
Method Article
Analysis of Group IV Viral SSHHPS Using In Vitro and In Silico Methods
In This Article
Summary
We present a general protocol for identifying short stretches of homologous host-pathogen protein sequences (SSHHPS) embedded in the viral polyprotein. SSHHPS are recognized by viral proteases and direct the targeted destruction of specific host proteins by several Group IV viruses.
Abstract
Alphaviral enzymes are synthesized in a single polypeptide. The nonstructural polyprotein (nsP) is processed by its nsP2 cysteine protease to produce active enzymes essential for viral replication. Viral proteases are highly specific and recognize conserved cleavage site motif sequences (~6-8 amino acids). In several Group IV viruses, the nsP protease(s) cleavage site motif sequences can be found in specific host proteins involved in generating the innate immune responses and, in some cases, the targeted proteins appear to be linked to the virus-induced phenotype. These viruses utilize short stretches of homologous host-pathogen protein sequences (SSHHPS) for targeted destruction of host proteins. To identify SSHHPS the viral protease cleavage site motif sequences can be inputted into BLAST and the host genome(s) can be searched. Cleavage initially can be tested using the purified nsP viral protease and fluorescence resonance energy transfer (FRET) substrates made in E. coli. The FRET substrates contain cyan and yellow fluorescent protein and the cleavage site sequence (CFP-sequence-YFP). This protease assay can be used continuously in a plate reader or discontinuously in SDS-PAGE gels. Models of the bound peptide substrates can be generated in silico to guide substrate selection and mutagenesis studies. CFP/YFP substrates have also been utilized to identify protease inhibitors. These in vitro and in silico methods can be used in combination with cell-based assays to determine if the targeted host protein affects viral replication.
Introduction
Evidence of horizontal gene transfer from virus to host, or host to virus, can be found in a variety of genomes1,2,3,4. Examples of viral endogenization are the CRISPR spacer sequences found in bacterial host genomes4. Recently, we have found evidence of host protein sequences embedded in the nonstructural polyproteins of (+)ssRNA Group IV viruses. These sequences within the coding regions of the viral genome can be propagated generationally. The short stretches of homologous host-pathogen protein sequences (SSHHPS) are found in the virus and host5,6. SSHHPS are the conserved cleavage site motif sequences recognized by viral proteases that have homology to specific host proteins. These sequences direct the destruction of specific host proteins.
In our previous publication6, we compiled a list of all of the host proteins that were targeted by viral proteases and found that the list of targets was non-random (Table 1). Two trends were apparent. First, the majority of the viral proteases that cut host proteins belonged to Group IV viruses (24 of 25 cases involved Group IV viral proteases), and one protease belonged to the (+)ssRNA Group VI retroviruses (HIV, human immunodeficiency virus)7. Second, the host protein targets being cut by the viral proteases were generally involved in generating the innate immune responses suggesting that the cleavages were intended to antagonize the host's immune responses. Half of the host proteins targeted by the viral proteases were known components of signaling cascades that generate interferon (IFN) and proinflammatory cytokines (Table 1). Others were involved in host cell transcription8,9,10 or translation11. Interestingly, Shmakov et al.4 have shown that many CRISPR protospacer sequences correspond to genes involved in plasmid conjugation or replication4.
Group IV includes, among others, Flaviviridae, Picornaviridae, Coronaviridae, Calciviridae, and Togaviridae. Several new and emerging pathogens belong to Group IV such as the Zika virus (ZIKV), West Nile (WNV), Chikungunya (CHIKV), severe acute respiratory syndrome virus (SARS) and Middle East respiratory syndrome virus (MERS). The (+)ssRNA genome is essentially a piece of mRNA. To produce the enzymes necessary for genome replication, the (+)ssRNA genome first must be translated. In alphaviruses and other Group IV viruses, the enzymes necessary for replication are produced in a single polyprotein (i.e., nsP1234 for VEEV). The nonstructural polyprotein (nsP) is proteolytically processed (nsP1234 nsP1, nsP2, nsP3, nsP4) by the nsP2 protease to produce active enzymes12 (Figure 1). Cleavage of the polyprotein by the nsP2 protease is essential for viral replication; this has been demonstrated by deletion and site-directed mutagenesis of the active site cysteine of the nsP2 protease13,14. Notably, the translation of viral proteins precedes genome replication events. For example, nsP4 contains the RNA-dependent RNA polymerase needed to replicate the (+)ssRNA genome. Genome replication can produce dsRNA intermediates; these intermediates can trigger the host's innate immune responses. Thus, these viruses may cleave host innate immune response proteins early in infection in order to suppress their effects15,16,17.
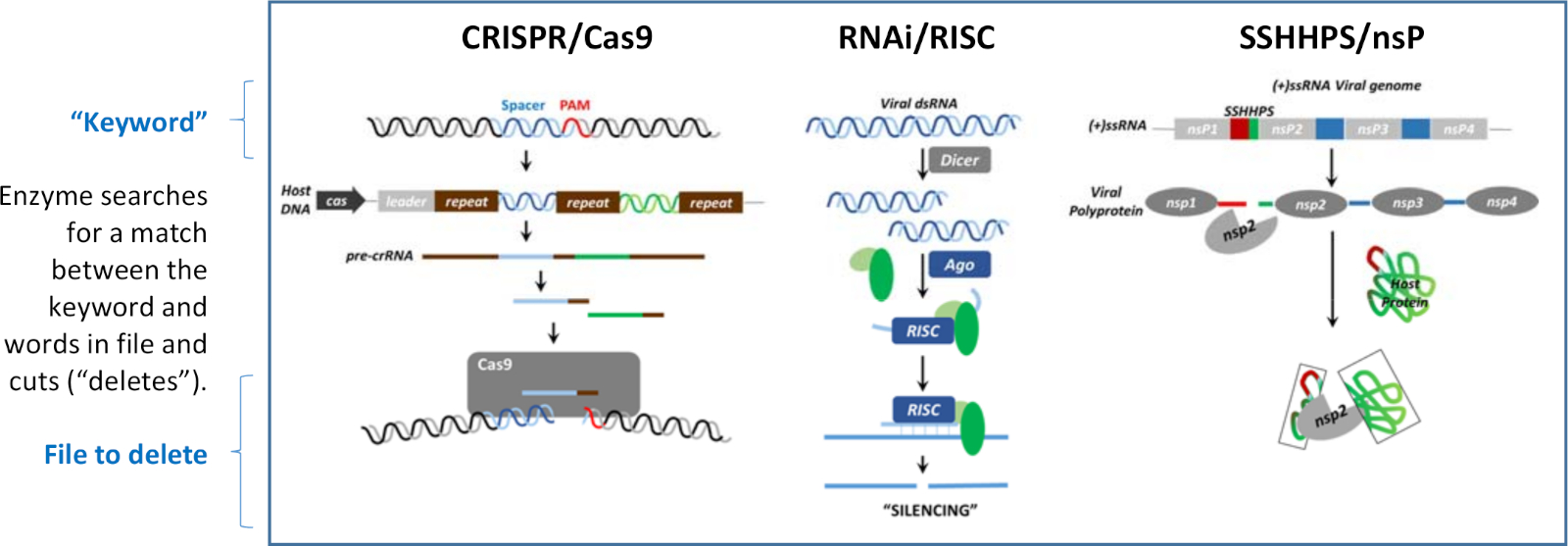
Silencing can occur at the level of DNA, RNA, and protein. What is common to each of the silencing mechanisms shown in Figure 1 is that short foreign DNA, RNA, or protein sequences are used to guide the destruction of specific targets to antagonize their function. The silencing mechanisms are analogous to "search and delete" programs that have been written in three different languages. The short cleavage site sequence is analogous to a "keyword". Each program has an enzyme that recognizes the match between the short sequence (the "keyword") and a word in the "file" that is to be deleted. Once a match is found, the enzyme cuts ("deletes") the larger target sequence. The three mechanisms shown in Figure 1 are used to defend the host from viruses, or to defend a virus from a host's immune system.
Viral proteases recognize short cleavage site motif sequences between ~2-11 amino acids; in nucleotides, this would correspond to 6-33 bases. For comparison, CRISPR spacer sequences are ~26-72 nucleotides and RNAi are ~20-22 nucleotides18,19. While these sequences are relatively short, they can be recognized specifically. Given the higher diversity of amino acids, the probability of a random cleavage event is relatively low for a viral protease recognizing protein sequences of 6-8 amino acids or longer. The prediction of SSHHPS in host proteins will largely depend upon the specificity of the viral protease being examined. If the protease has strict sequence specificity requirements the chance of finding a cleavage site sequence is 1/206 = 1 in 64 million or 1/208 = 1 in 25.6 billion; however, most proteases have variable subsite tolerances (e.g., R or K may be tolerated at the S1 site). Consequently, there is no requirement for sequence identity between the sequences found in the host versus the virus. For viral proteases that have looser sequence requirements (such as those belonging to Picornaviridae) the probability of finding a cleavage site in a host protein may be higher. Many of the entries in Table 1 are from the Picornaviridae family.
Schechter & Berger notation20 is commonly used to describe the residues in a protease substrate and the subsites to which they bind, we utilize this notation throughout. The residues in the substrate that are N-terminal of the scissile bond are denoted as P3-P2-P1 while those that are C-terminal are denoted as P1'-P2'-P3'. The corresponding subsites in the protease that bind these amino acid residues are S3-S2-S1 and S1'-S2'-S3', respectively.
To determine which host proteins are being targeted, we can identify SSHHPS in the viral polyprotein cleavage sites and search for the host proteins that contain them. Herein, we outline procedures for identifying SSHHPS using known viral protease cleavage site sequences. The bioinformatic methods, protease assays, and in silico methods described are intended to be used in conjunction with cell-based assays.
Sequence alignments of the host proteins targeted by viral proteases have revealed species-specific differences within these short cleavage site sequences. For example, the Venezuelan equine encephalitis virus (VEEV) nsP2 protease was found to cut human TRIM14, a tripartite motif (TRIM) protein6. Some TRIM proteins are viral restriction factors (e.g., TRIM5α21), most are thought to be ubiquitin E3 ligases. TRIM14 lacks a RING (really interesting new gene) domain and is not thought to be an E3 ligase22. TRIM14 has been proposed to be an adaptor in the mitochondrial antiviral signalosome (MAVS)22, but may have other antiviral functions23. Alignment of TRIM14 sequences from various species shows that equine lack the cleavage site and harbor a truncated version of TRIM14 that is missing the C-terminal PRY/SPRY domain. This domain contains a polyubiquitination site (Figure 2). In equine, these viruses are highly lethal (~20-80% mortality) whereas in humans only ~1% die from VEEV infections24. Cleavage of the PRY/SPRY domain may transiently short circuit the MAVS signaling cascade. This cascade can be triggered by dsRNA and leads to the production of interferon and pro-inflammatory cytokines. Thus, the presence of the SSHHPS may be useful for predicting which species have defense systems against specific Group IV viruses.
In Group IV viruses, IFN antagonism mechanisms are thought to be multiply redundant25. Host protein cleavage may be transient during infection and concentrations may recover over time. We found in cells that TRIM14 cleavage products could be detected very early after transfection (6 h) with a plasmid encoding the protease (cytomegalovirus promoter). However, at longer periods, the cleavage products were not detected. In virus-infected cells, the kinetics were different and cleavage products could be detected between 6-48 h6. Others have reported the appearance of host protein cleavage products as early as 3-6 h post infection9,11.
Proteolytic activity in cells is often difficult to catch; the cleavage products can vary in their solubility, concentration, stability, and lifetime. In cell-based assays, it cannot be assumed that cleavage products will accumulate in a cell or that the band intensities of cut and uncut protein will show compensatory increases and decreases as the cut protein may be degraded very quickly and may not be detectable in a Western blot at an expected molecular weight (MW) (e.g., the region containing the epitope could be cleaved by other host proteases or could be ubiquitinated). If the substrate of the viral protease is an innate immune response protein, its concentration may vary during infection. For example, some innate immune response proteins are present prior to viral infection and are induced further by interferon26. The concentration of the target protein may therefore fluctuate during infection and comparison of uninfected vs. infected cell lysates may be difficult to interpret. Additionally, all cells may not be uniformly transfected or infected. In vitro protease assays using purified proteins from E. coli on the other hand have fewer variables for which to control and such assays can be done using SDS-PAGE rather than immunoblots. Contaminating proteases can be inhibited in the early steps of the protein purification of the CFP/YFP substrate, and mutated viral proteases can be purified and tested as controls to determine if the cleavage is due to the viral protease or a contaminating bacterial protease.
One limitation of in vitro protease assays is that they lack the complexity of a mammalian cell. For an enzyme to cut its substrate, the two must be co-localized. Group IV viral proteases differ in structure and localization. For example, the ZIKV protease is embedded in the endoplasmic reticulum (ER) membrane and faces the cytosol, whereas the VEEV nsP2 protease is a soluble protein in the cytoplasm and nucleus27. Some of the cleavage site sequences found in the ZIKV SSHHPS analysis were in signal peptides suggesting that cleavage might occur co-translationally for some targets. Thus, the location of the protease and the substrate in the cell also needs to be considered in these analyses.
Cell-based assays can be valuable for establishing a role for the identified host protein(s) in infection. Methods that aim to halt viral protease cleavage of host proteins such as the addition of a protease inhibitor6 or a mutation in the host target16 can be used to examine their effects on viral replication. Overexpression of the targeted protein also may affect viral replication28. Plaque assays or other methods can be used to quantify viral replication.
Protocol
1. Bioinformatics: Identification of SSHHPS in the Host Genome Using BLAST
NOTE: Protein BLAST can be found at blast.ncbi.nlm.nih.gov/Blast.cgi.
- Input ~20 amino acids surrounding the scissile bond in the viral polyprotein. Select non-redundant protein sequences and type in the host genome to be searched (e.g., Homo sapiens).
- If needed, select PHI-BLAST. Type in a pattern sequence (e.g., for the 25 residues of V12 shown below enter the pattern "AG" without quotes).

NOTE: In PHI-BLAST, square brackets [XY] indicate that amino acid X or Y can be at the subsite position (e.g., AG[AC][GAY]). - Inspect the BLAST results and identify the hits that have high sequence identity to residues that are conserved in the polyprotein cleavage sites (e.g., tripartite motif protein 14) (Figure 3).
NOTE: For serine proteases higher conservation of the P1 residue is expected, while for cysteine proteases higher conservation of the P2 residue is expected. - Color the residues that are identical to a cleavage site sequence and are in sequential order (no gaps). Color the residues tolerated at the subsite, but present in a different cleavage site in a second color.
NOTE: Residues that represent conservative substitutions (e.g., Leu vs. Val) that are not present in a viral cleavage site also may be found and may or may not be recognized by the viral protease. - Rank order the BLAST hits based upon the number of consecutive identical or tolerated residues that match a cleavage site sequence. From the list, select the proteins containing ≥6 identical or similar residues for analysis in protease assays.
- Repeat the procedure for the other cleavage sites (nsP2/3, nsP3/4, etc.) and gradually strengthen the prediction by adding more highly conserved residues to the PHI-BLAST pattern.
- If needed, select PHI-BLAST. Type in a pattern sequence (e.g., for the 25 residues of V12 shown below enter the pattern "AG" without quotes).
2. In Vitro Assays: Designing and Preparing Protease Substrates
- Construct a plasmid encoding the cyan fluorescent protein (CFP), ≤25 amino acids of the cleavage site sequence, followed by the yellow fluorescent protein (YFP, also known as Venus29).
NOTE: The plasmid can be constructed using sequence and ligation independent cloning (SLIC)30 or commercial gene synthesis. A pet15b plasmid containing the sequence shown in Figure 4 was synthesized commercially and was used here.- To optimize the substrate length, construct additional variable length FRET substrates containing 12-25 amino acids of the natural viral polyprotein cleavage site sequences using a 2-fragment SLIC reaction. Analyze cleavage using the SDS-PAGE gel-based assay or by measuring steady state kinetic parameters using the methods below.
NOTE: In some cases, cleavage sites can be identified by homology to known cleavage sites31. If cleavage of the substrates containing the polyprotein junction sequences is not observed, there may be a requirement for additional residues or a structural motif (e.g., an alpha helix32). Alternatively, the purified viral protease may be inactive. Confirm cleavage of the viral polyprotein sequences before pursuing SSHHPS analysis. The number of residues in the substrate was optimized for the VEEV protease using variable length substrates (12 to 25 amino acids) followed by analysis of Vmax and Km32,33. The Zika viral ns2B/nsB protease cleavage sites used in the examples have been published34,35.
- To optimize the substrate length, construct additional variable length FRET substrates containing 12-25 amino acids of the natural viral polyprotein cleavage site sequences using a 2-fragment SLIC reaction. Analyze cleavage using the SDS-PAGE gel-based assay or by measuring steady state kinetic parameters using the methods below.
- Prepare the CFP/YFP substrates by freshly transforming 8-20 µL of BL-21(DE3) E. coli competent cells with the CFP-V12-YFP plasmid according to manufacturer's directions and plate on Luria Bertani (LB) agar plates containing 50 µg/mL Ampicillin (37 °C).
- Autoclave four 4 L flasks containing LB media (1.5 L media per flask) and 100 mL of LB in a 250 mL flask. Cap each flask with aluminum foil.
- Inoculate the 100 mL culture with a colony of the freshly transformed bacteria and grow at 37 °C with shaking (200 rpm) overnight.
- To make the CFP/YFP substrate, inoculate four 4 L flasks with 25 mL of an overnight culture. Begin shaking the cultures at 37 °C and monitor growth by UV-vis spectroscopy at 600 nm hourly.
- When the bacteria reach an absorbance of ~1.0 at 600 nm (approximately ~3-4 h of growth) induce protein expression by adding 0.5 mL of 1 M isopropyl-β-D-thiogalactoside (IPTG) per flask. After adding IPTG, lower the temperature of the shaking incubator to 17 °C and allow expression to continue overnight for 17-20 h.
- Pellet the bacteria using a high-speed centrifuge at 7,000 x g for 10 min (4 °C) and retain the pellets. Remove and discard liquid media. Store the pellets at -80 °C or lyse immediately.
- Prepare 100 mL of lysis buffer containing 50 mM Tris pH 7.6, 500 mM NaCl, 35 mL of bacterial protein extraction reagent, 30 mg of lysozyme, 25 U of DNase, and 1 protease inhibitor tablet. Resuspend the pellets in lysis buffer with a pipette and transfer ~25-35 mL into 50 mL disposable conical tubes.
- Place the tubes in a plastic beaker containing ice water. Insert the sonicator tip into the tubes so that the tip is ~1 cm from the bottom of the tube and sonicate the lysates 10-20 times on level 5 for 15 s intervals until the lysate becomes fluid and liquefied.
NOTE: Use hearing protection during sonication. - Transfer the lysate to high speed centrifuge tubes and centrifuge at 20,500 x g for 30 min at 4 °C. After the spin, retain the supernatant (~100 mL) and transfer it to a clean bottle. Discard the pellets.
- Prepare 1 L of Buffer A (50 mM Tris pH 7.6, 500 mM NaCl). Prepare 300 mL of Buffer B (50 mM Tris pH 7.6, 500 mM NaCl, 300 mM Imidazole).
- Equilibrate a 100 mL nickel column using 3 column volumes of Buffer A and a flow rate of 5 mL/min.
- Load the lysate onto the nickel column using a flow rate of 2 to 5 mL/min. Wash the column with 2 column volumes of Buffer A, followed by ~5 column volumes of 20% Buffer B. During the 20% Buffer B wash, the absorbance at 280 nm (A280) will increase as contaminants elute from the column during the wash. Continue washing the column until the A280 of the eluate has returned to baseline values.
- Elute the protein with 2-3 column volumes of 100% Buffer B using a flow rate of 2-5 mL/min and collect 10 mL fractions. Measure the A280 of each fraction.
- Combine and concentrate fractions containing A280 > 0.1 using a 15 mL centrifugal ultrafiltration unit. Spin the ultrafiltration units at 5,000 x g for 15 min and continue to add fractions until the volume has been reduced to ~50-75 mL.
- Cut a 14 inch piece of dialysis tubing with a molecular weight cut-off (MWCO) of 6-8 kDa. Hydrate the dialysis tubing by boiling it fully submerged in 300 mL of water for 10 min. Tie a secure knot at one end of the membrane. Fill the bag with dialysis buffer to ensure that no cracks or leaks are present. Remove the buffer from the bag and keep the bag submerged in the dialysis buffer.
- Transfer the concentrated protein from 2.2.13 into the dialysis bag with a plastic pipette. Remove any air bubbles from the bag. Close the bag with a second knot or a dialysis clip. Dialyze the protein against 500 mL of 50 mM Tris pH 7.6, 5 mM EDTA (ethylenediaminetetraacetic acid), 250 mM NaCl in a 500 mL graduated cylinder overnight at 4 °C.
- Dialyze the protein a second time against 500 mL of 50 mM Tris pH 7.6 at 4 °C for 2 h.
- For the anion exchange column prepare 500 mL of Buffer A (50 mM Tris pH 7.6) and 500 mL Buffer B (50 mM Tris pH 7.6, 1.0 M NaCl). Equilibrate a 30 mL anion exchange column with 3 column volumes of Buffer A (2-5 mL/min).
- Remove the protein from the dialysis bag and transfer to a bottle. Keep the bottle on ice. Load the dialyzed protein onto the column (2-5 mL/min).
NOTE: The CFP/YFP protein will bind the column and will be yellow in appearance. - Wash the column with Buffer A until the A280 returns to baseline (5 mL/min). Elute the protein using a gradient (0-50% Buffer B, 100 mL) and collect 10 mL fractions.
- Inspect the column fractions using SDS-PAGE. Combine those that are >95% pure.
- Concentrate the protein to an A280 ~10-20 using a 15 mL centrifugal ultrafiltration unit. Spin the concentrator at 4,500 x g for 10 min at 4 °C and continue to add protein until all of the protein-containing fractions have been combined.
- Remove the protein from the dialysis bag and transfer to a bottle. Keep the bottle on ice. Load the dialyzed protein onto the column (2-5 mL/min).
- Carefully remove the protein from the concentrator with a pipette. Aliquot the protein into 1.5 mL microcentrifuge tubes and flash freeze in liquid nitrogen for long term storage at -80 °C. Buffer exchange the protein at room temperature using a PD-10 column equilibrated with the appropriate assay buffer prior to use.
- Using Beer's law calculate the protein concentration using the A280 and a calculated extinction coefficient (e.g., for the V12 substrate the ɛ = 47,790 M-1 cm-1).
NOTE: The extinction coefficient (ɛ) can be calculated from the protein sequence in Figure 4 using the Expasy ProtParam program (https://web.expasy.org/protparam/).
3. Preparation of the Alphaviral nsP2 Cysteine Protease
- Design and construct a plasmid encoding the protease. For cysteine proteases, use the pet32 plasmid to construct a thioredoxin (Trx) fusion protein.
NOTE: The pet32 plasmid encodes a thrombin cleavage site (LVPRGS) for removal of the thioredoxin and His-tag (Figure 5). Thioredoxin will help maintain the active site cysteine in a reduced state during expression. For serine proteases, the thioredoxin is not needed and steps involving its removal by thrombin can be omitted. The VEEV nsP2 protease sequence was incorporated into a pet32b plasmid that was prepared commercially to avoid handling Select agents.- Freshly transform the plasmid DNA into BL21(DE3)pLysS E. coli according to manufacturer's directions. Plate the bacteria on LB agar plates containing Ampicillin.
NOTE: Chloramphenicol is only used for E. coli strains carrying the pLysS plasmid and is omitted if BL21(DE3) cells are used. It is not necessary to include chloramphenicol on the LB agar plate in this step. - Autoclave four 4 L flasks of 1.5 L of LB media (6 L total volume) and 100 mL of LB in a 250 mL flask. Cap each flask with aluminum foil.
- Inoculate a 100 mL overnight culture of LB/Ampicillin with a colony from the plate and grow in a shaking incubator (200 rpm) at 37 °C.
- Inoculate the 4 L flasks with 25 mL of the overnight culture and add the appropriate antibiotics.
NOTE: The media for the BL21(DE3) pLysS cells carrying the pet32 plasmid should have final concentrations of 25 µg/mL chloramphenicol and 50 µg/mL Ampicillin. - Induce protein expression by adding 0.5 mL of IPTG to the culture when the absorbance at 600 nm reaches 1.0. Lower the temperature of the shaking incubator to 17 °C. Allow expression to continue overnight (~17 h).
- Pellet the cells by centrifugation (7,000 x g for 10 min at 4 °C). Remove and discard the liquid media.
NOTE: The pellets can be stored at -80 °C for months or lysed immediately. - Prepare 100 mL of lysis buffer (50 mM Tris pH 7.6, 500 mL NaCl, 2 mM beta mercaptoethanol (BME), 30 mg lysozyme, 5% glycerol, 25 U DNase, 35 mL bacterial protein extraction reagent). Open bottles of BME in a chemical hood when adding. Keep the bacterial lysate on ice or at 4 °C for this and all subsequent steps.
NOTE: For cysteine proteases, 2 mM BME is included to keep the nucleophilic cysteine reduced. The columns can be run at room temperature using chilled buffers. Buffers should be made with cold deionized water cooled to 4 °C. - Resuspend the bacterial pellets in ~25 mL of lysis buffer and transfer ~25 mL of the lysate into 4 x 50 mL disposable conical tubes. Place the tubes into plastic beakers containing ice water. Sonicate the lysate 10 times on level 5 for 15 s intervals.
- Transfer the lysate into high speed centrifuge tubes. Clarify the lysate by centrifugation (30 min, 20,500 x g at 4 °C).
- Prepare 0.5 L of Buffer A (50 mM Tris pH 7.6, 500 mM NaCl, 5% glycerol, 2 mM BME) and chill to 4 °C.
- Prepare 250 mL of Buffer B (50 mM Tris pH 7.6, 500 mM NaCl, 5% glycerol, 2 mM BME, 300 mM imidazole) and chill to 4 °C.
- Equilibrate a 50 mL nickel column with 3 column volumes of Buffer A. Load the clarified lysate onto the column at 2-5 mL/min and discard the pellets.
- Wash the column (2-5 mL/min) with 2 column volumes of Buffer A followed by 5 column volumes of Buffer A containing 20% Buffer B (60 mM Imidazole). Elute the protein (5 mL/min) with 100% Buffer B and collect 10 mL fractions.
- Combine and concentrate fractions containing the protease that have A280 ≥ 0.1 using a 15 mL centrifugal ultrafiltration unit and 15 min spins at 5,000 x g at 4 °C. After the volume has been reduced to ~5 mL, buffer exchange the protein in the concentration unit by adding fresh dialysis buffer to the protein (50 mM Tris pH 7.6, 250 mM NaCl, 5 mM dithiothreitol (DTT), 1 mM EDTA, 5% Glycerol). Spin again at 5,000 x g at 4 °C for 15 min; repeat the buffer exchange step 2-3 times. Add thrombin to the protein (20 µL of 1 unit/µL) prior to dialysis to remove the thioredoxin and His-tag.
- Transfer the protein into a dialysis bag and dialyze against 500 mL of the dialysis buffer (4 °C) in a 500 mL graduated cylinder overnight.
NOTE: The FPLC (fast protein liquid chromatography) system and the nickel column should be thoroughly cleaned with stripping buffer (2 M NaCl, 50 mM EDTA) before proceeding to the anion exchange column. Any residual nickel in the FPLC lines will turn the buffer solutions containing DTT brown when mixed. Wash the nickel column and FPLC system with 4 column volumes of water. Pump wash the FPLC system thoroughly with water. The nickel column can be regenerated by flowing 2 column volumes of 0.2 M nickel sulfate over the resin for subsequent purifications.
- Freshly transform the plasmid DNA into BL21(DE3)pLysS E. coli according to manufacturer's directions. Plate the bacteria on LB agar plates containing Ampicillin.
- For the anion exchange column prepare 1 L of Buffer A (50 mM Tris pH 7.6, 5 mM DTT, 5% glycerol).
- Prepare 0.5 L of Buffer B (50 mM Tris pH 7.6, 5 mM DTT, 5% glycerol, 1.25 M NaCl).
- Equilibrate a 30 mL anion exchange column with Buffer A (3 column volumes, 2-5 mL/min). Place the tubes in the fraction collector for collection of the flow through.
NOTE: The VEEV protease has a calculated isoelectric point (pI) of 8.7 and will bind cation-exchange columns but will flow through anion exchange columns. The pI can be calculated from the protein sequence using the Expasy ProtParam program (https://web.expasy.org/protparam/). - Dilute the dialyzed protein 1:3 with Buffer A, and then load the protein (5 mL/min). Collect the flow-through in 10 mL fractions.
- Remove the anion exchange column from the FPLC system. Connect a cation exchange column to the FPLC system. Equilibrate a 30 mL cation exchange column with 3 column volumes of Buffer A (5 mL/min).
- Load the flow through of the anion exchange column onto the cation exchange column at 2-5 mL/min. Wash the column with Buffer A until the A280 returns to baseline level. Elute the protein with a 100 mL gradient (0-50% Buffer B) and collect 10 mL fractions.
NOTE: The VEEV protease will elute at around 0.6 M NaCl. - Inspect the column fractions using SDS-PAGE. Combine fractions that are >95% pure and concentrate to an A280 ≈ 2 using 15 mL centrifugal ultrafiltration units. The enzyme can be flash-frozen in liquid nitrogen and stored at -80 °C.
- Load the flow through of the anion exchange column onto the cation exchange column at 2-5 mL/min. Wash the column with Buffer A until the A280 returns to baseline level. Elute the protein with a 100 mL gradient (0-50% Buffer B) and collect 10 mL fractions.
4. Assaying the Enzyme Continuously Using a Plate Reader
- Prepare 50 mL of assay buffer (50 mM HEPES pH 7.0).
- As the alphaviral proteases have relatively low kcat values, dilute the enzyme in the assay buffer to 4.7 µM (this will roughly correspond to an A280 = 0.2 for the VEEV protease without Trx).
- To measure the activity of the enzyme, prepare a stock of substrate in the assay buffer with a concentration of 185 µM; this will roughly correspond to an A280 = 9. In 8 microcentrifuge tubes, prepare the reaction mixes shown in Table 2 by combining the appropriate volumes of the 185 µM substrate stock and buffer. In a black half-area 96-well plate pipet 45 µL of the reaction mixes into 3 wells (columns 1, 2, 3). Row A should contain the [S] = 5 µM reaction mix, and Row H should contain the [S] = 140 µM reaction mix.
- Set the plate reader to detect simultaneously fluorescence at two wavelengths with a fixed photomultiplier tube (PMT) setting (e.g., low):
Wavelength 1 excitation = 434 nm,emission = 527 nm
Wavelength 2 excitation = 434 nm,emission = 470 nm - Set the read time to 20 min (measuring 1 read per minute) and select the wells to be read. Insert the plate into the plate reader and measure the spontaneous rate of hydrolysis for 20 min. Monitor the emission ratios (emission at 527/emission at 470) over time.
- Run an endpoint read of the plate containing the "UNCUT" substrate.
NOTE: These values will be used in subsequent data calculations. The average of the emission ratios from 3 wells will be the values of the "UNCUT" substrate at t = 0 in Table 3. - Remove the plate and pipet 5 µL of enzyme into each well. Read the plate again for 20 min with 1 read per minute. Set the plate reader to output absolute values.
NOTE: For this assay, the slopes will be negative. Each well will contain a total volume of 50 µL. - At the end of the read, seal the plate with film to prevent evaporation. Leave the plate at room temperature overnight to allow the enzyme to cut the substrate completely.
- After ~24 h, remove the sealing film and perform an endpoint read of the plate using the same PMT as in the prior plates. Average these emission ratios and input into Table 3 under "CUT". Confirm the cleavage of the substrate using the SDS-PAGE discontinuous assay described below (Step 5.1.).
- Export the data to a spreadsheet. Output the fluorescence units at each time point for the 2 wavelengths (Table 4).
- Calculate the nmol of substrate that have been cut at time t using equation (1) where X is the emission ratio (527 nm/470 nm) at a given time point, neg is the emission ratio of the "UNCUT" substrate at t = 0, and pos is the emission ratio of the completely "CUT" substrate measured after 24 h of cutting (Table 3).

NOTE: Representative fluorescence data are shown for one well (well E7) containing 80 µM substrate (4 nmol of S per well) in Table 4. The calculations were performed for each well in the plate. - For each well, plot nmol vs. time (min) and obtain the initial velocities (slopes) by fitting the data to y = mx + b. For the data collected in 4.1.5, plot nmol vs. time (min) for each well. The slope will equal the nmol product produced per minute. Subtract the spontaneous rates of hydrolysis measured in 4.1.5 from the enzyme-catalyzed reaction rates (Table 5).
NOTE: The first read can be clipped from the data if it is artifactually high due to movement of the plate into the plate reader. - Calculate the amount of enzyme in mg that was added to each well (e.g., 0.0009 mg). A unit is defined as a µmol of product produced per minute (µmol/min). Divide the nmol/min by the mg of enzyme present in the well to obtain mU/mg; divide by 1,000 to obtain U/mg.
- Plot [S] µM on the x-axis and U/mg on the y-axis and fit the data to the Michaelis-Menten equation to obtain Vmax and Km. This can be done in the software (e.g., GraFit).
5. Assaying the Enzyme Discontinuously Using SDS-PAGE Analysis
- Prepare a 50 µL reaction containing 10 µM substrate and buffer in place of enzyme and label as "UNCUT."
NOTE: The volumes of substrate and buffer are shown in Table 2. If the continuous assay has been run, the samples can be used directly from the 96-well plate.- Prepare a 50 µL reaction containing 10 µM substrate and 5 µL enzyme and label as "CUT". Start the timer when the enzyme is added to the substrate.
NOTE: Inhibitors can be added to additional tubes containing enzyme and substrate. Adjust the volume of added buffer to compensate for the added volume of inhibitor. Concentrations of DMSO should not exceed 2%. - Incubate the reactions for ~15-24 h at room temperature (22 ± 3 °C). Stop the reactions by adding 50 µL of 2x Laemelli buffer. After stopping the reaction boil, each tube for 3-10 min.
- Assemble the gel tank according to the manufacturer's directions. Insert a 17-well pre-cast 12% polyacrylamide gel cassette and a buffer dam on the other side. Fill the interior reservoir of the cell with 1x SDS running buffer until the buffer reaches the top of the cassette. Fill the external reservoir half-full with the same buffer.
- To analyze cleavage using the discontinuous assay, load 5 µL of each reaction mixture into a lane of an SDS-PAGE gel beginning with the "UNCUT" reaction. Include a molecular weight marker in the first or last lane.
- Attach the electrodes of the gel tank to the power supply and separate the products at 110 V for 60 min. Remove the gel from the cassette by inserting the cracking tool in between the plates. Place the gel in a plastic tray and submerge the gel in 5-10 mL of gel staining solution; bands will be visible within 30 min. After 1-24 hours remove the excess stain, submerge the gel in water and use a gel imager to take a picture of the gel.
- Prepare a 50 µL reaction containing 10 µM substrate and 5 µL enzyme and label as "CUT". Start the timer when the enzyme is added to the substrate.
6. Docking Substrate Peptides to the VEEV-nsP2 Cysteine Protease
- Download the coordinate file for the VEEV cysteine protease from the PDB (https://www.rcsb.org/). The PDB code is 2HWK. Save the file as 2HWK.pdb.
- Prepare the protein structure using MOE (https://www.chemcomp.com/). Load the protein PDB file into MOE. Click the Select and Solvent on the right hand side bar and delete the solvent.
- Open the Structure Preparation panel from the top menu bar Protein. Automatically correct all structural items by clicking on Correct and protonate the structure by clicking on Protonate3D. Add partial charges to the protein by opening Partial Charges panel and selecting Amber 99 and Adjust hydrogens and Lone pairs as required. Finally, save the structure file as "2HWK_dock.pdb".
- Build the structure for the substrate peptides (nsP12, nsP23, nsP34) and TRIM14 using MOE. Open the Protein Builder panel, enter the substrate sequence, set the Geometry as Extended, and click on Build. The structure will be shown in MOE window.
- Minimize the peptide structure by clicking Minimize on the panel. Save the structure as a PDB file (Figure 6).
- Dock the substrate peptides to VEEV-nsP2 using PyRx/AutoDock 4.2 (http://autodock.scripps.edu/). Open the PyRx Tool, edit the preference setting, inactivate all torsions for Ligand Preparation. Load the substrate molecule, right click the molecule name on the Navigator panel, select Make ligand to prepare the ligand docking file. Load the protein 2HWK_clean.pdb, and select Make macromolecule to prepare the pdbqt docking file (Figure 7).
- Start the AutoDock Wizard on the docking panel at the bottom. Select the prepared ligand and protein files. Define the protein binding pocket by manually adjusting the grid dimension which is centered at the catalytic residue Cys-477. Using the default spacing parameter 0.375 Å. Click on Run AutoGrid to generate grid maps.
- Run AutoDock and select the Lamarckian Genetic Algorithm (LGA) method. Click on the Docking Parameters and set the Number of GA runs to 50. Use the default parameters for others. Click on Forward to start the docking run.
- Open the Analyze Results panel. Inspect all predicted binding poses. Select the best model with the lowest predicted binding energy and reasonable binding interactions between the Cys-477 and substrate on the cleavage site. Save the binding model as PDB file for further MD simulations.
7. MD Simulations of Docked VEEV-substrate Complexes
- Prepare the input files using Amber (http://ambermd.org/). Following the standard protocol, MD simulations are performed for the predicted substrate binding models using the AMBER package and the ff99SB force field.
NOTE: The solvated systems are subjected to a thorough energy minimization prior to MD simulations. Periodic boundary conditions are applied to simulate a continuous system. The particle mesh Ewald (PME) method was employed to calculate the long-range electrostatic interactions. The simulated system was first subjected to a gradual temperature increase from 0 K to 300 K over 100 ps, and then equilibrated for 500 ps at 300 K, followed by production runs of 2 ns length in total.- Run the simulation job at a high performance computing facility. Our simulations were run on the Biowulf cluster (https://hpc.nih.gov/) (Figure 8).
- Visualize the trajectory output using the VMD program (http://www.ks.uiuc.edu/Research/vmd/). Analyze the binding interactions and conformational changes of the substrates and TRIM14 within the active site of nsP2 (Figure 9).
Results
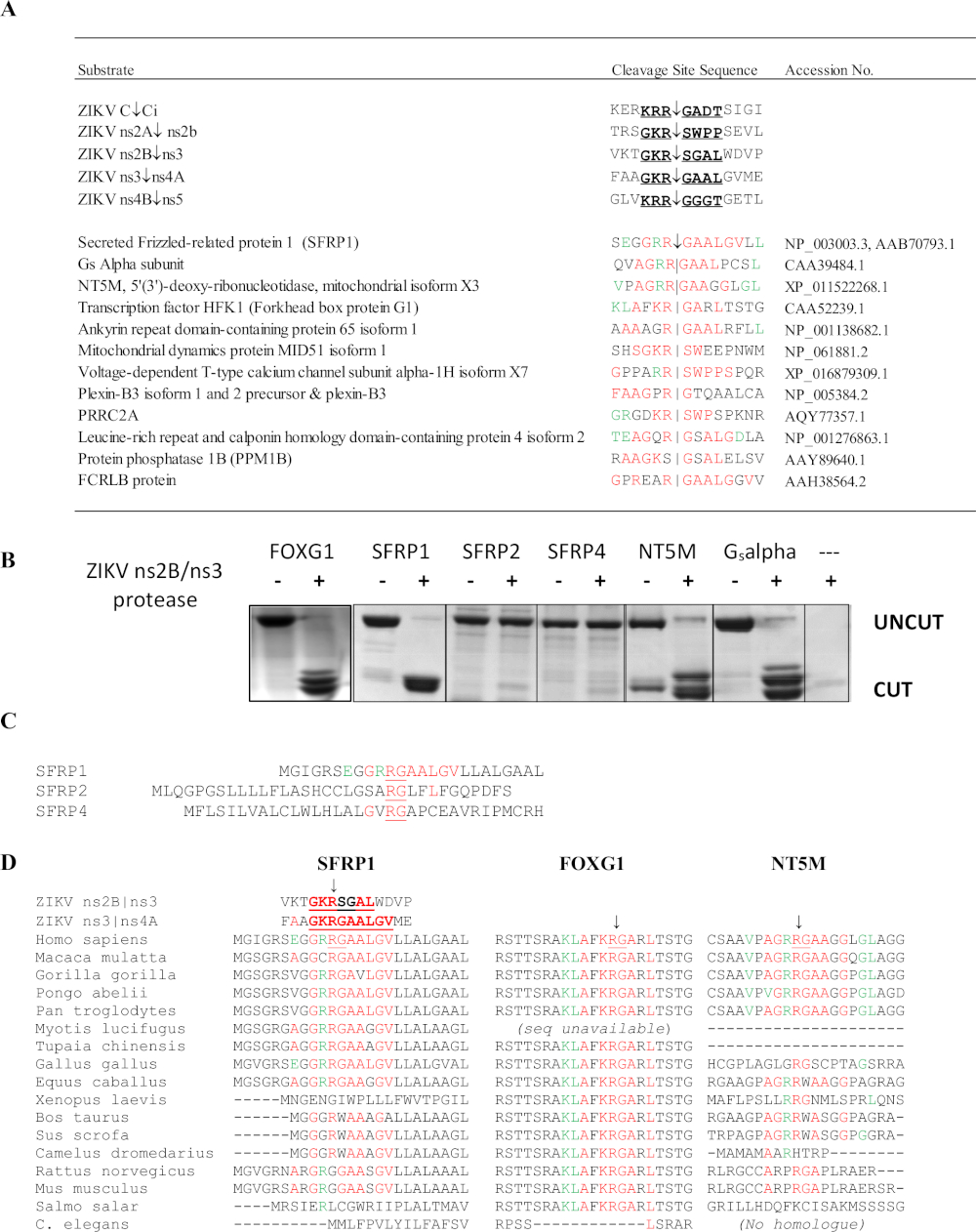
SSHHPS analysis of the ZIKV ns2B/3 protease identified 4 host protein targets: FOXG1, SFRP1, a Gs alpha subunit from a retinal cDNA library, and the NT5M mitochondrial 5',3'-nucleotidase (Figure 10)6. Notably, no other method predicted these proteins as potential targets of the ZIKV protease. Mutations in the FOXG1 gene have been linked to a congenital syndrome characterized by impaired development and structural brain abnormalities such as microcephaly. SFRP1 is a secreted frizzled-related protein (SFRP); these soluble receptors can competitively bind Wnt ligands to antagonize and inhibit Wnt signaling. The Wnt signaling pathway is involved in the regulation of the IFN response during Flavivirus infection36. The cleavage of SFRP1 would be expected to enhance flavivirus replication. SFRP1 is also involved in Th17-cell differentiation37. Sequence alignments of the SSHHPS showed species-specific differences in the cleavage site sequences (Figure 10D). The cleavage site sequence in SFRP1 was identical in humans and chickens; ZIKV can induce mortality and microcephaly in chicken embryos38. In rodents, the highly conserved P1 residue (K/R)R↓G is substituted by a glycine (RGG). Immunocompetent strains of mice are generally resistant to ZIKV infection and disease39.
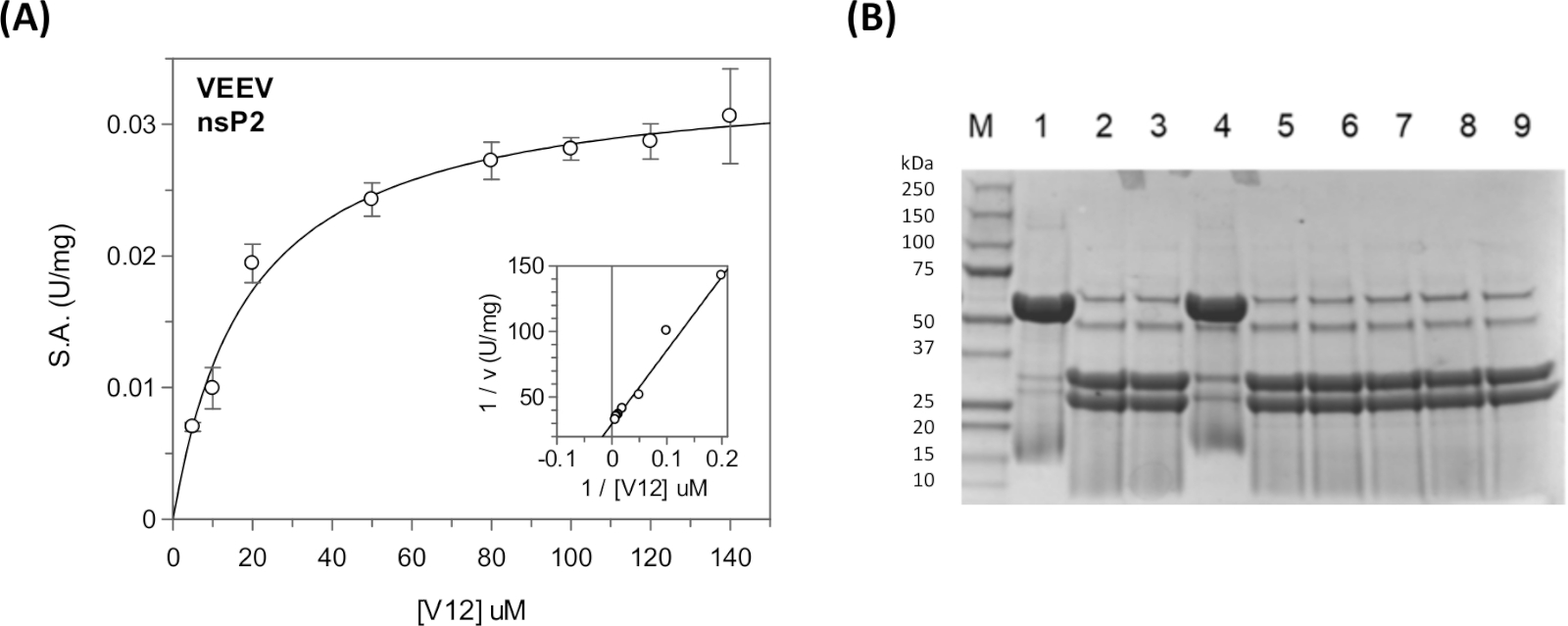
Steady state kinetic parameters and inhibition constants can be measured for the viral polyprotein sequences and for the host protein sequences using the continuous assay in a plate reader31,40,41 (Figure 11A). For qualitative cleavage information, such as cleavage of a particular sequence or the inhibition of the protease by various compounds, the discontinuous assay can be used (Figure 11B).
Optimization of the number of residues in between CFP and YFP may be required. A substrate-bound model can be made using the in silico methods. A representative docked model of the nsP1/nsP2 junction is shown in Figure 9. For the VEEV nsP2 protease, cleavage of a 12-amino acid Semliki Forest Virus (SFV) sequence had been reported (Km = 0.58 mM)33. Lengthening the substrate sequence to 19, 22, and 25 residues and reducing the ionic strength of the buffer led to a significant reduction in Km. Examination of the VEEV nsP2 crystal structure and crystal packing also showed that a portion of one of the junctions was packed against the protease domain and was helical. Thus, the longer VEEV substrates may bind better due to the recognition of a secondary structural motif.
For TRIM14, we obtained a Km = 21 µM6,33. The Km for the substrate carrying the host protein sequence was comparable to the Km values of the substrates containing the viral polyprotein cleavage site sequences (Km(V12) = 12 µM and Km(V34) = 21 µM). The cleavage site sequences at the nsP1/nsP2, nsP2/nsP3, and nsP3/nsP4 junctions were cut with different efficiencies. In the cell, this is thought to allow for sequential cleavage of the polyprotein42.
Caution should be taken in interpreting negative results. If no cleavage occurs, the cleavage site may be too short or the purified protease may be inactive. For substrates that are cut, additional experiments are needed to confirm cleavage of the full length protein or cleavage in virus-infected cells. Appropriate follow-on experiments should be chosen. The effects of overexpression or silencing of the target protein on viral replication also can be tested.

Figure 1: Three mechanisms of silencing. Silencing can occur at the level of DNA, RNA, or protein. These "search and delete" algorithms each use a "keyword" to direct the cleavage of a file containing the word. This figure has been modified from Morazzani et al.32 and the references therein. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Species-specific differences in cleavage site sequences. The C-terminal PRY/SPRY domains of TRIM14 homologues are shown in the alignment. The PRY/SPRY domain can be identified by the conserved motifs highlighted in gray. Human TRIM14 is cut at QEAGA↓G by the VEEV nsP2 cysteine protease. The SSHHP sequence is shown in color. The residue in green is the P1' residue; in blue is the P4 residue, and in red are other conserved residues within the cleavage site motif sequence. Equine harbor a truncated version of TRIM14 lacking the PRY/SPRY domain. The lysine highlighted in cyan is poly-ubiquitinated and is important for the assembly of the MAVS signalosome. The C-terminal PRY/SPRY domain may be transiently cut by the nsP2 protease to impair the host's antiviral response intracellularly during an acute viral infection. In equine, this domain is always absent. This suggests that the PRY/SPRY domain of TRIM14 may have a protective function against VEEV infections. This figure has been reproduced from Morazanni et al.6 Please click here to view a larger version of this figure.
{kind=link}

Figure 3: SSHHPS identification using BLAST. The cleavage site motif sequence at the VEEV nsP1/nsP2 junction is aligned with the SSHHP sequence in the host protein TRIM14. The residue colored in green is the P1' residue; in blue is the P4 residue and in red are other conserved residues of the cleavage site motif sequence. Most alignments contained homology to regions outside of the conserved cleavage site motif or did not include the P1/P1' scissile bond residues. TRIM14 showed a match to 6 residues in sequential order that included P1 and P1'. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Protein and DNA sequences of the CFP-V12-YFP substrate for the VEEV nsP2 cysteine protease. The NdeI (CATATG) and XhoI (CTCGAG) restriction sites are shown in capital letters. In red is the cleavage site sequence from the viral polyprotein that is in between nsP1 and nsP2. The residue in green is the P1' residue and in blue is the P4 residue of the cleavage site. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Protein sequence of the Trx-VEEV-nsP2 cysteine protease construct. Thioredoxin (Trx) is shown in yellow. The thrombin cleavage site and His-tag are shown in cyan. The Cys-His dyad are labeled in red. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Peptide structures in MOE. Please click here to view a larger version of this figure.
{kind=link}

Figure 7: Docking of substrate peptide using PyRx/AutoDock. Please click here to view a larger version of this figure.
{kind=link}

Figure 8: Jobs running on the Biowulf cluster. Please click here to view a larger version of this figure.
{kind=link}

Figure 9: Model of the VEEV P12 substrate containing the cleavage site sequence at the nsP1/nsP2 junction. The Cys-477/His-546 catalytic dyad is shown in blue. Figure was made using Pymol (https://pymol.org). Please click here to view a larger version of this figure.
{kind=link}

Figure 10: SSHHPS Analysis of the Zika virus ns2B/ns3 protease. (A) Predicted host protein targets of the ZIKV ns2B/ns3 protease. Residues in red match a single cleavage site sequence. Residues in green are tolerated at the subsite and match residues at other cleavage sites. SFRP1 had the highest number of identical residues in consecutive order. (B) CFP-substrate-YFP proteins (~50-60 kDa) were expressed and purified containing the predicted SSHHP sequence from each host protein (human). The ZIKV protease cut human FOXG1, SFRP1, NT5M and a Gsalpha subunit isolated from a retinal cDNA library. The cleavage products are approximately 28-30 kDa. The substrate sequences are available in Morazzani et al.6 (C) While the ns2B/ns3 protease cut SFRP1, it did not cut its homologues (SFRP2 and SFRP4). (D) Alignment of the cleavage sites from different animal species may be useful in selecting an animal model for a Group IV virus. Note that the conserved R↓G sequence differs between humans and rodents in SFRP1. Figure reproduced from Morazzani et al.6 Please click here to view a larger version of this figure.
{kind=link}

Figure 11: Steady state kinetic analysis using the continuous and discontinuous assays. (A) The kinetic data shown in Table 5 was plotted in GraFit. The inset shows the Lineweaver-Burk plot. (B) SDS-PAGE gel showing the cleavage products of the CFP-V12-YFP substrate. In lane 1 is the "UNCUT" substrate (48 kDa). In lane 2 is the "CUT" substrate (31 kDa and 27 kDa). In lanes 3-9 different compounds were included to test their inhibitory activity. Lane 4 contains the E64d covalent inhibitor. These reactions were run overnight for ~17 h at room temperature. Boiling of the samples was required to achieve the sharp banding pattern. The nsP2 protease is visible (56 kDa) in the reactions containing enzyme, but not in lane 1. Lane 1 is the "no enzyme" control. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Sequence-specific destruction of a protein or a nucleic acid guided by a foreign sequence is only seen in a few cases in biology. The mechanisms shown in Figure 1 are defensive mechanisms that protect a host from a virus, or a virus from a host.
Using bioinformatic methods we can identify the targets that are destroyed by these systems. In our analyses of SSHHP sequences, we discovered that many of them could be found in proteins needed to generate innate immune responses. Some had obvious roles such as MAVS and TRIF (TIR-domain-containing adapter-inducing interferon-β), while others were related to immunity though more complex mechanisms (e.g., Histone H3, SFRP1, FOXG1)8,9. The target information stored in the SSHHP sequence has the potential to identify pathways that have antiviral effects against these viruses. Antiviral responses in vivo are often virus-specific26,43. For example, subsets of TRIM proteins have antiviral effects on different viruses43,44,45, some are viral restriction factors (e.g., HIV and TRIM5α). The specificity of TRIM proteins (~70 have been identified) still is being examined44,45. The information within SSHHPS may contribute to our understanding of how these viruses evade the innate immune responses. Other patterns and correlations may be uncovered as more SSHHPS are examined.
Species-specific differences were apparent in our analyses (Figure 2, Figure 10). These viruses are known to affect some species more than others. Information about host range, host susceptibility, and host defenses may be present within SSHHPS. For example, equine, the most susceptible species to equine encephalitis viruses, lacked the region of human TRIM14 that was transiently cut by the VEEV nsP2 protease. Humans rarely die from VEEV infections but can be infected24. The human TRIM14 protein carried an nsP2 protease cleavage sequence6. The presence of the cleavage site suggest that humans have a defense mechanism against these viruses. Birds have been thought to be potential reservoirs of these viruses46. The corresponding SSHHP sequence in the TRIM14 protein from chickens differed from the sequences found in humans and other species. Subtle differences like these may make a target host protein uncleavable or more readily cleaved. Aguirre et al.16 showed that an uncleavable mutated STRING protein induced higher levels of IFN after Dengue virus infection and that mice naturally carry a version of STING that is not cut by the Dengue ns2B3 protease. The murine STING protein was not cut by the ZIKV protease47. In our SSHHPS analysis, we also observed differences in the ZIKV protease cleavage site sequences when we compared the human proteins with those of rodents6 (Figure 10D). Reproducing the species-specific proteolytic cleavages of host proteins may be important in animal models used for Group IV viruses. The inhibition of host protein cleavage also has implications with regards to the development of Group IV protease inhibitors. In our previous publication, we showed that we could inhibit TRIM14 cleavage by the VEEV nsP2 protease using CA074 methyl ester6. This result suggests that small molecule inhibitors of these proteases may be able to modulate the innate immune responses that are capable of suppressing the infection6,31.
Genetic variation within a species also has the potential to produce differences in proteolytic cleavage. Subtle differences in codon usage could affect ribosome pausing48. Since some Group IV viral proteases are embedded in the ER membrane, differences in these pauses could affect cleavage of a target if cleavage occurs co-translationally. Some of the cleavage sites that we identified were in predicted signal peptide sequences (e.g., SFRP1) while others were internal.
SSHHPS analysis can produce information that differs from other methods of host protein analyses. SSHHPS analysis was inexpensive and easy to employ. The use of a bacterial expression system allowed testing of short segments (~25 amino acids) of mammalian sequences without the use of mammalian cell culture. We found that the CFP-YFP substrates were able to tolerate all of the tested human protein sequences; however, yields varied. In similar assays, substrates containing human protein sequences as long as 63 amino acids were successfully expressed, purified, and utilized for kinetic analyses and inhibitor screening49,50,51. Since only small amounts of the substrate are needed for the discontinuous assay, a large number of targets can be explored. One advantage of the system is that the CFP/YFP substrates can be used for SDS-PAGE analyses and for more elaborate kinetic analyses (i.e., IC50, Ki, Km, Vmax). For drug discovery, inhibitory compounds can produce artifacts in fluorescent assays. Thus, the discontinuous assay in combination with a continuous assay allows one to confirm cleavage or inhibition of cleavage. The samples for the discontinuous SDS-PAGE assay can be taken directly out of the 96-well plates. CFP/YFP substrates have been used for compound library screening52. However, additional analyses are required to determine if a substrate is suitable for high throughput screening such as the calculation of a Z-factor53.
One challenge in designing a substrate is identifying the region around the scissile bond that is bound and recognized by the protease. In the examples shown here, we began with 12 residue sequences that were centered around the scissile bond. After analyzing sequence alignments of the cleavage sites homology to the residues N-terminal of the scissile bond was found for the VEEV protease, whereas for the ZIKV protease homology to several of the C-terminal residues was found. An in silico model of the docked substrate can be used to design site-directed mutagenesis experiments that probe the binding sites of the substrate. Since the substrate and enzyme sequences are on plasmids, either can be mutated to test the in silico models or subsite tolerances. This can be advantageous if a crystal structure of the bound substrate(s) is not available.
SSHHPS analysis may also yield new information about the mechanisms by which virus-induced phenotypes are produced by viral enzymes. One of the ZIKV targets, SFRP1, is part of the Wnt signaling pathway and has roles in both brain and eye development and in immune responses36,37,54,55,56,57. We found that the other protein sequences that could be cut by the ZIKV ns2B/ns3 protease were also in proteins involved in brain and eye development; abnormalities in both have been observed in congenital Zika syndrome and are thought to be part of the virus-induced phenotype58.
The predictability of host-pathogen interactions could be exploited for a variety of applications: target-specific oncolytic viral therapies; de-risking live virus vaccines; refinement, prediction or selection of animal models; prediction of host-range or susceptibility; prediction of zoonotic events; and prediction of host-defenses. Since the methods described are sequence-based, they may be of value to incorporate into software in the future.
Disclosures
The opinions expressed here are those of the authors and do not represent those of the U. S. Navy, U. S. Army, U. S. Department of Defense, or the U. S. government.
Acknowledgements
This work was supported by Defense Threat Reduction Agency (DTRA) project numbers CB-SEED-SEED09-2-0061 and CBCall4-CBM-05-2-0019, and in part by the Intramural/Extramural research program of the NCATS, NIH (XH) and Naval Research Laboratory base funds.
Materials
| Name | Company | Catalog Number | Comments |
| 250 mL Erlenmeyer Flask | VWR | 89000-362 | |
| 2-mercaptoethanol | Acros Organics (Fisher) | 125472500 | Danger: Acutely Toxic. Open bottle in hood to avoid inhaling the fumes. |
| 4 L Pyrex wide-mouth graduated Erlenmeyer flask with screw-cap | Millipore Sigma | CLS49954L-1EA | |
| AKTA Prime Plus | GE Healthcare | 17-0729-01 | |
| AKTA XK 16/20 Column | GE Healthcare | 28988937 | |
| Amicon Ultra-0.5 Centrifugal Filter Unit | Millipore Sigma | UFC501096 | |
| Amicon Ultra-15 Centrifugal Filter Unit | Millipore Sigma | UFC901096 | |
| Amicon Ultra-4 Centrifugal Filter Unit | Millipore Sigma | UFC801024 | |
| Ampicillin | Sigma | A0166 | Danger: Allergic reactions (skin or breathing). |
| Chelating Sepharose Fast Flow | GE Healthcare | 17-0575-02 | Once the resin is equilibrated with 0.2 M Nickel Sulfate it is refered to as a Nickel Column in the text. Column will have a green color after washing with water. The column will have a blue color after equilibrating with buffer. |
| Chloramphenicol | RPI | C61000 | Danger: May cause cancer. |
| Corning 50 mL centrifuge tubes | Corning | 430828 | Suggestion: Polypropylene tubes are less likely to crack during sonication than Polyethylene tubes |
| Corning 96 Well Half-Area Microplate, Non-Binding Surface | Corning | 3993 | |
| Dialysis Tubing Clips | Fisher Scientific | PI68011 | |
| Disposable PD-10 Desalting Column | GE Healthcare | 17-0851-01 | |
| DNAse | Sigma | DN25-1G | |
| DTT (DL-Dithiothreitol) | RPI | D11000-50.0 | Warning: Acute Oral Toxicity; skin and eye irritation |
| EDTA | Fisher Scientific | S311-500 | |
| Fisherbrand Petri Dishes with Clear Lid | Fisher Scientific | FB0875712 | |
| Glycerol | Acros Organics (Fisher) | 15892-0010 | |
| HEPES | Millipore Sigma | H4034-1KG | |
| Imidazole | Acros Organics | 301870010 | Danger: Toxic, Irritant |
| IPTG (Isopropyl β-D-thiogalactopyranoside) | Calbiochem (Millipore Sigma) | 420291 | Do not breathe dust. Avoid contact with eyes and skin. |
| Laemmli Sample Buffer | BIO-RAD | 1610737 | |
| Luria Bertani Agar | Fluka (Millipore Sigma) | L3027-1KG | Suggestion: Autoclave with magnetic stirrer in the liquid, and stir while cooling. Wait to add antibiotic until you can hold your hands on the bottle without pain for 30 seconds. |
| Luria Bertani Media | Fisher Bioreagents | BP1426-2 | |
| Lysozyme | Sigma | L4919-5g | |
| Mini-PROTEAN Tetra Vertical Electrophoresis CellGel Box | BIO-RAD | ||
| Nalgene Oak Ridge High-Speed PPCO Centrifuge Tubes | Nalgene (Thermo Scientific) | 3119-0050 | |
| Nanodrop | Thermo Fisher | ||
| New Brunswick Innova 42R Shaker Incubator | Eppendorf | M1335 | |
| Nickel Sulfate Hexahydrate (Crystalline/Certified ACS), Fisher Chemical | Fisher Scientific | N73-500 | Danger: Harmful if swallowed or inhaled, skin and eye irritation |
| One Shot BL21(DE3) Chemically Competent E. coli | Invitrogen (Thermo Fisher) | C600003 | May be harmful if inhaled or swallowed. May cause skin and eye irritation with susceptible people. |
| One Shot BL21(DE3) pLysS Chemically Competent E. coli | Invitrogen (Thermo Fisher) | C606003 | May be harmful if inhaled or swallowed. May cause skin and eye irritation with susceptible people. |
| pet15b plasmid DNA | Novagen (Millipore Sigma) | 69661 | GenScript Inc. was used for commerical DNA synthesis. The pet15b plasmid was used for the CFP/YFP substrates. |
| pet32b | Novagen (Millipore Sigma) | 69016-3 | The pet32b plasmid was used for the cysteine protease construct. |
| Pierce Protease Inhibitor Mini Tablets, EDTA-free | Thermo Fisher | A32955 | Warning: Skin corrosion/irriation; eye damage |
| Plate Reader | Molecular Devices | Model M5 | |
| Precision Plus Protein All Blue Prestained Protein Standard | BIO-RAD | 161-0373 | |
| Protein Extraction Reagent | Novagen (Millipore Sigma) | 70584-4 | BugBuster or Bper (Catalog # 78248, ThermoFisher) |
| Q-Sepharose Fast Flow | G.E. Healthcare | 17-0510-01 | Anion exchange resin |
| RunBlue (12%) 17-well PAGE gels | Expedeon | BCG01227 | Any 12% pre-cast polyacrylamide gel can be used |
| RunBlue 20x SDS Running Buffer | Expedeon | NXB50500 | Dilute 50 mL with 950 mL deionized water to obtain 1x |
| RunBlue Instant Blue Gel Stain | Expedeon | ISB1L | Do not dilute, use as directed |
| Sodium Chloride | Fisher Chemical | S271-10 | |
| Sonifier Cell Disrupter 450 Sonicator | Branson Ultrasonics (VWR) | Model No. 101-063-346R | Sonicator was used on level 5 |
| Spectra/Por 6-8 kD MWCO | Spectrum Labs | 132645T | Dialysis Tubing |
| SP-Sepharose Fast Flow | G.E. Healthcare | 17-0729-01 | Cation exchange resin |
| Thrombin from bovine plasma | Sigma | T6634-500UN | |
| Tris Base | Fisher Scientific | BP152-500 | Caution: Eye/Skin Irritant |
References
- Liu, H., et al. Widespread Horizontal Gene Transfer from Double-Stranded RNA Viruses to Eukaryotic Nuclear Genomes. Journal of Virology. 84 (22), 11876-11887 (2010).
- Hagai, T., Azia, A., Babu, M. M., Andino, R. Use of host-like peptide motifs in viral proteins is a prevalent strategy in host-virus interactions. Cell Reports. 7 (5), 1729-1739 (2014).
- Gorbalenya, A. E. Host-related sequences in RNA viral genomes. Seminars in Virology. 3, 359-371 (1992).
- Shmakov, S. A., et al. The CRISPR Spacer Space Is Dominated by Sequences from Species-Specific Mobilomes. MBio. 8 (5), 1-18 (2017).
- Legler, P. M., Morazzani, E., Glass, P. J., Compton, J. R. Proteome Editing System and A Biomarker of Veev Infection. United States patent application. , (2018).
- Morazzani, E. M., et al. Proteolytic cleavage of host proteins by the Group IV viral proteases of Venezuelan equine encephalitis virus and Zika virus. Antiviral Research. 164, 106-122 (2019).
- Alvarez, E., Castello, A., Menendez-Arias, L., Carrasco, L. HIV protease cleaves poly(A)-binding protein. Biochemical Journal. 396 (2), 219-226 (2006).
- Falk, M. M., et al. Foot-and-mouth disease virus protease 3C induces specific proteolytic cleavage of host cell histone H3. Journal of Virology. 64 (2), 748-756 (1990).
- Grigera, P. R., Tisminetzky, S. G. Histone H3 modification in BHK cells infected with foot-and-mouth disease virus. Virology. 136 (1), 10-19 (1984).
- Li, W., Ross-Smith, N., Proud, C. G., Belsham, G. J. Cleavage of translation initiation factor 4AI (eIF4AI) but not eIF4AII by foot-and-mouth disease virus 3C protease: identification of the eIF4AI cleavage site. FEBS Letters. 507 (1), 1-5 (2001).
- Kuyumcu-Martinez, M., et al. Calicivirus 3C-like proteinase inhibits cellular translation by cleavage of poly(A)-binding protein. Journal of Virology. 78 (15), 8172-8182 (2004).
- Pietila, M. K., Hellstrom, K., Ahola, T. Alphavirus polymerase and RNA replication. Virus Research. 234, 44-57 (2017).
- Hardy, W. R., Strauss, J. H. Processing the nonstructural polyproteins of sindbis virus: nonstructural proteinase is in the C-terminal half of nsP2 and functions both in cis and in trans. Journal of Virology. 63 (11), 4653-4664 (1989).
- Strauss, E. G., De Groot, R. J., Levinson, R., Strauss, J. H. Identification of the active site residues in the nsP2 proteinase of Sindbis virus. Virology. 191 (2), 932-940 (1992).
- Wang, D., et al. Foot-and-mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. Journal of Virology. 86 (17), 9311-9322 (2012).
- Aguirre, S., et al. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathogens. 8 (10), e1002934(2012).
- Barral, P. M., Sarkar, D., Fisher, P. B., Racaniello, V. R. RIG-I is cleaved during picornavirus infection. Virology. 391 (2), 171-176 (2009).
- Elbashir, S. M., Lendeckel, W., Tuschl, T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes & Development. 15 (2), 188-200 (2001).
- Deveau, H., Garneau, J. E., Moineau, S. CRISPR/Cas system and its role in phage-bacteria interactions. Annual Review of Microbiology. 64, 475-493 (2010).
- Schechter, I., Berger, A. On the size of the active site in proteases I. Papain. Biochemical and Biophysical Research Communications. 27 (2), 157-162 (1967).
- Bieniasz, P. D. Intrinsic immunity: a front-line defense against viral attack. Nature Immunology. 5 (11), 1109-1115 (2004).
- Zhou, Z., et al. TRIM14 is a mitochondrial adaptor that facilitates retinoic acid-inducible gene-I-like receptor-mediated innate immune response. Proceedings of the National Academy of Sciences of the U S A. 111 (2), E245-E254 (2014).
- Wang, S., et al. TRIM14 inhibits hepatitis C virus infection by SPRY domain-dependent targeted degradation of the viral NS5A protein. Scientific Reports. 6, 32336(2016).
- Zacks, M. A., Paessler, S. Encephalitic alphaviruses. Veterinary Microbiology. 140 (3-4), 281-286 (2010).
- Hollidge, B. S., Weiss, S. R., Soldan, S. S. The role of interferon antagonist, non-structural proteins in the pathogenesis and emergence of arboviruses. Viruses. 3 (6), 629-658 (2011).
- Carthagena, L., et al. Human TRIM gene expression in response to interferons. PLoS One. 4 (3), e4894(2009).
- Montgomery, S. A., Johnston, R. E. Nuclear import and export of Venezuelan equine encephalitis virus nonstructural protein 2. Journal of Virology. 81 (19), 10268-10279 (2007).
- Nenasheva, V. V., et al. Enhanced expression of trim14 gene suppressed Sindbis virus reproduction and modulated the transcription of a large number of genes of innate immunity. Immunologic Research. 62 (3), 255-262 (2015).
- Nagai, T., et al. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nature Biotechnology. 20 (1), 87-90 (2002).
- Li, M. Z., Elledge, S. J. SLIC: a method for sequence- and ligation-independent cloning. Methods in Molecular Biology. 852, 51-59 (2012).
- Hu, X., et al. Kinetic, Mutational, and Structural Studies of the Venezuelan Equine Encephalitis Virus Nonstructural Protein 2 Cysteine Protease. Biochemistry. 55 (21), 3007-3019 (2016).
- Morazzani, E. M., et al. Proteolytic cleavage of host proteins by the Group IV viral proteases of Venezuelan equine encephalitis virus and Zika virus. Antiviral Research. 164, 106-122 (2019).
- Zhang, D., Tozser, J., Waugh, D. S. Molecular cloning, overproduction, purification and biochemical characterization of the p39 nsp2 protease domains encoded by three alphaviruses. Protein Expression and Purification. 64 (1), 89-97 (2009).
- Lei, J., et al. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science. 353 (6298), 503-505 (2016).
- Shiryaev, S. A., et al. Characterization of the Zika virus two-component NS2B-NS3 protease and structure-assisted identification of allosteric small-molecule antagonists. Antiviral Research. 143, 218-229 (2017).
- Smith, J. L., Jeng, S., McWeeney, S. K., Hirsch, A. J. A MicroRNA Screen Identifies the Wnt Signaling Pathway as a Regulator of the Interferon Response during Flavivirus Infection. Journal of Virology. 91 (8), (2017).
- Lee, Y. S., et al. The Wnt inhibitor secreted Frizzled-Related Protein 1 (sFRP1) promotes human Th17 differentiation. European Journal of Immunology. 42 (10), 2564-2573 (2012).
- Goodfellow, F. T., et al. Zika Virus Induced Mortality and Microcephaly in Chicken Embryos. Stem Cells and Development. 25 (22), 1691-1697 (2016).
- Morrison, T. E., Diamond, M. S. Animal Models of Zika Virus Infection, Pathogenesis, and Immunity. Journal of Virology. 91 (8), (2017).
- Morazzani, E. M., et al. Books of Abstracts, 254th American Chemical Society National Meeting. , Washington, D.C. (2017).
- Compton, J. R., Mickey, M. J., Hu, X., Marugan, J. J., Legler, P. M. Mutation of Asn-475 in the Venezuelan Equine Encephalitis Virus nsP2 Cysteine Protease Leads to a Self-Inhibited State. Biochemistry. 56 (47), 6221-6230 (2017).
- Vasiljeva, L., et al. Regulation of the sequential processing of Semliki Forest virus replicase polyprotein. Journal of Biological Chemistry. 278 (43), 41636-41645 (2003).
- Uchil, P. D., Quinlan, B. D., Chan, W. T., Luna, J. M., Mothes, W. TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathogens. 4 (2), e16(2008).
- Ozato, K., Shin, D. M., Chang, T. H., Morse, H. C. 3rd TRIM family proteins and their emerging roles in innate immunity. Nature Reviews Immunology. 8 (11), 849-860 (2008).
- van Tol, S., Hage, A., Giraldo, M. I., Bharaj, P., Rajsbaum, R. The TRIMendous Role of TRIMs in Virus-Host Interactions. Vaccines (Basel). 5 (3), (2017).
- Molaei, G., et al. Dynamics of Vector-Host Interactions in Avian Communities in Four Eastern Equine Encephalitis Virus Foci in the Northeastern U.S. PLoS Neglected Tropical Diseases. 10 (1), e0004347(2016).
- Ding, Q., et al. Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proceedings of the National Academy of Sciences of the U S A. 115 (27), E6310-E6318 (2018).
- Angov, E., Legler, P. M., Mease, R. M. Adjustment of codon usage frequencies by codon harmonization improves protein expression and folding. Methods in Molecular Biology. 705, 1-13 (2011).
- Ruge, D. R., et al. Detection of six serotypes of botulinum neurotoxin using fluorogenic reporters. Analytical Biochemistry. 411 (2), 200-209 (2011).
- Hu, X., et al. Structural insight into exosite binding and discovery of novel exosite inhibitors of botulinum neurotoxin serotype A through in silico screening. Journal of Computer-Aided Molecular Design. 28 (7), 765-778 (2014).
- Dunning, F. M., et al. Detection of botulinum neurotoxin serotype A, B, and F proteolytic activity in complex matrices with picomolar to femtomolar sensitivity. Applied and Environmental Microbiology. 78 (21), 7687-7697 (2012).
- Nguyen, T. G., et al. Development of fluorescent substrates and assays for the key autophagy-related cysteine protease enzyme ATG4B. Assay and Drug Development Technologies. 12 (3), 176-189 (2014).
- Zhang, J. H., Chung, T. D., Oldenburg, K. R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. Journal of Biomolecular Screening. 4 (2), 67-73 (1999).
- Bovolenta, P., Esteve, P., Ruiz, J. M., Cisneros, E., Lopez-Rios, J. Beyond Wnt inhibition: new functions of secreted Frizzled-related proteins in development and disease. Journal of Cell Science. 121 (Pt 6), 737-746 (2008).
- Esteve, P., et al. SFRPs act as negative modulators of ADAM10 to regulate retinal neurogenesis. Nature Neuroscience. 14 (5), 562-569 (2011).
- Garcia-Hoyos, M., et al. Evaluation of SFRP1 as a candidate for human retinal dystrophies. Molecular Vision. 10, 426-431 (2004).
- Marcos, S., et al. Secreted frizzled related proteins modulate pathfinding and fasciculation of mouse retina ganglion cell axons by direct and indirect mechanisms. Journal of Neuroscience. 35 (11), 4729-4740 (2015).
- Moore, C. A., et al. Characterizing the Pattern of Anomalies in Congenital Zika Syndrome for Pediatric Clinicians. JAMA Pediatrics. 171 (3), 288-295 (2017).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved