
Method Article
Antibiotic Efficacy Testing in an Ex vivo Model of Pseudomonas aeruginosa and Staphylococcus aureus Biofilms in the Cystic Fibrosis Lung
In This Article
Summary
This workflow can be used to perform antibiotic susceptibility testing using an established ex vivo model of bacterial biofilm in the lungs of individuals with cystic fibrosis. Use of this model could enhance the clinical validity of MBEC (minimal biofilm eradication concentration) assays.
Abstract
The effective prescription of antibiotics for the bacterial biofilms present within the lungs of individuals with cystic fibrosis (CF) is limited by a poor correlation between antibiotic susceptibility testing (AST) results using standard diagnostic methods (e.g., broth microdilution, disk diffusion, or Etest) and clinical outcomes after antibiotic treatment. Attempts to improve AST by the use of off-the-shelf biofilm growth platforms show little improvement in results. The limited ability of in vitro biofilm systems to mimic the physicochemical environment of the CF lung and, therefore bacterial physiology and biofilm architecture, also acts as a brake on the discovery of novel therapies for CF infection. Here, we present a protocol to perform AST of CF pathogens grown as mature, in vivo-like biofilms in an ex vivo CF lung model comprised of pig bronchiolar tissue and synthetic CF sputum (ex vivo pig lung, EVPL).
Several in vitro assays exist for biofilm susceptibility testing, using either standard laboratory medium or various formulations of synthetic CF sputum in microtiter plates. Both growth medium and biofilm substrate (polystyrene plate vs. bronchiolar tissue) are likely to affect biofilm antibiotic tolerance. We show enhanced tolerance of clinical Pseudomonas aeruginosa and Staphylococcus aureus isolates in the ex vivo model; the effects of antibiotic treatment of biofilms is not correlated with the minimum inhibitory concentration (MIC) in standard microdilution assays or a sensitive/resistant classification in disk diffusion assays.
The ex vivo platform could be used for bespoke biofilm AST of patient samples and as an enhanced testing platform for potential antibiofilm agents during pharmaceutical research and development. Improving the prescription or acceleration of antibiofilm drug discovery through the use of more in vivo-like testing platforms could drastically improve health outcomes for individuals with CF, as well as reduce the costs of clinical treatment and discovery research.
Introduction
Chronic biofilm infections affect individuals whose normal immune defenses are compromised. Groups at risk include those with the genetic condition cystic fibrosis (CF)1. Colonization of the abnormally thick, adhesive mucus in the respiratory tract in early infancy leads to intractable biofilm infections of the bronchioles2,3. The growth of bacteria as extensive matrix-encapsulated biofilms is one factor that distinguishes chronic infections of immunocompromised people from acute infections of healthy hosts and the biofilm state both protects bacteria from antibiotic exposure (due to reduced diffusion through the matrix) and decreases their antibiotic susceptibility (e.g., through induction of quiescence or upregulation of efflux pumps)4,5. However, disease-specific alterations in host tissue physiology and chemistry further alter bacterial physiology from that observed in acute infections or in standard laboratory growth conditions. Key examples in CF include the use of unusual carbon sources, such as fatty acids and amino acids released from lung surfactant and produced by microbial degradation of mucin, the release of micronutrients, such as iron from damaged tissues, and microaerobiosis6,7,8.
The specific physicochemical conditions in a particular biofilm infection context can therefore influence responses to antibiotics. First, the structure and depth of the extracellular matrix depends on local environmental conditions, such as nutrients or shear forces. Second, environmental cues can trigger expression of specific antibiotic resistance genes. For example, the CF pathogen Pseudomonas aeruginosa shows increased expression of a beta-lactamase and reduced expression of porins in CF sputum versus in vitro9, while another CF pathogen, Burkholderia cenocepacia, upregulates beta-lactamases and efflux pumps when grown in CF sputum10. Third, in-host conditions can cue a physiological or genetic switch to antibiotic-tolerant phenotypes, which are hard to recapitulate in vitro. These include small colony variants of the CF pathogen Staphylococcus aureus11,12.
All of these data indicate that when diagnostic labs isolate individual clones from pathogenic biofilm and perform AST on planktonic or agar-plate grown cultures in standard laboratory media (broth microdilution, disk diffusion or Etest), the results often do not predict which antibiotics will actually work in vivo. Even if in vitro biofilm assays are used, they may not cue an in vivo-like biofilm phenotype due to differences in the medium and attachment surface used, so assays using flow cells or high-throughput microplate platforms can over-estimate antibiotic sensitivity13. The same problem applies to researchers in academia and industry seeking to develop new antibiofilm agents: testing drug potential using in vitro platforms like flow cells, microtiter plates, or Center for Disease Control biofilm reactors may set the biofilm efficacy bar too low and produce false positives in the research, development pipeline.
The poor correlation between AST results and clinical outcome after antibiotic treatment in CF is well known. Many clinicians simply ignore diagnostic lab AST as there are no uniform, CF-specific guidelines for interpreting these results and instead make case-by-case decisions for prescribing. Attempts have been made to improve CF AST by using the Calgary biofilm device, which uses biofilms grown on the surface of plastic pegs set within the wells of a microplate containing standard AST medium (e.g., cation-adjusted Muller-Hinton broth)14,15. This assay does no better at predicting which antibiotics will work in vivo than standard planktonic AST16. The impact on patients with CF is stark. Despite repeated antibiotic administration (regular inhaled antibiotics and a median of 27 days/year receiving intravenous antibiotics for individuals with CF in the United Kingdom)17, frequent and unpredictable episodes of acute pulmonary exacerbation lead to progressive lung damage and, in approximately 90% of cases, death from respiratory failure. In a recent analysis, bacterial lung infection was the strongest predictor of medication costs in CF, adding on average €3.6K/patient/year to direct healthcare costs18,19.
For acute infections of otherwise healthy individuals, current research and policy focusing on rapid AST based on, for example, point-of-care genomic prediction is ideal20. But in the case of chronic CF infections, it is clear that a different approach is needed: the implementation of AST in host-mimicking models that better recapitulate the in vivo environment and pathogen metabolic state and allow for the formation of realistic biofilm structure.
We have previously developed a CF biofilm model that comprises sections of pig bronchiole incubated in synthetic CF sputum and infected with P. aeruginosa or S. aureus. Uninfected EVPL retains normal histopathology for 7 days but lab or clinical isolates of P. aeruginosa and S. aureus reproducibly form in vivo-like aggregates around the tissue, mimicking the etiology of CF infection21,22,23. We present a protocol for using this high-validity, high-throughput model as a tailored biofilm AST platform for CF and present exemplary results showing the high tolerance of pathogen biofilms to clinically-used antibiotics when grown in the model. The model could readily be incorporated into research, development pipelines for the management or prevention of biofilm formation and potentially into diagnostic AST. Most equipment used (see Table of Materials) may readily be found in a typical microbiology laboratory, although a bead beater is essential, and we have found from work with collaborators that a suitable ultraviolet germicidal cabinet may also need to be procured. As the lungs are sourced from commercial butchers or abattoirs, the model presents no ethical concerns.
Protocol
This protocol uses pig lungs sourced from a commercial abattoir that supplies meat for human consumption. Under UK legislation, using leftover tissue from animals slaughtered for meat does not require ethical approval; we advise readers to check relevant local laws and institutional guidelines before starting work.
1. Preparation of Synthetic CF Sputum Media (SCFM)
- To make SCFM for use with EVPL tissue, follow the recipe outlined by Palmer et al.24 with the modification that glucose is removed from the recipe.
NOTE: Palmer et al.'s recipe contains free amino acids, cations, anions and lactate at concentrations representative of the average concentrations found in a selection of sputum samples from CF patients. It has been shown to cue comparable carbon-usage pathways and expression of quorum sensing signals by P. aeruginosa PA14 to growth in medium made from lyophilised patient sputum24. A recipe for 1 L modified SCFM is supplied in Table S1. - Filter sterilize the SCFM immediately after preparation and store at 4 °C for up to 1 month.
2. Dissection and infection of ex vivo pig lung (EVPL) tissue
- Prior to dissection, prepare an agar plate/s of required bacterial strain/s for infection using whatever agar is standard in the lab for P. aeruginosa/S. aureus (e.g., lysogeny broth + 1.2% agar).
- Calculate how many porcine bronchiolar tissue pieces are required for the experiment, including uninfected control tissue pieces. Multiply this number by two to repeat the experiment in two replicate lungs to confirm repeatability of results.
- Multiply the total number of tissue pieces required by 0.5 to determine the volume of SCFM agarose (mL) needed to make agarose pads to make enough medium for 400 µL/tissue piece plus spare SCFM agar to account for any pipetting errors or evaporation during preparation.
- Add 0.12 g of agarose to every 15 mL of SCFM required to make the desired total volume of SCFM with 0.8% weight/volume agarose.
- Heat the SCFM agarose solution until the agarose is fully dissolved. A domestic microwave on low power is recommended. The time required depends on the wattage of the microwave. Allow the agarose to cool to approximately 50 °C (warm to the touch but comfortable to hold). Do not allow to cool any further.
- Using a pipette, add 400 µL of the SCFM agarose to one well of a 24-well plate per tissue piece needed.
- Sterilize the SCFM agarose-containing 24-well plate/s under ultraviolet light for 10 min.
- Prepare three replicate washes for every intact lung being dissected using 20 mL of sterile Dulbecco's modified Eagle medium (DMEM) plus 20 mL of sterile Roswell Park Memorial Institute (RPMI) 1640 supplemented with 50 µg/mL ampicillin.
- Make an aliquot of 40 mL SCFM as a final wash for every intact lung being dissected. All washes can be stored overnight at 4 °C or used immediately.
- Obtain lungs from the designated source as soon as possible after slaughter, ensuring they are kept cold by transporting to the laboratory in a domestic coolbox.
NOTE: Lungs closer to the day of slaughter show less bruising from storage, but tissue kept on cold storage for up to 4 days from slaughter can also be used. As the coolbox needs to be taken into the butcher's shop or abattoir, it must be decontaminated following local lab guidelines after each use and stored outside the microbiology lab when not in use, to reduce the risk of contamination and a breach of containment. - Working on a sterilized surface and under a flame, place the lungs on a clean plastic chopping board covered with autoclaved aluminum foil. Check that the bronchioles remain intact. If there has been any damage at the abattoir or during transport the lungs are not suitable for use.
- Heat a palette knife under a flame and very briefly touch the knife to the area of the lung surrounding the bronchiole to sterilize the surface of the tissue.
- Cut away the surface tissue surrounding the bronchiole using a sterile mounted razor blade. Make incisions parallel to the bronchiole to prevent any damage.
- Once the bronchiole has been exposed, make a cross-sectional incision through the bronchiole at the highest point visible to free the bronchiole.
- Using sterile forceps, lightly hold the free end of the bronchiole and cut away any remaining unwanted tissue using a sterile mounted razor blade. Make a final cross-sectional incision across the bronchiole before any branching is visible to remove the bronchiole from the lungs.
- Place the bronchiole in the first DMEM/RPMI 1640 wash. Leave the bronchiole in the wash and repeat steps 2.11-2.14 to harvest additional sections of bronchiole from the same lung as required to yield sufficient tissue sections for the planned experiment.
- Place any additional bronchiolar sections from the same lung into the wash (step 2.16). Leave in the wash for at least 2 min.
- Remove the bronchioles from the first DMEM/RPMI 1640 wash and place the samples in a sterile Petri dish.
- Hold each bronchiole lightly using sterile forceps, making sure not to damage the tissue. Remove as much remaining soft tissue as possible and cut the tissue into ~5 mm wide strips using sterile dissection scissors.
- Place all of the bronchiolar tissue strips into the second DMEM/RPMI 1640 wash. Leave in the wash for at least 2 min.
- Remove the tissue strips from the second wash using sterile forceps, taking care not to damage the tissue. Place the tissue in a clean, sterile Petri dish.
- Remove any remaining soft tissue attached to the bronchiole and cut the strips into squares (~5 mm x 5 mm) using sterile dissection scissors.
- Add the third DMEM/RPMI 1640 wash into the Petri dish. Lightly mix the tissue pieces in the wash by swirling the dish.
- Pour the third wash out of the Petri dish without removing the tissue pieces.
- Add the final SCFM wash to the tissue-containing Petri dish, ensuring that all of the tissue pieces are covered.
- Sterilize the tissue pieces in SCFM under UV light for 5 min.
- Use sterile forceps to transfer each sterilized bronchiolar tissue piece into individual wells of a 24-well plate/s containing SCFM agarose pads.
- To infect each tissue piece with the desired bacterial strain, touch a colony grown on an agar plate with the tip of a 29 G needle attached to a sterile 0.5 mL insulin syringe. Then touch the colony onto the tissue piece, gently pricking the tissue surface.
NOTE: Using an insulin syringe equipped with a 29 G needle allows the needle to be held accurately and comfortably while keeping fingers a safe distance from the both needle and lung tissue. It is possible to perform this step using 29 G needles that are not attached to a syringe, but this requires greater dexterity and increases the risk of a needlestick injury. Insulin syringes are readily available. - For the uninfected controls, gently prick the surface of each of the tissue piece with the tip of a 29 G needle attached to a sterile 0.5 mL insulin syringe.
- Use a pipette to add 500 µL of SCFM to each well.
- Sterilize a breathable sealing membrane for each 24-well plate under ultraviolet light for 10 min (Table of Materials).
- Remove the lid/s from the 24-well plate/s and replace with the breathable membrane.
- Incubate the plates at 37 °C for the desired incubation (infection) time without shaking. Check that there is no visible growth of the inoculated pathogen on the uninfected control pieces (contamination control).
NOTE: If desired, ampicillin may be added to the SCFM agarose pads in step 2.5 and covering SCFM in step 2.30 to a final concentration of 20 µg/mL. This will suppress the growth of most endogenous bacteria on the lungs without affecting P. aeruginosa or S. aureus growth but, as the presence of ampicillin may affect susceptibility to other antibiotics, the reader is left to make this choice depending on the strains and antibiotics they wish to test.
3. Determination of antibiotic efficacy
NOTE: A schematic detailing the steps of this assay is provided in Figure S1.
- To measure antibiotic tolerance of biofilms formed on EVPL, replicate sets of lung pieces, from at least two independent lungs, must be set up during the dissection and infection. One set of pieces is required for a negative control (no antibiotic treatment), and one set is required for each concentration of antibiotic to be tested.
- After 48 hours of incubation, visually inspect the uninfected tissue pieces. Some growth of bacteria endogenous to the pig lung may have occurred, leading the SCFM around these sections to be turbid. If growth typical of the selected study species are observed (e.g., blue-green pigmentation diagnostic of P. aeruginosa), re-start the experiment with fresh lungs.
- If the uninfected tissue sections show no or only minimal bacterial growth, prepare one 24-well wash plate and one 24-well treatment plate, each containing 500 µL of fresh SCFM without antibiotics or with the antibiotic of interest per well per lung tissue piece.
- Remove each infected tissue piece from the incubation plate with flame sterilized forceps, swirl briefly in a fresh well of the wash plate to remove any non-biofilm associated bacterial cells, and transfer to the appropriate well of the treatment plate.
- Seal the treatment plates with fresh breathable membrane.
- Incubate the treatment plate/s at 37 °C without shaking for 18-24 h.
- Using flame sterilized forceps, remove each lung piece from the 24-well plate and put in a sterile 2 mL homogenization tube containing 1 mL of phosphate-buffered saline (PBS) and 1 g of metal beads (Table of Materials).
- Bead beat for 40 seconds at 4 m/s.
NOTE: Bead beating with the specific beads and homogeniser suggested in the Table of Materials does not cause significant lysis of bacteria, but each lab using the protocol should check the effects of their chosen beads and homogeniser prior to commencing AST assays. - Serially dilute the lung homogenate using PBS and plate on Lysogeny Broth (LB) agar to determine the colony forming units (CFU) in individual untreated and antibiotic-treated tissue pieces according to standard plating methods.
NOTE: Optional: Prepare duplicate plates on selective media to confirm colony identities; e.g., using mannitol salt agar for S. aureus.
Results
The EVPL model provides a high throughput assay platform, making it possible to screen a large number of bacterial isolates for antibiotic susceptibility at one time (Figures 1 and 2) or to screen strains against a range of antibiotic concentrations in one experiment (Figure 3). With practice, we have found that approximately 200 bronchiolar tissue sections can be prepared from lungs in 2 hours. The entire experiment for AST can be completed within normal working hours. Growth of Pseudomonas aeruginosa and Staphylococcus aureus isolates and the establishment of 48 h biofilm in the model is reliable and, when monitored by viable cell count, produces consistent bacterial loads (Figures 1 and 2). Images of tissue-associated biofilms of Pseudomonas aeruginosa and Staphylococcus aureus grown in EVPL may be found, along with protocols for preparation for light microscopy and histological staining, in our publications21,23. However, the reproducibility of CFU counts varies for different bacterial species. This can be quantified using standard repeatability calculations after ANOVA25; we have found that there is typically greater variation between CFU in replicate lung samples for S. aureus than for P. aeruginosa. We recommend that, on adoption of the model by a laboratory, repeat calculations are conducted on pilot experiments to optimize experimental techniques and to determine samples sizes to be used in final experiments (an example of this may be found in the data supplement for Sweeney et al26).
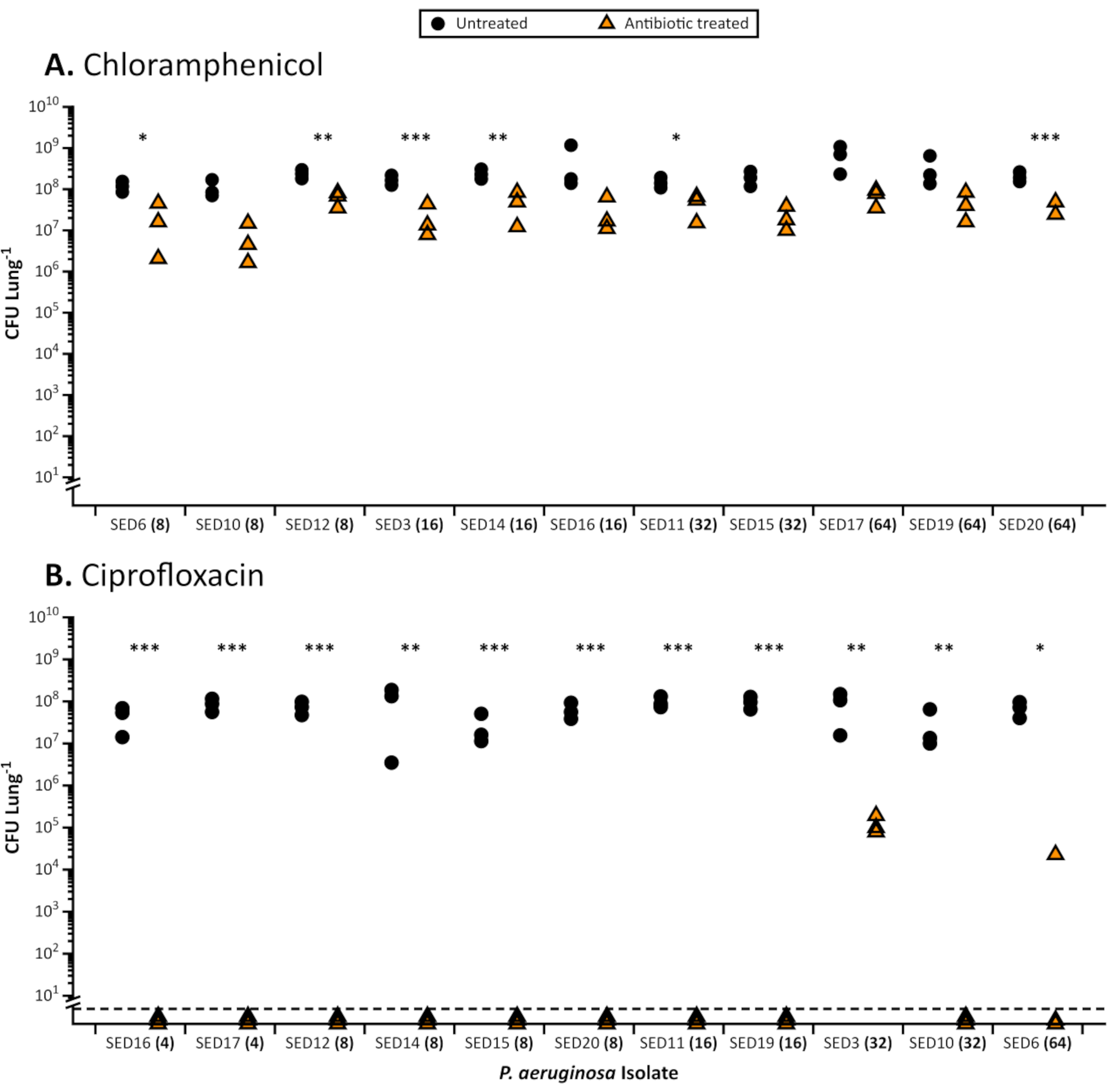
When grown in the EVPL, biofilms of P. aeruginosa and S. aureus demonstrate increased tolerance to antibiotics compared to susceptibility in standard, industry approved broth MIC (Figure 1) and disc assays using standard media (Figure 2). The various effects of different antibiotics on EVPL established biofilm are distinguishable, for example P. aeruginosa killing is achieved in EVPL with 4-16X MIC ciprofloxacin but not with 4-8X MIC chloramphenicol (Figure 1). A twice daily dose of 600 mg linezolid achieves a serum concentration above the MIC90 for susceptible pathogens (4 µg/mL)27 and is regarded as adequate exposure without adverse side effects28. Data presented in Figure 2 shows that S. aureus populations, susceptible to linezolid in the disc assay, are able to survive target serum concentrations, and higher (12 µg/mL), in EVPL. There is no clear correlation between MIC and antibiotic effects on EVPL-grown biofilms for P. aeruginosa (Figure 1). Gaining a more accurate measure of in vivo antibiotic tolerance is important because sub-optimal dosing of antibiotics could increase the risk of selection for resistance in chronic infection.
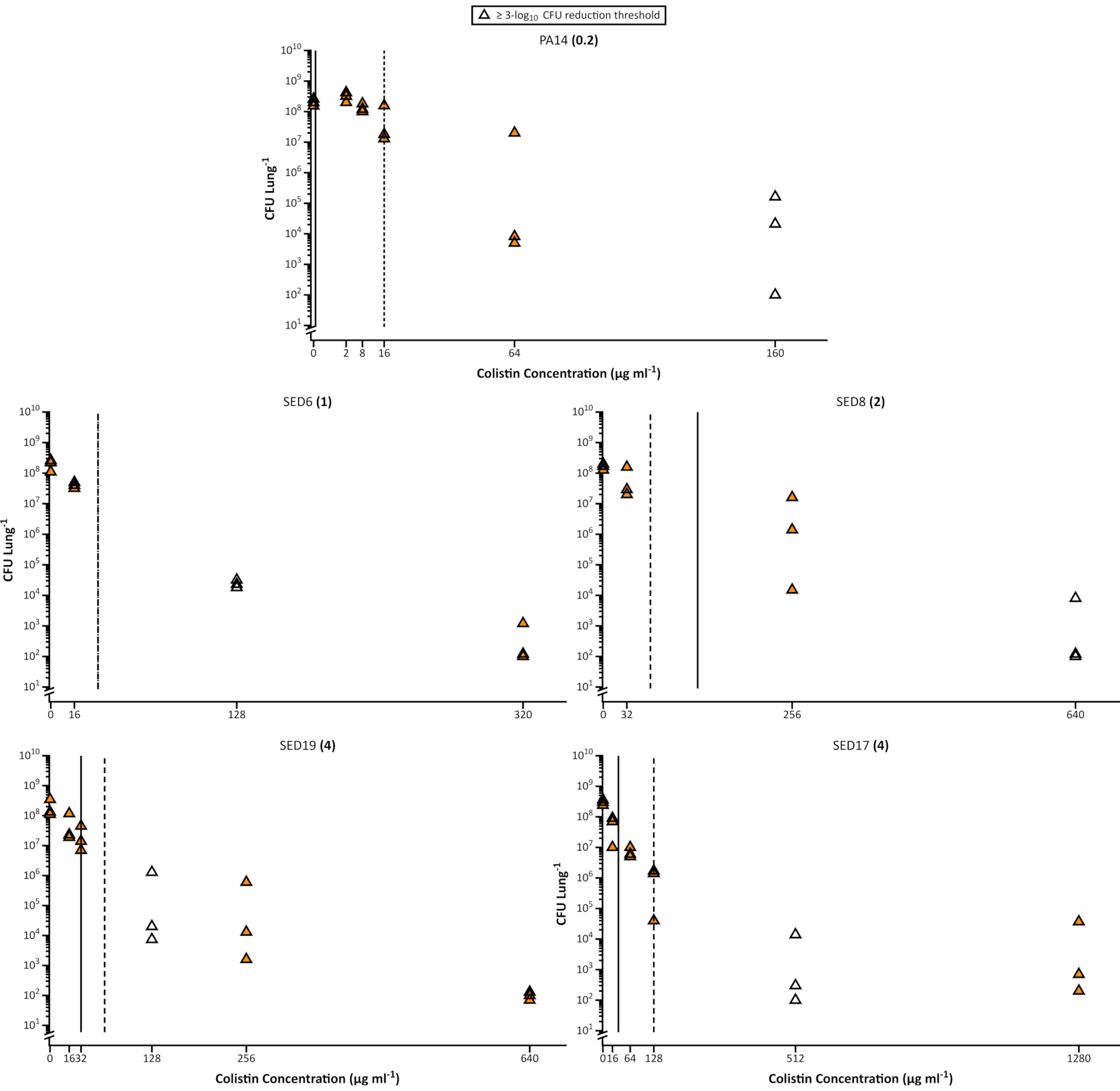
It is well known that the biofilm mode of growth can significantly reduce bacterial susceptibility to antibiotics. This has led to the development of many in vitro biofilm assays and the use of minimum biofilm eradication concentration (MBEC)14,15 instead of MIC as a more accurate predictor of susceptibility in chronic infection. The use of SCFM (in varying formulations) has also been recommended for use in MIC or MBEC testing29. Here we show that even an optimized in vitro assay cannot accurately predict P. aeruginosa susceptibility to colistin in the EVPL. The amount of antibiotic required to achieve 3 log10 killing of EVPL-grown bacteria is often significantly higher than the MIC or the MBEC calculated from standard in vitro assays, even when SCFM is used for these assays (Figure 3). This is consistent with a Cochrane review that reported that current implementations of in vitro biofilm susceptibility testing do not provide any increased predictive power for antibiotic prescribing in CF compared to standard susceptibility testing16.
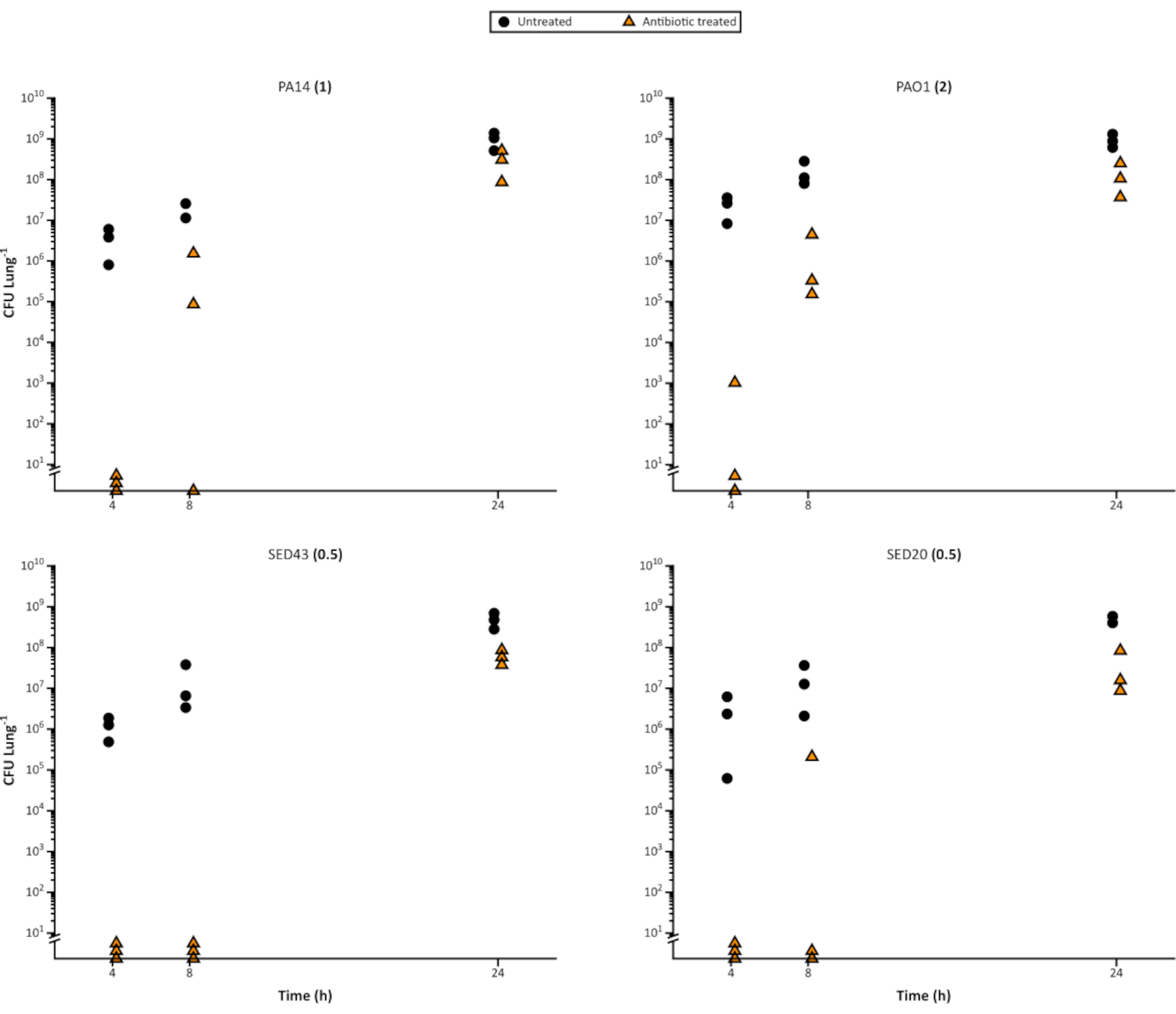
It is also simple to use the model to assess the impact of antibiotics on biofilm bacteria over time, as sufficient replica pieces of lung can be inoculated to allow destructive sampling. In addition to distinguishing differences between antimicrobial agents, the model can highlight changes in susceptibility at different bacterial growth stages or age of biofilm and for different antibiotic dosing intervals. Figure 4 illustrates the increasing tolerance of P. aeruginosa biofilms to meropenem as they mature. This could be useful to determine the efficacy of novel agents, for example whether they are more effective during rapid cell division. It may also be an important consideration when setting the constraints of an experiment, as it may be necessary to standardize and validate biofilm age to avoid the age having an influence on results.
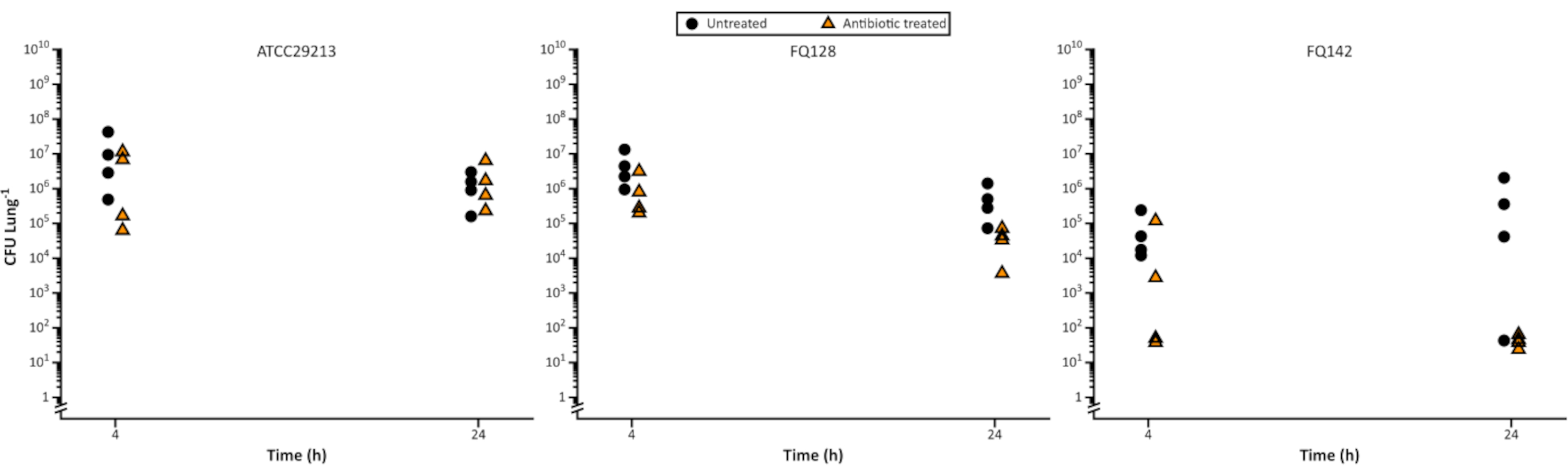
In Figure 5, S. aureus survival was measured at 4 h and 24 h post exposure to flucloxacillin and it was possible to observe differences in the reduction for bacterial cell counts across time and between isolates. This may be useful for drug development, for instance when defining pharmacokinetic and pharmacodynamic parameters or when elucidating the mode of action of a novel agent.
Variations in bacterial load often increase with extended culture times. This can be seen in the untreated control in Figure 5 following 48 h biofilm development and a further 24 h exposure to account for antibiotic dosing interval. Variation is intrinsic to the model; each lung sample is independent from others and reflects the natural variation of lungs. It is, therefore, important to ensure that a sufficient number of replicates is included to allow for validation and an accurate interpretation of results. We refer the reader back to our recommendation to conduct repeat calculations on the data to enable the selection of robust sample sizes.
For simplicity, we have presented representative data taken from replicate tissue sections acquired from a single pair of lungs in each experiment, but in practice it is necessary to perform repeat experiments on tissue sections taken from replicate animals. This should be done in order to account for any biological variation between individual pigs, and we refer the reader to our published work for examples of how consistent the results can be between tissues taken from replicate pigs and how this variation is accounted for in statistical analysis of data using analysis of variance (ANOVA)/general linear models (GLM)21,26.

Figure 1. Total CFU of 11 CF Pseudomonas aeruginosa clinical isolates recovered from the EVPL model following treatment with antibiotics. Representative results of antibiotic treatment of P. aeruginosa in the EVPL model. Each strain was grown on EVPL tissue for 48 h then transferred to antibiotic (triangles) or PBS as a control (circles) for 18 h and the CFU/lung determined. The MIC for the appropriate antibiotic determined in standard cation-adjusted MHB is shown in brackets next to each strain (x-axis). The strains are ordered by increasing MIC values. Data were analyzed using t-tests when appropriate and Mann-Whitney U tests for non-parametric datasets. Significant differences between antibiotic treated and untreated tissues are denoted by asterisks (P < 0.05). A. Recovered viable counts from P. aeruginosa biofilms grown in the EVPL model and treated with 64 µg/mL chloramphenicol (highest MIC value recorded). For each isolate, the standardized mean difference in CFU between chloramphenicol-treated and untreated tissue sections was calculated using Cohen's d. There was no correlation between MIC value in the standard test and the decrease in viable cell numbers in the EVPL model as measured by Cohen's d (Spearman's rank correlation, rs = 0.45, p = 0.16) B. Results of P. aeruginosa biofilms grown in the EVPL model and treated with 64 µg/mL ciprofloxacin (highest MIC value recorded). Values below the dashed line were below the limit of detection. Please click here to view a larger version of this figure.
{kind=link}

Figure 2. Total CFU of 8 Staphylococcus aureus CF clinical isolates recovered from the EVPL model following treatment with linezolid. Each strain was grown on EVPL tissue for 48 h then transferred to linezolid (triangles) for 24 h or were untreated as a control (circles). All strains were found to be sensitive to linezolid using the standard disk diffusion assay following EUCAST guidelines30 (zone of inhibition > 21 mm). Data were analyzed using t-tests when appropriate and Mann-Whitney U tests for non-parametric datasets (P < 0.05). No significant differences between antibiotic treated and untreated were found for any of the strains. Values below the dashed line were below the limit of detection. A. Results of S. aureus biofilms in the EVPL model treated with 4 µg/mL linezolid (clinical breakpoint for sensitive/resistant according to EUCAST classification31). B. Results of S. aureus biofilms in the EVPL model treated with 12 µg/mL linezolid (data reproduced from Sweeney et al23). Please click here to view a larger version of this figure.
{kind=link}

Figure 3. Viable Pseudomonas aeruginosa cell counts of the laboratory strain PA14 and 4 CF clinical isolates recovered from the EVPL model following treatment with increasing concentrations of colistin. Each strain was grown on EVPL tissue for 48 h then exposed to colistin for 18 h. The MIC determined in standard cation-adjusted MHB medium is shown in brackets next to each strain name. The vertical lines show the MBEC value determined in MHB (solid) and SCFM (dashed), with the exception of SED6, in which the value was the same in both media. The unfilled data points represent the lowest concentration of colistin tested that resulted in ≥ 3-log10 reduction in CFU/lungcompared to the untreated samples (0 µg/mL colistin) (data reproduced from Sweeney et al26). Please click here to view a larger version of this figure.
{kind=link}

Figure 4. Representative viable Pseudomonas aeruginosa cell counts from a time course of growth on the EVPL model over 24 h, and subsequent treatment with 64 µg/mL meropenem. The laboratory strain P. aeruginosa PA14 and 3 CF clinical isolates were grown on EVPL tissue for the time shown on the x-axis, then transferred to meropenem (triangles) for 24 h or left untreated as a control (circles). The CFU/lung was then determined. The MIC determined in cation-adjusted MHB medium is shown in brackets next to each strain name. Please click here to view a larger version of this figure.
{kind=link}

Figure 5. Representative viable Staphylococcus aureus cell counts following growth on the EVPL model then treated with 5 µg/mL flucloxacillin over a 24 h time course. The control strain ATCC29213 and two CF clinical isolates were grown on EVPL tissue for 48 h then transferred to flucloxacillin (triangles) or left untreated as a control (circles) for 4 h and 24 h, before CFU/lung was determined (data reproduced from Sweeney et al23). Please click here to view a larger version of this figure.
{kind=link}
Figure S1. Please click here to download this figure.
Discussion
The ex vivo lung model is high throughput and inexpensive and, because it uses post-consumer waste from the meat industry, it presents no ethical concerns. It is designed to mimic chronically-infected human CF airways better than currently available, in vitro AST platforms. Results presented here show that it may more accurately predict antibiotic susceptibility under these circumstances.
Critical steps in the protocol, that will ensure, reliable and reproducible results include the following:
- Use consistent time and storage methods between slaughter, collection, and processing of lung samples. It is important to use lungs as soon as possible after slaughter and to keep the potential for contamination to a minimum. Differences in the ability of experimental cultures to grow in lungs if they are not as fresh as possible have been observed.
- Maintaining sterility in the production of SCFM and dissection of lung pieces is essential. Healthy lungs are not sterile and so the presence of commensal bacteria may reflect a 'natural' environment for chronic infection. Nevertheless, as previously noted, bacterial interactions within multispecies populations may alter results and susceptibility to antibiotics, so contamination should be avoided and lungs should be sterilized before use. We advocate the use of UV sterilization, as it does not appear to cause changes tin tissue integrity and, if necessary, additional antibiotic washes. However, antibiotics should be used with caution, as they may influence results by introducing selective pressures and may alter gene expression in test bacterial populations.
- Use mock-infected, negative control tissue samples and cell count plates grown on a non-selective, rich medium to highlight the growth of any contaminant or commensal bacteria that have not been removed during sterilization. This is essential to mitigate for any impact of these bacteria on AST. It is also helpful to produce duplicate, selective agar, cell count plates specific to the organism of interest, as duplicate plates speed up colony identification and cell enumeration.
- Conduct pilot experiments when first using the model and when using it with new strains or genotype of bacteria to assess biofilm CFU variations between tissue sections, allowing the selection of optimal experimental sample sizes (e.g., how many replicate tissue sections to acquire from how many replicate lungs) through the use of power calculations.
- The assay uses a non-standardized inoculum, as this allows rapid inoculation after 48 hours of incubation and the formation of relatively consistent biofilms loads (especially for P. aeruginosa). To assay antibacterial efficacy in early biofilm growth stages, consider inoculating with a standardized CFU of colony-grown bacteria suspended in ASM. We do not recommend inoculating with planktonic bacteria: early pilot experiments showed that this leads to acute, invasive growth not reliable biofilm formation.
This protocol produces a robust prototype model for use with P. aeruginosa, with a great potential for development for use with S. aureus, but it does have some limitations that will need to be addressed for certain applications in the future. Tissue was inoculated from single colonies to allow the development of clonal populations. The results show that, for P. aeruginosa, this has little impact on cell numbers at 48 h. However, greater variability in bacterial load was observed for S. aureus and, given that different bacteria may grow differently within the model, a standardized starting inoculum and rigorous production of tissue samples of identical size and weight may be dependent on the organism of study. There may also be differences between labs due to differences in precise dissection/infection techniques or local pig breed/landrace. To assess the reproducibility of bacterial populations for individual implementations of the model, we suggest the use of repeatability calculations as part of the statistical analysis of results25 and the use of repeatability/power calculations based on pilot experiments to calculate the optimal sample size for their use in final experiments.
One of the key advantages of EVPL over traditional plate assays is that, rather than testing for bacteria growing planktonically or on abiotic surfaces, it allows for the spatial structuring of bacterial biofilms within a host environment and with cell differentiation. This has important implications for considering the impact of physiochemical and nutrient gradients on the activity of antimicrobial agents as well as the delivery and availability of active therapies at different microenvironments within a chronic infection and cell-cell interaction between bacteria. This latter point is particularly significant, as multispecies infections are routinely observed in CF and are becoming increasingly important to infections associated with other respiratory conditions, such as asthma and chronic obstructive pulmonary disease. There is potential to develop this model for the AST for individualized patient sputum sampling in the clinical diagnostics. An analogous trial is already underway using a wound-mimicking in vitro model for growth and AST of debrided biofilm from chronic wounds (Southwest Regional Wound Care Center in Lubbock, Texas, Dr. R. Wolcott).
Furthermore, the model uses post-mortem tissue, so the influence of the host immune response on antibiotic susceptibility is limited. Current in vitro models also do not account for host immune responses, so we do not see this as a barrier to the future use of the model in AST applications. However, the immune response is taken into consideration when pharmacokinetic and pharmacodynamic parameters and antibiotic dosing guidelines are determined. Although our studies have shown evidence of residual immune cells and responses within the tissue23 (and S. Azimi, personal communication), this is a prime area for further optimization and development of the model if a greater match to in vivo conditions is desired.
Providing more clinically valid AST for CF will help meet a key recommendation of the UK Health, Social Care Act 2008 that "procedures should be in place to ensure prudent prescribing and antimicrobial stewardship." We believe the EVPL is an ideal candidate model to help meet this need.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We thank all of our co-authors on the original papers from which we have taken exemplary results. The work was funded by an MRC New Investigator Research Grant (grant number MR/R001898/1) awarded to FH; by PhD studentships from the BBSRC Midlands Integrative Biosciences Training Partnership (MIBTP) awarded to NEH and IA; and by the University of Warwick Undergraduate Research Support Scheme's award to FA to conduct a summer vacation research project. We thank Steve Quigley, Sons (Cubbington, Warwickshire) and John Taylor, Son (Earlsden, Coventry) for supplying lungs. We would also like to acknowledge the help of the Media Preparation Facility in the School of Life Sciences at the University of Warwick, with special thanks to Cerith Harries and Caroline Stewart, and the help of Anita Catherwood at Warwick Antimicrobial Screening Facility.
Materials
| Name | Company | Catalog Number | Comments |
| 0.5 mL insulin syringes with 29G needle attached | |||
| 24-well culture plates | |||
| 70% ethanol or similar for surface sterilizaton and flamin gof dissection equipment | |||
| Agar plates to prepare streaks of P. aeruginosa/S. aureus (any suitable medium) | |||
| Agarose | |||
| Aluminum foil - pre-sterilised by autoclaving - to cover the chopping board on whcih you wil dissect lungs. | |||
| Bead beater designed to take 2 mL tubes | MP Biomedicals | 116004500 | FastPrep-24 Classic bead beating grinder and lysis system |
| Breathe-easy or Breathe-easier sealing membrane for multiwell plates | Diversified Biotech | BEM-1 or BERM-2000 | |
| Bunsen burner | |||
| Chopping board - we recommend a plastic board to allow for easy decontamination with alcohol. | |||
| Coolbox to transport lungs to lab | |||
| Dissection scissors in different sizes | |||
| Dulbecco’s modified Eagle medium (DMEM) | |||
| Fisherbrand 2 mL reinforced tubes | Thermo Fisher | 15545809 | |
| Fisherbrand 2.38 mm metal beads | Thermo Fisher | 15505809 | |
| Germicidal UV cabinet | |||
| Insulin syringes - 0.5 mL with 29G needle attached. | VWR | BDAM324892 | |
| Large pallet knife | |||
| LB agar plates to assess CFU in lung biofilm homogenate | |||
| Mounted razor blades | |||
| Nalgene RapidFlow PES 75 mm x 0.1 µm x 500 ml sterile filter unit | Thermo Fisher | 10474415 | For filter-sterilizing SCFM |
| Petri dishes | |||
| Phosphate-buffered saline | |||
| Plastic chopping board and aluminium foil to create a sterile and cleanable dissection surface | |||
| Roswell Park Memorial Institute (RPMI) 1640 medium | |||
| SCFM ingredients as listed in Table S1 | |||
| Selection of forceps (blunt tips recommended) | |||
| Selective agar plates to specifically assess P. aeruginosa / S. aureus CFU in lung biofilm homogenate, if required. | |||
| Suitable containers for disposing of contaminated sharps and pig ung tissue, according to your institution's health & safety policies. |
References
- Elborn, J. S. Cystic fibrosis. The Lancet. 388 (10059), 2519-2531 (2016).
- Høiby, N., et al. Diagnosis of biofilm infections in cystic fibrosis patients. APMIS. 125, 339-343 (2017).
- Bjarnsholt, T., et al. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatric Pulmonology. 44 (6), 547-558 (2009).
- Høiby, N., Bjarnsholt, T., Givskov, M., Molin, S., Ciofu, O. Antibiotic resistance of bacterial biofilms. International Journal of Antimicrobial Agents. 35 (4), 322-332 (2010).
- Penesyan, A., Gillings, M., Paulsen, I. Antibiotic discovery: Combatting bacterial resistance in cells and in biofilm communities. Molecules. 20 (4), 5286 (2015).
- Son, M. S., Matthews, W. J., Kang, Y., Nguyen, D. T., Hoang, T. T. In vivo evidence of Pseudomonas aeruginosa nutrient Acquisition and pathogenesis in the lungs of cystic fibrosis patients. Infection and Immunity. 75 (11), 5313-5324 (2007).
- Flynn, J. M., Niccum, D., Dunitz, J. M., Hunter, R. C. Evidence and role for bacterial mucin degradation in cystic fibrosis airway disease. PLoS Pathogens. 12 (8), 1005846 (2016).
- Stites, S. W., Plautz, M. W., Bailey, K., O'Brien-Ladner, A. R., Wesselius, L. J. Increased concentrations of iron and isoferritins in the lower respiratory tract of patients with stable cystic fibrosis. American Journal of Respiratory and Critical Care Medicine. 160 (3), 796-801 (1999).
- Cornforth, D. M., et al. Pseudomonas aeruginosa transcriptome during human infection. Proceedings of the National Academy of Sciences. 115 (22), 5125-5134 (2018).
- Drevinek, P., et al. Gene expression changes linked to antimicrobial resistance, oxidative stress, iron depletion and retained motility are observed when Burkholderia cenocepacia grows in cystic fibrosis sputum. BMC Infectious Diseases. 8, 121 (2008).
- Goerke, C., Wolz, C. Regulatory and genomic plasticity of Staphylococcus aureus during persistent colonization and infection. International Journal of Medical Microbiology. 294 (2-3), 195-202 (2004).
- Wolter, D. J., et al. Staphylococcus aureus small-colony variants are independently associated with worse lung disease in children with cystic fibrosis. Clin Infect Dis. 57 (3), 384-391 (2013).
- Roberts, A. E. L., Kragh, K. N., Bjarnsholt, T., Diggle, S. P. The limitations of in vitro experimentation in understanding biofilms and chronic infection. Journal of Molecular Biology. 427, 3646-3661 (2015).
- Ceri, H., et al. The Calgary biofilm device: New technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. Journal of Clinical Microbiology. 37 (6), 1771-1776 (1999).
- Moskowitz, S. M., Foster, J. M., Emerson, J., Burns, J. L. Clinically feasible biofilm susceptibility assay for isolates of Pseudomonas aeruginosa from patients with cystic fibrosis. Journal of Clinical Microbiology. 42 (5), 1915-1922 (2004).
- Smith, S., Waters, V., Jahnke, N., Ratjen, F. Standard versus biofilm antimicrobial susceptibility testing to guide antibiotic therapy in cystic fibrosis. Cochrane Database of Systematic Reviews. (6), (2020).
- Trust, C. F. . Annual Data Report 2019. , (2019).
- Angelis, A., et al. Social and economic costs and health-related quality of life in non-institutionalised patients with cystic fibrosis in the United Kingdom. BMC Health Services Research. 15 (1), 428 (2015).
- Eidt-Koch, D., Wagner, T. O. F., Mittendorf, T., von der Schulenburg, J. M. G. Outpatient medication costs of patients with cystic fibrosis in Germany. Applied Health Economics and Health Policy. 8 (2), 111-118 (2010).
- Harrington, N. E., Sweeney, E., Harrison, F. Building a better biofilm - Formation of in vivo-like biofilm structures by Pseudomonas aeruginosa in a porcine model of cystic fibrosis lung infection. Biofilm. 2, 100024 (2020).
- Harrison, F., Diggle, S. An ex vivo lung model to study bronchioles infected with Pseudomonas aeruginosa biofilms. Microbiology. 162, 1755-1760 (2016).
- Sweeney, E., et al. An ex vivo cystic fibrosis model recapitulates key clinical aspects of chronic Staphylococcus aureus infection. Microbiology. , (2020).
- Palmer, K. L., Aye, L. M., Whiteley, M. Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. Journal of Bacteriology. 189 (22), 8079-8087 (2007).
- Nakagawa, S., Schielzeth, H. Repeatability for Gaussian and non-Gaussian data: A practical guide for biologists. Biological Reviews. 85 (4), 935-956 (2010).
- Sweeney, E., Sabnis, A., Edwards, A. M., Harrison, F. Effect of host-mimicking medium and biofilm growth on the ability of colistin to kill Pseudomonas aeruginosa. Microbiology. 166 (12), 1171-1180 (2020).
- Stalker, D. J., Jungbluth, G. L., Hopkins, N. K., Batts, D. H. Pharmacokinetics and tolerance of single- and multiple-dose oral or intravenous linezolid, an oxazolidinone antibiotic, in healthy volunteers. Journal of Antimicrobial Chemotherapy. 51 (5), 1239-1246 (2003).
- Ager, S., Gould, K. Clinical update on linezolid in the treatment of Gram-positive bacterial infections. Infection and Drug Resistance. 5, 87-102 (2012).
- Kirchner, S., et al. Use of Artificial Sputum Medium to Test Antibiotic Efficacy Against Pseudomonas aeruginosa in Conditions More Relevant to the Cystic Fibrosis Lung. Journal of Visualized Experiments: JoVE. (64), e3857 (2012).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved