzionare come il relè di protoni necessario nel centro catalitico di FAHD115. Un enomio intermedio così formato potrebbe quindi essere spento da una triade E33-H30-H20 sotto la tardizzazione del substrato15. La forma a 2 cheto sarebbe stata sotto il controllo conformazionale del Q109, e l'idrossile formante avrebbe attaccato il carbonio acilo. La discussione riassunta implica un controllo di FAHD1 su una molecola d'acqua per passare tra acido e base attraverso l'interazione di residui di cavità-formazione.
Applicazioni future o indicazioni del metodo
Le applicazioni future dei metodi qui descritti sono numerose. Una pletora di membri prokaryotici della superfamiglia FAH attende ancora la caratterizzazione funzionale. Anche le informazioni disponibili sulle attività catalitiche dei membri noti della superfamiglia FAH sono scarse e, nella maggior parte dei casi, basate su ipotesi teoriche piuttosto che su dati sperimentali. L'applicazione dei metodi qui descritti per i membri della superfamiglia procastica FAH dipende dagli interessi specifici di ricerca in batteriologia. D'altra parte, la recente dimostrazione che i membri della superfamiglia faH eucariota svolgono ruoli essenziali in vari compartimenti cellulari (ad esempio, citosol contro mitocondri) evidenzia la necessità di caratterizzare meglio queste proteine (tre delle quali sono state identificati finora), in particolare perché i dati attuali suggeriscono che alcune proteine non caratterizzate possono svolgere diverse funzioni nel contesto della biologia mitocondriale, della ricerca sull'invecchiamento e della ricerca sul cancro. Si propone che la caratterizzazione molecolare e fisiologica completa di questi membri della superfamiglia eucanotica FAH possa fornire importanti informazioni sui principali campi della ricerca contemporanea nel settore biomedico. Ulteriori ricerche sui meccanismi di FAHD1 (e relativi enzimi) sono necessari per comprendere meglio i meccanismi alla base della bi-funzionalità di FAHD1, che non è ancora completamente chiarito. Ulteriori studi con mutanti FAHD1, indagini NMR, e studi strutturali sui complessi inibitori possono aiutare a risolvere i veri scenari meccanicistici per i quali FAHD1 sembra essere competente. Inoltre, progettazione computerizzata di enol mimis in grado di legarsi al Mg-cofattore alla fine porterà a potenti inibitori di FAHD1.
Divulgazioni
Gli autori non hanno nulla da rivelare e non dichiarano interessi finanziari concorrenti. H. G. è CEOCSO presso MoleculeCrafting.HuGs e.U. e ha fornito acylpyruvates per questo studio tramite sintesi personalizzata. Il lavoro nel laboratorio di P. J. D. è stato sostenuto dal Fondo scientifico austriaco (FWF): numero di progetto P 31582-B26. Le spese di pubblicazione per questo manoscritto sono state in parte coperte dal Fondo scientifico austriaco (FWF) con il numero di progetto P 31582-B26. A. N. e B. R. sono sostenuti dal Fondo scientifico austriaco (FWF) nell'ambito del progetto P28395-B26.
Riconoscimenti
Gli autori sono molto grati per l'assistenza tecnica esperta di Annabella Pittl e per lo sviluppo del metodo pilota di Haymo Pircher.
Materiali
| Name | Company | Catalog Number | Comments |
| BL21(DE3) pLysS competent E. coli | Promega | L1195 | High-efficiency protein expression from gene with T7 promoter and ribosome binding site |
| pET E. coli T7 Expression Vectors | MERCK | - | http://www.merckmillipore.com/AT/de/life-science-research/genomic-analysis/dna-preparation-cloning/pet-expression-vectors/qFSb.qB.mLQAAAFA6.VkiQ0G,nav |
| 0.45 µm filter units | MERCK | SLHP033NS | Millex-HP, 0.45 µm, PES 33 mm, not steril |
| 0.22 µm filter units | MERCK | SLGP033RS | Millex-HP, 0.22 µm, PES 33 mm, not steril |
| Eppendof tubes 1.5 mL | VWR | 525-1042 | microcentrifugal tubes; autoclaved |
| 15 mL Falcon | VWR | 734-0451 | centrifugal tubes |
| 50 mL Falcon | VWR | 734-0448 | centrifugal tubes |
| PS Cuvettes Spectrophotometer Semi-Micro | VWR | 30622-758 | VIS transparent cuvettes |
| UV Cuvettes Spectrophotometer Semi-Micro | VWR | 47727-024 | UV/VIS transparent cuvettes |
| isopropyl-β-D-thiogalactopyranosid (IPTG) | ROTH | 2316 | chemical used for induction of protein expression with the DE3/pET system |
| imidazole | ROTH | X998 | chemical used for elution of polyhistidine (6xHis) sequences from a nickel-charged affinity resin |
| Glass Econo-Column Columns | Bio-Rad | - | http://www.bio-rad.com/de-at/product/glass-econo-column-columns?ID=2cfb1c6e-32e8-4c72-b532-dd39013d707d&pcp_loc=catprod |
| chloramphenicol | Sigma-Aldrich | C0378 | antibiotic for bacterial growth selection; resistance endióded in pLysS plasmid of BL21(DE3) E. coli; 25 µg/mL final concentration |
| kanamycin | Sigma-Aldrich | 60615 | antibiotic for bacterial growth selection; to be used if this resistance is encoded in the employed pET vector; 50 µg/mL final concentration |
| ampicillin | Sigma-Aldrich | A1593 | antibiotic for bacterial growth selection; to be used if this resistance is encoded in the employed pET vector; 100 µg/mL final concentration |
| Ultra-15, MWCO 10 kDa | Sigma-Aldrich | Z706345 | centrifigal filters for protein enrichment; https://www.sigmaaldrich.com/catalog/product/sigma/z706345?lang=de®ion=AT |
| Ultra-0.5 Centrifugal Filter Units | Sigma-Aldrich | Z677108 | centrifigal filters for protein enrichment; https://www.sigmaaldrich.com/catalog/product/ALDRICH/Z677108?lang=de®ion=AT&cm_sp=Insite-_-prodRecCold_xviews-_-prodRecCold5-2 |
| oxaloacetic acid | Sigma-Aldrich | O4126 | TCA metabolite |
| sodium oxlalate | Sigma-Aldrich | 71800 | a competitive inhibitor of FAH superfamily enzymes |
| Dialysis tubing cellulose membrane | Sigma-Aldrich | D9277 | https://www.sigmaaldrich.com/catalog/product/sigma/d9277; or comparable |
| Ni-NTA agarose | Thermo-Fischer | R90101 | a nickel-charged affinity resin that can be used to purify recombinant proteins containing a polyhistidine (6xHis) sequence |
| 96-Well UV Microplate | Thermo-Fischer | 8404 | UV/VIS transparent flat-bottom 96 well plates |
| PageRuler Prestained Protein Ladder, 10 to 180 kDa | Thermo-Fischer | 26616 | https://www.thermofisher.com/order/catalog/product/26616?SID=srch-hj-26616 |
| ÄKTA FPLC system | GE Healthcare Life Sciences | - | using the FPLC system by GE Healthcare; different custom versions exist; this work used the "ÄKTA pure" system |
| HiTrap Phenyl HP column | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/it/shop/chromatography/prepacked-columns/hydrophobic-interaction/hitrap-phenyl-hp-p-05630 |
| Mono S 10/100 GL | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/ch/shop/chromatography/prepacked-columns/ion-exchange/mono-s-cation-exchange-chromatography-column-p-00723 |
| Mono Q 10/100 GL | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/ch/shop/chromatography/prepacked-columns/ion-exchange/mono-q-anion-exchange-chromatography-column-p-00608 |
| HiLoad Superdex column 75 pg (G75) | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/ch/shop/chromatography/prepacked-columns/size-exclusion/hiload-superdex-75-pg-preparative-size-exclusion-chromatography-columns-p-05800 |
| HiLoad Superdex column 200 pg (G200) | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/ch/shop/chromatography/prepacked-columns/size-exclusion/hiload-superdex-200-pg-preparative-size-exclusion-chromatography-columns-p-06283 |
| TECAN microplate reader | TECAN Life Sciences | - | https://lifesciences.tecan.com/microplate-readers |
| acetylpyruvate | MoleculeCrafting.HuGs e.U. | - | custom synthesis |
| benzoylpyruvate | MoleculeCrafting.HuGs e.U. | - | custom synthesis |
| VDX™ plate (24 wells) | Hampton | HR3-142 | 24 well plates used for crystallization via Hanging Drop Vapor Diffusion |
| paraffin oil | Hampton | HR3-411 | used for crystallization via Hanging Drop Vapor Diffusion |
| coverslips (22 mm) | Karl Hecht KG | 14043 | coverslips used for crystallization via Hanging Drop Vapor Diffusion |
| Luria broth (LB) medium | self-prepared | - | a general growth medium for E. coli: 5 g/L yeast extract; 10 g/L peptone from casein; 10 g/L sodium chloride; 12 g/L agar-agar |
| NZCYM medium | self-prepared | - | a better growth medium for E. coli, used for amplification: 10 g/L NZ amine; 5 g/L NaCl; 5 g/L yeast extract; 1 g/L casamino acids; 2 g/L MgSO4; adjust pH to 7.4 |
| Luria broth (LB) agarose plates | self-prepared | - | autoclaved agarose plates containing LB-medium and antibiotics for bacterial groth selection; https://www.addgene.org/protocols/pouring-lb-agar-plates/ |
| Ni-NTA running buffer | self-prepared | - | 20 mM Tris-HCl pH 7,4; 50-300 mM NaCl; 10-200 mM imidazole; ranges: optimal value varies among FAHD proteins |
| Ni-NTA elution buffer | self-prepared | - | 20 mM Tris-HCl pH 7,4; 50-300 mM NaCl; 200-500 mM imidazole; ranges: optimal value varies among FAHD proteins |
| HIC running buffer | self-prepared | - | 44 mM NaH2PO4; 6 mM Na2HPO4; 100 mM NaCl; 20 mM DTT; adjust to pH 7 |
| HIC running buffer AS | self-prepared | - | HIC running buffer saturated with ammonium sulfate (AS); adjust to pH 7: 70 g ammonium sulfate + 90 mL buffer, stirred overnight in the cold room; adjust to pH 7.0 |
| Mono S low salt buffer | self-prepared | - | 44 mM NaH2PO4; 6 mM Na2HPO4; 10-300 mM NaCl; ranges: optimal value varies among FAHD proteins |
| Mono S high salt buffer | self-prepared | - | 44 mM NaH2PO4; 6 mM Na2HPO4; 1-2 M NaCl; ranges: optimal value varies among FAHD proteins |
| Mono Q low salt buffer | self-prepared | - | 20 mM Tris-HCl; 15 mM NaCl; adjust to pH 8.0 |
| Mono Q high salt buffer | self-prepared | - | 20 mM Tris-HCl; 1 M NaCl; 10 % glycerol; adjust to pH 8.0 |
| G75 / G200 running buffer | self-prepared | - | 15 mM Tris-HCl; 300 mM NaCl; adjust to pH 7.4 |
| enzyme assay buffer | self-prepared | - | 50 mM Tris-HCl pH7.4; 100 mM KCl; 1 mM MgCl2 |
| protein crystallization buffer | self-prepared | - | G75 / G200 running buffer with 1 mM DTT |
| reservoir solution for crystallization | self-prepared | - | 100 mM Na-HEPES pH 7.5; 5-20 % (w/v) PEG4k; 10 mM-200 mM MgCl2 |
Riferimenti
- Brouns, S. J. J., et al. Structural Insight into Substrate Binding and Catalysis of a Novel 2-Keto-3-deoxy-d-arabinonate Dehydratase Illustrates Common Mechanistic Features of the FAH Superfamily. Journal of Molecular Biology. 379, 357-371 (2008).
- Timm, D. E., Mueller, H. A., Bhanumoorthy, P., Harp, J. M., Bunick, G. J. Crystal structure and mechanism of a carbon-carbon bond hydrolase. Structure (London, England: 1993). 7, 1023-1033 (1999).
- Weiss, A. K. H., Loeffler, J. R., Liedl, K. R., Gstach, H., Jansen-Dürr, P. The fumarylacetoacetate hydrolase (FAH) superfamily of enzymes: multifunctional enzymes from microbes to mitochondria. Biochemical Society Transactions. 46, 295 (2018).
- Guimarães, S. L., et al. Crystal Structures of Apo and Liganded 4-Oxalocrotonate Decarboxylase Uncover a Structural Basis for the Metal-Assisted Decarboxylation of a Vinylogous β-Keto Acid. Biochemistry. 55, 2632 (2016).
- Zhou, N. Y., Fuenmayor, S. L., Williams, P. A. nag genes of Ralstonia (formerly Pseudomonas) sp. strain U2 encoding enzymes for gentisate catabolism. Journal of Bacteriology. 183, 700 (2001).
- Izumi, A., et al. Structure and Mechanism of HpcG, a Hydratase in the Homoprotocatechuate Degradation Pathway of Escherichia coli. Journal of Molecular Biology. 370, 899-911 (2007).
- Manjasetty, B. A., et al. X-ray structure of fumarylacetoacetate hydrolase family member Homo sapiens FLJ36880. Biological Chemistry. 385, 935-942 (2004).
- Tame, J. R. H., Namba, K., Dodson, E. J., Roper, D. I. The crystal structure of HpcE, a bifunctional decarboxylase/isomerase with a multifunctional fold. Biochemistry. 41, 2982-2989 (2002).
- Ran, T., et al. Crystal structures of Cg1458 reveal a catalytic lid domain and a common catalytic mechanism for the FAH family. The Biochemical Journal. 449, 51-60 (2013).
- Ran, T., Wang, Y., Xu, D., Wang, W. Expression, purification, crystallization and preliminary crystallographic analysis of Cg1458: A novel oxaloacetate decarboxylase from Corynebacterium glutamicum. Acta Crystallographica Section F: Structural Biology and Crystallization Communications. 67, 968-970 (2011).
- Pircher, H., et al. Identification of human Fumarylacetoacetate Hydrolase Domain-containing Protein 1 (FAHD1) as a novel mitochondrial acylpyruvase. Journal of Biological Chemistry. 286, 36500-36508 (2011).
- Pircher, H., et al. Identification of FAH domain-containing protein 1 (FAHD1) as oxaloacetate decarboxylase. Journal of Biological Chemistry. 290, 6755-6762 (2015).
- Petit, M., Koziel, R., Etemad, S., Pircher, H., Jansen-Dürr, P. Depletion of oxaloacetate decarboxylase FAHD1 inhibits mitochondrial electron transport and induces cellular senescence in human endothelial cells. Experimental Gerontology. 92, 7-12 (2017).
- Etemad, S., et al. Oxaloacetate decarboxylase FAHD1 – a new regulator of mitochondrial function and senescence. Mechanisms of Ageing and Development. 177, 22-29 (2019).
- Weiss, A. K. H., et al. Structural basis for the bi-functionality of human oxaloacetate decarboxylase FAHD1. Biochemical Journal. 475, 3561-3576 (2018).
- Taferner, A., et al. FAH domain-containing protein 1 (FAHD-1) Is required for mitochondrial function and locomotion activity in C. elegans. PLoS ONE. 10, 1-15 (2015).
- Mizutani, H., Kunishima, N. Purification, crystallization and preliminary X-ray analysis of the fumarylacetoacetase family member TTHA0809 from Thermus thermophilus HB8. Acta Crystallographica Section F Structural Biology and Crystallization Communications. 63, 792-794 (2007).
- Bateman, R. L., Bhanumoorthy, P., Witte, J. F., McClard, R. W., Grompe, M., Timm, D. E., et al. Mechanistic Inferences from the Crystal Structure of Fumarylacetoacetate Hydrolase with a Bound Phosphorus-based Inhibitor. Journal of Biological Chemistry. 276, 15284-15291 (2001).
- Zeng, F., et al. Efficient strategy for introducing large and multiple changes in plasmid DNA. Scientific Reports. 8, 1714 (2018).
- Higuchi, R., Krummel, B., Saiki, R. K. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Research. 16, 7351-7367 (1988).
- Jansen-Duerr, P., Pircher, H., Weiss, A. K. H. The FAH Fold Meets the Krebs Cycle. Molecular Enzymology and Drug Targets. 2, 1-5 (2016).
- Rupp, B. Origin and use of crystallization phase diagrams. Acta Crystallographica Section F Structural Biology Communications. 71, 247-260 (2015).
- Rupp, B. . Biomolecular Crystallography: Principles, Practice, and Application to Structural Biology. , (2010).
- Flint, D. H., Nudelman, A., Calabrese, J. C., Gottlieb, H. E. Enol oxalacetic acid exists in the Z form in the crystalline state and in solution. The Journal of Organic Chemistry. 57, 7270-7274 (1992).
- Pogson, C. I. I., Wolfe, R. G. G. Oxaloacetic acid tautomeric and hydrated forms in solution. Biochemical and Biophysical Research Communications. 46, 1048-1054 (1972).
- Kost, T. A., Condreay, J. P., Jarvis, D. L., Kost, A. T. Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nature Biotechnology. 23, 567-575 (2005).
- Steinberger, R., Westheimer, F. H. Metal Ion-catalyzed Decarboxylation: A Model for an Enzyme System 1. Journal of the American Chemical Society. 73, 429-435 (1951).
- Tate, S. S., Grzybowski, A. K., Datta, S. P. The stability constants of the magnesium complexes of the keto and enol isomers of oxaloacetic acid at 25. Journal of Chemical Society. , 1381-1389 (1964).
- Tate, S. S., Grzybowski, A. K., Datta, S. P. The acid dissociations of the keto and enol isomers of oxaloacetic acid at 25. Journal of Chemical Society. 1372, 1380 (1964).
- Brecker, L., et al. Synthesis of 2,4-diketoacids and their aqueous solution structures. New Journal of Chemistry. 23, 437-446 (1999).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati