
Method Article
Dissection and Isolation of Murine Glia from Multiple Central Nervous System Regions
In This Article
Summary
Here we present a protocol for in vitro isolation of multiple glial cell populations from a mouse CNS. This method allows for the segregation of regional microglia, oligodendrocyte precursor cells, and astrocytes to study the phenotypes of each in a variety of culture systems.
Abstract
The methods presented here demonstrate laboratory procedures for the dissection of four different regions of the central nervous system (CNS) from murine neonates for the isolation of glial subpopulations. The purpose of the procedure is to dissociate microglia, oligodendrocyte progenitor cells (OPCs), and astrocytes from cortical, cerebellar, brainstem, and spinal cord tissue to facilitate further in vitro analysis. The CNS region isolation procedures allow for the determination of regional heterogeneity among glia in multiple cell culture systems. Rapid CNS region isolation is performed, followed by the mechanical removal of meninges to prevent meningeal cell contamination of glia. This protocol combines gentle tissue dissociation and plating on a specified matrix designed to preserve cell integrity and adherence. Isolating mixed glia from multiple CNS regions provides a comprehensive analysis of potentially heterogenous glia while maximizing the use of individual experimental animals. Additionally, following dissociation of regional tissue, mixed glia are further divided into multiple cell types including microglia, OPCs, and astrocytes for use in either single cell type, cell culture plate inserts, or co-culture systems. Overall, the demonstrated techniques provide a comprehensive protocol of broad applicability for careful dissection of four individual CNS regions from murine neonates and includes methods for the isolation of three individual glia cell types to examine regional heterogeneity in any number of in vitro cell culture systems or assays.
Introduction
Glia are necessary for proper neuronal function in the CNS. They are composed of three major subpopulations, astrocytes, oligodendrocytes, and microglia, each with a different, yet indispensable role1. Without the proper glial cell diversity and activity, neuronal function would be severely impacted, leading to CNS impairment. Glia are capable of influencing neurotransmission, and each cell type does so in a unique manner. Glial cells in the brain have the capacity to communicate amongst themselves, as well as with neuronal cells, in order to facilitate proper CNS function2. Oligodendrocytes increase the speed of electrical transmission through the formation of a myelin sheath, which facilitates the clustering of ion channels at the nodes of Ranvier, the sites of neuronal action potential generation3. Microglia are critical for the pruning of synapses by monitoring synaptic transmission and “rewiring” neuronal connections following injury4. In addition, microglia are the most abundant resident immune cell of the CNS, acting as the primary form of host defense against pathogens5. Astrocytes can regulate synaptic transmission between neurons by modifying the concentration of extracellular potassium6. They also have roles in controlling local blood flow7, releasing and taking up neuromodulatory elements8, and have a key role in blood-brain barrier maintenance9. Thus, each glial subtype is critical for CNS function, as defects in any type have long been associated with a wide variety of pathological states, including psychiatric diseases, epilepsy, and neurodegenerative conditions10.
The greatest obstacle in the study of CNS pathobiology is the inability to investigate human cells in the context of their microenvironmental niche. Human biopsy tissue is most collected post-mortem and cells can easily be damaged or lost during the extraction and processing. Furthermore, it is a challenge to keep human cells alive and viable in vitro for any length of time without deriving immortalized cell lines from tumors, at which point they no longer accurately reflect their normal physiological properties11,12. Additionally, there is a significant amount of regional heterogeneity among individual glia cell types13,14,15, and obtaining regional CNS samples from individual patients is nearly impossible. As such, it is necessary to develop alternative models to study the contribution of regional glia in specific CNS disorders.
Here, we describe an in vitro system using mouse CNS region-specific isolation of multiple glial subpopulations, allowing for the manipulation and quantification of microglia, oligodendrocyte precursor cells (OPCs), which give rise to mature oligodendrocytes, and astrocytes. Each population can be independently isolated and subjected to a wide variety of experimental techniques including drug or molecule treatment, immunocytochemistry, protein/RNA extraction and analysis, and other co-culture systems depending on experimental necessity. Additionally, this isolation technique yields high cell number, allowing for the characterization and investigation of each glial population in a high-throughput manner. It also enables the study of CNS cell differentiation, growth, and proliferation in response to a wide variety of microenvironmental stimuli in a controlled manner in order to avoid confounding factors which are typically present in an in vivo setting. Lastly, this cell isolation technique facilitates the manipulation of glial cell populations within different CNS regions to investigate how regional glia interact with each other and respond to varying stimuli, allowing for precision and reproducibility.
Protocol
NOTE: All animal studies were authorized and approved by the Cleveland Clinic Lerner Research Institute Institutional Animal Care and Use Committee.
1. Prepare media and supplies for dissection
NOTE: All buffer and media recipes are provided in Table 1. This procedure is done under sterile conditions in a tissue culture designated biosafety cabinet.
- Prepare and sterile filter mixed glia media (MGM). This can be done the day before and stored at 4 °C.
- Dilute fibronectin at a 1:100 concentration with sterile H2O. The volume of diluted fibronectin will depend of the number of pups and CNS region used for the experiment. Use one T25 flask for 1 cortex, one T25 flask for 2-4 cerebella, one T25 flask for 2-4 brain stems and one T25 flask for 2-4 spinal cords.
NOTE: Combining tissue from the same region using the criteria listed above will allow adequate dissociation without altering the protocol described below. Combining more tissue than described in Step 1.2 will require further optimization of dissociation reagent concentrations. - Pipette 3 mL of diluted fibronectin into T25 flasks at room temperature and allow to sit for 2 min.
- Aspirate the fibronectin and allow flasks to dry overnight with flask lid loosened.
2. Cortex, cerebellum, brainstem, and spinal cord dissection
NOTE: This procedure can be done on the benchtop and requires a dissection scope. Use strict aseptic technique for all steps of the procedure and minimize tissue exposure to the room air. Keep all media chilled on ice during dissection to ensure maximal tissue preservation. Alternatively, this procedure could be done in a hood that allows the use of an internal dissection scope.
- Wipe all areas with 70% ethanol.
- Pipette 10 mL of PBS/antibiotic solution (PAS) into a 10 cm Petri dish. Prepare one Petri dish for each pup to be dissected. Set on ice to keep the solution chilled.
- In a disinfected tissue culture hood, pipette 9 mL of DMEM (with no additives) into 4 separate 15 mL conical tubes, one for each CNS region, replace tube caps, and put on ice. Place ice bucket with tubes within reaching distance from the dissection scope.
- Place 10 cm Petri dish prepared in step 2.2 on disinfected dissection scope stage.
- Anesthetize and euthanize mouse pup according to institutional protocol and remove the head by rapid decapitation with sharp scissors.
NOTE: Postnatal day (P) 3-5 pups are used. Older pups have a more developed CNS and may not be an adequate source of expanding glial cells. Younger pups (P) 0-2 may yield a higher number of glia and can be used; however, spinal cord tissue is very difficult to dissect at this young age due to size. - Clean the pup’s skin using 70% ethanol.
- Using fine scissors, cut the cutaneous layer along the midline of the head of the animal, starting caudally and moving rostral, until reaching the snout. Avoid cutting deeply into the skull to avoid any tissue damage.
- Angling the head down, pull cutaneous layer to each side of the skull and using spring scissors, make an incision along the skull midline, starting at the foramen magnum, again cutting caudal to rostral.
- With fine-tipped forceps, pull the skull halves to the right and left sides, exposing the cortex, cerebellum, and brainstem.
- Once exposed, gently lift the brain out of the skull and into the 10 cm Petri dish prepared in Step 2.2. Ensure the brain remains undamaged to preserve anatomical structure, with the hindbrain attached.
- Using fine, curved-tipped forceps with the point of the forceps facing upward, pinch off the cerebellum. Remove the meninges and place in designated 15 mL conical tube in ice bucket.
- Ensure that the brainstem is directly ventral to the cerebellum and is visible after removal of the cerebellum. Remove it with fine-tipped forceps, remove meninges, and place in designated 15 mL conical tube in ice bucket.
- Separate the midbrain from the cortex, remove cortical meninges, and place in designated 15 mL conical tube in ice bucket.
- To remove the spinal cord, place the decapitated mouse pup in a supine position (lying face upward) with the severed vertebral column elevated towards the investigator.
- Spray again with 70% ethanol.
- Cut along the lateral sides of the vertebral column, in a rostral to caudal direction, through the rib cage until reaching the hind limbs. While cutting, push back the internal organs until the vertebral column is visible.
- Cut along each lateral side of the vertebral column until it is isolated and place in the 10 cm Petri dish prepared in Step 2.2.
- With ventral side up, using fine spring scissors, alternate cutting the right and left sides of each vertebrae until reaching the lumbar region to expose the spinal cord tissue.
- Gently remove the spinal cord and the meninges with fine-tipped forceps under the dissecting microscope. Place in designated 15 mL conical tube in ice bucket.
- Repeat steps 2.5.-2.19 for each pup, combining tissue from the same region to fit the criteria outlined in step 1.2 for each prepared T25 flask.
NOTE: Remove the meninges as completely as possible. If a significant amount of meninges remain, the fibroblast-like phenotype of meningeal cells will outgrow and overwhelm the cell culture. Multiple spinal cords, of equal phenotype, can be combined in order to generate a specific cell culture. Take care to ensure that overcrowding of cells does not occur, which may lead to glial apoptosis and differential phenotypes.
3. Tissue dissociation
NOTE: All the following procedures are carried out in a sterile tissue culture designated biosafety cabinet using aseptic technique and sterile materials.
- Add 1 mL of 0.05% trypsin containing 0.53 mM EDTA to each 15 mL conical tube of 9 mL DMEM and tissue to begin tissue lysis.
NOTE: DMEM contains a high amount of calcium chloride, which can act as an inhibitor of trypsin. If dissociation is not complete following the outlined steps, Hanks’ Balanced Salt Solution without calcium or magnesium can be used. After trypsinization, the enzyme can be neutralized by adding a trypsin inhibitor, although calcium should be added back to the solution as it is a cofactor for DNase I, which is used in subsequent steps. - Triturate with a 10 mL pipette approximately 20x.
- Transfer the cell suspensions to empty 50 mL conical tubes.
- Incubate the solution at 37 °C, 5% CO2 for 15 min, gently agitating the lysates after 8 min.
- Add 5 mL of MGM and 200 µL (5 mg/mL) DNase I to each tube for a final concentration of 50 µg/mL.
- Triturate each lysate with a 10 mL pipette 10x.
- Let the cell suspensions sit for 3 min at room temperature to allow non-dissociated tissue to settle at the bottom of the tubes.
- Transfer the cell suspensions to new 50 mL conical tubes, leaving behind the non-dissociated tissue.
NOTE: The lysis and trituration steps described above significantly limits the amount of non-dissociated tissue. - Centrifuge the tubes at 300 x g for 3 min at 4 °C without brake.
- Aspirate the supernatant and resuspend the remaining cell pellets in 5 mL of MGM.
- Triturate the pellet with a 5 mL pipette 20x.
- Plate the 5 mL cell suspensions on coated T25 flasks.
- Incubate the cells at 37 °C, 5% CO2 and change media initially after 24 h to remove cell debris.
NOTE: Some protocols recommend an initial media change after 72 h. Optimization of this step may be required. - Perform a 100% media change with MGM every 48-72 h until cells are 80% confluent (approximately 5-7 days).
NOTE: All media must be warmed to 37 °C before media changes.
4. Microglia isolation
- Once mixed glia cultures have reached 80% confluency, prepare flasks for shaking by tightening lids and sealing with paraffin film.
- To remove microglia from mixed glial cultures, secure flasks horizontally on an orbital shaker inside of a 37 °C incubator. Shake flasks at 15 x g for 1 h.
- Remove media and pipette into a 15 mL conical tube. Rinse flasks twice with 3 mL warm MGM, adding wash to the 15 mL conical tubes. These are the microglia.
- Add 5 mL of warm, fresh MGM to the culture flasks.
- Reseal flasks with paraffin film, secure flasks horizontally on shaker, and shake at 15 x g at 37 °C for 15 h for separation of OPCs from astrocytes.
NOTE: This step can be done overnight, but the 15 h shake time is critical as excess time may result in cell death. - Centrifuge the supernatant from step 4.3. at 300 x g for 3 min and culture microglia according to standard protocols16 or use for a biological assay.
5. Oligodendrocyte precursor cell isolation
NOTE: When plating OPCs following initial isolation, they must be plated on a poly-D-lysine-coated surface (sterile plate or cover slip). Prepare these materials prior to the completion of this section.
- Following 15 h shake, remove the supernatant from flasks and plate on sterile 100 mm Petri dish.
- Incubate the supernatant at 37 °C, 5% CO2 for 30 min, swirling after 15 min to remove remaining microglia, as these will very quickly adhere to the dish. Non-tissue culture-treated Petri dishes may be used for this step.
- Remove non-adherent cell supernatant, count, and plate on a poly-D-lysine-coated surface. Typically, 7,500-10,000 OPCs are plated/cm2.
- Incubate at 37 °C, 5% CO2 for at least 1 h (up to 6 h), then gently aspirate 95% of media, and slowly add warm OPC Media, pipetting media against the wall of the well to minimize disruption of OPCs. Change media every 48 h until cells are ready for use.
NOTE: It is critical that only one well is changed at a time. OPCs are sensitive and are especially intolerant to dry conditions. The addition of PDGF-AA in OPC media is to delay OPC maturation into oligodendrocytes. This factor may be excluded from culture media if the experimental focus is mature oligodendrocytes.
6. Astrocyte isolation
- Following 15 h shake, remove supernatant and rinse flasks 2x with warm 1x PBS.
- Add 4 mL of 0.05% trypsin containing 0.53 mM EDTA and incubate at 37 °C, 5% CO2 for 5 min or until cells have lifted. To ensure astrocytes have lifted, visualize them using a standard wide-field microscope. Astrocytes will appear spherical following trypsinization.
- Once astrocytes have detached, stand flask vertically and add 4 mL MGM. Triturate to mix.
- Pipette astrocytes into a 15 mL conical tube, centrifuge at 300 x g for 5 min.
- Resuspend astrocytes in Astrocyte Media and plate on fibronectin-coated surface as described in step 1.2. or use for biological assay.
NOTE: 20 ng/mL murine fibroblast growth factor can be added during the first media change to help establish the astrocyte culture. Additionally, standard gelatin coating may be a low-cost alternative to fibronectin.
7. Identification and isolation of microglia, OPCs, astrocytes, and mature oligodendrocytes using immunocytochemistry
- Once plated cells have reached appropriate confluency, gently remove media and fix adherent cells using 4% paraformaldehyde (PFA) for 10 min. Do this step in a biosafety cabinet.
NOTE: When aspirating or adding solutions, pipette with gentle pressure to prevent cell detachment. - Slowly aspirate PFA then wash cells 3x with 1x PBS for 5 min.
- Prepare appropriate blocking solution using 10% serum and 0.1% Triton X-100 in 1x PBS.
NOTE: Serum source reflects the host animal in which the secondary antibody was raised. For example, if the secondary antibody is goat anti-rabbit, the appropriate blocking serum is normal goat serum. Likewise, if the secondary antibody is donkey anti-rabbit, the blocking serum should be normal donkey serum. - Add blocking solution until cells are completely covered. Block for 1 h at room temperature.
- Prepare an antibody diluent solution (9 mL of 1x PBS, 0.01 g bovine serum albumin, 30 µL Triton X-100). Alternatively, antibodies can be diluted in the blocking solution described in step 6.3.
- Dilute primary antibodies specific for the following in antibody diluent or blocking solution:
- Microglia: ionized calcium binding adaptor molecule 1 (Iba1) at a 1:250 dilution (2.4 μg/mL).
- OPCs: neural/glial antigen 2 (NG2) at a 1:200 dilution (5 μg/mL).
- Mature oligodendrocytes: myelin basic protein (MBP) at a 1:400 dilution (concentration is lot-dependent and optimization may be necessary).
- Astrocytes: glial fibrillary acidic protein (GFAP) at a 1:400 dilution(0.25 μg/mL)17.
NOTE: Do not mix primary antibodies raised in the same species.
NOTE: GFAP will reliably label white matter astrocytes. For grey matter astrocytes, an alternative marker may need to be used.
- Incubate primary antibody overnight at 4 °C (with gentle agitation is preferable).
- Wash 3x with 1x PBS for 5 min to remove the primary antibody.
- Incubate cells with appropriate secondary antibodies at a 1:400 dilution in antibody diluent or blocking solution protected from light.
NOTE: Secondary antibodies are conjugated to a fluorophore which may be interchanged depending on available microscope parameters. Once fluorescent secondary antibodies have been applied, protect from light as much as possible. - Incubate secondary antibody for 1 h at room temperature, limiting exposure to light.
- Wash 3 times with 1x PBS for 5 min to remove excess secondary antibody.
- To label nuclei, use a 1:1,000 DAPI/PBS solution and incubate cells for 5 min at room temperature in the dark.
- Wash 3x with 1x PBS for 5 min in the dark.
- To mount, apply mounting media and allow to dry in the dark before imaging.
NOTE: Mounted slides can be stored at room temperature for 2-3 days. For long term storage, move slides to 4 °C. Image within one week for maximal signal. All representative images have been imaged on a confocal microscope; however, inverted fluorescent microscopes are also recommended.
Results
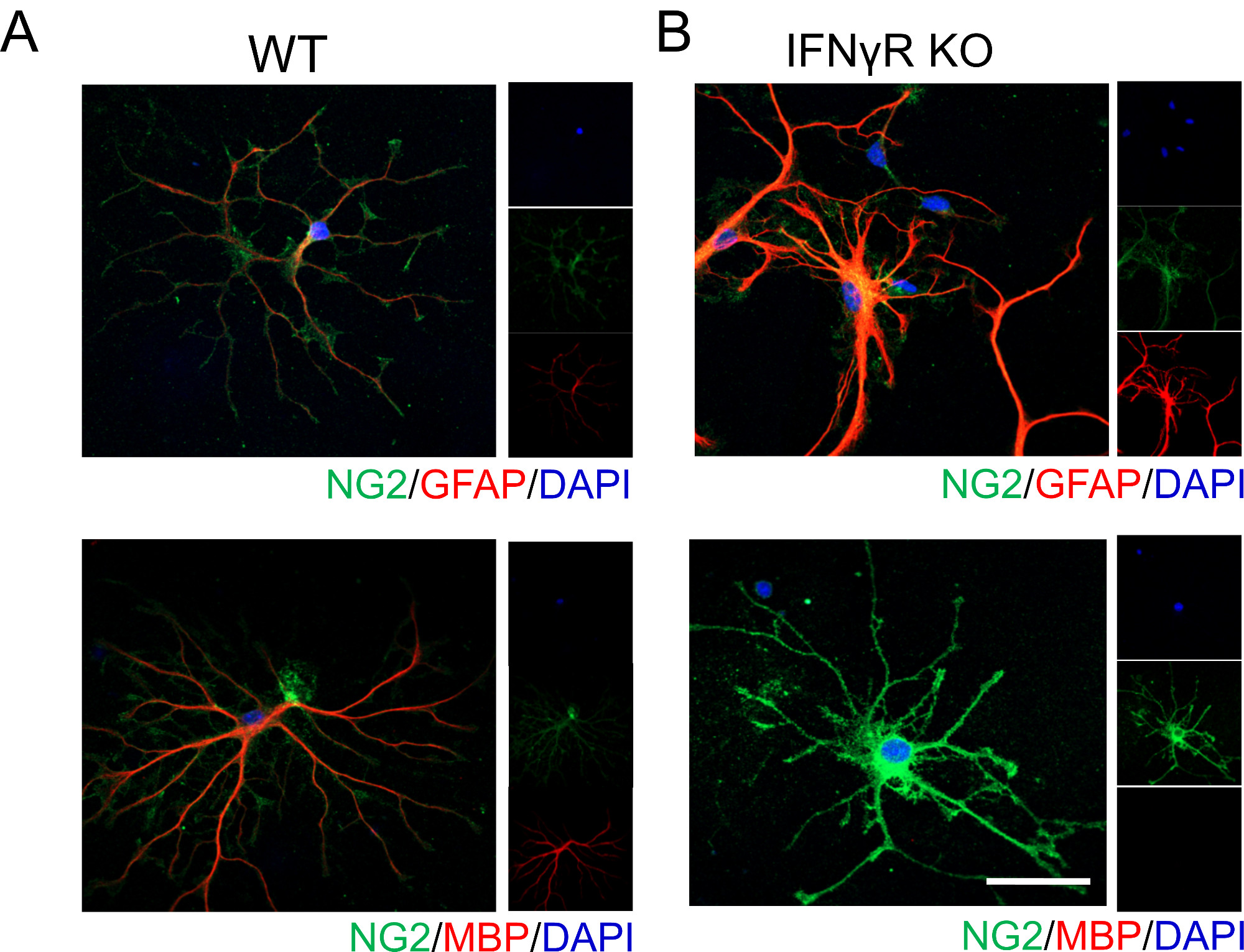
Representative data shown below illustrates that IFNγ signaling influences OPC differentiation and maturation. Without the presence of IFNγ receptor (IFNγR), cortical OPCs do not differentiate into mature myelinating oligodendrocytes as readily, which is evidenced by the absence of MBP staining (Figure 1). Since oligodendrocytes and astrocytes are derived from a common progenitor, we analyzed GFAP expression, which labels astrocytes. We found that IFNγR-deficient cells strongly express GFAP suggesting that they may be adopting an astrocytic phenotype, corroborating earlier reports19.
Additional evidence for regional heterogeneity in CNS cells is evidenced by varying astrocyte morphology as seen in astrocytes from the cortex, cerebellum, brainstem, and spinal cord (Figure 2). Of note, astrocytes from the same region may also exhibit morphological heterogeneity, supporting the notion that this glial subtype is highly dynamic. The differences in cellular architecture is suggestive of functional diversity and thus the ability to isolate glial populations is necessary to study phenotypic responses in the absence and presence of microenvironmental stimuli.
Oligodendrocytes are critical for the myelination of neuronal axons and are necessary for proper CNS repair and function. OPCs give rise to their mature counterparts, making it critical to understand the biology behind their ability to differentiate. Cytokine signaling significantly influences stem and immune cell behavior. Thus, it is important to understand how regional responses of OPCs may vary to differential cytokine stimulation (Figure 3), which may impact their ability to differentiate into mature myelinating oligodendrocytes.

Figure 1: Representative data showing OPC differentiation in WT and IFNγR-/- mice in the presence of exogenous IFNγ. Cortical OPCs were isolated from (A) WT and (B) IFNγR-/- P4 mouse pups following the procedure outlined above. Cells were treated with 1 ng/mL IFNγ for 48 h, then fixed and stained to delineate cell differentiation. OPCs were labeled for NG2 and GFAP to identify those which were adopting an astrocytic phenotype. Likewise, OPCs were also labeled for NG2 and MBP to identify those that were differentiating into mature oligodendrocytes. Scale bar = 20 μM. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Representative data demonstrating regional heterogeneity in astrocyte morphology. Astrocytes from cortex, cerebellum, brainstem, and spinal cord were isolated from P4 mouse pups using the protocol described above and labeled for GFAP (green) by immunocytochemistry following 48 h in culture. Scale bar = 20 μM. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Representative data demonstrating differential responses of regional OPCs to cytokines. OPCs were isolated from the brainstem and spinal cord of P4 mouse pups using the protocol described above. Cells were treated with increasing concentrations (1-10 ng/mL) of (A) IFNγ or (B) interleukin (IL)-17 in order to investigate the differential influence of cytokine on the ability of regional OPCs to differentiate into myelinating oligodendrocytes. Following a 48 h incubation with specified cytokines, OPCs were fixed and labeled for NG2 (green) and MBP (red). Scale bar = 20 μM. Data represent means ± SEM. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 by 2-way ANOVA. Please click here to view a larger version of this figure.
{kind=link}
| PBS/Antibiotic Solution (PAS): | ||
| 1.0 mL | 100X Antibiotic/Antimycotic containing 10,000 units/mL penicillin and 10,000 mg/mL streptomycin | |
| 25 mg/mL | Amphotericin B | |
| 99 mL | 1X PBS | |
| Mixed Glia Media (MGM) | ||
| 88 mL | 1X DMEM (high glucose, w/L-glutamine, w/Na pyruvate) | |
| 10 mL | Heat-inactivated FBS | |
| 1.0 mL | L-Glutamine (100X) | |
| 1.0 mL | Antibiotic/Antimycotic | |
| OPC Media (50mL) | ||
| 49 mL | Neurobasal media | |
| 1.0 mL | B27 supplement (50X) | |
| 10 ng/ml | PDGF-AA | |
| NOTE: PDGF-AA is added fresh prior to each media change. | ||
| Astrocyte Media (1 L) | ||
| 764 mL | MEM with Earle's salts containing glutamine | |
| 36 mL | Glucose (use 100 mg/mL stock for final concentation of 20 mM) | |
| 100 mL | Heat-inactivated FBS | |
| 100 mL | Heat-inactivated horse serum | |
| 10 mL | Glutamine (use 200 mM stock if not included in stock medium) | |
| OPTIONAL: 10 ng/mL recombinant mouse epidermal growth factor | ||
| NOTE: Sterile filter all media and store at 4°C until used. | ||
Table 1: Buffer and Media Recipes.
Discussion
In this protocol, we describe the isolation of the three major glial cell subpopulations from mouse CNS: microglia, OPCs, and astrocytes. A major setback for the investigation of neurodegenerative and neuroinflammatory CNS diseases is the lack of primary human cells and tissues, particularly those that are regional and from the same patient. In most instances, human CNS cell lines are derived from transformed, immortalized cancer cells which may not be accurate representations of their normal physiological behavior20,21,22. Thus, alternative methods are necessary to study CNS cell phenotypes in a controlled manner. Furthermore, the diversity of neurological glial cell populations makes it necessary to investigate each subtype both independently of one another, as well as in co-culture conditions in order to recapitulate both their cell autonomous and non-autonomous functions. Glial cells have a wide variety of critical functions in the CNS ranging from neuronal support23, learning/cognition24,25, and CNS immunological responses26. As such, it is necessary to understand the molecular and cellular functions of each glial subpopulation in a physiological and pathological context. In order to do so, we provide here a reliable method for the extraction and isolation of viable glia subtypes. Due to practical and ethical constraints in human subject’s research, animal models are currently the most relevant surrogates for human glial cell biology. In particular, mice are ideal model animals as their genome can be manipulated and analyzed to further dissect particular molecular mechanisms underlying health and disease. Therefore, the successful removal and separation of murine microglia, OPCs, and astrocytes is a key tool to investigate the functions of glia during physiological, neurodegenerative, or neuroinflammatory conditions.
This protocol can be optimized to explore CNS cell regional heterogeneity. It is becoming increasingly clear that glia exhibit regional heterogeneity in form and function. Astrocytes are regionally diverse and display distinct morphology depending on their location within the CNS27. Furthermore, the density of astrocytes and their mitotic index can define anatomical regions, supporting the hypothesis that regional astrocyte heterogeneity may reflect molecular and functional differences based on their location within the CNS28. Microglial regional heterogeneity is also under active investigation, although the underlying mechanisms and functional consequences of microglia diversity in CNS development or behavior are currently unclear. However, it is known that adult microglia display diversity in cell number, cell and subcellular structures, and molecular signatures29. Moreover, recent advances in multiplexed mass cytometry have further defined the regional heterogeneity of microglia, analyzing cellular phenotype from five different CNS regions of nine human donors, allowing for large-scale immunophenotyping of human microglia30. Currently, such approaches are in their nascent stages, making animal studies a viable solution for the study of regional glia in CNS disease development. Finally, regional heterogeneity has also recently been described in oligodendrocytes. Single-cell RNA sequencing on 5072 individual cells from 10 regions of juvenile and adult CNS identified 13 distinct subpopulations across different stages of differentiation31. Importantly, it was also found that as oligodendrocytes matured from OPCs, their transcriptional profiles diverged and their functional phenotypes changed, highlighting oligodendrocyte heterogeneity within the CNS31.
Thus, understanding regional heterogeneity of the various resident CNS cells in the context of their diverse neighboring neurons and other glia may provide important rationale for the future development of novel therapies to treat neuroinflammatory and neurodegenerative disorders. While this protocol focuses on the extraction, isolation, and identification of glial subpopulations, it provides a convenient starting point for the examination of their function. Furthermore, it can be adapted and combined with transgenic mouse models in order to study genetic mechanisms associated with glial cell biology. It can also be used to examine the responses of glial cells to each other in co-culture assays. The outlined steps represent a cost-efficient and high-throughput method of extracting and isolating different CNS glial populations which can then be adapted to a wide variety of experimental parameters. It should be noted; however, that the method described here utilizes neonates due to the lower levels of myelination and high density of proliferating glia. For these reasons, it is technically more feasible to isolate viable glia from neonates compared to adult animals. The phenotypic differences in neonatal glia compared to adult glia should thus be considered during experimental design and data interpretation.
Disclosures
The authors have conflicts of interest to disclose.
Acknowledgements
We thank Morgan Psenicka for manuscript editing and discussion and Dr. Grahame Kidd for assistance in figure formatting. This work was supported by NIAID K22 AI125466 (JLW).
Materials
| Name | Company | Catalog Number | Comments |
| 0.05% Trypsin and 0.53 mM EDTA | Gibco | 25300054 | Tissue dissociation |
| 12-Well Plates | Greiner Bio-One | 665 180 | Cell culture plate |
| 1X PBS pH 7.4 | Gibco | 10010031 | Standard reagent |
| 32% Paraformaldehyde | Electron Microscopy Sciences | 15714-S | Fixative |
| 50 mL, 25 cm2 cell culture flask | Greiner Bio-One | 690 175 | Cell culture (T25) flask |
| Antibiotic-Antimycotic 100X | Gibco | 15240-096 | Media component |
| B-27 Supplement 50X | Gibco | 17504-044 | Media component |
| Bovine serum albumin | Sigma | A9647-50G | Antibody diluent |
| Confocal Microscope | Zeiss | LSM 800 | Confocal for imaging |
| DAPI | ThermoFisher | D1306 | Nuclear stain |
| DMEM (1X), high glucose with Na pyruvate | Gibco | 11995040 | Media component |
| Dnase I | Sigma | 10104159001 | Tissue dissociation |
| Fetal bovine serum heat inactivated | Gibco | A3840001 | Media component |
| Fibronectin from bovine plasma | Sigma | F1141-1MG | Cell adherent |
| Fine stitch Scissors | Sklar | 64-3260 | Dissection tools |
| Goat anti-rabbit IgG Alexa Fluor 488 | Invitrogen | A11008 | Secondary staining antibody |
| Goat anti-rat IgG Alexa Fluor 555 | Invitrogen | A21434 | Secondary staining antibody |
| Hanks' Balanced Salt Solution (w/o Ca or Mg) | ThermoFisher | 14170120 | Tissue dissociation |
| L-glutamine, 200mM | Gibco | 20530081 | Media component |
| Murine epidermal growth factor | ThermoFisher | PMG8044 | Media component |
| Murine IFN-γ | Peprotech | 315-05-20UG | Media component |
| Murine PDGF-AA | Peprotech | 315-17 | Media component |
| Neurobasal | Gibco | 21103-049 | Media component |
| Normal goat serum | Sigma | G9023 | Blocking solution component |
| Operating Scissors | Surgi-OR | 95-272 | Dissection tools |
| Poly-D-Lysine 12 mm #1 German Glass Coverslip | Corning Biocoatt | 354086 | Cell adherent |
| Prolong Gold Antifade Reagent | Cell Signaling Technology | 9071S | Mounting Media |
| Rabbit anti-Iba1 | Wako | 019-19741 | Primary antibody |
| Rabbit anti-NG2 Chondroitin Proteoglycan | Millipore | ab5320 | Primary antibody |
| Rat anti-GFAP | ThermoFisher | 13-0300 | Primary antibody |
| Rat anti-myelin basic protein | Abcam | ab7349 | Primary antibody |
| Sharp Tip Scissors | Surgi-OR | 95-104 | Dissection tools |
| Stereo Microscope | Leica | S4 E Stereo Zoom Microscope | Microscope for dissection |
| Tissue Forceps | Sklar | 66-7644 | Dissection tools |
| Triton X-100 | Fisher Bioreagents | BP151-100 | Cell permabilization |
| Trypsin Inhibitor (from chicken egg white) | Sigma | 10109878001 | Tissue dissociation |
References
- Fields, R. D., et al. Glial Biology in Learning and Cognition. The Neuroscientist. 20 (5), 426-431 (2014).
- Nuriya, M., Hirase, H. Involvement of astrocytes in neurovascular communication. Progress in Brain Research. 225, 41-62 (2016).
- Nave, K. A. Myelination and support of axonal integrity by glia. Nature. 468 (7321), 244-252 (2010).
- Wake, H., Moorhouse, A. J., Miyamoto, A., Nabekura, J. Microglia: actively surveying and shaping neuronal circuit structure and function. Trends in Neurosciences. 36 (4), 209-217 (2013).
- Kierdorf, K., Prinz, M. Microglia in steady state. The Journal of Clinical Investigation. 127 (9), 3201-3209 (2017).
- Hertz, L., Chen, Y. Importance of astrocytes for potassium ion (K(+)) homeostasis in brain and glial effects of K(+) and its transporters on learning. Neurosciences and Biobehavior Reviews. 71, 484-505 (2016).
- Gordon, G. R., Howarth, C., MacVicar, B. A. Bidirectional Control of Blood Flow by Astrocytes: A Role for Tissue Oxygen and Other Metabolic Factors. Advances in Experimental Medicine and Biology. 903, 209-219 (2016).
- Schneider, J., Karpf, J., Beckervordersandforth, R. Role of Astrocytes in the Neurogenic Niches. Methods Mol Biol. 1938, 19-33 (2019).
- Sharif, Y., et al. Blood brain barrier: A review of its anatomy and physiology in health and disease. Clinical Anatomy. 31 (6), 812-823 (2018).
- von Bernhardi, R., Eugenin-von Bernhardi, J., Flores, B., Eugenin Leon, J. Glial Cells and Integrity of the Nervous System. Advances in Experimental Medicine and Biology. 949, 1-24 (2016).
- Gordon, J., Amini, S., White, M. K. General overview of neuronal cell culture. Methods in Molecular Biology. 1078, 1-8 (2013).
- Spaethling, J. M., et al. Primary Cell Culture of Live Neurosurgically Resected Aged Adult Human Brain Cells and Single Cell Transcriptomics. Cell reports. 18 (3), 791-803 (2017).
- Bayraktar, O. A., Fuentealba, L. C., Alvarez-Buylla, A., Rowitch, D. H. Astrocyte development and heterogeneity. Cold Spring Harbor Perspectives in Biology. 7 (1), 020362 (2015).
- Liu, R., et al. Region-specific and stage-dependent regulation of Olig gene expression and oligodendrogenesis by Nkx6.1 homeodomain transcription factor. Development. 130 (25), 6221-6231 (2003).
- Tan, Y. L., Yuan, Y., Tian, L. Microglial regional heterogeneity and its role in the brain. Molecular Psychiatry. 25 (2), 351-367 (2020).
- Witting, A., Moller, T. Microglia cell culture: a primer for the novice. Methods in Molecular Biology. 758, 49-66 (2011).
- Williams, J. L., Patel, J. R., Daniels, B. P., Klein, R. S. Targeting CXCR7/ACKR3 as a therapeutic strategy to promote remyelination in the adult central nervous system. Journal of Experimental Medicine. 211 (5), 791-799 (2014).
- Domingues, H. S., Portugal, C. C., Socodato, R., Relvas, J. B. Oligodendrocyte, Astrocyte, and Microglia Crosstalk in Myelin Development, Damage, and Repair. Frontiers in Cell and Developmental Biology. 4, 71 (2016).
- Tanner, D. C., Cherry, J. D., Mayer-Proschel, M. Oligodendrocyte progenitors reversibly exit the cell cycle and give rise to astrocytes in response to interferon-gamma. Journal of Neuroscience. 31 (16), 6235-6246 (2011).
- Stansley, B., Post, J., Hensley, K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer's disease. Journal of Neuroinflammation. 9 (1), 115 (2012).
- Spaethling, J. M., et al. Primary Cell Culture of Live Neurosurgically Resected Aged Adult Human Brain Cells and Single Cell Transcriptomics. Cell Reports. 18 (3), 791-803 (2017).
- Timmerman, R., Burm, S. M., Bajramovic, J. J. An Overview of in vitro Methods to Study Microglia. Frontiers in Cellular Neuroscience. 12 (242), (2018).
- Stevens, B. Glia: much more than the neuron's side-kick. Current Biology. 13 (12), 469-472 (2003).
- Fields, R. D., et al. Glial biology in learning and cognition. Neuroscientist. 20 (5), 426-431 (2014).
- Yamamuro, K., Kimoto, S., Rosen, K. M., Kishimoto, T., Makinodan, M. Potential primary roles of glial cells in the mechanisms of psychiatric disorders. Frontiers in Cellular Neuroscience. 9 (154), (2015).
- Hartenstein, V., Giangrande, A. Connecting the nervous and the immune systems in evolution. Communications Biology. 1 (1), 64 (2018).
- Emsley, J. G., Macklis, J. D. Astroglial heterogeneity closely reflects the neuronal-defined anatomy of the adult murine CNS. Neuron Glia Biology. 2 (3), 175-186 (2006).
- Chaboub, L. S., Deneen, B. Developmental Origins of Astrocyte Heterogeneity: The Final Frontier of CNS Development. Developmental Neuroscience. 34 (5), 379-388 (2012).
- Tan, Y. L., Yuan, Y., Tian, L. Microglial regional heterogeneity and its role in the brain. Molecular Psychiatry. 25 (2), 351-367 (2020).
- Bottcher, C., et al. Human microglia regional heterogeneity and phenotypes determined by multiplexed single-cell mass cytometry. Nature Neurosciences. 22 (1), 78-90 (2019).
- Marques, S., et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science. 352 (6291), 1326-1329 (2016).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved