
Method Article
Spot Variation Fluorescence Correlation Spectroscopy for Analysis of Molecular Diffusion at the Plasma Membrane of Living Cells
* These authors contributed equally
In This Article
Summary
This article aims to present a protocol on how to build a spot variation Fluorescence Correlation Spectroscopy (svFCS) microscope to measure molecular diffusion at the plasma membrane of living cells.
Abstract
Dynamic biological processes in living cells, including those associated with plasma membrane organization, occur on various spatial and temporal scales, ranging from nanometers to micrometers and microseconds to minutes, respectively. Such a broad range of biological processes challenges conventional microscopy approaches. Here, we detail the procedure for implementing spot variation Fluorescence Correlation Spectroscopy (svFCS) measurements using a classical fluorescence microscope that has been customized. The protocol includes a specific performance check of the svFCS setup and the guidelines for molecular diffusion measurements by svFCS on the plasma membrane of living cells under physiological conditions. Additionally, we provide a procedure for disrupting plasma membrane raft nanodomains by cholesterol oxidase treatment and demonstrate how these changes in the lateral organization of the plasma membrane might be revealed by svFCS analysis. In conclusion, this fluorescence-based method can provide unprecedented details on the lateral organization of the plasma membrane with the appropriate spatial and temporal resolution.
Introduction
The complexity of plasma membrane organization
The current understanding of cell membrane organization has to take into account several aspects1. First, a complex lipid composition varies not only between cell types, but also within a single cell (membrane organelles/plasma membrane). Besides, associated or intrinsic membrane proteins are mostly organized in dynamic multimeric complexes, with large domains extending outside of the membrane, accounting for a significantly larger area than that of the transmembrane domains alone. Moreover, membrane-associated proteins exhibit specific lipid-binding or lipid-interacting capacities that play roles in regulating protein function. These depend directly on the local composition and accessibility of the lipids2.
Finally, a significant level of asymmetry is observed between two membrane leaflets due to the intrinsic asymmetric structure of membrane proteins and the distribution of lipids. Indeed, a lipid metabolic balance between synthesis and hydrolysis, combined with lipid flip-flop between the leaflets, generates such asymmetric distribution. As any transport across the bilayer is constrained by the free energy required to move the polar head group through the hydrophobic interior of the membranes, it is usually assisted by selective transporters. For each cell type, asymmetry tends to be firmly maintained. Altogether, these factors contribute to lateral inhomogeneity or compartmentalization of the plasma membrane3,4.
We enrich this representation of the plasma membrane by taking into account the intrinsic molecular diffusion within and across the bilayer, which contributes to the dynamic lateral heterogeneity on a scale of tenths to hundreds of nanometers and microseconds to seconds. For instance, lipid-dependent membrane nanodomains—the so-called lipid rafts, defined as cholesterol, and sphingolipid-rich signaling platforms—contribute to the compartmentalization of the plasma membrane5,6. However, the current view of membrane organization is not restricted to lipid rafts alone. Membrane nanodomains are more complex and heterogeneous in composition, origin, and function. Still, their presence at the plasma membrane has to be tightly coordinated, and dynamic interactions between proteins and lipids seem to be important in the spatial distribution and chemical modification of membrane nanodomains1,3,7,8.
The svFCS principle and its application to probe the organization of the plasma membrane
Although much progress has been made in the analysis of membrane domains, mainly through biophysical techniques, the determinants that dictate the local organization of the plasma membrane need to be refined with appropriate spatial and temporal resolution. Determinants based on tracking individual molecules provide excellent spatial precision and allow the characterization of different modes of motion9,10,11,12, but have a limited temporal resolution with classical low camera frame rates and require more experimental effort to record a significant number of trajectories. Alternatively, the diffusion coefficient of membrane components can be evaluated by Fluorescence Recovery After Photobleaching (FRAP)13 or Fluorescence Correlation Spectroscopy (FCS)14. The latter has received more attention, mainly because of its high sensitivity and selectivity, microscopic detection volume, low invasiveness, and wide dynamic range15.
The conceptual basis of FCS was introduced by Magde and colleagues about 50 years ago16,17. It is based on recording the fluctuation of fluorescence emission with a high temporal resolution (from µs to s)18. In its modern version, measurements in living cells are performed by a small confocal excitation volume (~0.3 femtoliters) positioned within a region of interest (e.g., at the plasma membrane); the fluorescence signal generated by diffusing fluorescent molecules going in and out of the observation volume is collected with very high temporal resolution (i.e., the time of arrival of each photon on the detector). Then, the signal is computed to generate the autocorrelation function (ACF), from which the average time td (diffusion time) for which a molecule stays within the focal volume is extracted, together with the mean number of particles, (N), present in the observation volume, which is inversely proportional to the amplitude of the ACF. This last parameter might be useful information on the molecule concentration within the observation volume.
Since then, a growing number of FCS modalities have been implemented thanks to rapidly developing instrumentation in biophotonics, allowing the description of dynamic phenomena occurring in living systems. Still, a molecular species would experience a more overlapping distribution of the diffusion coefficient values, which is usually reflected by an anomalous diffusion characteristic, in which molecules diffuse with a nonlinear relationship in time19, and difficulty in identifying the biological meaning of this anomalous subdiffusion. In the past, this difficulty has been somewhat overcome by recording the molecular diffusion by FRAP from areas of various sizes, rather than from just one area, thereby providing additional spatial information. This enabled, for instance, the conceptualization of membrane microdomains20,21,22.
A translation of this strategy to FCS measurements (i.e., the so-called spot variation Fluorescence Correlation Spectroscopy (svFCS)) was established by varying the size of the focal volume of observation, allowing the fluctuation in fluorescence to be recorded on different spatial scales23. Thus, the svFCS approach provides indirect spatial information allowing for the identification and determination of molecular diffusion modes and type of membrane partitioning (isolated versus contiguous domains24) of studied molecules. By plotting the diffusion time td as a function of the various spatial scales defined by the waist (ω) value, which corresponds to the detection beam radius size in this case23,25, one can characterize the diffusion law of a given molecule in a given physiological condition. The svFCS is, therefore, a perfect analog to single-particle tracking in the time domain26. Under the Brownian diffusion constraint, one should expect a strictly linear relationship between the diffusion time td and the waist ω (Figure 1)23,25. The origin of the deviation of the diffusion law from this scheme can be attributed to nonexclusive reasons, such as cytoskeleton meshwork, molecular crowding, dynamic partitioning in nanodomains, or any combination of these and other effects (Figure 1), and needs to be tested experimentally25.
Here we provide all necessary control checkpoints for the daily use of a custom-made svFCS optical system built from scratch, which complements our previous protocol reviews27,28 on that experimental approach. Further, as a proof of concept, we give guidelines regarding the calibration of the setup, the preparation of cells, data acquisition, and analysis for the establishment of svFCS diffusion law (DL) for Thy1-GFP, a plasma membrane glycosylphosphatidylinositol-anchored protein, which is known to be localized in lipid-raft nanodomains29. Finally, we demonstrate how the partial destabilization of lipid-raft nanodomains by cholesterol oxidase treatment impacts the diffusion properties of Thy1-GFP. Additionally, a detailed description of building a svFCS setup from scratch is provided in Supplementary material.
Protocol
1. Setting specification for assembling a custom-made svFCS setup
NOTE: The simplicity of the proposed svFCS setup allows easy installation, operation, and maintenance at a low cost while ensuring efficiency in photon recovery. For more details, see Supplementary material.
- Experimental room and safety
- Install the system in a room stabilized at around 21 °C.
- Avoid direct airflow on the passive (or active) optical table and follow the laser safety rules for optical alignment.
- Hardware and software
NOTE: Supplementary material details the installation steps depicted in Figure 2.- Write the main acquisition and control software in LabVIEW using a state machine and event structure architecture where a multifunction acquisition board drives most of the controllers.
NOTE: The correlator, laser, and power meter are controlled or monitored by their own software. - Adapt the hardware and software installation procedures according to the hardware used.
- Write the main acquisition and control software in LabVIEW using a state machine and event structure architecture where a multifunction acquisition board drives most of the controllers.
- Optical setup
NOTE: Figure 3 illustrates the optical bench modules used in the following sections to control the quality of the optical alignments. All of the optical element specifications are listed in Table of materials. The procedure to build the setup is widely detailed in Supplementary material. This system comprises a continuous wave laser, a motorized inverted microscope equipped with an immersion water objective, an avalanche photodiode detector coupled to a single photon counting module, and a hardware correlator. A microscope incubation chamber with vibration-free heaters has been specially designed to control the temperature for experiments on living cells. By convention, the XY axis corresponds to the optical path's perpendicular plane, and the Z-axis corresponds to the optical path.
2. Daily checkpoint before running the experiment
- Control the excitation path (Figure 3,
 &
&  ).
).
- Open all the iris diaphragms.
- Measure the laser power with the power meter, keeping the first iris fully open.
- Turn the half-wave plate (HWP) to find the maximum power.
- Check the alignment using the irises if the laser power is lower than usual, and move L1 and M1 alternately, if necessary.
- Note the power value in the experiment laboratory notebook.
- Control the detection path (Figure 3,
 &
&  ).
).
- Place the water, a coverslip, and a droplet of a 2 nM rhodamine 6G (Rh6G) solution on the objective.
- If the fluorescence signal (count number on the APD, recorded with the LabVIEW software) is lower than usual, remake the Rh6G solution, check the positioning and the coverslip's number on the objective lens, or eliminate bubbles, if any.
- If the fluorescence signal is still lower than usual, place the power meter inside the optical path to block the beam.
- Turn off the APD (hereafter, APD refers to the APD and the single photon counting module).
- Remove the sample.
- Clean and replace the objective lens with a reflective target.
- Check the laser beam on the reflective target by removing the power meter from the light path. Make sure that the target's beam is centered, and the back reflection reaches the first iris on the line (Figure 3).
- If not, adjust the center positioning with M2 or the back reflection with the dichroic mirror.
- If microscope coupling is correct, push the objective lens back, add a drop of water, a coverslip, and a droplet of a more concentrated Rh6G solution (i.e., 200 nM), and set a lower laser power than for the classical measurements (few µW).
- Turn on the APD and optimize APD and pinhole alignment, alternately, with their respective XYZ adjustment screws while monitoring the intensity signal (LabVIEW software).
- Change the coverslip and add a lower concentration of Rh6G (2 nM). Move the pinhole along the Z-axis to find a position where the molecular brightness ratio increases, and the waist is minimum.
- Close the iris until the signal drops down: the laser beam size reaches the back aperture size of the objective (i.e., the minimal waist size, see Supplementary material).
- Launch the correlator software and record data (see section 7 for data recording).
- Check the ACF, which should display a low amount of noise, give a small waist size, and a high count-rate per molecule per second (see section 7 for data analysis and waist size evaluation).
3. General considerations for svFCS data recording and analysis
- Record and analyze the fluorescence data following this general scheme (see sections 7, 8, and 9): (1) fluorescence recording and ACF generation (correlator software), (2) unexpected discarding of data, an average of retained data, fitting with the appropriate model (with homemade Igor Pro software), (3) diffusion law plot (homemade MATLAB software 1), and (4) optional diffusion law comparison (homemade MATLAB software 2). The different software programs are available upon request.
NOTE: The hardware correlator has a minimum sampling time of 12.5 ns (i.e., a sampling frequency of 80 MHz). It provides a temporal resolution that is at least 1,000 lower than the typical resident time of freely diffusing small molecule in solution and 106 smaller than the diffusion time of membrane proteins within a confocal observation volume.
4. Cell culture and transfection
- Seed the Cos7 cells in 8-well chambered coverglass with #1.0 borosilicate glass bottom at a density of 10,000 cells/well using complete Dulbecco's Modified Eagle Medium (DMEM) supplemented with 5% fetal bovine serum, penicillin (100 U/mL), streptomycin (100 U/mL), and L-glutamine (1 mM).
- Culture the cells at 37 °C in a humidified atmosphere containing 5% CO2 for 24 h.
- Remove the medium, add 300 µL of the fresh complete culture medium per well, and preincubate the cells for 30 min at 37 °C.
- Dilute 0.5 µg of the plasmid DNA encoding Thy-1 protein fused with eGFP25 in 50 µL of serum-free DMEM. Vortex briefly to mix.
- Dilute 1.5 µL of the DNA transfection reagent in 50 µL of serum-free DMEM, and mix the solution well.
- Add the diluted transfection reagent directly into the prepared DNA solution, and mix the compounds immediately.
- Incubate the prepared mixture for 10 to 15 min at room temperature.
- Add 10 µL of the combined DNA/transfection reagent complexes dropwise onto the medium in each well, and homogenize by gently swirling the plate.
- Incubate the cells at 37 °C with 5% CO2 for 3 h.
- After the incubation, replace the medium containing DNA/transfection reagent complexes with 400 µL of fresh complete DMEM, and culture the cells for 16 h before the svFCS experiment.
5. Preparation of cells for svFCS measurements
- Remove the culture medium.
- Wash the cells gently two to three times with serum-free Hank’s balanced salt solution (HBSS) buffer containing Ca2+ and Mg2+ supplemented with 10 mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES), pH 7.4 (HBSS/HEPES).
- Maintain the cells in HBSS/HEPES buffer during all svFCS acquisitions.
6. Pharmacological treatment
- Remove the culture medium, and wash the cells two to three times with serum-free HBSS supplemented with 10 mM HEPES, pH 7.4 (HBSS/HEPES).
- Incubate the cells with 1 U/mL of cholesterol oxidase (COase) solution in HBSS/HEPES buffer for 1 h at 37 °C.
- Remove the solution, and maintain the cells in the presence of 0.1 U/mL of COase in HBSS/HEPES buffer while performing the svFCS measurements.
7. Spot size calibration
- Prewarm the microscope chamber at 37 °C.
- Prepare a standard 2 nM solution of Rh6G by serial dilution.
- Drop 200 µL of 2 nM Rh6G solution on a glass coverslip placed on the water-immersion objective.
- Start all the hardware and software.
- Measure and adjust the 488 nm laser beam power to 300 µW. Depending on the brightness and the photo-stability of the fluorescent probe used, adapt this power according to (1) the fluorescence intensity (on the LabVIEW software), which should be stable, (2) the ACF shape (on the correlator software), which should have a constant shape over the time, and (3) the fitting parameters giving a small waist size and a high count rate per molecule (photons per molecule per second, typically few tens to hundreds of photons per molecule per second).
NOTE: The amplitude of the ACF (called G(0)) is inversely proportional to the number of the molecule (i.e., the concentration of the fluorescent probe). For the waist size calibration, this is a good quality control candidate parameter. Therefore, G(0) should be similar for the same concentration from day to day as it links the waist size and concentration. For cell measurements, as FCS is more accurate for low concentration, G(0) should be high for the proper parameter fitting extraction. - Set the svFCS illumination/detection microscope port with the LabVIEW software.
- Turn on the APD.
- Close the iris until the signal drops down to obtain the minimal waist size, or close it for bigger waist size.
- Record several ACFs of selected duration (namely a run) to improve statistical reproducibility, typically 10 runs lasting for 20 s each with the correlator software.
- Turn off the APD.
- Use the Igor Pro software to check and discard the runs with strong fluctuations due to molecular aggregates. Perform this step manually— it should be user-independent after users have been trained.
- Fit the average of the retained ACFs with a 3D diffusion model.
- Extract from the fitting parameters the average diffusion time
 and save it into a “.txt” file (the file format is dictated by the Igor Pro software).
and save it into a “.txt” file (the file format is dictated by the Igor Pro software). - Check the count-rate per molecule per second (a good performance indicator) by dividing the average intensity (extracted from the fluorescence trace) by the number of molecules (extracted from the ACF).
NOTE: Make sure that this value is high and stable from day to day for the same acquisition parameters. - Knowing the diffusion coefficient of Rh6G in aqueous solution at 37 °C (D) and (see 7.13), calculate the experimental waist size ω according to:
 .
. - Apply the procedure for every waist size modification required to plot the FCS diffusion law and before any new experimental series of svFCS data acquisition.
8. svFCS data acquisitions on cells
- Measure and adjust the 488 nm beam power between 2 and 4 µW. Depending on the brightness and the photostability of the fluorescent probe used, adapt this power to allow a high count rate per molecule (typically several thousand of photons per molecule per second), while keeping the photobleaching low (i.e., a stable intensity trace on the LabVIEW software).
- Equilibrate samples for 10 min at 37 °C before starting the measurements.
- Set the epi-fluorescence illumination microscope with the LabVIEW software.
- Choose a cell with an appropriate fluorescent probe location and (low) fluorescence signal intensity.
NOTE: The lower the fluorescence is, the better the FCS measurements are (see step 8.1). - Set the svFCS illumination/detection microscope port with the LabVIEW software.
- Turn on the APD.
- Perform an xy-scan of the selected cell with the LabVIEW software.
- Perform a z-scan and locate the confocal spot at the maximal fluorescence intensity by choosing the plasma membrane at the top and start the data acquisition. To maximize the separation between the two membranes, preferably perform the scan in the nuclear area of the cell.
- Record one series of 20 runs lasting for 5 s, each with the correlator software.
NOTE: Make sure that the duration of each run is long enough to obtain ACFs with reduced noise. Long acquisitions are susceptible to photobleaching or unexpected substantial variations (e.g., aggregates). Adapt the number of runs, their duration, and the number of series to the samples, but make sure that they remain constant within the same bulk of experiments for reproducibility. - Turn off the APD.
- Discard unexpected runs with the Igor Pro software.
- Fit the average ACF with a 2-species 2D diffusion model. Adapt this model to the type of diffusion behavior of the target molecule.
- Save the fitting parameters into the previous file (see step 7.13).
- Perform 10 to 15 series of recordings on at least 10 different cells, and reproduce steps 8.3 to 8.13. Check that the single file obtained contains the waist size information and the fitting parameters of the 10–15 recordings.
- To establish a single diffusion law, analyze at least four waist sizes varying between 200 and 400 nm. This range is defined by the diffraction optical limit, but is objective- (numerical aperture) and laser (wavelength)-dependent.
NOTE: As the waist size calibration is not absolute and has some degree of uncertainty, a dedicated MATLAB software28 accounting for the x and y error (namely ω2 and td) was built to fit the diffusion law. - Start the MATLAB software 1 and select a folder containing all the “.txt” files corresponding to at least four waist size experiments.
- Plot <td> versus <ω2>, namely the diffusion law. Two major parameters can be extracted: the y-axis intercept (t0) and the effective diffusion coefficient (Deff, inversely proportional to the slope).
9. Diffusion laws of different experimental condition comparison
NOTE: If necessary, reproduce sections 7 and 8 for different experimental conditions. A dedicated software (MATLAB software 2) was developed to determine whether these diffusion laws are similar or not according to the t0 and Deff values28. It tests two hypotheses: the two values are different, or the two values are not different at a threshold set above a probability of false alarm (PFA). An arbitrary PFA value of 5% (T = 3.8) is considered the upper limit of significance between two parameters (t0 or Deff), indicating that there is only 5% chance that the two values are identical.
- Create an “.xls” file containing the characteristic diffusion law values of each condition to compare (i.e., a file containing the t0, t0 error, Deff and Deff error for the non-treated (NT) and treated (COase) conditions as a table).
- Start the MATLAB software 2.
- Select the “.xls” file.
- Analyze the generated color-coded 2D plot, where the t0 and Deff statistical tests are to be plotted on the x- and y-axes, respectively (Figure 4). The higher T is, the greater is the difference between the compared values.
10. Cholesterol concentration measurements
- Cell treatment and lysis
- Seed the Cos7 cells in triplicate in 6-well plates at 4 × 105 cells/well and incubate in 2 mL of complete DMEM at 37 °C with 5% CO2 overnight to allow the cells to attach to the plate.
- Remove the culture medium and wash the cells three times with phosphate-buffered saline (PBS).
- Add 1 mL of HBSS/HEPES buffer containing (or not, for controls) 1 U/mL of Coase, and incubate for 1 h at 37 °C with 5% CO2.
- Replace the medium with 1 mL of HBSS/HEPES containing 0.1 U/mL of Coase, and incubate for 1 h at 37 °C with 5% CO2.
- Remove the solution and harvest the cells.
- Wash the cells three times with PBS, and centrifuge at 400 × g for 5 min at room temperature.
- Lyse the cells with radioimmunoprecipitation assay buffer (25 mM HEPES, pH 7.4, 150 mM NaCl, 1% NP40, 10 mM, MgCl2, 1 mM ethylenediamine tetraacetic acid, 2% glycerol, protease and phosphatase inhibitor cocktail) for 30 min on ice.
- Centrifuge the lysates at 10000 × g for 10 min at 4 °C and collect the supernatant.
- Quantify total protein concentration for each sample by modified Bradford's protein assay using the working solution according to the manufacturer's recommendations.
- Cholesterol concentration measurement
- To determine total cellular cholesterol level enzymatically, use the appropriate kit (e.g., Amplex Red Cholesterol Assay Kit) according to the manufacturer's recommendations.
- For each reaction, mix the sample containing 5 µg of protein with Amplex Red reagent/horseradish peroxidase/cholesterol oxidase/cholesterol esterase working solution, and incubate for 30 min at 37 °C in the dark.
- Measure the fluorescence using excitation of 520 nm, and detect the emission at 560–590 nm using a microplate reader.
- Subtract the background from the final value, and determine cholesterol concentration using a standard curve.
- Calculate the final cholesterol content in ng of cholesterol per µg of protein.
Results
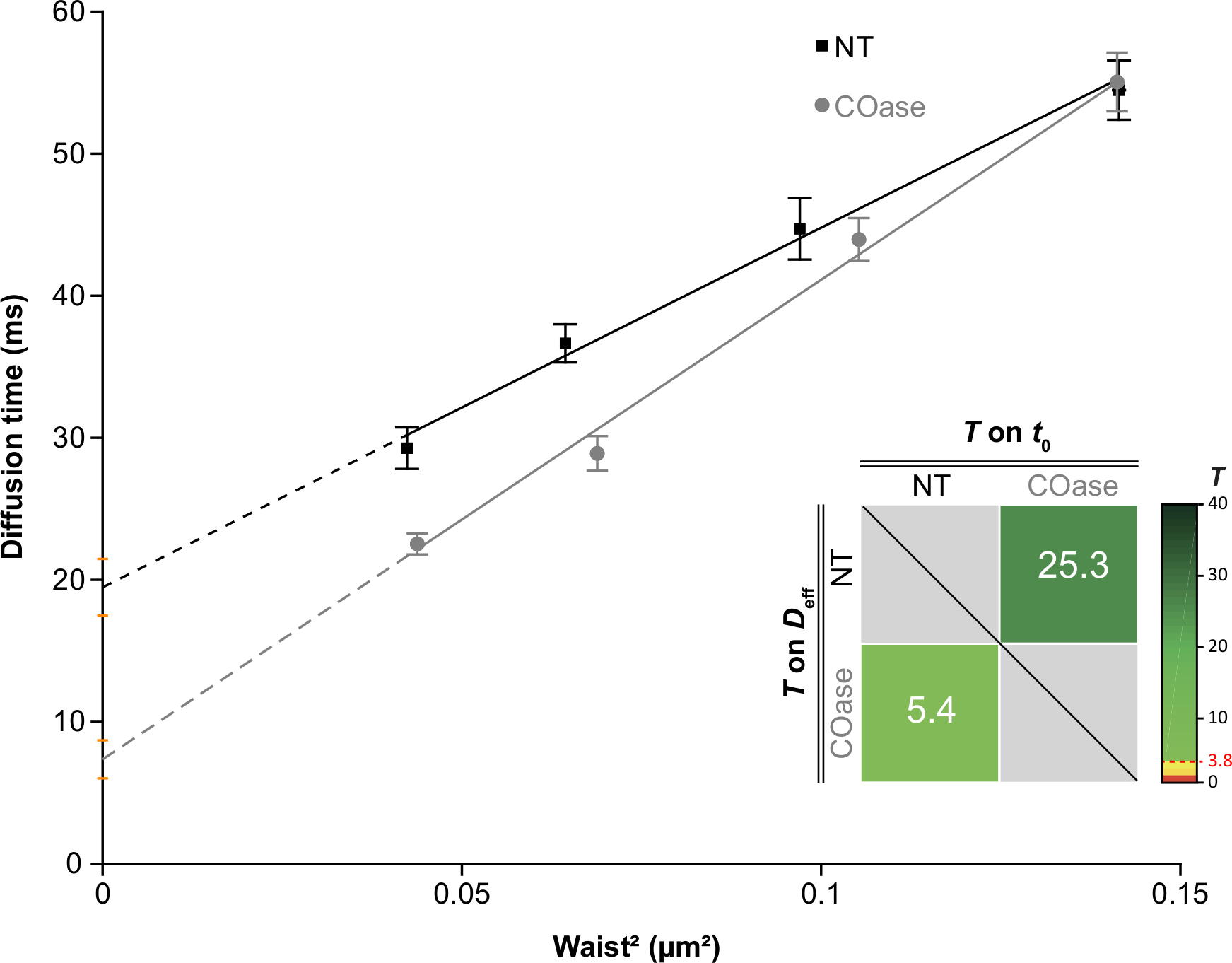
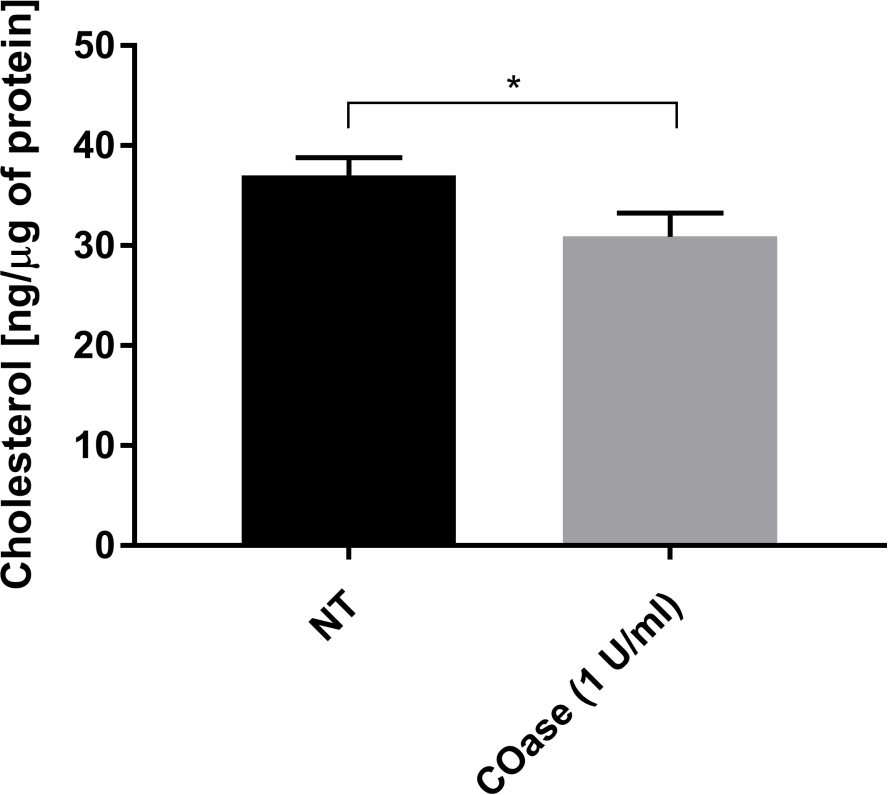
We generated a DL for Thy1-GFP expressed in Cos-7 cells (Figure 4, black squares). The diffusion law has a positive t0 value (19.47 ms ± 2 ms), indicating that Thy1-GFP is confined in nanodomain structures of the plasma membrane. The cholesterol oxidase treatment of the cells expressing Thy1-GFP resulted in the shift of the DL t0 value to 7.36 ± 1.34 ms (Figure 4, gray squares). This observation confirms that the nature of Thy1-GFP confinement depends on the cholesterol content and is associated with lipid raft nanodomains. These two diffusion laws are shown to be different according to the statistical test described above (see step 9.1.3) in terms of t0 and Deff values. In addition, we assessed the concentration of total cellular cholesterol in non-treated Cos-7 cells versus the cells treated with COase. A small, but significant, decrease in total cholesterol content is observed upon COase treatment (Figure 5). As this enzyme acts only on the cholesterol pool accessible at the outer leaflet of the plasma membrane, we assume that the observed decrease in cholesterol is associated only with the plasma membrane and results in the destabilization of lipid raft nanodomains.

Figure 1: Simulated fluorescence correlation spectroscopy (FCS) diffusion laws established by spot-variation FCS for different forms of membrane organization. (Upper panels) Schematic representation of membrane organization—(A) free diffusion, (B) meshwork barriers, and (C) trap/domain confinements—with the trajectory drawn for a single molecule (red). Blue circles denote the intersection of the membrane and laser beam of waist ω. (Lower panels) FCS diffusion laws represented by plotting the diffusion time td as a function of the squared radius ω2. Diffusion law projection (green dashed line) intercepts the time axis at (A) the origin (t0 = 0) in the case of free diffusion; (B) in negative axis (t0 < 0) when there are meshwork barriers, or (C) in positive axis (t0 > 0) when there are traps and domains (lipid rafts). D is the lateral diffusion coefficient for Brownian motion; Deff, the effective diffusion coefficient; Dmicro, the microscopic diffusion coefficient inside the meshwork traps; Din, the diffusion coefficient inside domains; Dout, the diffusion coefficient outside domains; L, the size of the side of a square domain; and rD, the radius of a circular domain. This figure has been modified from He and Marguet6. Please click here to view a larger version of this figure.
{kind=link}

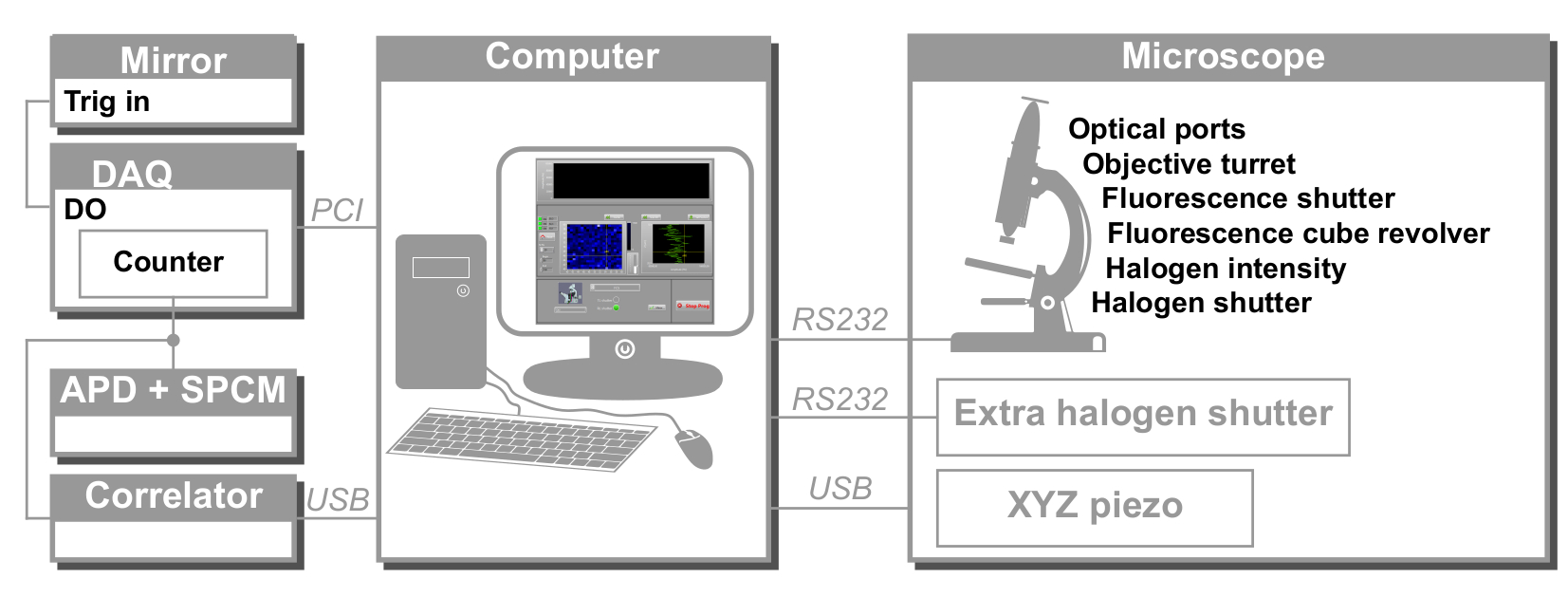
Figure 2: Schematic view of svFCS hardware control. The computer controls all the devices through different communication protocols: serial (microscope, external shutter), USB (XYZ piezoelectric stage, correlator), and PCI (acquisition board). DAQ: data acquisition board, APD: avalanche photodiode, SPCM: single-photon counting module, DO: digital output. Please click here to view a larger version of this figure.
{kind=link}

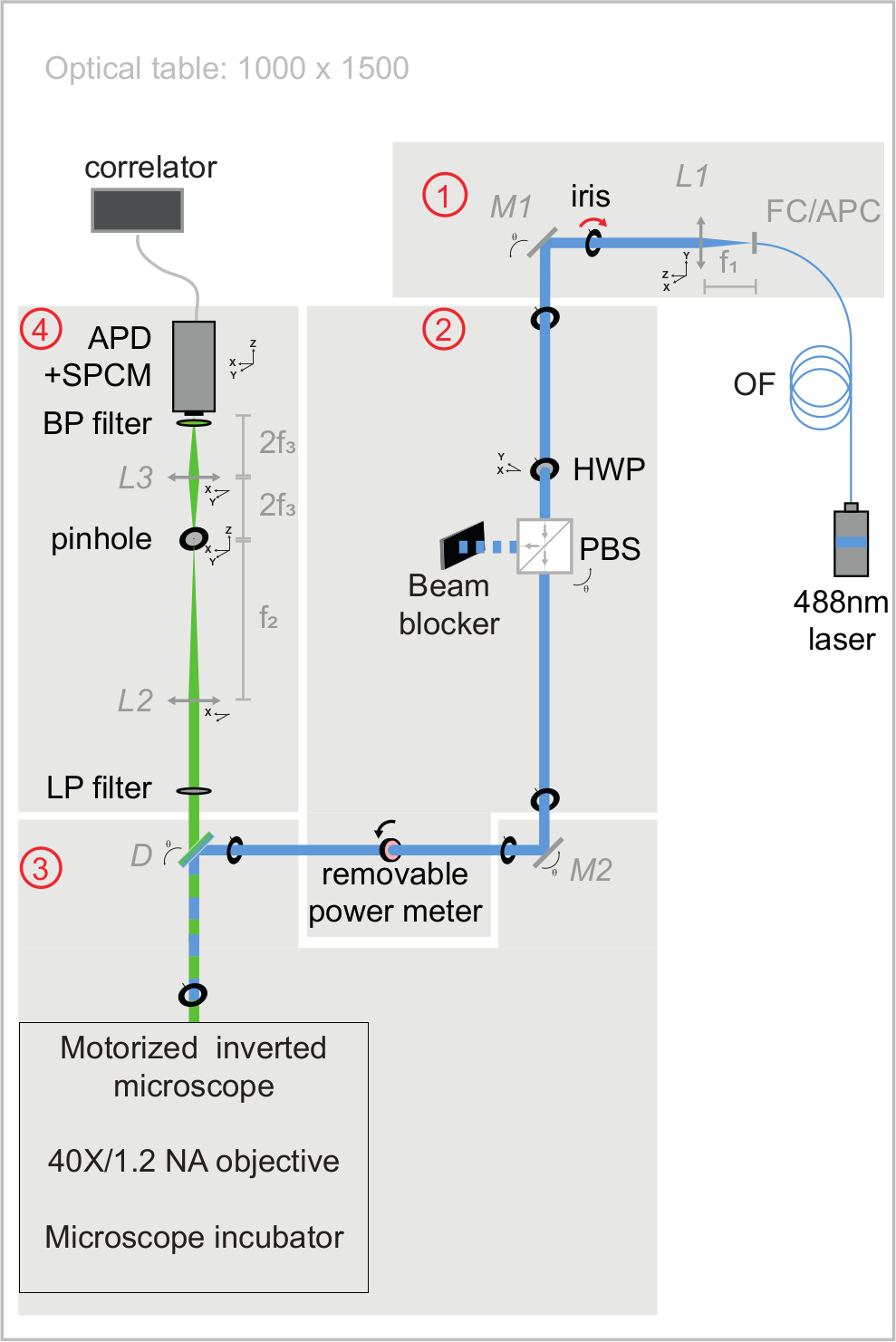
Figure 3: Schematic view of excitation and emission optical paths of the svFCS setup. The svFCS setup contains four modules: (1) the output of a fibered 488 nm laser is collimated, (2) a combination of a half-wave plate and polarizing beam-splitter sets the optical power, (3) the laser beam focused on the sample after traveling through a tube-lens free motorized microscope, and (4) the fluorescence is detected through a confocal-like detection path onto an avalanche photodiode coupled to a single photon counting module, which delivers a signal to a hardware correlator. Simplicity gives the system its sensitivity, robustness, and ease of use (widely commented in Supplementary material). Please click here to view a larger version of this figure.
{kind=link}

Figure 4: The svFCS diffusion laws generated from diffusion analysis of Thy1-GFP expressed in Cos-7. svFCS diffusion laws of Cos-7 cells without treatment (NT, black squares) and after cholesterol oxidase treatment (COase, gray circles). The insert in the graph represents statistical testing of a significant difference between the two presented svFCS diffusion laws (according to Mailfert et al.28). The test value (T) should be above the threshold set at 3.8 when both diffusion laws are different. The higher it is, the greater is the difference between the diffusion laws. The value of T is color-coded. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Comparison of total cholesterol content in Cos-7 cells. Cos-7 cells were either non-treated (NT) or treated with 1 U/mL of cholesterol oxidase (COase) for 1 h. The data represent an example of one experiment in triplicate. A two-tailed, unpaired t-test was used to assess the statistical difference (α=0.05). Please click here to view a larger version of this figure.
{kind=link}
Table of materials: The list of optical elements required for the svFCS setup.
Supplementary material: This document describes the building of a svFCS setup from scratch. Please click here to download this file.
Discussion
Here, we have described the implementation of the svFCS module on a standard fluorescent microscope, a powerful experimental approach to decipher the dynamics of the plasma membrane organization in living cells thanks to the FCS diffusion law analysis. Conceptually, the svFCS is based on a simple principle: correlation measurements of fluorescence in the time domain while varying the size of the illumination area23. This strategy has been instrumental in deducing nanoscopic information from microscopic measurements, which helps decipher the main physicochemical elements contributing to the plasma membrane organization in steady state25 and physiological processes30,31,32,33. Altogether, these svFCS analyses unambiguously demonstrate the existence of lipid-dependent nanodomains in various cell types and their direct implication in tuning different signaling events.
Within this framework, there are some optical aspects that need to be considered while building the svFCS setup to optimize the photon budget and minimize optical aberrations. Thus, we recommend using a microscope from which the tube lens can be removed when the svFCS measurement is performed. Moreover, a single iris plays a key role in the svFCS setup: it changes the beam size at the back aperture of the objective, thus directly varying the effective waist size (i.e., the effective excitation volume). The beam diameter should fit the objective back pupil to obtain the smallest waist size34. This option, which helps tune the waist size, ensures optimization of the photon budget and is easy to implement. Finally, a minimal number of optical parts are used along the light path; the less complex the system, the fewer the photons that are lost. All of these options significantly improve the robustness of svFCS experiments.
Regarding the protocol itself, a few critical steps have to be considered. The most important is an appropriate alignment of the optical paths that is crucial for successful svFCS measurements (protocol, section 2). This is easy to check by analyzing the fluorescence signal from a 2 nM Rh6G solution, which should be ~200 kHz under 300 µW laser illumination. All irises should be opened, and the ACFs should have an important amplitude (typically G0~1.5–2.0). Another critical point concerns the cells and their preparation for svFCS analysis (protocol, sections 4–8). Their density has to be adapted so that isolated cells to be observed are available for analysis. Non-adherent cells have to be immobilized on a chambered coverglass by using poly-L-lysine solution. The fluorescence signal from cell labeling should not be too strong, or it will result in very flat ACFs that are difficult to fit, and the fit parameters are burdened with an important error. Additionally, nonhomogeneous labeling and fluorescence aggregates in cells make the svFCS measurements extremely difficult to interpret. Finally, cholesterol oxidase treatment affects cell viability, and the svFCS analysis should not exceed one hour after the treatment. It is also better to record the fluorescence fluctuations from the upper plasma membrane as it is not attached to the support, and there is no risk of hindered diffusion of molecules due to the physical interactions with the support.
There have been enough advances in the svFCS technique for its use in different approaches owing to the diversity of modalities for adjusting the detection volume, making it possible to study various biological processes in living cells. An alternative to adjusting the size of the excitation volume is to use a variable beam expander35. It is also possible to simply modulate the size of the illumination area by recording the fluorescent signal from the intercept of the plasma membrane along the z direction36. This can be done on a standard confocal microscope for which a theoretical framework has been developed to derive the diffusion law37,38.
Although the svFCS method offers spatio-temporal resolution, which is necessary for the characterization of the inhomogeneous lateral organization of the plasma membrane, the geometrical modes of confinement are not mutually exclusive. A deviation of t0 in one direction or the other exclusively reveals a dominant mode of confinement25. Moreover, another important limitation of the present svFCS method results from the classical optical diffraction limit (~200 nm). This is unquestionably greater than the domains confining the molecules within the cell plasma membrane. Therefore, the analysis of the confinement is inferred from the t0 value, extrapolated from the diffusion law.
This drawback has been overcome by implementing alternative methods. Initially, using metallic films drilled with nanoapertures offered the possibility of illuminating a very small membrane area (i.e., below the optical diffraction limit of single nanometric apertures of radii varying between 75 and 250 nm)39. The transition regime predicted from the theoretical diffusion law for isolated domain organization was thus reported, and it allowed a refinement of the characteristic size of the nanometric membrane heterogeneities and a quantitative estimate of the surface area occupied by lipid-dependent nanodomains39. Alternatively, nanometric illumination has also been developed using near-field scanning optical microscopy40 or planar optical nanoantennas41. More recently, combining stimulated emission depletion (STED) and FCS has provided a powerful and sensitive tool to document the diffusion law with very high spatial resolution. This STED-FCS gives access to molecular diffusion characteristics on a nanoscale occurring within a short period of time, allowing the study of the dynamic organization of lipid probes at the plasma membrane42,43. However, the incomplete suppression of fluorescence in the STED process challenges the analysis of the auto-correlation curves in FCS.
A new fitting model has been developed to overcome this difficulty, improving the accuracy of the diffusion times and average molecule numbers measurements44. Finally, for slow molecular diffusion at the plasma membrane, the svFCS principle can be applied to data recorded by image correlation spectroscopy45. Recently, it has been demonstrated that combining atomic force microscopy (AFM) with imaging total internal reflection-FCS (ITIR-FCS) contributes to the refinement of the nature of the mechanism hindering molecular diffusion at the plasma membrane, especially near the percolation threshold membrane configuration because of a high density of nanodomains46.
In conclusion, establishing diffusion law by svFCS has provided the experimental evidence to infer local heterogeneity created by dynamic collective lipids and membrane proteins’ associations. As stated by Wohland and co-workers46, “the FCS diffusion law analysis remains a valuable tool to infer structural and organizational features below the resolution limit from dynamic information”. Still, we need to develop new models to refine the interpretation of the diffusion law that should allow for a better understanding of the dynamics of the molecular events occurring at the plasma membrane.
Disclosures
The authors have nothing to disclose.
Acknowledgements
SB, SM and DM was supported by institutional funding from the CNRS, Inserm and Aix-Marseille University and program grants from the French National Research Agency (ANR-17-CE15-0032-01 and ANR-18-CE15-0021-02) and the French "Investissement d'Avenir" (ANR-10-INBS-04 France-BioImaging, ANR-11-LABX-054 labex INFORM). KW acknowledges "BioTechNan", a program of interdisciplinary environmental doctoral studies KNOW in the field of Biotechnology and Nanotechnology. EB acknowledges the financial support of the National Science Centre of Poland (NCN) under project no. 2016/21/D/NZ1/00285, as well as the French Government and the Embassy of France in Poland. MŁ acknowledges the financial support from the Polish Ministry of Development (CBR POIR.02.01.00-00-0159/15-00/19) and the National Center for Research and Development (Innochem POIR.01.02.00-00-0064/17). TT acknowledges financial support from the National Science Center of Poland (NCN) under project no. 2016/21/B/NZ3/00343 and from the Wroclaw Biotechnology Center (KNOW).
Materials
| Name | Company | Catalog Number | Comments |
| Aligment tool | Spanner Wrench for SM1-Threaded Retaining Rings | Thorlabs | SPW602 |
| Avalanche Photodiode and Single Photon Counting Module (SPCM) | Single-Photon Counting Module, Avalanche Photodiode | Excelitas | SPCM-AQRH-15 |
| BNC 50 Ω plug to 50 Ω plug lead 2 m | RS Components | 742-4315 | |
| Coaxial cable 415 Cinch Connectors, RG-316, 50 Ω With connector, 1.22 m, RoHS2 | RS Components | 885-8172 | |
| Tee 50Ω RF Adapter BNC Plug to BNC Socket 0 → 1GHz | RS Components | 546-4948 | |
| Brennenstuhl 2.5 m, 8 Socket Type E – French Extension Lead, 230 V | RS Components | 768-5500 | |
| Mascot, 6W Plug In Power Supply 5V dc, 1.2A, 1 Output Switched Mode Power Supply, Type C | RS Components | 452-8394 | |
| Crystek CCSMACL-MC-24 Reference Oscillator Power Cable RF Adapter | RS Components | 792-4079 | |
| Fluorescence filtering | 535/70 ET Bandpass, AOI 0° Chroma Diameter 25 mm | AHF filter | F47-539 |
| Laser Beamsplitter zt488 RDC, AOI 45° Chroma 25.5 x 36 x 1 mm | AHF filter | F43-088 | |
| 496/LP BrightLine HC Longpass Filter, AOI 0° Chroma Diameter 25 | AHF filter | F37-496 | |
| Hardware correlator | 80 MHz Digital Correlator | Correlator.com | Flex02-12D |
| Laser | LASER LASOS LDM-XT fiber coupled, 488 nm, 65 mW | Lasos | BLD-XT 488100 |
| Laser safety | High-Performance Black Masking Tape, 1" x 180' (25 mm x 55 m) Roll | Thorlabs | T743-2.0 |
| Lens Tissues, 25 Sheets per Booklet, 5 Booklets | Thorlabs | MC-5 | |
| Laser Safety Glasses, Light Orange Lenses, 48% Visible Light Transmission | Thorlabs | LG3B | |
| Microscope | Zeiss Axiovert 200M Motorized Inverted Fluorescence Microscope Fine and coarse focusing, reflector turret rotation, objective nosepiece rotation, switching camera ports, and internal light shutters | Carl Zeiss | |
| C-Apochromat 40x/1,2 W Korr.selected for FCS (D=0.14-0.19 mm) (WD=0.28 mm at D=0.17 mm), UV-VIS-IR | Carl Zeiss | 421767-9971-711 | |
| Adapter W0.8 / M27x0.75 H "5" | Carl Zeiss | 000000-1698-345 | |
| Middle ring W0.8 - W0.8 H "5" | Carl Zeiss | 000000-1698-347 | |
| Optical path | D25.4mm Mirror, Protected Silver | Thorlabs | PF10-03-P01 |
| D25.4mm, F=60.0.mm, Visible Achromat | Thorlabs | AC254-060-A | |
| D25.4mm, F=35.0.mm, Visible Achromat | Thorlabs | AC254-035-A | |
| 25 µm mounted pinhole | Thorlabs | P25S I | |
| 25.4mm Mounted Zero, Order 1/2 Waveplate 488 nm | Thorlabs | WPH10M-488 (HWP) | |
| 20mm Polarizing Beamsplitter Cube 420-680 nm | Thorlabs | PBS201 | |
| Rotation Stage 56 mm x 26 mm Threaded ID | Thorlabs | RSP1/M | |
| 52 mm x 52 mm Kinematic Platform Mount | Thorlabs | KM100B/M | |
| Adjustable Prism Clamp | Thorlabs | PM3/M | |
| Beam block - active area 19 mm x 38 mm | Thorlabs | LB1/M | |
| Iris Diaphragm 1 mm to 25 mm Aperture | Thorlabs | ID25/M | |
| Left-Handed Kinematic Cylindrical Lens Mount | Thorlabs | KM100CL | |
| 1" Optic Holder, M4 Tap | Thorlabs | MFF101/M | |
| 1" Stackable Lens Tube | Thorlabs | SM1L03 | |
| Stackable Lens Mount for 1" optic-usable depth ½ | Thorlabs | SM1L05 | |
| Stackable Lens Mount For 1"Optic-usable Depth 2" | Thorlabs | SM1L20 | |
| Small Optical Rails 600mm, metric | Thorlabs | RLA600/M | |
| Small Optical Rails 75mm, metric | Thorlabs | RLA075/M | |
| Small Optical Rails 150mm, metric | Thorlabs | RLA150/M | |
| Rail Carrier, Counterbored Hole 1"x 1" | Thorlabs | RC1 | |
| Rail Carrier, Perpendicular Dovetail | Thorlabs | RC3 | |
| High Precision Translating Lens Mount for 1 inch | Thorlabs | LM1XY/M | |
| ½ " (12mm) Dovetail Translation Stage | Thorlabs | DT12/M | |
| Rail Clamps | Thorlabs | CL6 | |
| Metric XYZ Translation Stage (Includes PT102) | Thorlabs | PT3/M | |
| Black Rubberized Fabric | Thorlabs | BK5 | |
| Ball Driver kit/ 6 tools | Thorlabs | BD-KIT/M | |
| Adapter with External M6 x 1.0 Threads and External M4 x 0.7 Threads | Thorlabs | AP6M4M | |
| Mounting Base, 25 mm x 58 mm x 10 mm, 5 Pack | Thorlabs | BA1S/M-P5 | |
| Lens Mount for 25.4mm optic | Thorlabs | LMR1/M | |
| SM1 FC/APC Adapter | Thorlabs | SM1FCA | |
| Kinematic Mirror Mount For 1 inch Optics | Thorlabs | KM100 | |
| Silicon Power Head, 400-1100nm, 50mW | Thorlabs | S120C | |
| 12.7 mm Post Holders, Spring-Loaded Hex-Locking Thumbscrew, L=50 mm, 5 Pack | Thorlabs | PH50/M-P5 | |
| Post Holder with Spring-Loaded Hex-Locking Thumbscrew, L=20 mm | Thorlabs | PH20/M | |
| 12.7 mm x 50 mm Stainless Steel Optical Post, M4 Stud, M6-Tapped Hole, 5 Pack | Thorlabs | TR50/M-P5 | |
| 12.7 mm x 75 mm Stainless Steel Optical Post, M4 Stud, M6-Tapped Hole, 5 Pack | Thorlabs | TR75/M-P5 | |
| USB Power and Energy Meter Interface | Thorlabs | PM100USB | |
| 12.7 mm x 30 mm Stainless Steel Optical Post, M4 Stud, M6-Tapped Hole | Thorlabs | TR30/M | |
| 12.7 mm x 20 mm Stainless Steel Optical Post, M4 Stud, M6-Tapped Hole | Thorlabs | TR20/M | |
| 750 mm long Structural Rail (detection box) | Thorlabs | XE25L750/M | |
| 350 mm long Structural Rail (detection box) | Thorlabs | XE25L350/M | |
| Quick Corner Cube for 25 mm Rails | Thorlabs | XE25W3 | |
| Right-Angle Bracket for 25 mm Rails | Thorlabs | XE25A90 | |
| Black posterboard 20" x 30" (508 mm x 762 mm), 1/16" (1.6 mm) Thick, 5 Sheets | Thorlabs | TB5 | |
| Hinge for 25 mm Rail Enclosures | Thorlabs | XE25H | |
| Lid Stop for 25 mm Rail Enclosures | Thorlabs | XE25LS | |
| M4 Cap Screw Kit | Thorlabs | HW-KIT1/M | |
| M6 Cap Screw Kit and Hardware kit | Thorlabs | HW-KIT2/M | |
| Table Clamp, L-Shape, 5 Pack | Thorlabs | CL5-P5 | |
| SM1 Ring-Actuated Iris Diaphragm (Ø0.8 - Ø12 mm) | Thorlabs | SM1D12D | |
| Ø1" SM1-Mounted Frosted Glass Alignment Disk w/Ø1 mm Hole | Thorlabs | DG10-1500-H1-MD | |
| Honeycomb Optical Table Top, Standa | Standa | 1HB10-15-12 | |
| Optical Table support, Standa | Standa | 1TS05-12-06-AR | |
| Sample nano-positionning | Precision XYZ Nanopositioning | Physik Instrumente | PI P527-3.CD |
| Digital Multi-Channel Piezo Co, 3 Channels, -30 to 130 V Sub- D Connector(s), Capacitive Sensors, | Physik Instrumente | PI E727-3.CD | |
| Temperature chamber | Zeiss 200M Inverted Microscope Incubator System MATT BLK | Digital Pixel | |
| Dual Channel Microprocessor Temperature Controller | Digital Pixel | DP_MTC_2000_DUO | |
| Two Vibration Free Heater Modules | Digital Pixel | DP_150_VF | |
| PT100 Temperature Sensor | Digital Pixel | DP_P100_TS | |
| Biological Reagents and Materials | |||
| Cell culture and transfection | Cos7 cells | ATCC® | CRL-1651™ |
| 8- well Lab-Tek chambers | Thermo Fisher Scientific | 155411PK | |
| Dulbecco's Modified Eagle Medium (DMEM) | Thermo Fisher Scientific | 11965092 | |
| Fetal bovine serum | Thermo Fisher Scientific | 16000044 | |
| L-glutamine | Thermo Fisher Scientific | 25030081 | |
| PBS buffer | Thermo Fisher Scientific | 14190144 | |
| PenStrep | Thermo Fisher Scientific | 15140122 | |
| PolyJet Transfection Reagent | SignaGen Laboratories | SL100688 | |
| Cholesterol content measurement | Amplex Red Cholesterol Assay Kit | Thermo Fisher Scientific | A12216 |
| Protease Inhibitor Cocktail | Thermo Fisher Scientific | 87786 | |
| Phosphatase Inhibitor Cocktail | Thermo Fisher Scientific | 78420 | |
| ROTI Nanoquant Working Solution | Roth | K880 | |
| GloMax Discover Microplate Reader | Promega | GM3000 | |
| svFCS measurements | HBSS buffer | Thermo Fisher Scientific | 14025092 |
| Hepes buffer | Thermo Fisher Scientific | 15630080 | |
| Cholesterol oxidase | Sigma-Aldrich | C8868 | |
| Rhodamine 6G | Sigma-Aldrich | 83697-1G |
References
- Engelman, D. M. Membranes are more mosaic than fluid. Nature. 438 (7068), 578-580 (2005).
- Newton, A. C. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. The Biochemical Journal. 370, Pt 2 361-371 (2003).
- Marguet, D., Lenne, P. F., Rigneault, H., He, H. T. Dynamics in the plasma membrane: how to combine fluidity and order. The EMBO Journal. 25 (15), 3446-3457 (2006).
- Edidin, M. The state of lipid rafts: From model membranes to cells. Annual Review of Biophysics and Biomolecular Structure. , (2003).
- Lingwood, D., Simons, K. Lipid rafts as a membrane-organizing principle. Science. , (2010).
- He, H. T., Marguet, D. Detecting nanodomains in living cell membrane by fluorescence correlation spectroscopy. Annual Reviews of Physical Chemistry. 62, 417-436 (2011).
- Rossy, J., Ma, Y., Gaus, K. The organisation of the cell membrane: Do proteins rule lipids. Current Opinion in Chemical Biology. 20 (1), 54-59 (2014).
- Nicolson, G. L. The Fluid - Mosaic Model of Membrane Structure: Still relevant to understanding the structure, function and dynamics of biological membranes after more than 40 years. Biochimica et Biophysica Acta - Biomembranes. 1838 (6), 1451-1466 (2014).
- Fujiwara, T., Ritchie, K., Murakoshi, H., Jacobson, K., Kusumi, A. Phospholipids undergo hop diffusion in compartmentalized cell membrane. The Journal of Cell Biology. 157 (6), 1071-1081 (2002).
- Kusumi, A., et al. Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: high-speed single-molecule tracking of membrane molecules. Annual Review of Biophysics and Biomolecular Structure. 34, 351-378 (2005).
- Destainville, N., Salomé, L. Quantification and correction of systematic errors due to detector time-averaging in single-molecule tracking experiments. Biophysical Journal. 90 (2), 17-19 (2006).
- Wieser, S., Moertelmaier, M., Fuertbauer, E., Stockinger, H., Schütz, G. J. Un)confined diffusion of CD59 in the plasma membrane determined by high-resolution single molecule microscopy. Biophysical Journal. 92 (10), 3719-3728 (2007).
- Axelrod, D., Koppel, D. E., Schlessinger, J., Elson, E., Webb, W. W. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophysical Journal. , (1976).
- Petrášek, Z., Schwille, P. Precise measurement of diffusion coefficients using scanning fluorescence correlation spectroscopy. Biophysical Journal. , (2008).
- Elson, E. L. 40 Years of FCS: How it all began. Methods in Enzymology. , (2013).
- Elson, E. L., Magde, D. Fluorescence correlation spectroscopy. I. Conceptual basis and theory. Biopolymers. , (1974).
- Magde, D., Elson, E. L., Webb, W. W. Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers. , (1974).
- Schwille, P., Haupts, U., Maiti, S., Webb, W. W. Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one- and two-photon excitation. Biophysical Journal. , (1999).
- Bouchaud, J. P., Georges, A. Anomalous diffusion in disordered media: Statistical mechanisms, models and physical applications. Physics Reports. , (1990).
- Yechiel, E., Edidin, M. Micrometer-scale domains in fibroblast plasma membranes. Journal of Cell Biology. 105 (2), 755-760 (1987).
- Salomé, L., Cazeils, J. L., Lopez, A., Tocanne, J. F. Characterization of membrane domains by FRAP experiments at variable observation areas. European Biophysics Journal. 27 (4), 391-402 (1998).
- Niv, H., Gutman, O., Kloog, Y., Henis, Y. I. Activated K-Ras and H-Ras display different interactions with saturable nonraft sites at the surface of live cells. The Journal of Cell Biology. 157 (5), 865-872 (2002).
- Wawrezinieck, L., Rigneault, H., Marguet, D., Lenne, P. F. Fluorescence correlation spectroscopy diffusion laws to probe the submicron cell membrane organization. Biophysical Journal. 89 (6), 4029-4042 (2005).
- Saxton, M. J. Fluorescence corralation spectroscopy. Biophysical Journal. , (2005).
- Lenne, P. F., et al. Dynamic molecular confinement in the plasma membrane by microdomains and the cytoskeleton meshwork. EMBO Journal. 25 (14), 3245-3256 (2006).
- Ruprecht, V., et al. Cortical contractility triggers a stochastic switch to fast amoeboid cell motility. Cell. , (2015).
- Billaudeau, C., et al. Probing the plasma membrane organization in living cells by spot variation fluorescence correlation spectroscopy. Methods in Enzymology. 519, 277-302 (2013).
- Mailfert, S., Hamon, Y., Bertaux, N., He, H. T., Marguet, D. A user's guide for characterizing plasma membrane subdomains in living cells by spot variation fluorescence correlation spectroscopy. Methods in Cell Biology. 139, 1-22 (2017).
- Rege, T. A., Hagood, J. S. Thy-1, a versatile modulator of signaling affecting cellular adhesion, proliferation, survival, and cytokine/growth factor responses. Biochimica et Biophysica Acta - Molecular Cell Research. , (2006).
- Cahuzac, N., et al. Fas ligand is localized to membrane rafts, where it displays increased cell death-inducing activity. Blood. , (2006).
- Guia, S., et al. Confinement of activating receptors at the plasma membrane controls natural killer cell tolerance. Science Signaling. 4 (167), 21(2011).
- Blouin, C. M., et al. Glycosylation-dependent IFN-γR partitioning in lipid and actin nanodomains is critical for JAK activation. Cell. 166 (4), 920-934 (2016).
- Chouaki-Benmansour, N., et al. Phosphoinositides regulate the TCR/CD3 complex membrane dynamics and activation. Scientific Reports. , (2018).
- Wawrezinieck, L., Lenne, P. F., Marguet, D., Rigneault, H. Fluorescence correlation spectroscopy to determine diffusion laws: application to live cell membranes. Biophotonics Micro- and Nano-Imaging. , (2004).
- Masuda, A., Ushida, K., Okamoto, T. New fluorescence correlation spectroscopy enabling direct observation of spatiotemporal dependence of diffusion constants as an evidence of anomalous transport in extracellular matrices. Biophysical Journal. , (2005).
- Humpolíčková, J., et al. Probing diffusion laws within cellular membranes by Z-scan fluorescence correlation spectroscopy. Biophysical Journal. , (2006).
- Benda, A., et al. How to determine diffusion coefficients in planar phospholipid systems by confocal fluorescence correlation spectroscopy. Langmuir. , (2003).
- Ganguly, S., Chattopadhyay, A. Cholesterol depletion mimics the effect of cytoskeletal destabilization on membrane dynamics of the serotonin1A receptor: A zFCS study. Biophysical Journal. , (2010).
- Wenger, J., et al. Diffusion analysis within single nanometric apertures reveals the ultrafine cell membrane organization. Biophysical Journal. 92 (3), 913-919 (2007).
- Manzo, C., Van Zanten, T. S., Garcia-Parajo, M. F. Nanoscale fluorescence correlation spectroscopy on intact living cell membranes with NSOM probes. Biophysical Journal. , (2011).
- Regmi, R., et al. Planar optical nanoantennas resolve cholesterol-dependent nanoscale heterogeneities in the plasma membrane of living cells. Nano Letters. , (2017).
- Mueller, V., et al. FCS in STED microscopy: Studying the nanoscale of lipid membrane dynamics. Methods in Enzymology. , (2013).
- Sezgin, E., et al. Measuring nanoscale diffusion dynamics in cellular membranes with super-resolution STED-FCS. Nature Protocols. , (2019).
- Wang, R., et al. A straightforward STED-background corrected fitting model for unbiased STED-FCS analyses. Methods. , (2018).
- Veerapathiran, S., Wohland, T. The imaging FCS diffusion law in the presence of multiple diffusive modes. Methods. , (2018).
- Gupta, A., Phang, I. Y., Wohland, T. To hop or not to hop: exceptions in the FCS diffusion law. Biophysical Journal. , (2020).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved