Method Article
Expressão, purificação, cristalização e ensaios enzimáticos de proteínas contendo domínio de Fumarilacetoacetato de hidrolase
Neste Artigo
Resumo
Expressão e purificação de proteínas contendo domínio de fumarilacetoacetato hidrolase é descrita com exemplos (expressão em E. coli, FPLC). As proteínas purificadas são utilizadas para a cristalização e produção de anticorpos e empregadas para ensaios enzimáticos. Os ensaios fotométricos selecionados são apresentados para exibir a multifuncionalidade de FAHD1 como oxaloacetato descarboxilase e acylpyruvate hydrolase.
Resumo
As proteínas contendo domínio de fumarylacetoacetate hidrolase (Fah) (Fahd) são membros identificados da superfamília Fah em eucariontes. As enzimas desta superfamília geralmente exibem multifuncionalidade, envolvendo principalmente mecanismos de hidrolase e decarboxilase. Este artigo apresenta uma série de métodos consecutivos para a expressão e purificação de proteínas FAHD, principalmente FAHD proteína 1 (FAHD1) ortologues entre as espécies (humano, rato, nematodes, plantas, etc.). Os métodos cobertos são expressão protéica em e. coli, cromatografia de afinidade, cromatografia de troca iónica, filtração preparativa e analítica de gel, cristalização, difração de raios X e ensaios fotométricos. Proteína concentrada de altos níveis de pureza (> 98%) podem ser empregadas para a cristalização ou produção de anticorpos. Proteínas de qualidade semelhante ou inferior podem ser empregadas em ensaios enzimáticos ou usadas como antígenos em sistemas de detecção (Western-blot, ELISA). Na discussão deste trabalho, os mecanismos enzimáticos identificados de FAHD1 são esboçados para descrever mais detalhadamente sua bi-funcionalidade do hidrolase e do descarboxilase.
Introdução
A hidrolase de fumarilacetoacetato (Fah)1,2 superfamília de enzimas descreve um grupo de enzimas que compartilham o domínio catalítico altamente conservado do Fah3,4,5,6 , 7 anos de , 8 . º , 9 anos de , 10. apesar de seu centro catalítico comum, estas enzimas são multifuncionais, e a maioria são encontrados em procariontes, onde eles são usados para quebrar compostos recuperados de fontes de carbono complexas3. Somente três membros desta família foram identificados nos eucariotas até agora: o nome que dá Fah2, assim como a proteína de domínio-contendo do Fah 1 (FAHD1)11,12,13,14 ,15 e Fah proteína contendo domínio 2 (FAHD2). A depleção de FAHD1 tem sido associada à respiração mitocondrial prejudicada13,16 e associada a um tipo reversível de fenótipo de senescência celular14 que está ligado ao potencial intermediário deficiências no sistema de transporte de elétrons. Human FAHD1 e seus ortologues em sistemas modelo (mouse, nematódeo, linhas de células cancerosas, plantas, etc.), bem como variantes de mutação de ponto selecionadas, tornaram-se alvos druggable de interesse potencial. Para esta pesquisa, a proteína recombinante em altos níveis de pureza, bem como informações sobre os mecanismos catalíticos guiados por estruturas cristalinas e anticorpos seletivos são vitais.
Este manuscrito descreve métodos para expressão protéica de FAHD em E. coli, cromatografia de afinidade, cromatografia de troca iónica, precipitação de sulfato de amônio, filtração de gel preparativo e analítico, cristalização, difração de raios X e ensaios fotométricos. O objetivo dos métodos e protocolos descritos aqui é fornecer orientação para cientistas que trabalham em diversos campos, como bacteriologia, biologia vegetal, bem como estudos em animais e humanos, para caracterizar os membros da superfamília FAH, incluindo Membros da superfamília não caracterizados devem tornar-se relevantes em um determinado campo. Os protocolos aqui descritos podem fornecer suporte valioso para projetos que visem caracterizar outros membros da superfamília procariótica ou eucariótica.
A fundamentação subjacente aos métodos descritos aqui é o fato de que para a caracterização de proteínas mal descritas (em particular, enzimas metabólicas de relevância fisiológica desconhecida), a abordagem para iniciar com proteínas recombinantes purificadas permite que o desenvolvimento de ferramentas de pesquisa inestimáveis e de alta qualidade, como preparações enzimáticas ativas in vitro, anticorpos de alta qualidade e inibidores farmacológicos potentes e específicos para enzimas selecionadas. Os métodos descritos exigem a cromatografia líquida rápida da proteína (FPLC) e o crystallography do raio X. Os métodos alternativos (por exemplo, para expressar a proteína sem indução química, ou para indicar a purificação da proteína pela centrifugação após o tratamento térmico seguido pelo dessalinização e pela cromatografia da exclusão do tamanho), podem ser encontrados em outra parte17. Quando um espectro mais largo dos métodos está disponível para a expressão e a purificação de enzimas da superfamília de Fah2,7,9,17,18, este trabalho centra-se sobre a expressão e purificação das proteínas FAHD em particular.
Na seção de discussão deste manuscrito, os mecanismos catalíticos identificados para a proteína FAHD1 (hidrolase, decarboxilase)15 são descritos em mais detalhes, a fim de demonstrar o caráter químico das reações catalisadas. Os dados obtidos com base no trabalho anterior7,15,18 (PDB: 6fog, PDB: 6foh) implicam uma terceira atividade da enzima como keto-enol isomerase.
Protocolo
1. expressão de proteínas FAHD em E. coli competente
- Transformação de e. coli com vetores para expressão de proteína Fahd
Observação: as etapas discutidas na seção a seguir são resumidas no esboço na Figura 1a, B. O mesmo protocolo aplica-se para toda a proteína de FAHD, incluindo variações do ponto-mutante. Tais variantes podem ser obtidas por meio de mutagenese dirigida por local e técnicas de PCR19 (como a PCR20de dois lados) do cDNA do tipo Wild.
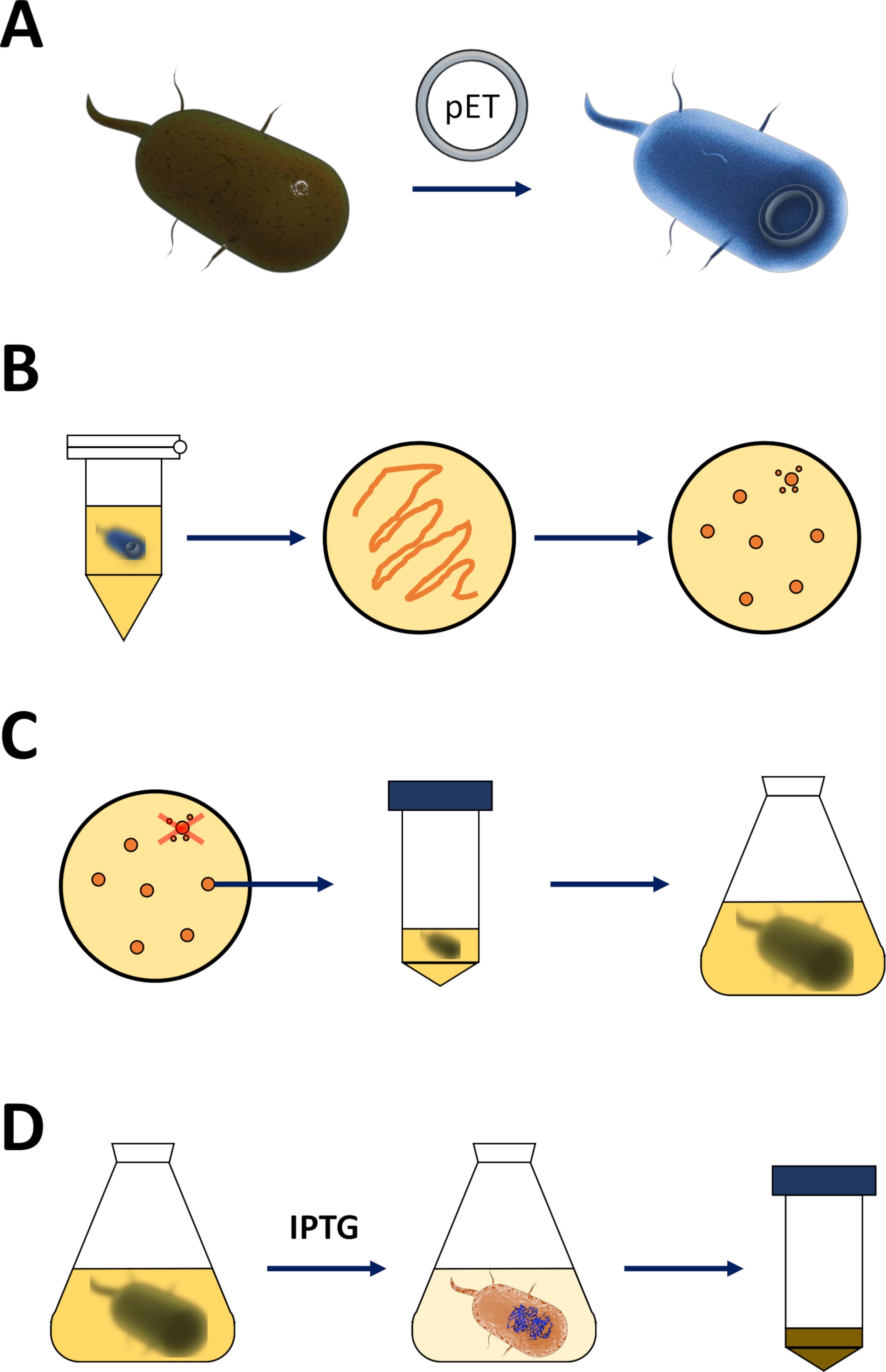
Figura 1 : Amplificação de e. coli competente e indução de expressão protéica.
(A) inserção do vetor Pet em bactérias BL21 competentes (de3) plyss E. coli , descritas na seção 1. (B) protocolo de choque térmico e chapeamento do animal de estimação transformado e. coli bactérias, descrita na etapa 1 do protocolo. As bactérias transformadas são chapeadas em placas de agar LB com antibióticos para seleção. (C) amplificação de bactérias de E. coli transformadas em animais de estimação , descritas na seção 1. As colônias são colhidas a partir de uma placa de agar LB e amplificadas em meio nutritivo (LB ou NZCYM) até que a densidade bacteriana atingiu o limiar empírico de 0,4. (D) indução da expressão protéica por meio do sistema de3-IPTG-pET , descrito na seção 1 e esboçado na Figura 2. A produção de proteínas é iniciada pela aplicação do produto químico IPTG. No final da seção 1, a pelota bacteriana contendo a proteína é colhida. Por favor clique aqui para ver uma versão maior desta figura.- Obter as bactérias BL21 (DE3) plyss e. coli competentes e um vetor de expressão pET (ver tabela de materiais). Preferivelmente escolha um vetor do animal de estimação que igualmente codifica um N-terminal his-tag ou a etiqueta relacionada da captação para a conveniência simplificar as seguintes etapas da purificação.
- Obter cDNA da proteína FAHD de escolha e inseri-lo no local de clonagem ativo do vetor de expressão pET, entre o promotor T7 e sites de terminador T7, respectivamente.
- Após a amplificação e verificação plasmídeo bem-sucedidas [via sequenciação por um fornecedor comercial (os primers T7 podem ser usados com o sistema pET para conveniência: promotor T7, iniciador dianteiro: TAATACGACTCACTATAGGG; T7 Terminator, primer reverso: GCTAGTTATTGCTCAGCGG)], insira 5 – 10 ng de plasmídeo em 100 μL de bactérias BL21 (DE3) plyss E. coli competentes no gelo. Não aspirar para cima e para baixo, mas levemente bata o tubo com a fim de misturar o conteúdo.
- Mantenha as bactérias no gelo por 30 min, tocando suavemente o tubo a cada poucos minutos.
- Aqueça um dispositivo de aquecimento ou um banho de água a 42 ° c (exato). Coloque o tubo contendo as bactérias no aparelho e mantê-los para 90 s (exato). Coloque-os no gelo imediatamente (Figura 1a).
- Após 5 – 10 min no gelo, adicione 600 μL de meio NCZYM (ver tabela de materiais) e coloque o tubo em uma incubadora de bactérias. Agitar o tubo a uma velocidade média orientada ao longo do sentido de agitação a 37 ° c durante 1 h.
- Placa 200 μL da cultura bacteriana em uma placa de 10 cm LB-Agar (ver tabela de materiais), contendo antibióticos de seleção de escolha [por exemplo, um específico para a resistência BL21 (de3) plyss (cloranfenicol), e um para a resistência codificado no vetor pET (canamicina ou ampicilina, Figura 1b)].
- Cultura das bactérias na placa de LB-Agar em uma incubadora bacteriana em 37 ° c durante a noite.
- Expressão de proteínas FAHD por indução de IPTG
Observação: os primeiros passos discutidos na seção a seguir são resumidos como um esboço na Figura 1C, D. O sistema da expressão T7 através da combinação da gaveta de3 bacteriana e do sistema do vetor do animal de estimação é sumariado na Figura 2.
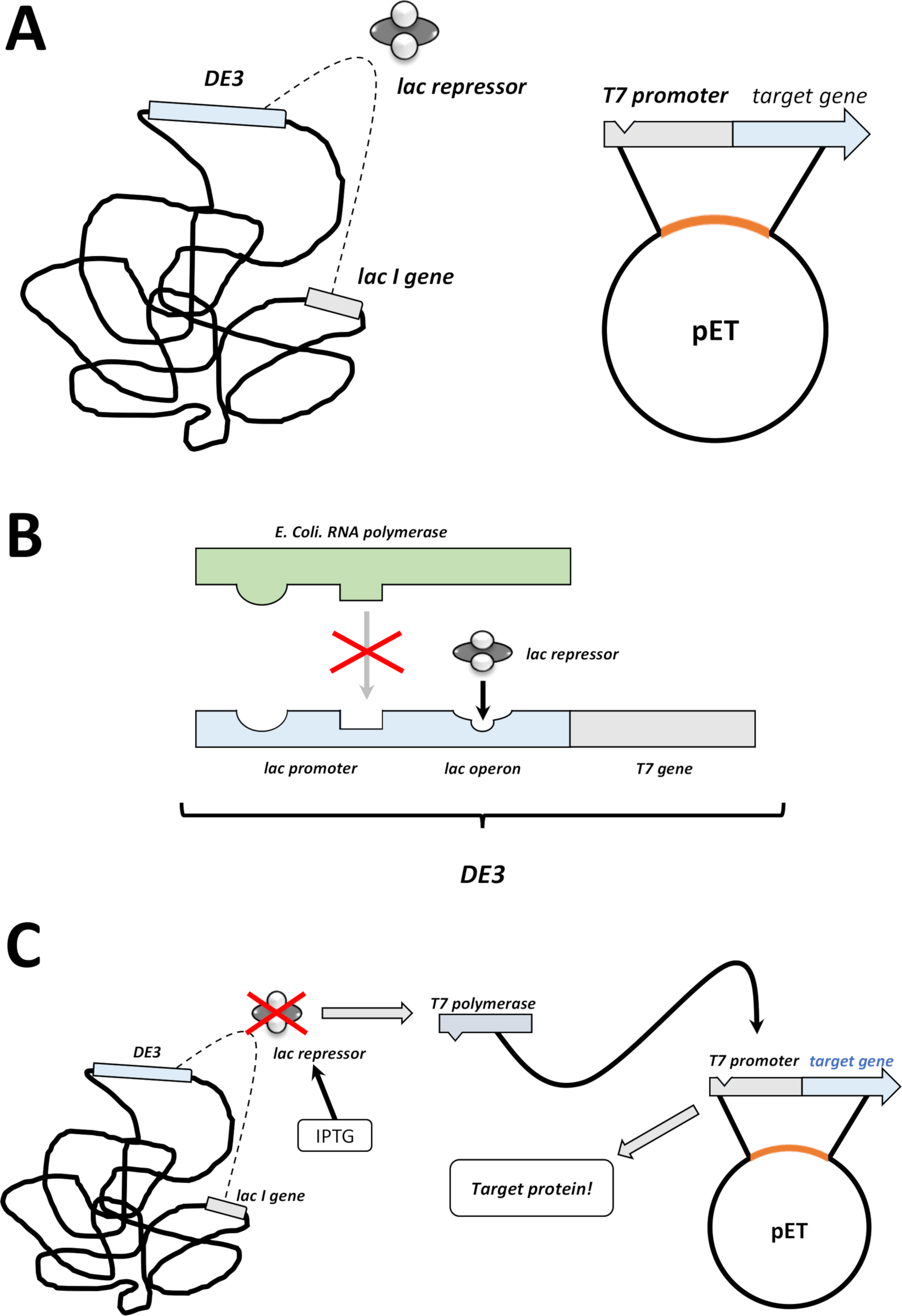
Figura 2 : O sistema duplo do vetor de de3 Cassette/pET explicou.
(A) o genoma esboçado de Pet vector transformado BL21 (de3) plyss E. coli bactérias. O genoma bacteriano nativo carrega uma gaveta DE3 (Veja o painel B), assim como um gene Lac que expresse constantemente unidades do repressor do Lac. O vetor não-nativo do animal de estimação carreg o gene da proteína introduzido entre um promotor do polymerase T7 e uma seqüência do terminador. Mais detalhes no painel B. (b) a gaveta de3 do genoma bacteriano nativo codifica a informação para o polymerase T7 nos termos de um operon do polymerase do RNA de E. coli . Esta proteína, entretanto, não é expressada porque a unidade repressor do Lac impede a proteína do polymerase do RNA da ligação. Daqui nenhuma polimerase T7 é expressada e nenhuma proteína exógena é expressada. (C) a aplicação do produto químico IPTG (tabela de materiais) distorça a estrutura das unidades repressor Lac e impede que elas se vinculem à gaveta de3. Em conseqüência, o polymerase do RNA pode agora ligar ao cassette, para que o polymerase T7 é expressado, como é proteína exógena eventualmente. Por favor clique aqui para ver uma versão maior desta figura.- Após a formação de colônia bem-sucedida, escolha uma única colônia (sem quaisquer colônias de satélites) e dispersar-a em 5 ml de nzcym ou meio lb com antibióticos, selecionados como antes (passo 1.1.7). Cultura na incubadora bacteriana a 37 ° c durante a noite (Figura 1C).
- Após o sucesso do crescimento bacteriano, amplificar as bactérias em 250 mL, 500 mL, ou 1 L lotes de médio, dependendo da demanda de quantidade de proteínas.
- Apropriado ao volume, aplique os antibióticos selecionados como feito na etapa 1.1.7 e adicione aproximadamente 1%-2% da pre-cultura bacteriana densa (isto é, 2.5-5.0 mL a 250 mL de volume do meio, etc.). Pegue uma amostra para ser usada na etapa 1.2.5 (1 mL ou mais) e verifique a densidade óptica (OD) em 600 nm. Bactérias da cultura na incubadora bacteriana em 37 ° c para 2 – 3 h (Figura 1C).
- Após 2 – 3 h, desenhe uma amostra para análise fotométrica. Se o OD em 600 nm atingiu 0,4, aplique 200 μM até 1 mM de isopropílico-β-D-tiogalactopyranosid (IPTG, ver tabela de materiais).
Nota: o valor real é empírico para cada variante de proteína FAHD ou mutação de ponto, onde 1 mM IPTG é o máximo que deve ser aplicado. Isso induz a expressão protéica (Figura 1D, Figura 2C). - Após 3 – 5 horas mais na incubadora bacteriana a 37 ° c, a expressão protéica está esgotada.
Observação: consulte a seção de discussão para comentários sobre controle de temperatura. Mais do que 5 h de agitação após a indução não é recomendado. Pegue uma amostra para uso na etapa 1.2.5 (1 mL ou mais) e verifique a densidade óptica (OD) em 600 nm.- Colha a pelota bacteriana através da centrifugação em 5.000 x g por 5 min. descarte o sobrenadante e congele o pellet em-80 ° c para maior armazenamento ou-20 ° c para breve armazenamento (Figura 1D).
- Verificar a indução através das duas amostras fotométricas recuperadas, que são rotuladas "-I" (antes da indução) e "+ I" (após a indução). Após a centrifugação e ressuspensão da pelota bacteriana, analise as duas amostras por SDS-PAGE carregando a mesma quantidade de proteína total.
Nota: a amostra "+ I" deve apresentar uma banda forte associada ao peso molecular da proteína escolhida, enquanto a amostra "-I" não deve conter esta banda. Um baixo nível de indução é um problema comum para a produção de proteínas, mas o nível de proteína expressa é muitas vezes suficiente para as seguintes etapas. Um alto nível de indução é uma vantagem, mas não é obrigatório.
{kind=link}
{kind=link}
2. lise de pelotas bacterianas e filtração de detritos
- Dependente de se a proteína escolhida é sua-etiquetada ou Untagged, selecione o amortecedor running de NI-NTA (His-etiquetado, vejaa tabela dos materiais) ou gelo-frio HIC que executa o amortecedor (untagged).
- Para cada 250 mL de suspensão bacteriana original, aplique 5 mL do tampão seleccionado à pelota bacteriana (5 mL para 250 mL, 10 mL para 500 mL, etc.). Adicionar 10 μL de β-mercaptoetanol (β-ME) por 5 mL de tampão aplicado. Use um Pipet de 10 mL Pasteur para forçar mecanicamente o pellet em suspensão por arranhões e pipetagem (Evite a formação de bolhas de ar durante a pipetagem). Eventualmente transferir toda a suspensão em 1 50 mL tubo.
- De preferência proceda (6x para 15 s em força média) a suspensão.
- Centrifugador por 30 minutos em alta velocidade (10.000 x g) a 4 ° c. Filtre o sobrenadante consecutivamente com unidades filtrantes (por exemplo, 0,45 μm, 0,22 μm) no gelo.
Nota: dependendo da etapa anterior da centrifugação, a filtração diretamente através de um tamanho pequeno do poro do filtro pode ser tedioso e exige geralmente a pre-filtração com um tamanho de poro maior. DNAse pode ser adicionado para melhores resultados. - Armazene a amostra no gelo e prossiga imediatamente com a seção 3 ou 4, dependendo se a proteína é sua-etiquetada ou untagged.
3. purificação de suasproteínas Fahd marcadas usando a cromatografia de afinidade NI-NTA
Nota: os íons Ni2 + estão ligados através de ácido nitrilotriacético (NTA) a uma resina de agarose que é utilizada em cromatografia de afinidade (cromatografia de íon metálico imobilizado, iMac, Figura 3a). As etiquetas do ácido aminado da poli-histidina ligam fortemente a este Ni-Chelate, e as proteínas his-etiquetadas podem ser separadas da maioria de proteínas permanecendo. Uma alternativa à preparação descrita das colunas NI-NTA está usando colunas NI-NTA pré-embaladas e um sistema FPLC.

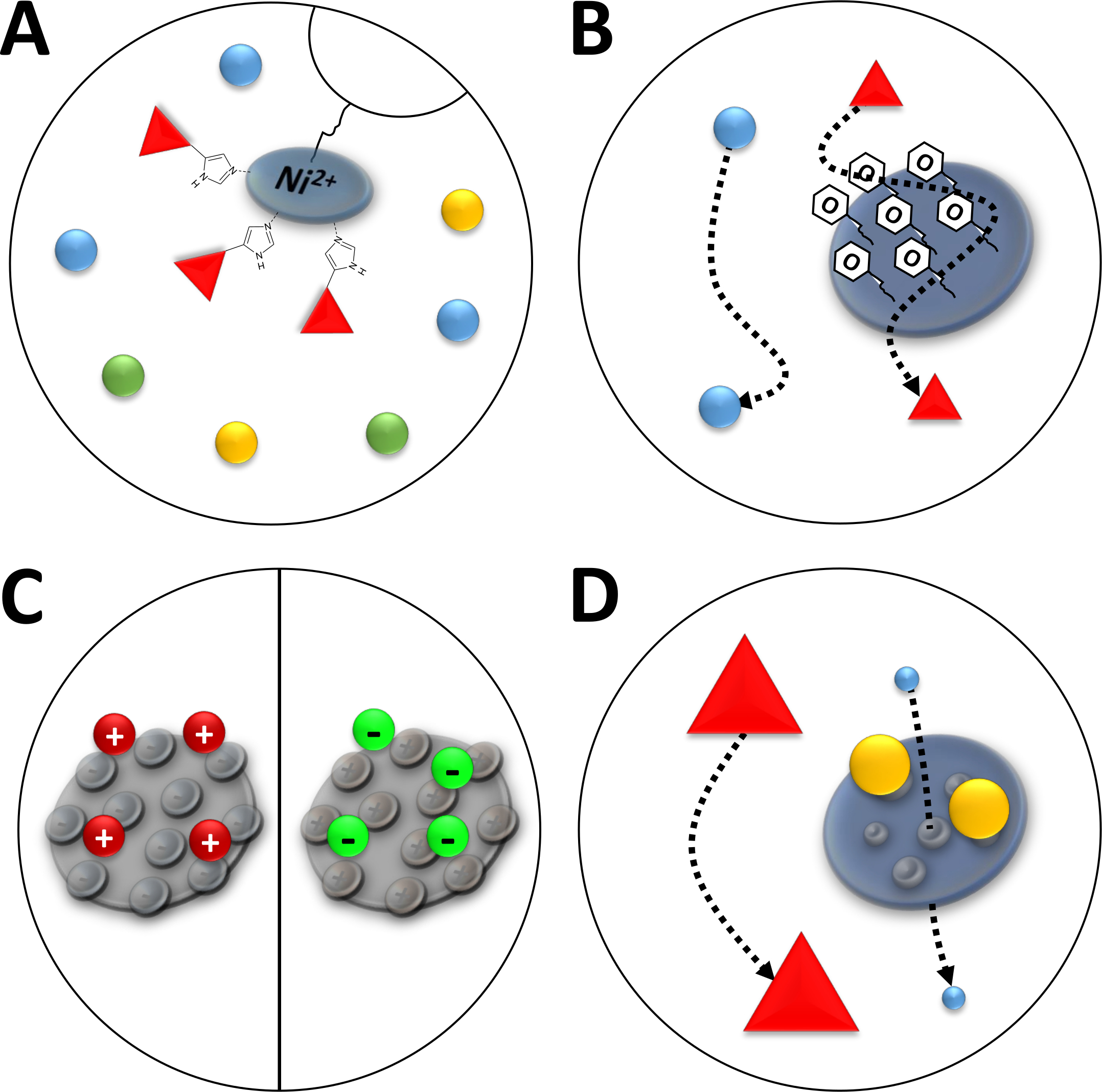
Figura 3 : Ilustrações esboçadas de tipos comuns de cromatografia.
(A) a resina de uma coluna NI-NTA. A NTA detém íons bivalentes de níquel que são usados em termos de cromatografia de afinidade de íons metálicos imobilizados (IMAC). Os Tag do poli-histidine ligam preferivelmente a este motivo e podem ser eluída pelo imidazole. (B) o revestimento típico de partículas de sílica em uma cromatografia de interação hidrofóbica à base de fenil (HIC-Phenyl). Proteínas hidrofóbicas interagem com o material de revestimento e são atrasadas em sua migração, enquanto outros não. (C) o revestimento típico de partículas de sílica na cromatografia de interação iónica. As proteínas polarizadas e carregadas interagem com o material de revestimento e são atrasadas em sua migração quando outro não forem. (D) a resina de um gel de sílica em cromatografia de exclusão de tamanho (SEC). Com base em poros definidos no material de sílica, as proteínas podem ser separadas pelo seu tamanho (em uma primeira aproximação correspondente à sua massa molecular). As proteínas pequenas permeiam o material poroso da coluna e são, quando as grandes proteínas migrarem mais rapidamente em torno das partículas porosas. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
- Proceder da etapa 2,5 (ou seja, a proteína está em NI-NTA running buffer e filtrada por 0,22 μm unidades de filtro no gelo).
- Prepare uma coluna de plástico ou vidro vazia lavando a coluna vazia e anexando-a a um retentor estável. Escolha o tamanho da coluna dependendo do volume da suspensão da proteína.
- Para cada 10 mL de suspensão proteica, aplique 500 μL de lama de agarose NI-NTA na coluna (agitar pesadamente antes da utilização). Aplique a pasta lentamente e gota gota no filtro inferior da coluna usando uma pipeta. Deixe a coluna resolver, o que demora alguns segundos.
- Encha a coluna completamente com o tampão running de NI-NTA, assegurando não interromper a resina do agarose. Deixe o tampão passar pela gravidade. O processo pode ser acelerado aplicando a pressão do polegar no líquido (usando uma tampa ou uma luva e uma pressão do polegar), mas tome o cuidado para não distorcer a resina do agarose.
- Aplique a suspensão proteica. Como antes, deixe a amostra correr pela gravidade. Acelerar este passo usando a pressão do polegar não é recomendado, como a ligação de proteínas para a coluna é reforçada se a taxa de fluxo é baixa. Colete o fluxo-através de um tubo (tabela de materiais).
- Após a amostra ter passado, preencha a coluna inteira novamente com o NI-NTA executando buffer. Tome cuidado para não interromper a resina de agarose. Deixe a amostra passar por gravidade, mas em contraste com a etapa anterior, acelerar o processo através da pressão do polegar é recomendado, como potenciais contaminações por causa de interações inespecíficas podem ser interrompidas desta forma. Colete a solução de lavagem em um tubo. Repita este passo.
- Coloque uma cubeta UV-transparente abaixo da coluna e aplique 1 mL do amortecedor da eluição do NI-NTA. Colete a amostra sem aplicar nenhuma pressão do polegar à resina.
- Verifique a densidade óptica (OD) da amostra em 280 nm vs. uma amostra em branco (i.e., tampão de eluição de NI-NTA). Otimamente, o exemplo exibe um OD de maior que 2,5. Um OD abaixo de 0,5 denota que nenhuma quantidade significativa de proteína está na amostra.
Nota: conforme descrito na secção de discussão, as concentrações de sal e imidazol do tampão de eluição podem ter de ser adaptadas para cada proteína FAHD individualmente. - Repita as etapas 3.1.7 e 3.1.8 até que o OD caia abaixo de 0,5. Piscina todas as amostras com maior OD em um tubo no gelo.
- Comece outra vez com etapa 3.1.4, usando o fluxo-através da etapa 3.1.5 como a entrada nova para esta repetição da etapa 3.1.5. Repita esse processo até que o primeiro exemplo coletado na etapa 3.1.6 exiba um OD abaixo de 0,5.
Nota: conforme descrito na parte de solução de problemas da seção de discussão, as proteínas his-Tagged podem se ligar insuficientemente à resina Ni2 +. Nesses casos, é necessária a repetição desta etapa ou métodos alternativos (por exemplo, cromatografia de troca iónica). - Pegue amostras de todas as frações intermediárias para análise de SDS-PAGE.
- As proteínas de FAHD no tampão da eluição de NI-NTA precipitarão em cima de congelar-se e thawing. Portanto, Dialize a proteína contra um tampão diferente (durante a noite no gelo, usando 1 μL de DTT por 100 ml de tampão de diálise). Use o tampão do baixo-sal baseado em que tipo de cromatografia da troca iónica deve ser executada após esta etapa. Use a tubulação comum da celulose com um corte de peso molecular típico de 14 kDa (tabela de materiais).
- Após a diálise durante a noite, opcionalmente concentrar a proteína usando unidades de filtro de ultra-centrifugação. Execute a análise de SDS-PAGE (12,5% gel running, gel de empilhamento de 4%) para verificar a perda potencial de proteína, a eluição insuficiente, e a pureza da proteína geralmente. Se tudo estiver bem, prossiga para a seção 5.
4. purificação de proteínas FAHD não marcadas via cromatografia de interação hidrofóbica (HIC)
Nota: fenil-grupos na superfície de revestimento de um gel de sílica em uma coluna HIC para FPLC (Figura 3B) permitem a separação de proteínas de acordo com o caráter hidrofóbico. As etapas descritas devem ser executadas com um sistema de FPLC equipado com um 5 mL da coluna de HIC-Phenyl. As colunas podem ser lavadas com NaOH de 1 M para serem reutilizadas para diferentes proteínas. Entretanto, as colunas usadas uma vez para um tipo de proteína de FAHD devem reúso para somente este tipo de proteína.
-
Sulfato de amônio (AS) precipitação
- Prossiga a partir do passo 2,5. A proteína está no gelo-frio HIC running buffer (tabela de materiais).
- Avalie o volume da solução de proteína preparada precisamente para o microlitro (Vinicial). Lentamente e gota-sábio adicione a solução de buffer running de HIC pre-cooled, até que um volume 35-% como a saturação esteja alcangado: Vcomo adicionado = vInitial * 0,538. Mexa suavemente a solução durante 30 min. centrifugador durante 15 min a alta velocidade (≥ 10.000 x g) a 4 ° c.
- Filtre o sobrenadante usando uma unidade de filtro de 0,22 μm no gelo. Opcionalmente, pegue uma amostra para análise de SDS-PAGE: diluir 1:4 e aquecer imediatamente a 95 ° c por 5 min ou então a amostra será protuberância. A amostra pode ser congelada neste ponto (-20 ° c), a fim de prosseguir mais um dia.
-
FPLC usando uma coluna HIC
- Configurar o sistema FPLC e equilibrar uma coluna de 5mL HIC-Phenyl com 5 volumes de coluna (CV) de 20% EtOH (em H2o) seguido por 5 CV de h2o.
- Misture 260 mL de HIC que executa o amortecedor (exato) com 140 mL de HIC que executa o amortecedor como (exato). Isso resulta em uma solução de 35 volume-% AS. Verifique o pH (7,0); Isso é buffer A. buffer B é 250 mL de buffer em execução. Adicionar 1 mM DTT para ambos os buffers a e B, em seguida, mantê-los no gelo.
- Equilibrar a coluna com 8 mL de buffer A, 8 mL de buffer B e 8 mL de buffer A nessa sequência. Aplique a amostra preparada na etapa de protocolo 4,1. Lave com o tampão A, até que a absorção ótica da linha de base em 280 nanômetro alcangue 1000 – 500 mAU.
- Aplique uma mistura de buffers A e B, de modo que a concentração de como é 33% (w/v). Lave com 1 CV, resultando em um platô no cromatograma. Configurar um gradiente de buffer B (até 100% buffer B ao longo do tempo): 1,5 mL de tampão B em 3,8 min (i.e., 5,7% tampão B com inclinação de 1% B/mL). Quando o sinal UV em 280 nm sobe, começar a recolher a fração e colocá-lo no gelo imediatamente.
- No final, lave a coluna com o tampão B. Pegue amostras de todas as frações para análise de SDS-PAGE. Congelar todas as amostras usando nitrogênio líquido, e armazená-los em-80 ° c.
- Realize a análise de SDS-PAGE (e Western blot) para detectar a proteína FAHD nas frações coletadas. As frações que contêm a proteína são agrupadas e aplicadas à purificação adicional, conforme descrito nas seguintes etapas do protocolo. Lave a coluna com H2o e 20% EtOH (em h2o).
5. purificação de proteínas FAHD via cromatografia de troca iónica
Nota: as moléculas com grupos funcionais carregados são ligadas a uma coluna de partícula de sílica para FPLC (Figura 3C). Isto permite a diferenciação de proteínas de acordo com seu caráter iónico, tal como a carga de superfície. As etapas descritas devem ser realizadas com uma máquina FPLC e know-how associado, respectivamente. O método descrito é o mesmo para cromatografia de troca catiônica ou aniónica, mas os buffers a serem usados são ligeiramente diferentes.
- Escolheu o sistema de cromatografia de troca catiônica ou aniónica. Esta escolha é empírica e pode variar entre as proteínas FAHD. Otimamente, ambos os métodos podem ser usados consecutivamente.
- Configurar o sistema FPLC e lavar a coluna com 5 CV de 20% EtOH (em H2o), seguido por 5 CV de h2o. equilibram a coluna com 1 CV de tampão de sal baixo, tampão de sal alto e, novamente, tampão de sal baixo nesta sequência.
- Aplique a amostra (dialyzed contra o buffer de sal baixo correto da etapa 3.1.11) na coluna. Recolha o fluxo-através de. Lave a coluna para 1 CV com tampão de sal baixo.
- Configurar uma eluição de gradiente: 100% tampão de sal alto em 30 min a uma taxa de fluxo de 1 mL/min, ou 60 min a uma taxa de fluxo de 0,5 mL/min. Isso pode ser reselecionado com base em um cromatograma FPLC já conhecido, a fim de otimizar a purificação. Colete todas as frações de pico.
Observação: as condições de alto sal podem variar entre as proteínas FAHD, conforme descrito na seção de discussão. - Após o gradiente ter terminado, execute com buffer de sal alto até que não mais picos são detectados ao longo do intervalo de 1 CV (recolher as frações).
- Pegue amostras de todas as frações coletadas e execute a análise de SDS-PAGE (12,5% gel running, gel de empilhamento de 4%). Congelar as amostras individuais em nitrogênio líquido e armazená-los em-80 ° c.
- Após a análise da SDS-PAGE ser concluída, o agrupamento das amostras contendo a proteína FAHD e descartar os outros. Opcionalmente, concentrar a proteína usando unidades de filtro de ultra-centrifugação.
- Aplique 1 mL de SDS de 25% em 0,5 M NaOH (ou outros detergentes) para limpar a coluna. Lave a coluna com H2o e 20% EtOH (em h2o).
- Opcionalmente, repita a seção 5 com a coluna alternativa (cromatografia de troca catiônica ou aniónica). A proteína obtida deste método é suficientemente pura para realizar ensaios de atividade básica ou pode ser usada em ensaios de triagem para cristalografia. Para aplicações avançadas, prossiga com a secção 6.
6. purificação de proteínas FAHD através de cromatografia de exclusão de tamanho (SEC)
Nota: partículas porosas em uma coluna de sílica gel para FPLC permitem a diferenciação de proteínas de acordo com o tamanho molecular, como raio hidrodinâmico (Figura 3D). As etapas descritas devem ser executadas com um sistema FPLC, usando colunas SEC.
- Escolha uma coluna SEC, dependente dos pesos moleculares das contaminações ainda presentes, como detectado através de SDS-PAGE e coloração de prata. O método descrito é adequado para ambas as colunas. Lave a coluna durante a noite com 400 ml de H2o e equilibrar com o tampão running SEC. Recomenda-se escrever um programa para o sistema FPLC para automatizar esta etapa.
- Adicionar 1 mM DTT para 300 mL de SEC buffer de execução e colocá-lo no gelo. Este é o buffer de execução. Aplique 60 mL desse buffer para a coluna.
- Centrifugue a amostra de proteína (10.000 x g por 10 min) para remover qualquer microprecipitação. Aplique o sobrenadante à coluna. Recomenda-se geralmente filtrar o sobrenadante antes do FPLC.
- Aplique o buffer em execução na coluna até que toda a proteína seja eluída. Colete todos os picos em frações de volume adequado (por exemplo, 2 mL). Pegue amostras para SDS-PAGE e congele todas as frações usando nitrogênio líquido. Armazene as frações congeladas a-80 ° c.
- Após a análise de SDS-PAGE (e Western blot), colete e pool todas as frações contendo a proteína FAHD. A mancha de prata é recomendada para detectar contaminações menores que podem ainda estar atuais.
- Use unidades filtrantes de ultra centrifugação para concentrar a proteína. Embora não seja obrigatório para proteínas FAHD, em geral, uma etapa de dessalinagem (por exemplo, por diálise) é recomendada para ensaios enzimáticos e cristalização.
- Repita as etapas 6,3 – 6,6 várias vezes com diferentes taxas de fluxo e concentrações de sal (empírica), a fim de aumentar a pureza da proteína FAHD. Lave a coluna durante a noite com H2o e 20% EtOH (em h2o).
7. ensaios básicos de atividade FAHD com substratos oxaloacetato e acetilpiruvato
Nota: a proteína 1 de Fahd (FAHD1) indica a atividade do oxaloacetato descarboxilase (ODX) e do acylpyruvate hidrolase (ApH). Isso é descrito em mais detalhes na seção de discussão. Por causa da destabilização por keto-enol carbeno na solução aquosa (isto é, enolization), o oxaloacetato decai por se ao longo do tempo (auto-descarboxylation) em função da concentração e do pH do cofator. Em torno de um pH de 7 e temperatura de 25 ° c, este efeito não é dramático, mas os ensaios devem ser anulados para ter em conta a auto-descarboxilação e concentração enzimática. O esquema de pipetagem está delineado na figura 4a. Em geral, recomenda-se o uso de pipetas bem calibradas para este ensaio, pois é bastante sensível a pequenos erros de pipetagem.

Figura 4 : Esquema de pipetagem esboçado para ensaios enzimáticos.
(A) esquema de pipetagem esboçado para ensaios enzimáticos básicos de proteína Fahd com base em substrato. Substrato em branco:-S/-E; amostra de substrato: + S/-E; enzima em branco:-S/+ E; amostra enzimática: + S/+ E (S: substrato, E: enzima). Consulte o protocolo passo 7 para obter mais detalhes. (B) um esquema de pipetagem esboçado para avaliar A cinética de Michaelis-Menten da proteína Fahd. Substrato em branco:-S/-E; amostra de substrato: + S/-E; enzima em branco:-S/+ E; amostra enzimática: + S/+ E (S: substrato, E: enzima). Consulte a seção 8 do protocolo para obter mais detalhes. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
- Iniciar um leitor de microplacas e equilibrar por 30 min a 25 ° c. Configurar um programa para leitura de 12 poços (conforme descrito na figura 4a) em 255 Nm. Recomenda-se usar 25 leituras múltiplos com atraso de tempo de 5 ms. Configurar um ciclo para medir 15x a cada 2 min (30 min total).
- Por predefinição, prepare um tampão de ensaio enzimático (ver tabela de materiais) com 1 mm MgCl2 a pH 7,4. As proteínas variante FAHD podem necessitar de diferentes cofactores ou níveis de pH. Mg2 + e Mn2 + são conhecidos cofatores para FAHD1,3,11,12,21.
- Criar uma solução de proteína de 1 μg/μL, diluindo com tampão de ensaio enzimático (tabela de materiais).
- Configurar 1 mL de solução de 20 mM de um substrato a ser testado (até agora, os substratos identificados de proteínas FAHD são listados em outros lugares3) no tampão do ensaio enzimático.
- De acordo com o esquema de pipetagem apresentado na figura 4a, prepare a enzima em branco e os poços amostrais: pipet 90 μL de tampão do ensaio enzimático (tabela de materiais) nos poços com 5 μL (5 μg) de solução enzimática.
- De acordo com o esquema de pipetagem apresentado na figura 4a, prepare o substrato em branco e os poços da amostra: pipet 95 μL de tampão do ensaio enzimático nos poços.
- Logo antes da medição, aplique 5 μL de tampão do ensaio enzimático nos seis poços em branco. Aplique 5 μL da solução de substrato de 20 mM aos poços amostrais. Recomenda-se a utilização de uma pipeta multicanal.
- Use uma pipeta multicanal com 50 μL de configurações para misturar suavemente todos os poços. Comece com os espaços em branco e prossiga com os poços de amostra. Tome cuidado para não criar bolhas. Insira a placa num leitor de microplacas e meça cada poço a 255 Nm (conforme descrito no passo 7,1).
- Realize a análise em uma planilha. Copie os dados brutos do fotômetro em uma planilha e escreva todas as configurações (ou seja, toda a documentação) em outra planilha. Média dos dados dos três poços de cada uma das quatro preparações. Subtrair o espaço em branco do exemplo. Também computar desvios padrão e somar os desvios de espaço em branco e amostra.
- Plotar esses dados (y: densidade óptica, x: tempo em min). Uma curva exponencialmente decrescente deve ser exibida. Dependente do tipo de substrato em uso, um aumento inicial dentro dos primeiros 10 min pode ser observado, após o qual o sinal diminui. Isto é atribuído ao carbeno do keto-enol do substrato, conforme descrito em mais detalhes na seção de discussão.
- Divida os dados do sinal óptico ao longo do tempo pelo valor máximo da plotagem, a fim de dimensionar os dados para baixo no intervalo [0,1] (um exemplo é fornecido na Figura 5a). Identifique o intervalo linear da curva, iniciando na diminuição inicial e calcule a inclinação negativa (1/min).
- O curso do tempo da diminuição no OD é associado ao substrato através de sua concentração inicial: 100 nmol/poço * inclinação. Usando a concentração proteica avaliada c0, a atividade específica é computada: 100 nmol/poço * inclinação * 1/c0. Expressando c0 em μg/bem, a atividade específica computada desta forma é expressa utilizando a unidade nmol/min/μg, que equivale a μmol/min/mg.
8. avaliando a cinética de Michaelis-Menten das proteínas FAHD
Nota: a avaliação da cinética de Michaelis-Menten das proteínas FAHD é tediosa, pois a atividade protéica específica depende tanto da concentração relativa de proteínas-substratos quanto do volume físico em que a reação está ocorrendo. A cinética do estado estacionário deve ser estabelecida com o intuito de obter resultados confiáveis. Um protocolo testado em uma placa transparente UV do poço 96 é esboçado nas seguintes etapas. Cada etapa precisa ser realizada com muito cuidado, como pequenos erros geralmente estragar o experimento. Recomenda-se dominar os ensaios descritos na secção 7 antes de tentar o ensaio mais complicado descrito abaixo.

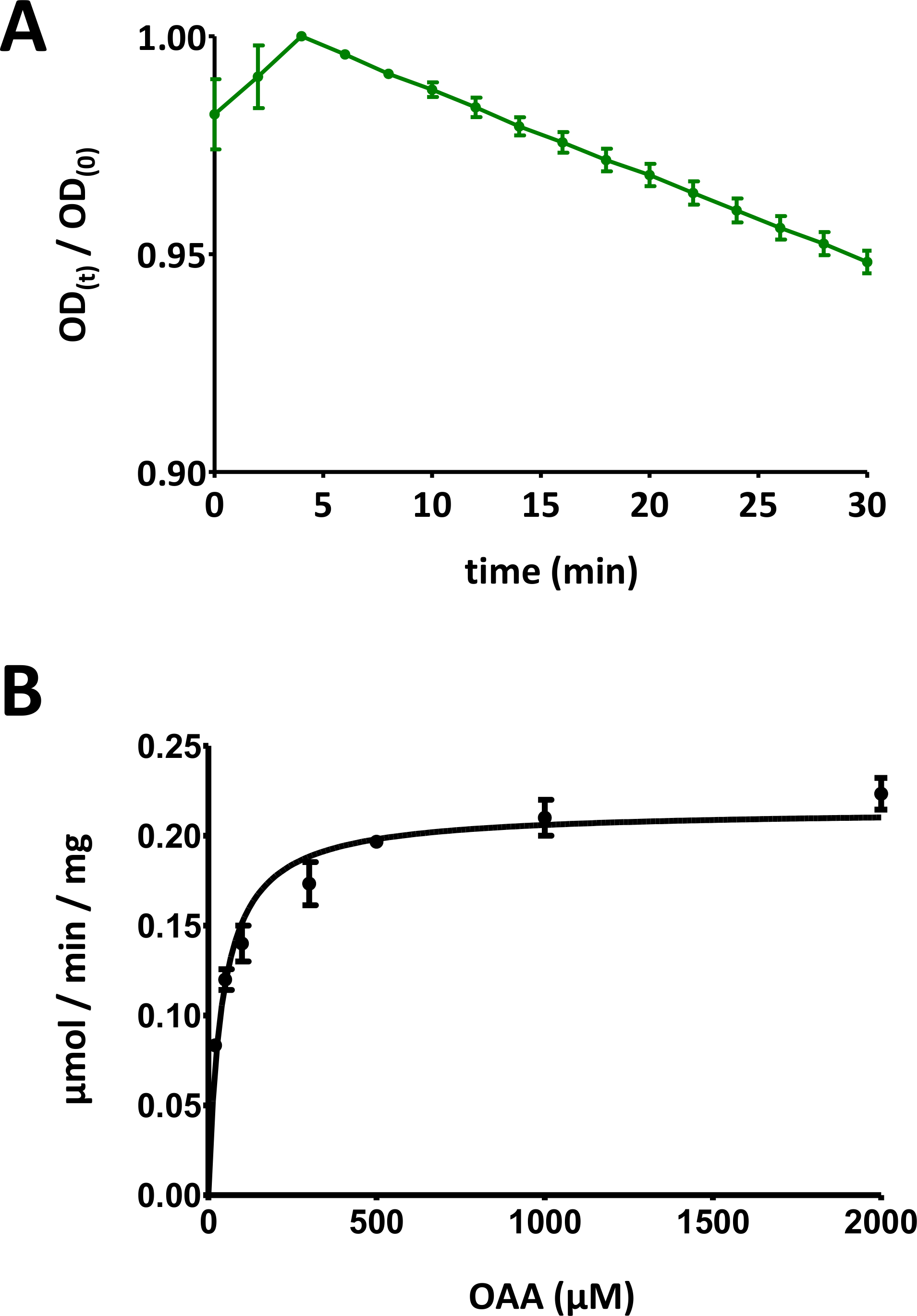
Figura 5 : Resultados exemplares de ensaios enzimáticos.
(A) uma curva de absorção UV exemplar obtida para ensaios enzimáticos básicos de proteína Fahd baseados em substrato (normalizados na faixa de 0 a 1) com desvio padrão. A relação de densidade óptica (OD) [OD (t)/OD (0)] em um determinado momento t [OD (t)] é normalizada para o OD inicial [t = 0; OD (0)]. Consulte a seção 7 do protocolo para obter mais detalhes. (B) cinética exemplar de Michaelis-Menten da proteína FAHD1 humana com desvio padrão. Consulte a seção 8 do protocolo para obter mais detalhes. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
- Iniciar um leitor de microplacas e equilibrar por 30 min a 25 ° c. Configure um programa para leitura de 72 poços (conforme descrito na Figura 4B) em 255 Nm. Recomenda-se usar 25 leituras múltiplos com um atraso de tempo de 5 ms. Configurar um ciclo para medir 15x cada 2 min (30 min total).
- Execute as etapas 7,2 e 7,3. Em seguida, configure 1 mL de solução de substrato de 100 mM no tampão do ensaio enzimático.
- Prepare diluições da solução de substrato no tampão do ensaio enzimático: 40 mM, 20 mM, 10 mM, 6 mM, 4 mM, 2 mM. O ensaio é realizado com concentrações de enzima/substrato emparelhwise ("ajustadas"). Para isso, prepare as seguintes diluições da solução enzimática no tampão do ensaio enzimático: 0,5 μg/μL, 0,4 μg/μL, 2,5 μg/μL, 2 μg/μL, 1,5 μg/μL, 1 μg/μL.
- Em todos os poços representados na Figura 4B aplique 180 μL de tampão do ensaio enzimático. Aplique 10 μL de tampão do ensaio enzimático em todos os poços para o substrato (em branco e amostra). Aplique 10 μL da série de diluição protéica preparada nos poços para a enzima (em branco e amostra). Aplique 10 μL de tampão do ensaio enzimático em todos os poços para poços para o substrato em branco e a enzima em branco.
- Logo antes da medição, aplique 10 μL da série de diluição de substrato preparada nos poços para a amostra de substrato e a amostra enzimática.
- Use uma pipeta multicanal com 50 μL de configurações para misturar suavemente todos os poços, começando com os espaços em branco, procedendo aos poços amostrais. Tome cuidado para não criar bolhas.
- Insira a placa em um leitor de microplacas e meça cada poço em 255 Nm, conforme descrito na etapa 8,1. Realize a análise em uma planilha. Copie os dados brutos do fotômetro em uma planilha, escreva todas as configurações (ou seja, toda a documentação) em outra folha.
- Realize a análise de dados individuais por ponto na série de diluição conforme descrito nas etapas 7,11. a 7,14. Eventualmente, obter todas as atividades específicas e plotar contra a concentração inicial do substrato: 2 mM, 1 mM, 0,5 mM, 0,3 mM, 0,2 mM, 0,1 mM.
- Exiba todos os pontos de dados com desvios padrão individuais. Cinética de Michaelis-Menten do computador através do encaixe não-linear da curva, ou através da análise de Lineweaver-Burk. Pode ser necessário remedir pontos individuais e adaptar as proporções individuais de concentração de proteínas/substrato-concentração nas etapas 8,5 e 8,6. O diagrama de Michaelis-Menten para o FAHD1 humano é fornecido na Figura 5b.
9. cristalização de proteínas FAHD
Nota: a cristalização das proteínas FAHD (FAHD1 humanas descritas anteriormente15) pode ser alcançada pelo método de difusão do vapor de gota suspenso em um formato de 24 poços (Figura 6a). Um protocolo passo a passo sobre a cristalização do FAHD1 humano usando esta técnica é apresentado abaixo de15. Uma descrição mais detalhada é fornecida na seção de discussão.

Figura 6 : Cristalização de proteínas de FAHD.
(A) placas de cristalização no padrão 24 bem ou 96 bem pegada SBS. Consulte a seção 9 para obter mais detalhes. (B) o processo básico de configuração da placa na cristalização das proteínas Fahd. Esta figura é redesenhada com permissão23. Consulte a seção 9 para obter mais detalhes. (C) cristais FAHD1 humanos e padrões de difração correspondentes (pastilhas pequenas). O espaçamento de treliça mais próximo é indicado nas pastilhas como medida para a qualidade de difração dos cristais. Números inferiores indicam maior resolução e, portanto, dados mais informativos. Consulte a seção 9 do protocolo para obter mais detalhes. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
- Assegure-se de que a proteína esteja diafiltrado de encontro ao buffer running SEC. A proteína FAHD1 deve estar disponível em concentrações elevadas (2 – 5 mg/mL). Em concentrações mais baixas, a proteína não pode cristalizar devido à falta da nucleação espontânea.
- Prepare ≥ 20 mL da solução do reservatório para a cristalização. Fazer três soluções de ações, utilizando água destilada ou desionizada como solvente: 1 M na-HEPES (mínimo de 25 mL, ajustado para pH 7,5), 50% (p/v) polietileno glicol 4000 (PEG4k) (mínimo 65 mL), e 1 M MgCl2 (10 ml).
- Configurar uma grelha de 4 x 6 (24 total) diferentes tubos de 15 mL. Rotulá-los de acordo com as posições correspondentes na placa (por exemplo, linha (A, B, C, D) vs. coluna (1 – 6) como "a1", "B5", "D6", etc.). Pipete 1 mL de na-HEPES de 1 M para cada tubo.
- Pipete 1 mL de 50% (p/v) PEG4k na carreira a dos tubos, 2 mL na carreira B, 3 mL na carreira C e 4 mL na carreira D. pipetas 100 μL de 1 M MgCl2 para a coluna 1 dos tubos, 250 μl na coluna 2 , 500 μL na coluna 3, 1,0 mL na coluna 4, 1,5 mL na coluna 5 e 2,0 mL na coluna 6.
- Encha todos os tubos até um volume de 10 mL com água destilada ou desionizada, onde a balança nos tubos é suficientemente precisa.
- Tome a amostra de proteína FAHD1 humana (~ 5 mg/mL) da geladeira (ou do gelo) e gire para baixo na velocidade máxima com uma centrífuga de tampo de mesa a 4 ° c por pelo menos 10 min. Se a cocristalização com oxalato for desejada, adicione oxalato de uma solução de estoque para que a amostra de proteína contenha uma concentração final de oxalato de 2 mM. Aplique 1 mM de DTT e armazene no gelo.
- Entretanto, descompacte uma placa de 24 cristalização bem, idealmente dentro de uma sala de temperatura controlada a 18 ° c. Distribuir uma fina camada de óleo de parafina sobre a borda em cima de cada poço da placa de 24 poços com a ajuda de um vidro fino ou haste de plástico. Adicionar 800 μL de cocktails de cristalização preparados (a1 a D6) em cada poço correspondente da placa de cristalização.
- Coloque as coberturas frescas de 22 mm sobre uma superfície limpa. Evite contaminar os deslizamentos da tampa com sujeira ou poeira. Se necessário, retire os detritos do deslizamento de cobertura usando ar comprimido ou um spray de espanador.
- Após a centrifugação estar completa, evite agitar a amostra de proteínas para que os agregados e detritos na parte inferior do tubo não flutuem novamente. Nas etapas a seguir, pipeta da amostra de proteína logo abaixo da superfície da solução, a fim de evitar a agitação de agregados e depósitos a partir do fundo.
- Para cada poço (ver Figura 6B) Pipetar 1 μL de solução proteica para o centro de um deslizamento de cobertura e adicionar 1 μl do respectivo cocktail de reservatório à gota proteica, evitando bolhas. Gire a lamínula de cabeça para baixo e coloque-a na parte superior do poço para que o óleo sele o poço com a lamínula hermética. Repita até que a placa de 24 poços esteja terminada.
- Conservar a placa a 18 ° c e observar as gotas numa programação progressiva com um microscópio adequado. Os cristais FAHD1 humanos geralmente aparecem durante a noite (ver Figura 6C).
Resultados
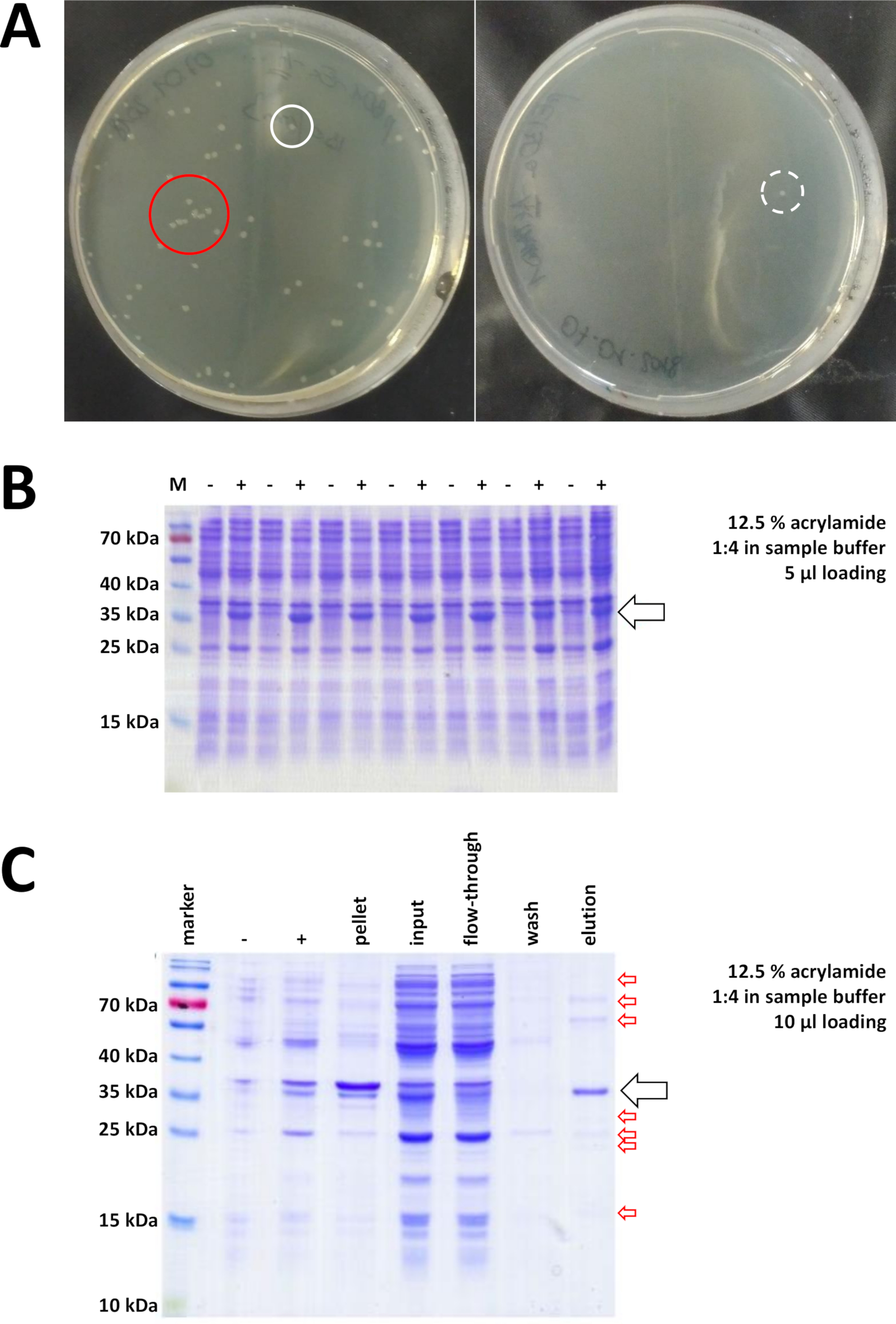
Começando com um vetor de clonagem preparado e comprado BL21 (DE3) plyss e. coli, o plasmídeo é inserido nas bactérias através de choque térmico ou qualquer método alternativo apropriado (Figura 1). Após um curto período de amplificação, as bactérias transformadas são chapeadas em placas de agar LB, a fim de crescer durante a noite. As placas neste ponto podem parecer diferentes, dependendo de uma variedade de fontes de erro potenciais. As placas podem estar vazias (ou seja, sem colônias), completamente cultivadas por bactérias, ou algo entre, respectivamente. Dois exemplos de placas de agar LB após a transformação ideal e não ideal são representados na Figura 7a. Demasiadas colônias bacterianas indicam ou que muitas bactérias foram chapeadas (provavelmente) ou que os antibióticos em uso podem ser expirados (improvável). Poucas colônias bacterianas podem indicar que não foi utilizado plasmídeo suficiente para a transformação (use mais próxima vez) ou que muitos antibióticos foram usados para selecionar as bactérias. Em qualquer caso, se as colônias estão presentes, eles devem estar bem, como o uso de dois antibióticos seletivos implica uma chance bastante insignificante de bactérias não transformadas para crescer. Nenhuma colônia, no entanto, indica que ou as bactérias perderam sua competência de transformação (por causa do armazenamento errado ou armazenamento em períodos mais longos, congelamento repetitivo e degelo, etc.), o calor-choque não foi bem sucedido (sem absorção de plasmídeo ou bactérias morte por muito calor), o vetor de clonagem está corrompido, ou por engano um conjunto errado de antibióticos seletivos foi usado (verificar o gene de resistência no vetor plasmídeo).

Figura 7 : Resultados representativos para a transformação de bactérias e iMac.
(A) placas de agar lb representativas com BL21 (de3) E. colitransformadas, obtidas seguindo a etapa de protocolo 1,1. Esquerda: uma placa com colônias bem distribuídas (exemplo positivo). Direita: uma placa com apenas uma única colônia (exemplo negativo). Círculos brancos marcam boas colônias. O círculo vermelho marca as colônias que estão crescendo demasiado perto de se e não deve ser escolhida contanto que as colônias isoladas estiverem disponíveis. (B) uma análise de SDS-PAGE de acrilamida a 12,5% de uma série de controles de indução ("-" indica antes da indução de IPTG; "+" indica após a indução de IPTG, antes da colheita da pelota), ajustado às quantidades iguais de proteína total. Isso é descrito na etapa 1,2. (C) um exemplar 12,5% acrilamida SDS-PAGE análise de NI-NTA purificação de his-Tagged FAHD1 proteína. Isso é descrito na seção 3 do protocolo. A cromatografia de afinidade produz proteínas de alta pureza (> 70%, seta preta), no entanto, várias pequenas contaminações também são observadas (setas vermelhas). Estas contaminações consistem em proteínas não-FAHD que se ligam à coluna, e de proteínas que se ligam à proteína FAHD. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
Colônias validadas são selecionadas e escolhidas. Após a amplificação em meio nutritivo, a expressão protéica é desencadeada pela aplicação do produto químico IPTG. A pelota bacteriana contendo a proteína expressa em quantidades de miligrama é colhida, e a expressão é verificada via SDS-PAGE (ver, por exemplo, a Figura 7B). Alguns problemas podem ocorrer durante este processo de outra forma simples. Em primeiro lugar, algumas proteínas formam corpos de inclusão, porque aparentemente de alguma forma interferem com o metabolismo natural das bactérias hospedeiras. Isto foi observado para algumas mutações do ponto de FAHD1 e de FAHD2 humanos. Nesses casos, outros sistemas de expressão como as células de insetos podem ser mais apropriados e devem ser considerados. Após a colheita de uma pelota de células de insetos, por exemplo, a purificação das proteínas segue os mesmos passos descritos neste protocolo. Em segundo lugar, o sistema DE3-pET é encontrado às vezes para ser "Leaky" (isto é, a proteína é expressada já em alguma extensão antes da indução de IPTG). O motivo potencial para isso não é bem compreendido, mas pode ajudar a expressar a proteína lentamente durante a noite em uma incubadora de sala fria. Em terceiro lugar, nenhuma proteína é expressa. Este é provavelmente o cenário do pior caso, como ele provavelmente indica um vetor de plasmídeo corrompido e, portanto, aconselhável para sequenciar o plasmídeo.
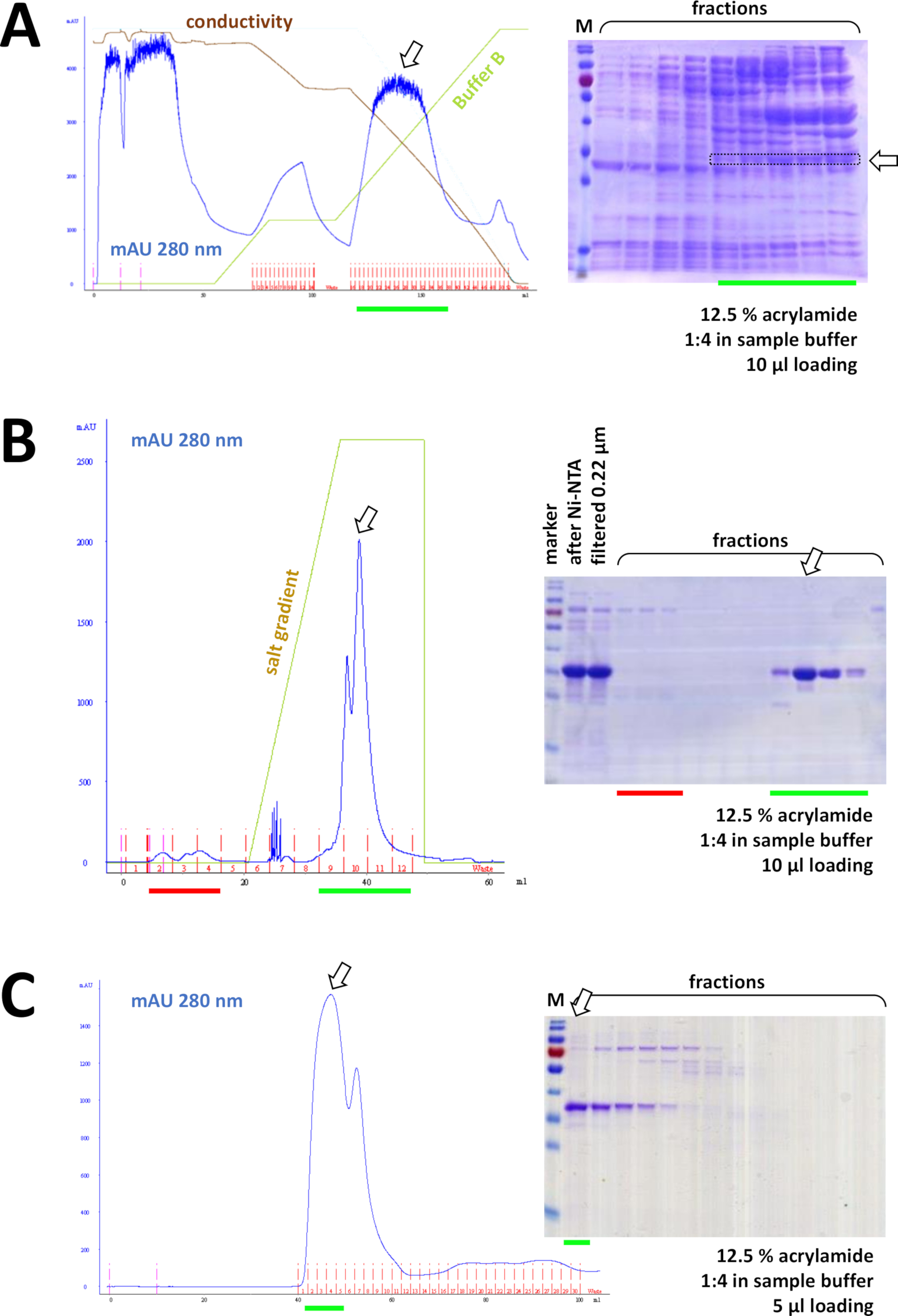
Se um seu-tag foi usado para etiquetar a proteína, a cromatografia de afinidade com o agarose de NI-NTA é um método fácil e barato da captação que elimina a maioria das contaminações (Figura 7C). Existem métodos semelhantes para outros sistemas de Tags (por exemplo, Strep-II). Se nenhuma tag foi usada, uma combinação de precipitação de sulfato de amônio e cromatografia de troca hidrofóbica consecutiva também pode separar a proteína da maioria das outras proteínas (figura 8a). No entanto, comparando os dois métodos (Figura 7C versus figura 8a), a superioridade dos métodos NI-NTA pode ser DEMONSTRADA pela análise de SDS-PAGE. A utilização da proteína his-Tagged é, portanto, aconselhada.

Figura 8 : Resultados representativos para experimentos de FPLC (HIC, troca iónica, SEC).
(A) um cromatograma típico e uma análise de 12,5% de ACRYLAMIDA SDS-PAGE da cromatografia de HIC-Phenyl após a precipitação do sulfato de amónio (como) da proteína FAHD1 Untagged, como descrito na seção 4 do protocolo. A linha verde reflete o gradiente do buffer B que não contém AS. Durante o processo como é gradualmente lavado para fora do sistema. Comparando este painel com a Figura 7C exibe o poder da cromatografia de afinidade NI-NTA em comparação com o método HIC-Phenyl, e a vantagem de usar um sistema his-tag para purificação de proteínas. (B) um cromatograma exemplar e 12,5% de ACRILAMIDA SDS-PAGE análise de cromatografia de troca catiônica de sua-TAGGED Fahd após apurificação de NI-NTA. Usando um gradiente de sal, a amostra aplicada é separada em proteínas individuais. (C) um cromatograma exemplar e uma análise de 12,5% de acrilamida SDS-PAGE de cromatografia de exclusão de tamanho G75 da sua-Tagged Fahd após cromatografia de troca catiônica. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
Consecutivamente, a proteína é ainda mais separada das contaminações remanescentes por cromatografia de troca catiónica/Anion (por exemplo, ver Figura 8B), seguida por cromatografia de exclusão de tamanho (por exemplo, ver Figura 8C). É aconselhável estabelecer uma estratégia de purificação inicial nesta ordem; no entanto, essas colunas devem ser usadas em combinação, subseqüentemente e em variação, até que a proteína seja suficientemente pura.
Ensaios de atividade simples, para testar as decisões "Sim ou não" sobre substratos ativos e/ou cofatores, podem ser realizados com proteínas marcadas após a purificação de NI-NTA ou proteínas não marcadas após a coluna de troca iónica. Atividades específicas e constantes cinéticas devem ser determinadas com proteínas de maior pureza. A cristalização pode ser tentada com proteínas após a coluna de troca iónica, mas a qualidade dos cristais quase sempre se correlaciona com a pureza da proteína. Anticorpos policlonais podem ser levantados contra proteínas em qualquer fase do protocolo de purificação; no entanto, aqui a qualidade também se correlaciona com a pureza da proteína.
Discussão
Etapas críticas
As proteínas FAHD são muito sensíveis às concentrações de sal. Em concentrações baixas de NaCl, as proteínas podem precipar-se em cima do descongelar, mas podem geralmente inteiramente ser reconstituídas em umas concentrações de sal mais elevadas. Ou seja, se uma proteína FAHD precipita por algum motivo, pode ser recuperada ou redobrada com maiores concentrações de sal (> 300 μM). Algumas proteínas mais hidrofóbicas, no entanto, não podem ser recuperados (por exemplo, FAHD2 humano), mas detergentes como CHAPS (máximo de 1%) ou glicerol (10%) pode ser usado para mantê-los em solução estável. Em qualquer caso, recomenda-se o congelamento de choque com nitrogênio líquido e armazenamento a-80 ° c, pois é um processo suave e lento de descongelação.
Alguns problemas inesperados podem ocorrer durante a purificação de NI-NTA na etapa 3.1.10. Da nota, um OD mais elevado na segunda amostra coletada do que na primeira amostra indica um volume demasiado elevado da resina do agarose (tome uma nota e use menos resina no experimento seguinte). Também, a resina do agarose conduz a um sinal do OD em 280 nanômetro (isto é, o rompimento da cama da resina do agarose dará sinais artificiais). Em caso de dúvida, é aconselhável usar outros métodos como um ensaio de Bradford ou BSA para determinar as concentrações de proteínas.
Em ensaios enzimáticos, há três aspectos críticos a serem considerados. Primeiro, avaliar a concentração proteica é fundamental para se obter as atividades específicas corretas. O nível de pureza da proteína está influenciando o resultado e precisa ser estimado. Em caso de proteína etiquetada, a massa da tag-parte tem que ser computada, e a atividade específica tem que ser corrigida correspondentemente. Para ensaios simples descritos na seção 7 do protocolo, a pureza de NI-NTA é suficiente para distinguir entre substratos ativos e inativos, cofatores, etc. No caso de uma cinética mais complexa de Michaelis-Menten, todas as concentrações de reagente e substrato devem ser corretamente determinadas. Especialmente ao usar oxaloacetato (que auto-descarboxylates ao longo do tempo) a parte enzimática da reação deve ser corrigida para auto-descarboxilação (sob a suposição de que ambas as reações ocorrem simultaneamente). Devem ser consideradas as alterações iniciais no sinal de densidade óptica dirigida à tautomerização de keto-enol do substrato. Em terceiro lugar, as concentrações e os volumes devem ser ajustados. Uma reação com concentrações definidas de enzima e substrato pode dar resultados diferentes dependentes do volume do ensaio. Se houver demasiada enzima por poço, a aderência do líquido pode, de facto, influenciar o resultado.
Para avaliar a cinética de Michaelis-Menten recomenda-se realizar experimentos iniciais em 100 μl, 200 μL e 300 μL de lotes, a fim de encontrar a combinação ideal. Aspectos semelhantes se aplicam à proporção de concentrações de substrato enzimático para ensaios cinéticos. Demasiada enzima por substrato ou muito substrato por enzima colocar o sistema fora da linha linear de estado estacionário Michaelis. Experimentos iniciais são necessários para otimizar essas condições. O ajuste exemplar para a proteína FAHD1 humana (Wild-Type) é fornecido na seção 8, resultando em diagramas cinéticos (como apresentado na Figura 5b, por exemplo).
Para a cristalização, uma gota de solução proteica é pipetada no centro de uma lamínula e misturada com uma gota de coquetel de cristalização, que geralmente é composta por um tampão (por exemplo, Tris-HCl, HEPES) e um precipitante (por exemplo, polietileno glicol, amônio sulfato de sódio). Uma gota da solução do inibidor para A cocristalização (tal como o oxalato neste protocolo) pode opcionalmente ser aplicada. O lamela é coloc então upside-down acima de um poço do reservatório que contem o cocktail da cristalização, selando o ar do poço firmemente com a ajuda do óleo do vedador (Figura 6B). Idealmente, nenhuma precipitação ocorre dentro da gota no início do experimento que significa que a proteína permanece em solução. Uma vez que a concentração precipitante no reservatório é maior do que na gota, a gota começa a perder água por evaporação na atmosfera do poço até que o equilíbrio com o reservatório seja atingido. A difusão da água no reservatório provoca uma diminuição lenta do volume da gota que, por sua vez, provoca um aumento da concentração de proteínas e precipitantes na gota. Se a solução proteica atinge o Estado requerido de supersaturação e, portanto, meta-estabilidade, a nucleação espontânea seguida de crescimento de cristais pode ocorrer. Atingir o estado supersaturado é uma condição necessária, mas não suficiente para a cristalização. A cristalização das proteínas necessita de condições termodinâmicas e cinéticas favoráveis, e depende muito das propriedades imprevisíveis da proteína a ser cristalizada22.
Modificações e solução de problemas
A expressão da proteína em E. coli pode ser ineficiente. Diferentes concentrações de IPTG, temperatura de expressão e tempo de amplificação, como temperatura ambiente por várias horas ou em sala fria durante a noite, podem precisar ser testadas para cada nova proteína para encontrar condições ideais. A precipitação da proteína em corpos de inclusão é observada às vezes para umas proteínas mais hidrofóbicas de FAHD. Nesses casos, recomenda-se expressão protéica em outros sistemas de modelo, como células de insetos, pois os corpos de inclusão são menos propensos a formar26.
Como proteínas Fahd são sensíveis às concentrações de sal e cofator, bem como pH, estratégias de purificação para diferentes homólogos, ortologues, e variantes de mutação de ponto podem diferir em configurações individuais. Os métodos de purificação descritos são desenvolvidos para o selvagem-tipo humano e rato FAHD1 proteína. As concentrações de substâncias químicas, como NaCl e imidazol, bem como o pH, podem ter que ser adaptadas para proteínas individuais com um ponto isoelétrico diferente (pI). Também da nota, não cada proteína his-etiquetada pode ligar bem a uma resina de NI-NTA. Se a ligação de proteínas à coluna NI-NTA for ineficiente, as concentrações adaptadas de NaCl e imidazol, bem como as condições de pH variáveis no tampão de corrida NI-NTA podem ajudar a melhorar a qualidade do resultado. Se não, pular a etapa NI-NTA e prosseguir para a etapa de cromatografia de troca iónica também pode levar a uma estratégia de purificação bem-sucedida. Se uma proteína se liga à coluna NI-NTA, mas não pode ser eluída da coluna, a adição de alguns mM EDTA pode ajudar a interromper o Ni2 + Complex.
Em relação ao processo de cristalização, é preciso entender que a autoorganização de moléculas de proteínas grandes e complexas em uma estrutura periódica regular é um processo inerentemente improvável que depende fortemente de parâmetros cinéticos de difícil controle. Mesmo pequenas mudanças na set-up usado para a cristalização pode alterar drasticamente o resultado e nenhum cristais irá formar. A pureza da proteína é geralmente da importância primordial. Como regra geral, um gel SDS-PAGE muito sobrecarregado não deve mostrar outras bandas. Além disso, a sequência na qual as etapas são executadas pode afetar o resultado. Como exemplo, para garantir a reprodutibilidade, muitas vezes é necessário manter a seqüência de pipetagem o mesmo, em seguida, primeiro adicionar a proteína, e, finalmente, adicionar precipitante para a gota de cristalização (ou vice-versa). Seja qual for o método usado, ele deve ser mantido o mesmo ao tentar reproduzir ou escalar experimentos. Se não forem observados cristais após este protocolo, a composição precipitante química, o pH, o tamanho da gota e a relação proteína-precipitada podem ser variados em pequenos incrementos. Paciência e observações consistentes das gotas são de virtude.
Observações aos mecanismos catalíticos de FAHD1
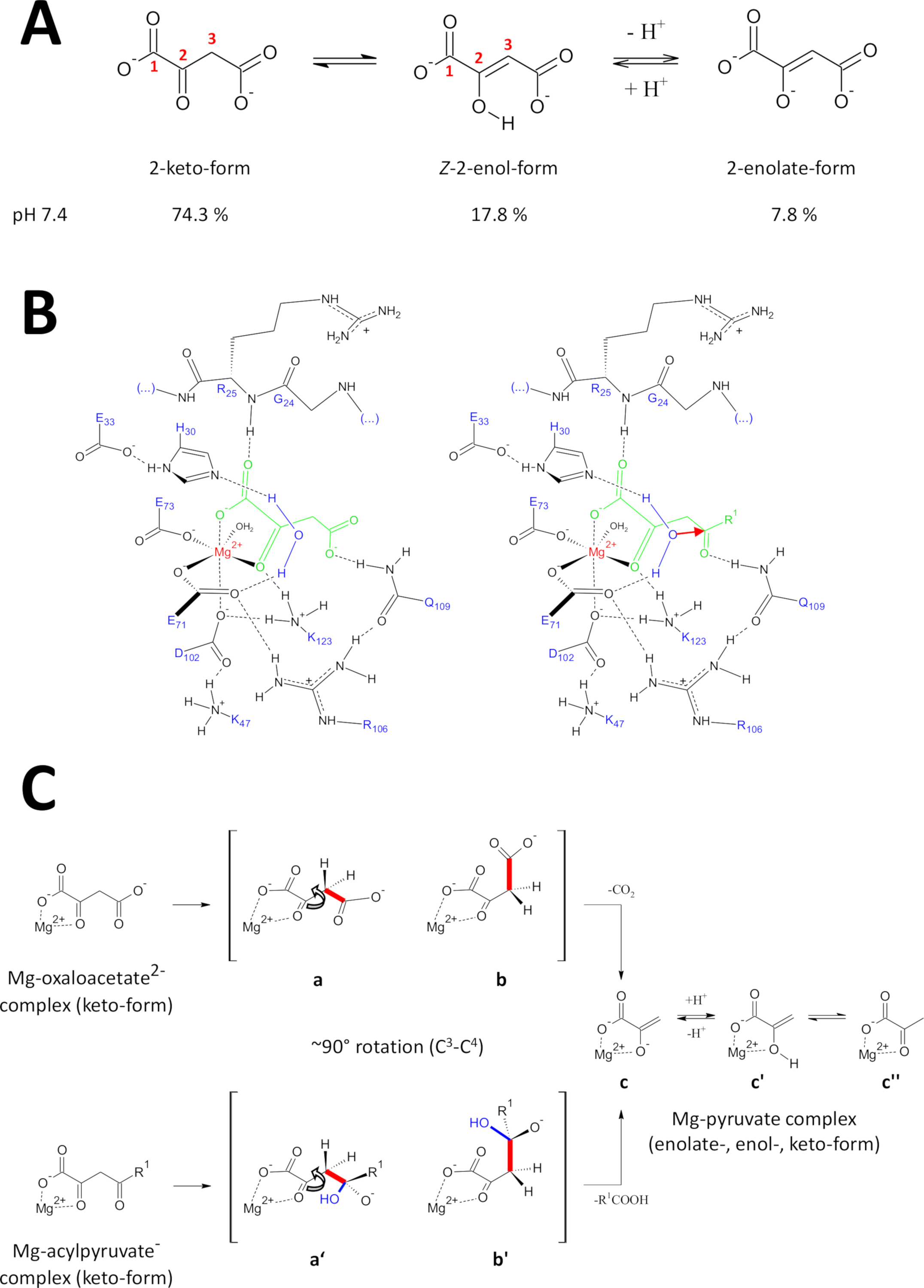
Os métodos apresentados foram desenvolvidos especificamente para obter FAHD1 proteínas de alta qualidade. Isto permitiu o crescimento de cristais FAHD1 assim como a engenharia dos cristais que contêm FAHD1 complexado a um inibidor (oxalate, PDB: 6fog). As estruturas de raios-X fornecem uma arquitetura 3D da cavidade catalítica da enzima. Estes resultados estabelecem uma descrição detalhada dos resíduos potencialmente importantes para os mecanismos catalíticos desta intrigante enzima. FAHD1 foi descrito pela primeira vez para ser capaz de Cleave acylpyruvates (acetilpiruvato, fumarilpiruvato)11. Mais tarde, verificou-se que a FAHD1 atua também como decarboxilase de oxaloacetato12. Embora os substratos acilpiruvato e oxaloacetato sejam diferentes moieties químicos, as transformações químicas compartilham mecanisticamente a clivagem estratégica de uma única ligação C3-c4 comum, facilitada energeticamente se o c3 -C4 orbitais Bond permanecer ortogonal aos π-orbitais do C2-carbonyl15. Tal conformação permite a estabilização da ressonância do C3-carbanion transientemente formado durante o processo de clivagem. Os substratos FAHD1 (oxaloacetato e acylpyruvates) são moléculas flexíveis e podem existir em tautoméricas (keto-enol), bem como em C2-formas hidratadas (Figura 9A). Os equilíbrios entre as diferentes espécies são determinados principalmente pela natureza da composição tampão utilizada, pH e presença de íons metálicos. A seguir, discutimos cenários mecanísticos hipotéticos inspirados na análise de estruturas de cristais de raios-X que divulgaram o centro catalítico de FAHD1.

Figura 9 : Detalhes sobre o mecanismo catalítico proposto de FAHD1.
(A) oxaloacetato existe em estado cristalino, bem como em solução neutra, principalmente na forma Z-enol24. Entretanto, o pH fisiológico-condiciona a forma 2-keto é a respresentação predominante25. (B) esboço químico da cavidade hFAHD115 com oxaloacetato de mg-Bound (esquerda) e acilpiruvato (direito, com R1 como repouso orgânico; a seta vermelha denota um ataque nucleofílico da molécula de água estabilizada adjacente) (ver discussão). (C) comparação de conformações favorecidas para clivagem c3-c4 em decarboxilase (b a c) e hidrolase (b ' a c) mecanismo de FAHD1: ambos os processos resultam em piruvato-enolato de mg-complexado (ver discussão). Os intermediários b e b ' são esperados para ser estabilizado por Q109, como esboçado no painel B (ver discussão). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
A atividade de decarboxylase de FAHD1
O oxaloacetato existe no estado cristalino assim como na solução neutra principalmente no formulário Z-enol24. Mas foi demonstrado que, condições de pH fisiológico (condições tampão em pH 7,4), a forma 2-keto é a representação predominante do oxaloacetato25 (Figura 9A), e que a enolização não é um pré-requisito para a descarboxilação27 . Da nota, os íons do magnésio2 + não têm nenhuma influência na relação da espécie do oxaloacetato em um pH de 7,4 ou abaixo de28. A transposição do keto de oxaloacetato para o centro catalítico de FAHD1 (guiada pelo oxalato acoplado na enzima complexada (APO: 6FOG15)) revelou resíduo Q109 como regulador conformacional do oxaloacetato de ligação15. Como descrito em outro artigo15, ligação de hidrogênio para o grupo carbamoil de Q109 estabiliza um oxaloacetato-conformação resultante da rotação em torno do c2-c3 Bond (Figura 9B, painel esquerdo). Como consequência dessa rotação, a ligação C3-c4 (a ser clivada) adota uma disposição próxima à ortogonal em relação aos π-orbitais do c2-carbonilo (Figura 9c). O dióxido de carbono pode ser liberado. O produto preliminar deste processo seria magnésio-enolate estabilizado ressonância do piruvato. É conhecido a partir de investigações de complexos de oxaloacetato-mg que o enolato forma o complexo mais estável28,29. Supondo uma estabilidade comparável para um enolato-complexo do magnésio-piruvato o cofator de FAHD1 poderia ser obstruído, mas o resíduo do lisina K123 pode protonate o PYRUVATE-enolate em um equilíbrio para proibir a perda do cofator15.
A interpretação dada sugere o piruvato enol como um intermediário distinto na função catalítica do ODX de FAHD1. Nesta etapa do modelo hipotético, os dados experimentais não fornecem nenhuma indicação adicional a respeito de porque a tampa fechada deve abrir para liberar o produto. Pode-se deduzir, no entanto, que o mecanismo proposto se parece com uma inibição enzimática pelo produto: a estrutura cristalina revela uma molécula de água conservada mantida em orientação direcional para o centro catalítico FAHD1 pelos resíduos h30 e e33 apresentados em um a hélice curta15, que é induzida em cima da ligação do ligante e do fechamento da tampa. Se o enol preliminar permanecer em um equilíbrio com o enolato, o enolato estabilizado ressonância poderia ser extinto ao piruvato pela molécula de água. O hidroxila resultante seria capaz de deslocar o piruvato do magnésio-cofactor em cima de que a tampa abriria. Finalmente, o centro catalítico seria restaurado no ambiente mitocondrial. Neste cenário hipotético, a molécula de água da cavidade operaria como um ácido, respectivamente.
Atividade de hydrolase de FAHD1
A atividade de hydrolase de uma enzima exige implicitamente a formação intermediária de um nucleophile do hidroxila. Este mecanismo é geralmente encontrado em combinação com a atividade catalítica ácida-base. O estado transitório da reacção tem de ser preparado através de um controlo conformacional por cadeias laterais críticas de aminoácidos na cavidade. Na analogia à discussão da função do descarboxilase, o acylpyruvate enzima-ligado no formulário 2-keto será põr o controle conformacional pela hidrogênio-ligação do oxigênio 4-carbonyl a Q109 (Figura 9B, painel direito). A estrutura cristalina do FAHD1 (PDB: 6FOG), de oxalato, revela uma molécula de água conservada mantida em orientação direcional para o centro catalítico FAHD1 pelos resíduos h30 e e33 apresentados em uma hélice curta15. O díade de E33-h30 é competente para deprotonar a água posicionada direcional e o hidroxila resultante está na disposição ideal para atacar o 4-carbonyl do acylpyruvate apresentado o controle conformacional por Q10915.
De notar, um mecanismo semelhante foi proposto para a FAH18. O ataque pelo nucleófilo do hidroxila é esperado conduzir a uma espécie do oxiânion, que seja estabilizado em cima da clivagem controlada orbital da ligação de c3-c4 (Figura 9c). Neste modelo, a rotação de vínculo C3-c4 (Figura 9c) acontece após o ataque nucleofílico pelo hidroxilo formado indicado na Figura 9B (isto é, prepara o acilpiruvato para a clivagem da ligação). Os produtos primários seriam o ácido acético e o enolato de mg-piruvato. Neste cenário hipotético, o ácido acético pode saciar o enol ao piruvato e, subsequentemente, auxiliar o deslocamento do produto. Acima de um pH de 7,5 e na presença de íons do magnésio, os acylpyruvates existem em um equilíbrio entre o keto-e enol-formulários, o último na preferência ligeira30. O mais provavelmente ambos os formulários são capazes de ligar ao cofator de FAHD1 o fechamento subseqüente da tampa. O processamento de substratos enólicos do acylpyruvate pela enzima é dificultado devido à estrutura lisa do enol-formulário. A clivagem c3-c4 resultaria em um carbânion vinílico sem estabilização de ressonância.
Conseqüentemente, nós propor uma etapa catalítica do ketonization para preparar-se para o ataque do nucleófilo do hidroxila no carbonyl do acil. Este processo de ketonização, no entanto, exigiria controle sobre transposições de prótons por resíduos de FAHD1, o que atribuiria uma atividade inerente de isomerase a FAHD1. Relatou-se que a acidez do hidrogênio de Enol mg-Bound revela um aumento 10000-fold comparado ao formulário un-complexed28. A desprotonação do mg vinculado enol-forma seria viável por un-protonated K123. A desprotonação de K123 pode ser assistida pelo carboxilato de D102. Uma rede de ligação de hidrogênio formada por resíduos D102-K47-K123 poderia operar como o relé de prótons necessário no centro catalítico de FAHD115. Um enolato intermediário tal-dado forma poderia então ser extinto por uma tríade de E33-h30-H20 o ketonization do substrato15. O formulário 2-keto viria o controle conformacional de Q109, e o hidroxila concomitantemente dado forma atacaria o carbonyl do acil. A discussão resumida implica um controle de FAHD1 sobre uma molécula de água para alternar entre ácido e base através da interação de resíduos de formação de cavidade.
Aplicações futuras ou direções do método
As aplicações futuras dos métodos descritos aqui são numerosas. Uma pletora de membros procarióticas da superfamília de Fah ainda espera A caracterização funcional. Mesmo as informações disponíveis sobre as atividades catalíticas de membros conhecidos da superfamília FAH são escassas e, na maioria dos casos, baseadas em pressupostos teóricos em vez de dados experimentais. A aplicação dos métodos descritos aqui para membros procarióticos da superfamília FAH depende dos interesses específicos de pesquisa em bacteriologia. Por outro lado, a recente demonstração de que os membros da superfamília eucariótica FAH desempenham papéis essenciais em vários compartimentos celulares (por exemplo, citosol vs. mitocôndria) destaca a necessidade de melhor caracterizar essas proteínas (três das quais foram identificados até agora), em particular porque os dados atuais sugerem que algumas proteínas não caracterizadas podem realizar funções diferentes no contexto da biologia mitochondrial, da pesquisa do envelhecimento, e da pesquisa do cancro. Propõe-se que a caracterização molecular e fisiológica completa desses membros da superfamília eucariótica da FAH possa fornecer uma visão importante dos principais campos da pesquisa contemporânea no setor biomédico. Mais pesquisas sobre os mecanismos de FAHD1 (e enzimas relacionadas) são necessárias para compreender melhor os mecanismos subjacentes à bi-funcionalidade do FAHD1, que ainda não está totalmente esclarecido. Estudos adicionais com mutantes FAHD1, NMR-investigações e estudos estruturais sobre complexos inibidores podem ajudar a resolver os verdadeiros cenários mecanísticos para os quais FAHD1 parece ser competente. Além disso, o desenho assistido por computador de Enol imita capaz de se vincular ao mg-cofactor acabará por levar a inibidores potentes da FAHD1.
Divulgações
Os autores não têm nada a divulgar e declarar nenhum interesse financeiro concorrente. H. G. é CEOCSO em MoleculeCrafting. HuGs e.U. e forneceu acylpyruvates para este estudo através da síntese feita encomenda. O trabalho no laboratório do P. J. D. foi apoiado pelo Austrian Science Fund (FWF): número de projeto P 31582-B26. As taxas de publicação para este manuscrito foram parcialmente cobertas pelo fundo austríaco de ciência (FWF) o número de projeto P 31582-B26. A. N. e B. R. são apoiados pelo Austrian Science Fund (FWF) o projeto P28395-B26.
Agradecimentos
Os autores são muito gratos pela assistência técnica especializada de Annabella Pittl e pelo desenvolvimento do método piloto por Haymo Pircher.
Materiais
| Name | Company | Catalog Number | Comments |
| BL21(DE3) pLysS competent E. coli | Promega | L1195 | High-efficiency protein expression from gene with T7 promoter and ribosome binding site |
| pET E. coli T7 Expression Vectors | MERCK | - | http://www.merckmillipore.com/AT/de/life-science-research/genomic-analysis/dna-preparation-cloning/pet-expression-vectors/qFSb.qB.mLQAAAFA6.VkiQ0G,nav |
| 0.45 µm filter units | MERCK | SLHP033NS | Millex-HP, 0.45 µm, PES 33 mm, not steril |
| 0.22 µm filter units | MERCK | SLGP033RS | Millex-HP, 0.22 µm, PES 33 mm, not steril |
| Eppendof tubes 1.5 mL | VWR | 525-1042 | microcentrifugal tubes; autoclaved |
| 15 mL Falcon | VWR | 734-0451 | centrifugal tubes |
| 50 mL Falcon | VWR | 734-0448 | centrifugal tubes |
| PS Cuvettes Spectrophotometer Semi-Micro | VWR | 30622-758 | VIS transparent cuvettes |
| UV Cuvettes Spectrophotometer Semi-Micro | VWR | 47727-024 | UV/VIS transparent cuvettes |
| isopropyl-β-D-thiogalactopyranosid (IPTG) | ROTH | 2316 | chemical used for induction of protein expression with the DE3/pET system |
| imidazole | ROTH | X998 | chemical used for elution of polyhistidine (6xHis) sequences from a nickel-charged affinity resin |
| Glass Econo-Column Columns | Bio-Rad | - | http://www.bio-rad.com/de-at/product/glass-econo-column-columns?ID=2cfb1c6e-32e8-4c72-b532-dd39013d707d&pcp_loc=catprod |
| chloramphenicol | Sigma-Aldrich | C0378 | antibiotic for bacterial growth selection; resistance endióded in pLysS plasmid of BL21(DE3) E. coli; 25 µg/mL final concentration |
| kanamycin | Sigma-Aldrich | 60615 | antibiotic for bacterial growth selection; to be used if this resistance is encoded in the employed pET vector; 50 µg/mL final concentration |
| ampicillin | Sigma-Aldrich | A1593 | antibiotic for bacterial growth selection; to be used if this resistance is encoded in the employed pET vector; 100 µg/mL final concentration |
| Ultra-15, MWCO 10 kDa | Sigma-Aldrich | Z706345 | centrifigal filters for protein enrichment; https://www.sigmaaldrich.com/catalog/product/sigma/z706345?lang=de®ion=AT |
| Ultra-0.5 Centrifugal Filter Units | Sigma-Aldrich | Z677108 | centrifigal filters for protein enrichment; https://www.sigmaaldrich.com/catalog/product/ALDRICH/Z677108?lang=de®ion=AT&cm_sp=Insite-_-prodRecCold_xviews-_-prodRecCold5-2 |
| oxaloacetic acid | Sigma-Aldrich | O4126 | TCA metabolite |
| sodium oxlalate | Sigma-Aldrich | 71800 | a competitive inhibitor of FAH superfamily enzymes |
| Dialysis tubing cellulose membrane | Sigma-Aldrich | D9277 | https://www.sigmaaldrich.com/catalog/product/sigma/d9277; or comparable |
| Ni-NTA agarose | Thermo-Fischer | R90101 | a nickel-charged affinity resin that can be used to purify recombinant proteins containing a polyhistidine (6xHis) sequence |
| 96-Well UV Microplate | Thermo-Fischer | 8404 | UV/VIS transparent flat-bottom 96 well plates |
| PageRuler Prestained Protein Ladder, 10 to 180 kDa | Thermo-Fischer | 26616 | https://www.thermofisher.com/order/catalog/product/26616?SID=srch-hj-26616 |
| ÄKTA FPLC system | GE Healthcare Life Sciences | - | using the FPLC system by GE Healthcare; different custom versions exist; this work used the "ÄKTA pure" system |
| HiTrap Phenyl HP column | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/it/shop/chromatography/prepacked-columns/hydrophobic-interaction/hitrap-phenyl-hp-p-05630 |
| Mono S 10/100 GL | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/ch/shop/chromatography/prepacked-columns/ion-exchange/mono-s-cation-exchange-chromatography-column-p-00723 |
| Mono Q 10/100 GL | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/ch/shop/chromatography/prepacked-columns/ion-exchange/mono-q-anion-exchange-chromatography-column-p-00608 |
| HiLoad Superdex column 75 pg (G75) | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/ch/shop/chromatography/prepacked-columns/size-exclusion/hiload-superdex-75-pg-preparative-size-exclusion-chromatography-columns-p-05800 |
| HiLoad Superdex column 200 pg (G200) | GE Healthcare Life Sciences | - | https://www.gelifesciences.com/en/ch/shop/chromatography/prepacked-columns/size-exclusion/hiload-superdex-200-pg-preparative-size-exclusion-chromatography-columns-p-06283 |
| TECAN microplate reader | TECAN Life Sciences | - | https://lifesciences.tecan.com/microplate-readers |
| acetylpyruvate | MoleculeCrafting.HuGs e.U. | - | custom synthesis |
| benzoylpyruvate | MoleculeCrafting.HuGs e.U. | - | custom synthesis |
| VDX™ plate (24 wells) | Hampton | HR3-142 | 24 well plates used for crystallization via Hanging Drop Vapor Diffusion |
| paraffin oil | Hampton | HR3-411 | used for crystallization via Hanging Drop Vapor Diffusion |
| coverslips (22 mm) | Karl Hecht KG | 14043 | coverslips used for crystallization via Hanging Drop Vapor Diffusion |
| Luria broth (LB) medium | self-prepared | - | a general growth medium for E. coli: 5 g/L yeast extract; 10 g/L peptone from casein; 10 g/L sodium chloride; 12 g/L agar-agar |
| NZCYM medium | self-prepared | - | a better growth medium for E. coli, used for amplification: 10 g/L NZ amine; 5 g/L NaCl; 5 g/L yeast extract; 1 g/L casamino acids; 2 g/L MgSO4; adjust pH to 7.4 |
| Luria broth (LB) agarose plates | self-prepared | - | autoclaved agarose plates containing LB-medium and antibiotics for bacterial groth selection; https://www.addgene.org/protocols/pouring-lb-agar-plates/ |
| Ni-NTA running buffer | self-prepared | - | 20 mM Tris-HCl pH 7,4; 50-300 mM NaCl; 10-200 mM imidazole; ranges: optimal value varies among FAHD proteins |
| Ni-NTA elution buffer | self-prepared | - | 20 mM Tris-HCl pH 7,4; 50-300 mM NaCl; 200-500 mM imidazole; ranges: optimal value varies among FAHD proteins |
| HIC running buffer | self-prepared | - | 44 mM NaH2PO4; 6 mM Na2HPO4; 100 mM NaCl; 20 mM DTT; adjust to pH 7 |
| HIC running buffer AS | self-prepared | - | HIC running buffer saturated with ammonium sulfate (AS); adjust to pH 7: 70 g ammonium sulfate + 90 mL buffer, stirred overnight in the cold room; adjust to pH 7.0 |
| Mono S low salt buffer | self-prepared | - | 44 mM NaH2PO4; 6 mM Na2HPO4; 10-300 mM NaCl; ranges: optimal value varies among FAHD proteins |
| Mono S high salt buffer | self-prepared | - | 44 mM NaH2PO4; 6 mM Na2HPO4; 1-2 M NaCl; ranges: optimal value varies among FAHD proteins |
| Mono Q low salt buffer | self-prepared | - | 20 mM Tris-HCl; 15 mM NaCl; adjust to pH 8.0 |
| Mono Q high salt buffer | self-prepared | - | 20 mM Tris-HCl; 1 M NaCl; 10 % glycerol; adjust to pH 8.0 |
| G75 / G200 running buffer | self-prepared | - | 15 mM Tris-HCl; 300 mM NaCl; adjust to pH 7.4 |
| enzyme assay buffer | self-prepared | - | 50 mM Tris-HCl pH7.4; 100 mM KCl; 1 mM MgCl2 |
| protein crystallization buffer | self-prepared | - | G75 / G200 running buffer with 1 mM DTT |
| reservoir solution for crystallization | self-prepared | - | 100 mM Na-HEPES pH 7.5; 5-20 % (w/v) PEG4k; 10 mM-200 mM MgCl2 |
Referências
- Brouns, S. J. J., et al. Structural Insight into Substrate Binding and Catalysis of a Novel 2-Keto-3-deoxy-d-arabinonate Dehydratase Illustrates Common Mechanistic Features of the FAH Superfamily. Journal of Molecular Biology. 379, 357-371 (2008).
- Timm, D. E., Mueller, H. A., Bhanumoorthy, P., Harp, J. M., Bunick, G. J. Crystal structure and mechanism of a carbon-carbon bond hydrolase. Structure (London, England: 1993). 7, 1023-1033 (1999).
- Weiss, A. K. H., Loeffler, J. R., Liedl, K. R., Gstach, H., Jansen-Dürr, P. The fumarylacetoacetate hydrolase (FAH) superfamily of enzymes: multifunctional enzymes from microbes to mitochondria. Biochemical Society Transactions. 46, 295 (2018).
- Guimarães, S. L., et al. Crystal Structures of Apo and Liganded 4-Oxalocrotonate Decarboxylase Uncover a Structural Basis for the Metal-Assisted Decarboxylation of a Vinylogous β-Keto Acid. Biochemistry. 55, 2632 (2016).
- Zhou, N. Y., Fuenmayor, S. L., Williams, P. A. nag genes of Ralstonia (formerly Pseudomonas) sp. strain U2 encoding enzymes for gentisate catabolism. Journal of Bacteriology. 183, 700 (2001).
- Izumi, A., et al. Structure and Mechanism of HpcG, a Hydratase in the Homoprotocatechuate Degradation Pathway of Escherichia coli. Journal of Molecular Biology. 370, 899-911 (2007).
- Manjasetty, B. A., et al. X-ray structure of fumarylacetoacetate hydrolase family member Homo sapiens FLJ36880. Biological Chemistry. 385, 935-942 (2004).
- Tame, J. R. H., Namba, K., Dodson, E. J., Roper, D. I. The crystal structure of HpcE, a bifunctional decarboxylase/isomerase with a multifunctional fold. Biochemistry. 41, 2982-2989 (2002).
- Ran, T., et al. Crystal structures of Cg1458 reveal a catalytic lid domain and a common catalytic mechanism for the FAH family. The Biochemical Journal. 449, 51-60 (2013).
- Ran, T., Wang, Y., Xu, D., Wang, W. Expression, purification, crystallization and preliminary crystallographic analysis of Cg1458: A novel oxaloacetate decarboxylase from Corynebacterium glutamicum. Acta Crystallographica Section F: Structural Biology and Crystallization Communications. 67, 968-970 (2011).
- Pircher, H., et al. Identification of human Fumarylacetoacetate Hydrolase Domain-containing Protein 1 (FAHD1) as a novel mitochondrial acylpyruvase. Journal of Biological Chemistry. 286, 36500-36508 (2011).
- Pircher, H., et al. Identification of FAH domain-containing protein 1 (FAHD1) as oxaloacetate decarboxylase. Journal of Biological Chemistry. 290, 6755-6762 (2015).
- Petit, M., Koziel, R., Etemad, S., Pircher, H., Jansen-Dürr, P. Depletion of oxaloacetate decarboxylase FAHD1 inhibits mitochondrial electron transport and induces cellular senescence in human endothelial cells. Experimental Gerontology. 92, 7-12 (2017).
- Etemad, S., et al. Oxaloacetate decarboxylase FAHD1 – a new regulator of mitochondrial function and senescence. Mechanisms of Ageing and Development. 177, 22-29 (2019).
- Weiss, A. K. H., et al. Structural basis for the bi-functionality of human oxaloacetate decarboxylase FAHD1. Biochemical Journal. 475, 3561-3576 (2018).
- Taferner, A., et al. FAH domain-containing protein 1 (FAHD-1) Is required for mitochondrial function and locomotion activity in C. elegans. PLoS ONE. 10, 1-15 (2015).
- Mizutani, H., Kunishima, N. Purification, crystallization and preliminary X-ray analysis of the fumarylacetoacetase family member TTHA0809 from Thermus thermophilus HB8. Acta Crystallographica Section F Structural Biology and Crystallization Communications. 63, 792-794 (2007).
- Bateman, R. L., Bhanumoorthy, P., Witte, J. F., McClard, R. W., Grompe, M., Timm, D. E., et al. Mechanistic Inferences from the Crystal Structure of Fumarylacetoacetate Hydrolase with a Bound Phosphorus-based Inhibitor. Journal of Biological Chemistry. 276, 15284-15291 (2001).
- Zeng, F., et al. Efficient strategy for introducing large and multiple changes in plasmid DNA. Scientific Reports. 8, 1714 (2018).
- Higuchi, R., Krummel, B., Saiki, R. K. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Research. 16, 7351-7367 (1988).
- Jansen-Duerr, P., Pircher, H., Weiss, A. K. H. The FAH Fold Meets the Krebs Cycle. Molecular Enzymology and Drug Targets. 2, 1-5 (2016).
- Rupp, B. Origin and use of crystallization phase diagrams. Acta Crystallographica Section F Structural Biology Communications. 71, 247-260 (2015).
- Rupp, B. . Biomolecular Crystallography: Principles, Practice, and Application to Structural Biology. , (2010).
- Flint, D. H., Nudelman, A., Calabrese, J. C., Gottlieb, H. E. Enol oxalacetic acid exists in the Z form in the crystalline state and in solution. The Journal of Organic Chemistry. 57, 7270-7274 (1992).
- Pogson, C. I. I., Wolfe, R. G. G. Oxaloacetic acid tautomeric and hydrated forms in solution. Biochemical and Biophysical Research Communications. 46, 1048-1054 (1972).
- Kost, T. A., Condreay, J. P., Jarvis, D. L., Kost, A. T. Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nature Biotechnology. 23, 567-575 (2005).
- Steinberger, R., Westheimer, F. H. Metal Ion-catalyzed Decarboxylation: A Model for an Enzyme System 1. Journal of the American Chemical Society. 73, 429-435 (1951).
- Tate, S. S., Grzybowski, A. K., Datta, S. P. The stability constants of the magnesium complexes of the keto and enol isomers of oxaloacetic acid at 25. Journal of Chemical Society. , 1381-1389 (1964).
- Tate, S. S., Grzybowski, A. K., Datta, S. P. The acid dissociations of the keto and enol isomers of oxaloacetic acid at 25. Journal of Chemical Society. 1372, 1380 (1964).
- Brecker, L., et al. Synthesis of 2,4-diketoacids and their aqueous solution structures. New Journal of Chemistry. 23, 437-446 (1999).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados