Method Article
Analisi a livello genomico della metilazione del DNA nel cancro gastrointestinale
In questo articolo
Riepilogo
Qui descriviamo una procedura per l'analisi a livello genomico della metilazione del DNA nei tumori gastrointestinali. La procedura è rilevante per studi che studiano le relazioni tra i modelli di metilazione dei geni e i fattori che contribuiscono alla carcinogenesi nei tumori gastrointestinali.
Abstract
La metilazione del DNA è un importante cambiamento epigenetico che è biologicamente significativo e un punto focale frequente della ricerca sul cancro. La metilazione del DNA a livello genomico è una misura utile per fornire un'analisi accurata dello stato di metilazione delle neoplasie maligne gastrointestinali (IG). Dati i molteplici potenziali usi traslazionali dell'analisi della metilazione del DNA, i medici praticanti e altri nuovi studi di metilazione del DNA devono essere in grado di capire passo dopo passo come vengono eseguite queste analisi a livello genomico. L'obiettivo di questo protocollo è fornire una descrizione dettagliata di come questo metodo viene utilizzato per l'identificazione del biomarcatore nelle neoplasie maligne gastrointestinali. È importante sottolineare che descriviamo tre passaggi critici necessari per ottenere risultati accurati durante l'analisi a livello genomico. Scritti in modo chiaro e conciso, questi tre metodi sono spesso carenti e non evidenti per quelli nuovi agli studi epigenetici. Abbiamo utilizzato 48 campioni di malignità gastrointestinale (cancro gastrico) per evidenziare praticamente come l'analisi della metilazione del DNA a livello genomico può essere eseguita per le neoplasie maligne gastrointestinali.
Introduzione
L'epigenetica si riferisce a cambiamenti ereditari nella funzione genica senza alterazione della sequenza del DNA1. Tali cambiamenti possono essere dovuti alla metilazione del DNA, in cui i gruppi metili su una base di DNA possono alterare l'espressione genica attraverso cambiamenti nell'imballaggio della cromatina. Lo sviluppo e la progressione del cancro possono verificarsi se questo effetto si traduce in un'espressione alterata dei geni soppressori deltumore 2. L'invecchiamento e l'infiammazione cronica sono entrambe cause di cancro e le principali ragioni dei cambiamenti nella metilazione del DNAnell'uomo 3,4,5. Di conseguenza, ciò consente l'utilizzo della metilazione del DNA come biomarcatore nella diagnosi del cancro e come obiettivo per il trattamento e la prevenzione. Per la diagnosi precoce e la prognosi del cancro, la metilazione del DNA viene misurata nei campioni di tumore, sangue, urina e feci 6 ,mentregli agenti demetilanti vengono ora utilizzati per trattare le leucemie come la sindrome mielodisplastica7.
L'analisi della metilazione del DNA a livello genomico utilizzando una piattaforma array per la valutazione complessa della metilazione del DNA in un singolo locus CpG nel genoma umano può essere utilizzata per esaminare lo stato di metilazione di oltre 450.000 siti CpG nel DNA genomico8,9, che consente l'esplorazione dell'epigenetica del cancro (vedi Tabella dei materiali). Le tecnologie di sequenziamento della bisolfito dell'intero genoma (WGBS) hanno cambiato i nostri approcci nel campo dell'epigenetica10,11. Tuttavia, ci sono alcuni svantaggi per le tecnologie in termini di costi e tempi di elaborazione sostanziali per l'analisi epigenetica di un gran numerodi campioni 10,11. Pertanto, la piattaforma array è più fattibile per una valutazione complessa della metilazione del DNA nel genoma umano. La disponibilità di approcci per le analisi di metilazione a livello genomico è migliorata negli ultimi anni e ci consente di espandere la nostra conoscenza di come la metilazione del DNA contribuisca allo sviluppo e alla progressione del cancro12. I recenti progressi negli approcci a piattaforma microarray ci forniscono la logica per l'analisi della metilazione a livello genomico per identificare una nuova firma epigenetica nei tumorigastrointestinali 13. L'obiettivo di questo protocollo è quello di fornire una descrizione dettagliata di come questo metodo viene utilizzato per l'identificazione del biomarcatore nelle neoplasie di ig.
Protocollo
Tutte le procedure seguite erano conformi agli standard etici del comitato etico per la ricerca umana delle istituzioni. Lo studio è stato approvato dall'Institutional Review Board presso l'ospedale Shizuoka dell'Università juntendo e il consenso informato scritto è stato revocato a causa del design retrospettivo.
1. Lavare le diapositive
- Preparare 10 μm di sezioni non macchiate di paraffina fissa formalina (FFPE).
- Posizionare le diapositive in un portasci di vetro: utilizzare circa 3-5 diapositive trasversali più grandi a meno che il tessuto non sia minimo e siano necessarie più diapositive.
- Riempire il supporto della diapositiva con xilene e assicurarsi che tutto il tessuto sullo scivolo sia sommerso. Lasciare riposare per 15 minuti.
- Dopo 15 minuti, versare lo xilene con le diapositive tenute con una punta di pipetta in modo che le diapositive non cadano.
- Versare più xilene allo stesso livello di prima. Lasciare riposare per altri 15 minuti.
- Dopo 15 minuti, versare di nuovo lo xilene.
- Riempire il porta scorrevole con etanolo al 100% (EtOH) e assicurarsi che tutto il tessuto sullo scivolo sia completamente sommerso. Lasciare riposare per 3 minuti.
- Versare l'EtOH tenendo con cura le diapositive. Ricarica allo stesso livello con più EtOH. Lasciare riposare per 2 minuti.
- Versare di nuovo l'EtOH e rimuovere la diapositiva. Posizionarli con cura a faccia in su un tovagliolo di carta pulito per asciugarli. Lasciare riposare per 10 minuti.
2. Raschiare le diapositive
- Preparare il tampone di digestione/lisi con 650 ml di acqua trattata con dietilpirrocarbonato (DEPC), 100 ml di acido etilendiamminatetraacetico (EDTA), 50 ml di tricloruro (Tris-HCL) 2 M pH 8,8 e 200 ml di dodecil solfato di sodio (SDS) (vedi tabella dei materiali).
- Riempire un tubo di polipropilene mono impiego da 1,5 ml con 80 μL di tampone di digestione/lisi (vedi Tabella dei materiali).
- Mettere una punta di pipetta pulita nel tampone.
- Identificare i tessuti tumorali dell'area tumorale più adatti alla macrodisezione secondo l'appropriata sezione macchiata di H&E.
NOTA: L'area tumorale per la macrodisezione deve essere identificata preferibilmente da due patologi qualificati. - Macrodissect tessuto tumorale basato sulla sezione macchiata H&E contrassegnata.
- Prendi una lama di rasoio pulita e raschia delicatamente il tessuto tumorale dal vetrino, cercando di tenerlo in un unico pezzo in modo che sia più facile lavorare.
- Togliere la punta della pipetta dal tubo di polipropilene mono impiego contenente tampone e utilizzarlo per trasferire il tessuto scartato nel flaconcino tampone (vedere Tabella dei materiali).
NOTA: La punta bagnata dovrebbe attirare il tessuto, il che rende il trasferimento più facile.
- Ripetere il passaggio 2.5 con il resto delle diapositive.
- Dopo che tutto il tessuto è in tubo di polipropilene mono-uso, utilizzare la punta per assicurarsi che il tessuto sia completamente sommerso e non sia attaccato alla parete del tubo.
- Aggiungere 20 μL di una proteasi serina correlata alla subtililina al flaconcino e scorrere delicatamente per mescolare (vedere Tabella dei materiali).
- Posizionare il flaconcino in un blocco termico di 55 °C per almeno 4 ore o durante la notte. Assicurarsi di vortice leggermente dopo 2 h.
3. Trattamento bisolfito
- Utilizzare 45 μL della soluzione tissutale digerita come campione.
- Eseguire il trattamento con bisolfito utilizzando reagenti in un kit di conversione della bisolfito secondo le istruzioni del produttore (vedere Tabella dei materiali).
- Aggiungere 5 μL di tampone di diluizione al campione di DNA e incubare a 37 °C per 15 minuti (vedi Tabella dei materiali).
- Durante l'incubazione dei campioni, preparare il reagente di conversione della bisulfite aggiungendo 750 μL di dH2O e 210 μL di tampone di diluizione a un tubo di reagente di conversione CT (vedi Tabella dei materiali). Mescolare i tubi con vortici per 10 minuti.
- Dopo l'incubazione di 15 minuti, aggiungere 100 μL del reagente di conversione CT preparato a ciascun campione e mescolare per inversione.
- Incubare i campioni al buio a 50 °C per 12-16 ore.
- Dopo l'incubazione, rimuovere i campioni e posizionare sul ghiaccio per 10 minuti.
- Aggiungere 400 μL di tampone di legame e mescolare ogni campione pipettando su e giù (vedere Tabella dei materiali). Caricare ogni campione in una colonna di rotazione e posizionare la colonna in un tubo di raccolta da 2 ml (vedere Tabella dei materiali).
- Centrifugare ogni campione a tutta velocità (10.000 x g)per 1 minuto e scartare il flusso.
- Aggiungere 200 μL di tampone di lavaggio a ogni colonna e ruotare a tutta velocità per 1 minuto, scartando il flusso (vedere Tabella dei materiali).
- Aggiungere 200 μL di tampone di desulfonazione a ciascuna colonna e lasciare che la colonna si trovi a temperatura ambiente per 15 minuti (vedi Tabella dei materiali). Dopo l'incubazione, ruotare le colonne a tutta velocità per 1 minuto e scartare il flusso.
- Aggiungere 200 μL di tampone di lavaggio a ciascuna colonna e ruotare a tutta velocità per 1 minuto (vedi Tabella dei materiali).
- Ripetere questo passaggio ancora una volta.
- Aggiungere 46 μL di dH2O a ciascuna colonna e posizionare ogni colonna in un nuovo tubo sterile in polipropilene monouso da 1,5 ml (vedi Tabella dei materiali). Girare ogni tubo per 2 minuti per elutare il DNA.
- Rimuovere ogni colonna di rotazione dal tubo di polipropilene mono impiego e scartare (vedere Tabella dei materiali). Il DNA è ora pronto per l'analisi.
4. Piattaforma array per la valutazione della metilazione del DNA in un locus CpG nel genoma umano
- Valutare la qualità del DNA (controllo qualità: QC) utilizzando il saggio QC FFPE su uno strumento di amplificazione e rilevamento PCR in tempo reale, con successiva analisi dei dati eseguita secondo le istruzioni dei produttori (vedi Tabella dei materiali).
NOTA: I campioni con ∆Cq inferiori al 5.0 consigliato vengono ulteriormente elaborati14. - Analizzare campioni con una piattaforma array per una valutazione complessa della metilazione del DNA per valutare lo stato di metilazione dei siti CpG >450.000 nel genoma (vedere Tabella dei materiali). La scheda informativa sul prodotto, la scheda tecnica e i file di prodotto per la piattaforma array sonodisponibili 15.
NOTA: Il test viene eseguito secondo le istruzioni del produttore per il materiale FFPE14. - Calcolare la metilazione locus alta e bassa in uno strumento di analisi dei dati per una piattaforma array come valore β, con un intervallo rispettivamente da 0 a 1 (vedere Tabella dei materiali). Utilizzare software disponibile in commercio (vedere Tabella dei materiali) per calcolare un β valore aggiunto.
- Importare i dati generati sulla piattaforma array per una valutazione complessa della piattaforma di metilazione del DNA nell'ambiente software R (R v.2.15.1) ed elaborarli utilizzando uno strumento per analizzare gli array dimetilazione 16,17.
NOTA: Sulla mappa termica, generata utilizzando uno strumento di analisi dei dati, le colonne vengono ordinate in base al clustering non supervisionato, mentre l'ordine delle righe si basa sul significato decrescente della statistica t per la metilazione differenziale dall'alto verso il basso. - Dividere la mappa termica in gruppi di metilazione alti e bassi utilizzando il primo divisore nel clustering non supervisionato.
NOTA: Per la convalida con PCR (qMSP) specifico della metilazione quantitativa, scegliere i geni basati su un valore di β più grande in relazione alle isole CpG nella regione del promotore e a causa della loro idoneità per la progettazione di primer e sonde per qMSP.
5. PCR quantitativo specifico per la metilazione (qMSP)
- Utilizzare il DNA modificato dalla bisolfito del passaggio 3.3 come modello per la PCR in tempo reale basata sulla fluorescenza in qMSP per valutare la metilazione della regione promotrice in ogni analisi genica.
- Eseguire qMSP utilizzando uno strumento PCR in tempo reale a 96 po' (vedere Tabella dei materiali).
- Verificare lo stato di metilazione del promotore del gene bersaglio sul DNA modificato con bisolfito usando primer in avanti da 200 nM, primer inverso da 200 nM e 80 sonde nM. Preparare il master mix con 16,6 mM (NH4)2SO4, 67 mM Tris pH 8.8, 10 mM β-mercaptoetanolo, 10 nM di fluoresceina, 0,166 mM di ogni trifosfato deossinucleotide e 0,04 U/μl di DNA polimerasi (vedi Tabella dei materiali). Il volume di reazione finale in ogni pozzo per tutti i saggi è di 25 μL.
- Eseguire il ciclismo di qMSP come segue: 95 °C per 5 minuti, seguito da 55 cicli di 95 °C per 15 s, 60 °C per 1 min e 72 °C per 1 min.
NOTA: Il gene target deve essere scelto in base ai criteri di avere valori beta più grandi, essere correlato alle isole CpG nella regione promotrice ed essere adatto per la progettazione di primer e sonde per qMSP.
- Utilizzare la genomica umana trattata con CpG Metilasi (M.SssI) come controllo positivo della metilazione (vedi Tabella dei materiali).
NOTA: La quantificazione finale della metilazione è definita come il valore relativo di metilazione (RMV) e calcolata come 2–ΔΔCt per ogni replica di rilevamento della metilazione rispetto alla T media per β-Actin (ACTB)18. Le sequenze primer e probe sono mostrate nella tabella 1. Un Ct di 100 viene utilizzato per repliche non rilevate, che dà un valore di 2–ΔΔCt vicino a zero. Viene usata la seguente formula: media 2–ΔΔCt (RMV) = (2–ΔΔ Ct_replicate_1 + 2–ΔΔ Ct_replicate_2 + 2–ΔΔ Ct_replicate_3)/318.
Risultati
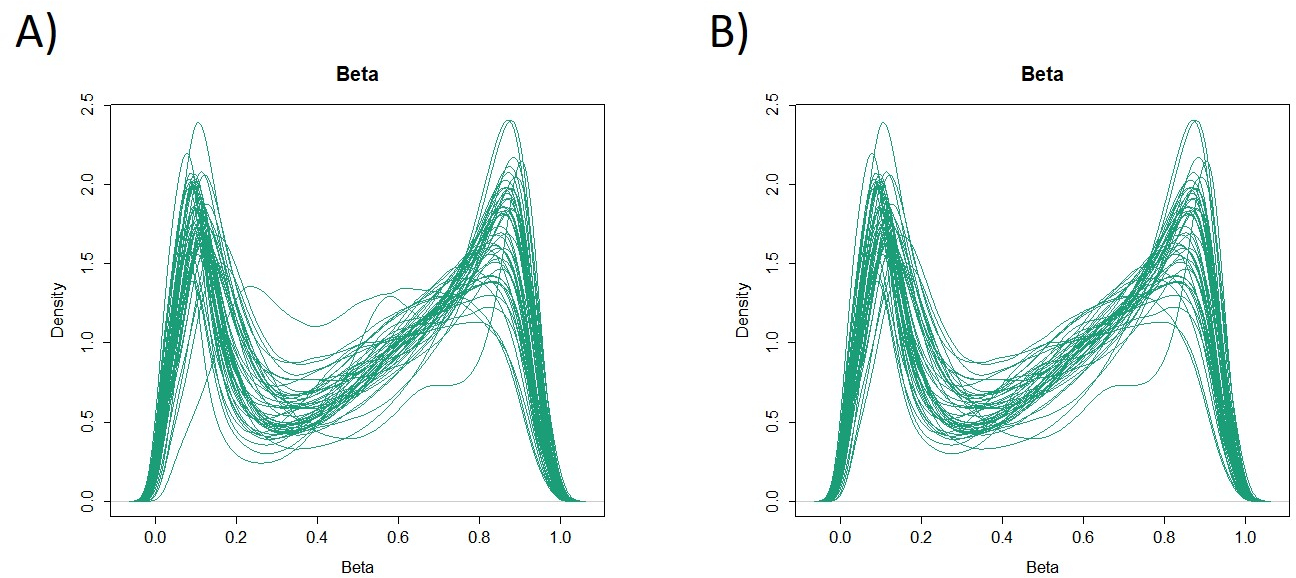
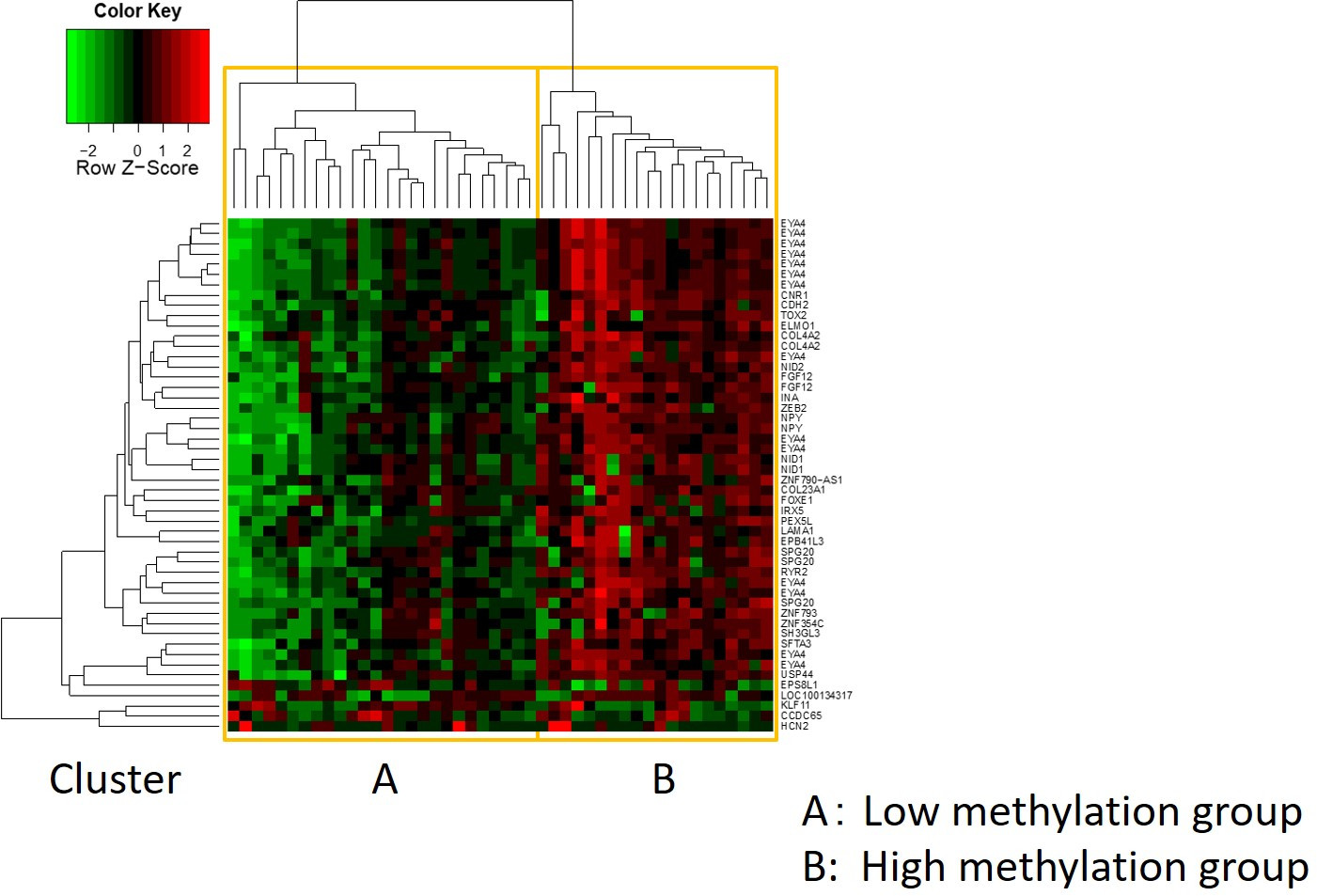
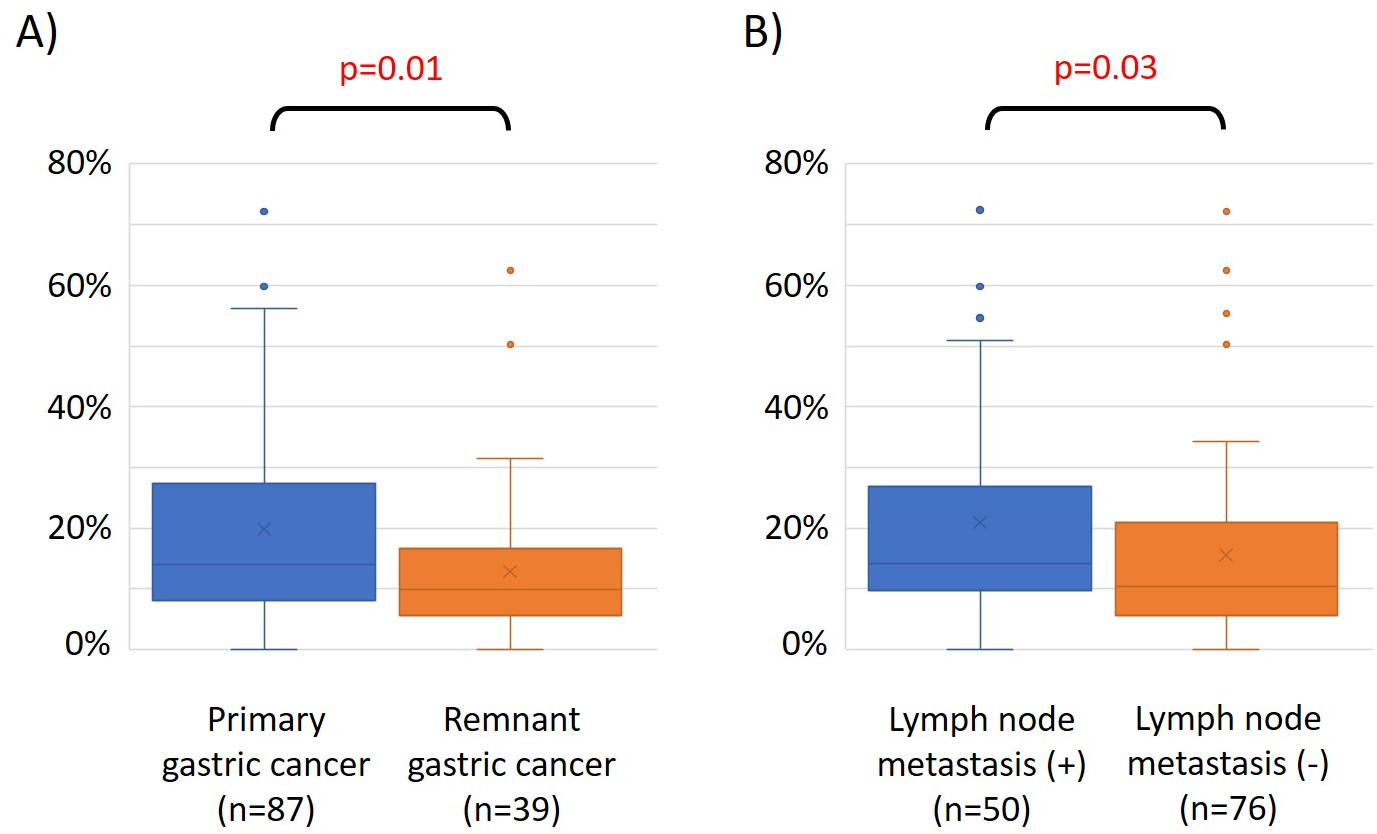
Le caratteristiche di 48 pazienti con cancro gastrico nella coorte di formazione sono le seguenti (Tabella 2): l'età media dei pazienti era di 74 anni (52-89 anni), e la coorte comprendeva 38 maschi (79,2%) e 10 femmine (20,8%). I pazienti sono stati 35 (72,9%) con tumore gastrico primario e in 13 pazienti (27,1%) con tumore gastrico residuo (cancro gastrico primario: prima comparsa di una malignità non metastatica nello stomaco; cancro gastrico residuo: cancro nello stomaco residuo che si è sviluppato più di 5 anni dopo la gastrectomia distale, indipendentemente dalla ragione della resezione originale19). I pazienti sono stati 23 (47,9%) con metastasi linfonodo e 25 pazienti (52,1%) Senza. In primo luogo, tutti i 48 campioni (la coorte di addestramento) sono stati caricati per l'identificazione degli outlier(figura 1A). Due campioni hanno dato picchi superiori a due deviazioni standard spostati dagli altri e questi campioni sono stati rimossi (Figura 1B). Pertanto, 46 campioni sono stati raggruppati dall'ipermetilazione del promotore del DNA. La mappa termica risultante è stata suddivisa in due gruppi basati su metilazione elevata e bassa (Figura 2). Questa mappa termica consente la visualizzazione delle prime 50 sonde entro 1.500 bp dal sito iniziale trascrizionale (TSS) nell'analisi della metilazione differenziale. I gruppi di metilazione alti e bassi differivano nei fattori clinicopatologici legati a un fenotipo maligno aggressivo. Cioè, il tipo di cancro (cancro gastrico primario: PGC) (p = 0,01, odds ratio = 9,09 (1,67-50,00)) e presenza di metastasi linfonodo (positiva) (p = 0,03, odds ratio = 6,82 (1,16-40,08)) sono emersi come fattori predittivi indipendenti significativi quando i fattori clinicopatologici sono stati utilizzati come covariati nell'analisi multivariata(tabella 3). Infine, la Corte ha identificato che il gene EPB41L320,21 (sequenze di primer e sonde mostrate nella tabella 1)è fortemente associato alla codifica della coorte di addestramento in gruppi di metilazione alti e bassi nell'analisi del microarray. Utilizzando qMSP, sono stati convalidati i risultati dell'analisi microarray per EPB41L3 nella coorte di prova (126 campioni). Le caratteristiche dei pazienti nella coorte di prova sono indicate nella tabella 4. Gli RMV di EPB41L3 nei tessuti PGC erano significativamente più alti di quelli del cancro gastrico residuo (RGC) nell'analisi univariata (p = 0,01) (Figura 3A). Analogamente, gli RMV nei campioni con metastasi linfonodo erano significativamente più alti di quelli senza metastasi linfonodo (p = 0,03) (Figura 3B). In questo modo, l'analisi a livello genomico della metilazione del DNA può aiutarci a identificare geni specifici per caratterizzare determinati stati clinici in pazienti con neoplasie maligne gastrointestinali.

Figura 1: Valori beta in 48 campioni (coorte di allenamento). Tutti i 48 campioni (coorte di addestramento) sono stati caricati e sono stati esaminati valori anomali (A). Due campioni avevano picchi che erano valori anomali e questi sono stati rimossi (B). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: La mappa termica risultante. I restanti 46 campioni sono stati raggruppati dall'ipermetilazione del promotore del DNA. La mappa termica era divisa in gruppi di metilazione alti e bassi. Questa mappa termica consente la visualizzazione delle prime 50 sonde entro 1.500 bp dal sito iniziale trascrizionale (TSS) nell'analisi della metilazione differenziale. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: Valori relativi di metilazione (RMV) per EPB41L3 nel cancro gastrico primario (PGC) rispetto al cancro gastrico residuo (RGC) e nei casi con e senza metastasi linfonodo. I risultati dell'analisi del microarray per EPB41L3 nella coorte di prova (126 campioni) sono stati convalidati utilizzando qMSP. (A) Nell'analisi univariata, gli RMV dell'EPB41L3 nei tessuti PGC erano significativamente più alti di quelli dell'RGC (p = 0,01). (B) Analogamente, gli RMV nei campioni con metastasi linfonodo erano significativamente più alti che in quelli senza metastasi linfonodo (p = 0,03). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
| Gene | Avanti 5' - 3' | Inverti 5' - 3' | Sonda |
| B-ACTIN | TAG GGA GTA TAT AGG TTG GGG AAG TT | AAC ACA CAA TAA CAA ACA CAA ATT CAC | \56-FAM\ TGT GGG GTG \ZEN\ GTG ATG GAG GAG GTT TAG \3IABkFQ\ |
| EPB41L3 | GGG ATA GTG GGG TTG ACG C | ATA AAA ATC CCG ACG AAC GA | AAA TTC GAA AAA CCG CGC GAC GCC GAA ACC A |
Tabella 1: Primer e sequenze di sonda.
| Fattori clinicopatologici | Variabili | |
| Età | 74 (52 - 89) * | |
| Genere | Maschio / Femmina | 38 (79.2%) / 10 (20.8%) |
| digitare | PGC / RGC | 35 (72.9%) / 13 (27.1%) |
| Metastasi linfonodo | (+) / (-) | 23 (47.9%) / 25 (52.1%) |
| PGC: Cancro gastrico primario, RGC: Cancro gastrico residuo | ||
| * Mediana (minimo massimo) | ||
Tabella 2: Caratteristiche di 48 pazienti con cancro gastrico nella coorte di formazione.
| Valore P | Rapporto quote | Intervallo di confidenza del 95% | |
| Tipo (PGC) | 0.01 | 9.09 | 1.67 – 50.00 |
| Metastasi linfonodo (+) | 0.03 | 6.82 | 1.16 – 40.08 |
| PGC: Cancro gastrico primario | |||
Tabella 3: Fattori predittivi per il gruppo ad alta metilazione (cluster B).
| Fattori clinicopatologici | Variabili | |
| Età | 71 (33 - 86) * | |
| Genere | Maschio / Femmina | 96 (76.2%) / 30 (23.8%) |
| digitare | PGC / RGC | 87 (69.0%) / 39 (31.0%) |
| Metastasi linfonodo | (+) / (-) | 50 (39.7%) / 76 (60.3%) |
| PGC: Cancro gastrico primario, RGC: Cancro gastrico residuo | ||
| * Mediana (minimo massimo) | ||
Tabella 4: Le caratteristiche in 126 pazienti con cancro gastrico nella coorte di prova.
Discussione
Ci sono tre fasi critiche per ottenere risultati accurati dall'analisi a livello genomico della metilazione del DNA. Il primo è la macrodisezione di un'area tumorale da parte di due patologi qualificati basati su sezioni colorate H&E rappresentative. La macrodisezione imprecisa può causare contaminazione con tessuti non cancerosi adiacenti, il che genera risultati inaffidabili; pertanto, è necessaria un'attenta macrodisezione. Il secondo è la valutazione della qualità del DNA (controllo di qualità: QC). I campioni che non riescono il QC (∆Cq > 5.0) possono fornire dati di scarsa qualità. Pertanto, i campioni con ∆Cq > 5.0 devono essere rimossi e altri utilizzati. Il terzo passo è il calcolo del valore di β, che è determinato da uno strumento di analisi dei dati per il software della piattaforma array come segnale metilato / il segnale totale (metilato + non metilato)17. Il β-valore varia da 0 a 1 (o 0%–100%), che è semplice da interpretare biologicamente17. Il problema principale di questo valore sono le sue scarse proprietà statistiche, poiché la sua elevata eterogeneità implica che la varianza tra i campioni agli estremi dell'intervallo di metilazione (β = 0 o β = 1) èaltamente ridotta 17. Inoltre, a causa della scarsa qualità del campione, i valori di β potrebbero non mostrare picchi bifasici riproducibili22e i campioni senza tali picchi dovrebbero essere esclusi da ulteriori studi. Inoltre, il gene bersaglio dovrebbe essere scelto in base ai criteri di avere valori beta più grandi, essendo correlato alle isole CpG nella regione promotrice ed essendo adatto per la progettazione di primer e sonde per qMSP.
La valutazione della metilazione del DNA in un locus CpG nel genoma umano viene eseguita utilizzando una tecnologia basata su microarray con un numero fisso di sonde per rilevamento di loci genomici specifici. È il metodo più utilizzato negli studi di associazione a livello di epigenoma (EWAS) a causa del suo basso costo, della piccola quantità di DNA richiesta e del tempo di elaborazione del campione notevolmente più breve, che consente l'elaborazione ad alta produttività di molti campioni clinici23. Tuttavia, una piattaforma array per la valutazione complessa della metilazione del DNA in un singolo locus CpG è limitata dal numero e dalla specificità delle sonde per loci alterati epigeneticamente, che impedisce l'esplorazione di alcune regioni genomiche. Il WGBS è generalmente visto come il metodo gold standard grazie al suo più ampio spettro di coperturagenomica 10,11. Tuttavia, questo metodo ha un costo sostanziale e un tempo di elaborazione relativamente lungo per l'analisi di un gran numerodi campioni 10,11. Pertanto, non è sempre fattibile. In confronto, la piattaforma array per la valutazione complessa della metilazione del DNA in un singolo locus CpG nel genoma umano è ragionevole per l'uso in termini di costi e copertura genomica. Recentemente, gli ultimi chip di perline aggiornati si sono preparati a utilizzare24. Questi test possono aiutarci ad analizzare siti CpG misurati quasi raddoppiati, che possono ottenere uno studio di associazione ideale a livello genomico (GWAS) per grandi popolazioni campione.
In sintesi, l'analisi a livello genomico della metilazione del DNA con la piattaforma array per la valutazione complessa della metilazione del DNA in un singolo locus CpG nel genoma umano può fornire importanti informazioni sui biomarcatori epigenetici nel cancro gastrointestinale. Rispetto a WGBS, questo metodo è conveniente e riduce i tempi di elaborazione dei campioni. Pertanto, questo metodo per il rilevamento della metilazione del DNA in un locus CpG è probabile che sia ampiamente utilizzato nella ricerca sui biomarcatori epigenetici.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Siamo particolarmente grati a tutti i membri del Dipartimento di Chirurgia, il Sidney Kimmel Comprehensive Cancer Center presso la Johns Hopkins University School of Medicine per utili discussioni e supporto tecnico. Ringraziamo anche Kristen Rodgers per la generosa guida tecnica sulle procedure per il trattamento bisolfito e qMSP.
Materiali
| Name | Company | Catalog Number | Comments |
| (NH4)2SO4 | Sigma-Aldrich | 14148 | Step 5.2. |
| 10% Sodium dodecyl sulfate (SDS) | Quality Biological | 351-032-721 EA | Step 2.1. |
| 100 % Ethanol | Sigma-Aldrich | 24194 | Step 1.7. |
| ABI StepOnePlus Real-Time PCR System | Applied BioSystems | 4376600 | Step 5.2. 96-well Real-Time PCR instrument |
| CT conversion reagent | Zymo Research | D5001-1 | Step 3.2.3. |
| Deoxynucleotide triphosphate (dNTP) | Invitrogen | 10297-018 | Step 5.2. |
| DEPC-treated water | Quality Biological | 351-068-131 | Step 2.1. |
| Ethylenediaminetetraacetic acid (EDTA) | Corning | 46-034-CL | Step 2.1. |
| EZ DNA Methylation Kit | Zymo Research | D5002 | Step 3.2. |
| Fluorescein | Bio-Rad | #1708780 | Step 5.2. |
| GenomeStudio | omicX | OMICS_00854 | Step 4.3. Data analysis tool for an array platform as a β-value, with a range from 0 to 1 |
| Human genomic DNA | New England Bio Labs | N4002S | Step 5.3. |
| Infinium HD FFPE QC Kit | Illumina | WG-321-1001 | Step 4.1. FFPE QC assay on a real-time PCR amplification |
| Infinium HumanMethylation450 assay | Illumina | WG-314 | Step 4.2. Array platform for complex evaluation of DNA methylation to assess the methylation status of >450,000 CpG sites in the genome |
| LightCycler 480 | Roche | 5015278001 | Step 4.1. |
| M-Binding Buffer | Zymo Research | D5002-3 | Step 3.2.6. |
| M-Desulphonation Buffer | Zymo Research | D5002-5 | Step 3.2.9. |
| M-Dilution Buffer | Zymo Research | D5002-2 | Step 3.2.1. |
| Minfi package | Bioconductor | N/A | Step 4.4. |
| M-Wash Buffer | Zymo Research | D5002-4 | Step 3.2.10. |
| Platinum Taq polymerase | ThermoFisher Scientific | 10966-034 | Step 5.2. |
| Proteinase K | New England Biolabs | P8107S | Step 2.8. |
| Single-use polypropylene (Eppendorf) tube | Eppendorf | 24533495 | Step 2.5.2. |
| Tris hydrochloride (Tris-HCL) 2 M pH 8.8 | Quality Biological | 351-092-101 | Step 2.1. |
| Xylene | Sigma-Aldrich | 214736 | Step 1.3. |
| Zymo Spin 1 Column | Zymo Research | C1003 | Step 3.2.6. |
| β-Mercaptoethanol | Sigma-Aldrich | M3148 | Step 5.2. |
Riferimenti
- Qiu, J. Epigenetics: unfinished symphony. Nature. 441 (7090), 143-145 (2006).
- Okugawa, Y., Grady, W. M., Goel, A. Epigenetic alterations in colorectal cancer: emerging biomarkers. Gastroenterology. 149 (5), 1204-1225 (2015).
- Ahuja, N., Li, Q., Mohan, A. L., Baylin, S. B., Issa, J. P. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Research. 58 (23), 5489-5494 (1998).
- Hsieh, C. J., et al. Hypermethylation of the p16INK4a promoter in colectomy specimens of patients with long-standing and extensive ulcerative colitis. Cancer Research. 58, 3942-3945 (1998).
- Ushijima, T., Okochi-Takada, E. Aberrant methylations in cancer cells: where do they come from. Cancer Science. 96 (4), 206-211 (2005).
- Coppedè, F. Epigenetic biomarkers of colorectal cancer: Focus on DNA methylation. Cancer Letters. 342 (2), 238-247 (2014).
- Vasilatou, D., Papageorgiou, S. G., Dimitriadis, G., Pappa, V. Epigenetic alterations and microRNAs: new players in the pathogenesis of myelodysplastic syndromes. Epigenetics. 8 (6), 561-570 (2013).
- Ma, X., Wang, Y. W., Zhang, M. Q., Gazdar, A. F. DNA methylation data analysis and its application to cancer research. Epigenomics. 5 (3), 301-316 (2013).
- Morris, T. J., Beck, S. Analysis pipelines and packages for Infinium HumanMethylation450 BeadChip (450k) data. Methods. 72, 3-8 (2015).
- Rauluseviciute, I., Drabløs, F., Rye, M. B. DNA methylation data by sequencing: experimental approaches and recommendations for tools and pipelines for data analysis. Clinical Epigenetics. 11 (1), 193 (2019).
- Cazaly, E. Making sense of the epigenome using data integration approaches. Frontiers in Pharmacology. 10, 126 (2019).
- Leal, A., Sidransky, D., Brait, M. Tissue and cell-free DNA-based epigenomic approaches for cancer detection. Clinical Chemistry. 66 (1), 105-116 (2020).
- Song, W., Ren, J., Wang, W. J., Wang, C. T., Fu, T. Genome-wide methylation and expression profiling identify a novel epigenetic signature in gastrointestinal pan-adenocarcinomas. Epigenomics. 12 (11), (2020).
- Wong, E. M., et al. Tools for translational epigenetic studies involving formalin-fixed paraffin-embedded human tissue: applying the Infinium HumanMethyation450 Beadchip assay to large population-based studies. BMC Research Notes. 8, 543 (2015).
- . Illumina Available from: https://jp.support.illumina.com/downloads/infinium_humanmethylation450_product_files.html (2020)
- Fackler, M. J., et al. Genome-wide methylation analysis identifies genes specific to breast cancer hormone receptor status and risk of recurrence. Cancer Research. 71 (19), 6195-6207 (2011).
- Dedeurwaerder, S., et al. A comprehensive overview of Infinium HumanMethylation450 data processing. Briefings in Bioinformatics. 15 (6), 929-941 (2014).
- Hulbert, A., et al. Early detection of lung cancer using DNA promoter hypermethylation in plasma and sputum. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 23 (8), 1998-2005 (2017).
- Shimada, H., Fukagawa, T., Haga, Y., Oba, K. Does remnant gastric cancer really differ from primary gastric cancer? A systematic review of the literature by the Task Force of Japanese Gastric Cancer Association. Gastric Cancer: Official Journal of the International Gastric Cancer Association and the Japanese Gastric Cancer Association. 19 (2), 339-349 (2016).
- Eijsink, J. J., et al. Detection of cervical neoplasia by DNA methylation analysis in cervico-vaginal lavages, a feasibility study. Gynecologic Oncology. 120 (2), 280-283 (2011).
- Sugimoto, K., et al. DNA methylation genome-wide analysis in remnant and primary gastric cancers. Gastric Cancer: Official Journal of the International Gastric Cancer Association and the Japanese Gastric Cancer Association. 22 (6), 1109-1120 (2019).
- Wang, Z., Wu, X., Wang, Y. A framework for analyzing DNA methylation data from Illumina Infinium HumanMethylation450 BeadChip. BMC Bioinformatics. 19, 115 (2018).
- Teh, A. L., et al. Comparison of methyl-capture sequencing vs. Infinium 450K methylation array for methylome analysis in clinical samples. Epigenetics. 11 (1), 36-48 (2016).
- Zhou, W., Laird, P. W., Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Research. 45 (4), 22 (2017).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati