
Method Article
Nano-Differential Scanning Fluorimetry for Screening in Fragment-based Lead Discovery
In This Article
Summary
Monitoring changes in the melting temperature of a target protein (i.e., thermal shift assay, TSA) is an efficient method for screening fragment libraries of a few hundred compounds. We present a TSA protocol implementing robotics-assisted nano-Differential Scanning Fluorimetry (nano-DSF) for monitoring intrinsic tryptophan fluorescence and light back-scattering for fragment screening.
Abstract
Thermal shift assays (TSAs) examine how the melting temperature (Tm) of a target protein changes in response to changes in its environment (e.g., buffer composition). The utility of TSA, and specifically of nano-Differential Scanning Fluorimetry (nano-DSF), has been established over the years, both for finding conditions that help stabilize a specific protein and for looking at ligand binding by monitoring changes in the apparent Tm. This paper presents an efficient screening of the Diamond-SGC-iNEXT Poised (DSi-Poised) fragment library (768 compounds) by the use of nano-DSF, monitoring Tm to identify potential fragment binding. The prerequisites regarding protein quality and concentration for performing nano-DSF experiments are briefly outlined followed by a step-by-step protocol that uses a nano-liter robotic dispenser commonly used in structural biology laboratories for preparing the required samples in 96-well plates. The protocol describes how the reagent mixtures are transferred to the capillaries needed for nano-DSF measurements. In addition, this paper provides protocols to measure thermal denaturation (monitoring intrinsic tryptophan fluorescence) and aggregation (monitoring light back-scattering) and the subsequent steps for data transfer and analysis. Finally, screening experiments with three different protein targets are discussed to illustrate the use of this procedure in the context of lead discovery campaigns. The overall principle of the method described can be easily transferred to other fragment libraries or adapted to other instruments.
Introduction
Drug discovery programs often start by screening chemical compounds for their ability to interact with and/or modify the function of drug targets, most often proteins. The so-called "hits" that are found in such screens, lay the basis for the discovery of novel leads and development candidates, and for most of the new drugs that are being licensed these days. The availability of high-throughput methods is therefore indispensable to screen an enormous number of available targets with a huge number of different compounds to quickly identify their tight binding or their ability to modulate a specific function of the target. After hits are identified, the most promising hit-target combinations are pushed into an extensive drug development pipeline using other, often expensive and time-consuming technologies, to understand "structure-activity relationships" (SAR).
Structural biology approaches, such as those offered by the EU-funded access programs "Infrastructure for NMR, EM, and X-rays for Translational Research" (iNEXT) and its current successor iNEXT-Discovery, are often used to study the interactions of numerous compounds in extreme detail while improving the affinity and pharmacological properties of the initial hits by typically several rounds of synthetic chemistry1. Lead compounds that emerge from these "from hit to lead" campaigns become development candidates and enter preclinical studies. The well-developed molecular screening methodology can be roughly categorized into two approaches, namely the ligand-based lead discovery (LBLD) and fragment-based lead discovery (FBLD). In LBLD campaigns, protein receptors are screened with either a few thousand hand-picked ligands (based on the structure of natural ligands or the structure of the target), or with many tens of thousands of compounds in drug-like ligand libraries that cover a large portion of chemical space.
Usually, the compounds are tested for their inhibitory activity in an activity assay, typically monitoring an enzymatic function. In FBLD campaigns2,3,4,5, however, some hundreds of compounds that are typically smaller than drugs (100-200 Dalton) are tested for their ability to bind the target directly, without the use of an activity assay. This binding could interfere with target activity and can be measured by many biophysical methods that report directly on the ability of fragments to bind to the target, or by structural methods such as X-ray crystallography6 and nuclear magnetic resonance spectroscopy7, and more recently, also cryo-electron microscopy. When fragments bind at different locations that are close to each other on the protein, the different, usually low-affinity binding fragments can be rationally combined chemically to create a small set of leads that can be studied in more detail. This frequently results in higher-affinity, more potent compounds, and this methodology has started to yield important molecules with clinical potential. The choice of an "ideal" fragment library that exploits chemical groups efficiently has been an active area of research for many years8,9,10.
While initial emphasis was on covering full chemical space, subsequent attention was focused on enabling the downstream chemical combination of fragment hits to produce lead compounds. Such research has led to the so-called "poised" libraries. These contain fragments with at least one functional group that allow rapid, cheap follow-up synthetic chemistry for efficient progress in studying SAR. One of the activities catalyzed by iNEXT was to update the poised library developed by researchers in the Diamond Light Source and Structural Genomics Consortium. This combined effort resulted in the DSi-Poised library11, which has also been validated within iNEXT12. Later, this library was aligned with the availability of compounds in the REAL Database of Enamine Ltd., a chemical research organization and producer of large collections of building blocks and compound libraries for screening. DSi-Poised is now available to anyone for purchase, but also available in many iNEXT-Discovery partner laboratories for supported fragment screening projects.
The high-end X-ray crystallography and NMR structural biology technologies both have their advantages and disadvantages for FBLD. Both require isolated target samples and yield the high-resolution atomic details that are required for FBLD. However, crystals are necessary for X-ray crystallography, and the fragments bind to cavities in the well-ordered protein regions that are not involved in building the three-dimensional crystal lattice. Solution NMR often yields different hits from X-ray crystallography, as it is unaffected by the crystal environment and is good in detecting binding also in partially ordered protein regions. However, while ligand-based NMR experiments are relatively fast, they still require a substantial amount of time and material and can routinely be done only for relatively small protein targets or domains. For the purpose of prioritizing compounds for crystallographic or NMR experiments, biophysical approaches have been used13,14,15.
As recent instrumentation and computational protocols allow efficient crystallographic screening for FBLD by determining structures and analyzing ~1,000 fragments very efficiently, this prioritization has become less essential in X-ray based research. For NMR however, it remains desirable to use cheaper and quicker experiments to prioritize library screening and save instrument time on highest-end equipment. At the same time, using a combination of essentially different technologies can provide independent confirmation of binding events, or even additional hits that are not picked up by employing only the crystallography or NMR method. Crystallographic and NMR techniques both require very expensive equipment and can often only be done in dedicated external facilities with help from local, highly skilled experts. In addition, the proper analysis of results also demands high expertise. While programs such as iNEXT and iNEXT-Discovery are democratizing access to such facilities16, it has been recognized that cheap, fast, and high-throughput FBLD screening by other methods can encourage drug-screening programs in a much wider range of laboratories. Such results can then be used as an indication to build collaborations with medicinal chemists, and to prioritize the most expensive screening experiments to the most promising compounds if NMR and crystallography facilities impose restrictions on the number of compounds that can be screened.
The TSA forms a quick, efficient, and relatively cheap and accessible biophysical method17 that can be used for FBLD screening. It has been used in multiple settings, from aiding to find stable protein conditions for crystallization trials18, to finding compounds that bind to specific targets in cells19. TSAs have also been used for measuring the dissociation constants for ligands binding target proteins, as ligand binding often leads to alterations in thermal stability. In all TSAs, the change in denaturation temperature of a protein (its stability) is measured as a function of a slow temperature increase. An efficient way to follow protein denaturation upon heating is by DSF or Thermofluor, which quantifies fluorescence quenching of a hydrophobic dye (typically Sypro Orange) upon interaction with exposed hydrophobic regions of protein that unfolds due to temperature increase.
Nano-DSF typically refers to measurement of the thermal stability of protein in the absence of external dyes. One of the first instruments that offered this possibility was the OPTIM1000 that measures a wide spectrum of light intensity as well as light scattering of a sample. This machine allowed the simultaneous measurement of protein unfolding (typically following tryptophan fluorescence) and protein aggregation (as formed nanoparticles result in an increase of light scattering at ~400 nm). Later, the Prometheus introduced the use of back-reflection for measuring aggregation and sensitive detection of the fluorescence signal, allowing screening of low protein concentrations with good sensitivity20. The following section describes how Prometheus was used to demonstrate a fragment screening protocol for detecting hits for different protein targets. A brief introduction about the expected protein quality and quantity is followed by a step-by-step protocol for preparing, performing, and analyzing the fragment screening experiments. Screening results for three proteins have been shown as example data obtained as part of iNEXT-Discovery collaborations.
Protocol
NOTE: The proteins used in nano-DSF experiments should be pure (>95%) and homogeneous as judged by sodium dodecylsulfate-polyacrylamide gel electrophoresis. Before performing the fragment screen, the stability of the proteins should be determined in various buffer conditions. A low ionic strength, low salt buffer that interferes minimally with the protein should be used so as not to affect its direct interaction with the fragments. The buffers typically used in this protocol for checking stability are shown in Supplemental Table S1. The concentration of the protein that needs to be used for this experiment as a stock solution is typically 0.2 mg mL-1. For screening the entire DSi-Poised library (768 compounds), a total of ~12 mL of protein of that concentration, a total of ~2.5 mg, is needed. The DSi-Poised library used in these experiments was supplied in 96-well format (Figure 1). The concentration of the fragments was adjusted to 100 mM in 20% v/v dimethylsulfoxide (DMSO). It should be noted that the mixing protocol described here results in low final DMSO concentrations of 0.4% v/v; although this is very unlikely to affect the stability of the protein, the effect of DMSO should be checked for each new protein.

Figure 1: The types of plates used for these experiments. (A) U-bottom plate. (B) 96-well plate. (C) A close-up view of 96-well plate showing the subwell 1. Please click here to view a larger version of this figure.
{kind=link}
1. Plate preparation
- Take out a fragment plate from the -20 °C/-80 °C freezer, and let it thaw at room temperature, with gentle shaking on a benchtop shaker. Centrifuge the plate at 500 × g for 30 s to collect any drops sticking to the side of the wells.
NOTE: As the fragment library is available dissolved in DMSO-d6 (melting point 19 °C), it is essential to make sure that each compound is completely thawed and solubilized. - Take an MRC 2-well crystallization plate (Figure 1A), and pipette 14.7 µL of the protein stock solution into each sub-well. To do this in a time-efficient manner, keep the protein in a reagent reservoir (Table of Materials), and use a multichannel pipette for dispensing (Figure 2A,B).
NOTE: For the DSi-Poised library, depending on the format, not all the wells of the 96-well plate contain compounds. Typically, rows A, H and columns 1, 12 are filled with DMSO. Thus, rows A, H and columns 1, 12 are not to be filled with protein, as these wells will not contain any fragments at the end of the next step; only DMSO will be transferred there by the robot. Please note that the empty rows and columns do differ in some plates.

Figure 2: Outline of the fragment screening procedure. (A) Using a multichannel pipette and reagent reservoir to dispense the protein. (B) Dispensing the protein in the 96-well plate. (C) Fragment dispensing by the dispenser robot. (D) Loading of the protein into the capillaries. (E) Drawer showing the capillary holder. (F) Close-up view of the capillary holder. Please click here to view a larger version of this figure.
{kind=link}

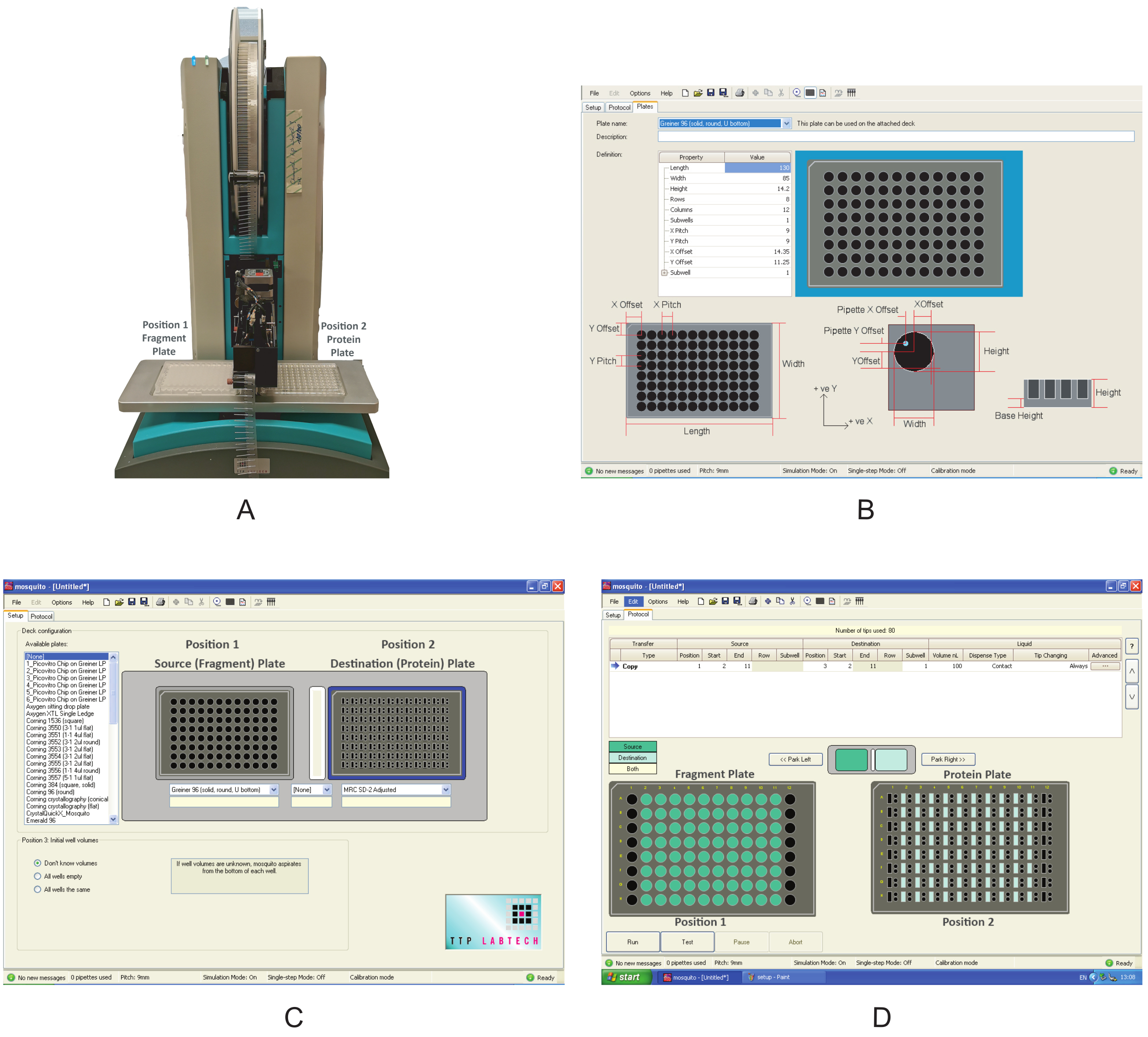
Figure 3: Overview of the equipment used in these experiments. (A) Nanodispenser robot used for fragment dispensing. Plate positions are indicated. (B) Program interface for defining a new plate. (C) Interface of the dispensing program. (D) The dispensing program used to dispense the fragments. Please click here to view a larger version of this figure.
{kind=link}
2. Fragment nano-dispensing by the Mosquito robot
- Check the type of plate in which the fragments are supplied (Table of Materials).
- Check fragment and protein plate definitions on the Mosquito.
- Switch on the nanodispenser (Figure 3A). Make sure that there are no obstacles around the moving components.
- Open the graphical user interface, click on the Setup tab, and under Deck Configuration, check whether the type of plate in which the compounds are supplied and the one in which the protein has been transferred in section 1 are already present in the list of the Available plates. If not, click Options|Plates and create a new plate definition by filling in the correct values for the Property type (Figure 3B).
NOTE: These values for the MRC 2-well plate and the 96-well plate used in this experiment are shown in Supplemental Table S2 and Supplemental Table S3, respectively.
- The dispensing program
- Under the Setup tab, specify the plate positions under Deck Configuration.
NOTE: The Mosquito used in this experiment has two plate positions on the deck. Position 1 is defined as the Source plate, and Position 2 is the Destination plate. - Using the dropdown menu, choose Greiner U bottom plate for Position 1 and MRC 2-well plate for Position 2 (Figure 2C). Save the protocol.
- Under the Setup tab, specify the plate positions under Deck Configuration.
- Fragment dispensing
- Place the fragment plate at Position 1 and the protein plate at Position 2 of the deck (Figure 3A).
- On the Protocol tab (Figure 3D), click on File and choose the protocol saved in section 2.3 for dispensing the fragments. Define the volume of the fragments to be dispensed; use 0.3 µL. For a typical plate, where columns 1 and 12 do not contain fragments, define the Start location to column 2 and the End location to column 11. In some plates, if columns 2 or 11 are empty, use columns 3 and 10 as start and end values, respectively.
NOTE: Because of the Mosquito setup, only columns can be skipped, not rows; for the rows, the mosquito will pipette DMSO. Make sure that the Tip Changing option is selected as Always, so as not to cross-contaminate the fragment library. - Click on Run to start the program. After dispensing, which takes ~2 min, is completed, remove the protein and the fragment plates from the robot, and seal them back with an adhesive sealing film. Briefly centrifuge the protein plate (500 × g, 30 s) to collect any drops sticking to the sides of the wells before proceeding to the next step.
3. Measurement of nano-DSF
NOTE: A detailed description about performing TSAs using the Prometheus NT.48 has been published previously20. Important points in the context of fragment screening are mentioned here.
- Plate inspection before the measurement
- Visually inspect the wells of the protein plate for precipitation that might have occurred due to the addition of the fragments. If precipitation is observed in many wells, reduce the concentration of the fragments, and repeat the experiment.
NOTE: It is recommended to keep the protein plate at room temperature; the fragments tend to become insoluble at lower temperatures.
- Visually inspect the wells of the protein plate for precipitation that might have occurred due to the addition of the fragments. If precipitation is observed in many wells, reduce the concentration of the fragments, and repeat the experiment.
- Preparing the Prometheus
- Switch on the Prometheus instrument. On the touchscreen, press Open Drawer to access the capillary loading module of the instrument. Remove the magnetic strip from the loading module, and clean the mirror with ethanol to remove any dust particles.
- Transfer from the protein-fragments plate to capillaries
NOTE: This step involves transferring the mixed protein/fragment sample from each well in the protein plate to the capillaries for use with the Prometheus. For this, although the Standard capillary type is typically used, it is possible to use the High Sensitivity capillaries for very low protein concentrations.- Place the protein plate and the capillaries next to the instrument to have easy access to the capillary loading module.
- Take one capillary, hold it at one end, and touch the solution in the protein plate with the far end of the capillary to transfer the sample by capillary action. Always wear gloves, and make sure not to touch the capillary in the middle, as impurities (e.g., dust particles) from the gloves would affect the measurement.
- Place the capillary in the designated position of the holder, making sure it is properly aligned and centered.
- Repeat steps 3.3.2 and 3.3.3, thereby filling up all positions in the loading module. Load all capillaries you will need for measurement.
NOTE: For a single run, a maximum of 48 capillaries can be loaded. - At the end, place the magnetic strip on top of the capillaries to hold them in place, and press Close Drawer to start the experiment.
- Perform the nano-DSF experiment
- Fluorescence scan
- Open the Prometheus application PR.ThermControl, and create a new project by clicking on Start New Session using the name ProteinName_ScreenName_PlateNumber. First, do a Discovery Scan to detect the fluorescence of the samples.
NOTE: The fluorescence levels should ideally be above 3,000 counts for each sample. Samples having fluorescence above the saturation limit of the instrument (20,000 counts) will not be measured. - Alter the fluorescence signal of the samples by adjusting the Excitation Power of the laser (Figure 4).
- Open the Prometheus application PR.ThermControl, and create a new project by clicking on Start New Session using the name ProteinName_ScreenName_PlateNumber. First, do a Discovery Scan to detect the fluorescence of the samples.
- Thermal denaturation
- Click on the Melting Scan tab. To perform the experiment, set the Start Temperature to 20 °C, the End Temperature to 95 °C, and the Temperature Slope to 1 °C min-1. Click on Start Measurement.
- Annotating the experiment
- After the experiments starts, click on the Annotation and Results tab. Annotate each capillary by giving it a unique plate- and well-number.
NOTE: For example, capillary 1B2 would correspond to Plate 1 and well B2. In this tab, extra columns can be added for the experiment, e.g., protein name, buffer, protein concentration. After the experiment is finished, the results are also displayed in this tab. This is a good time to pause at the end of the day; the experiments take place overnight, the results can be collected the next day.
- After the experiments starts, click on the Annotation and Results tab. Annotate each capillary by giving it a unique plate- and well-number.
- Data visualization and export
- After the experiment is finished, click on the Melting Scan tab to view the melting and scattering curves for the samples. To export the results in a spreadsheet, click on the Melting Scan tab, click on Export, and choose Export Processed Data from the drop-down menu.
- Fluorescence scan

Figure 4: Adjusting the excitation power and fluorescence signal. (A) At 80% excitation power, the fluorescence signal for most of the samples is beyond the saturation limit. (B) The fluorescence signal is reduced to measurable levels by decreasing the excitation power to 60%. Please click here to view a larger version of this figure.
{kind=link}
4. Iteration
- Repeat steps 1-3 to perform 16 runs on the entire Enamine library of 768 compounds (16 × 48 = 768).
- As the measurement of each plate takes ~1.5 h to complete, complete the annotation (3.4.3), and prepare the next protein-fragments plate by repeating steps in sections 1 and 2.
NOTE: In a typical working day, 4-6 plates can be measured, depending on experience. The screening of the entire DSi-library can be finished in a total of 3-4 days.
5. Data analysis
- Examining the generated data
- Once each run is completed, click on the Annotations and Results tab to display the results. For each sample, focus on two calculated values that are most important in this overview: 1) The scattering onset temperature (Onset #1 for Scattering), which indicates the temperature at the start of increased sample scattering events and is characteristic of aggregation; 2) the Tm (Inflection Point #1 for Ratio) value extracted from the ratio between fluorescence events at 330 and 350 nm-values that typically correspond to the maximal changes of tryptophan fluorescence upon changes in its environment.
- Be sure to check the scattering and the melting curves for each capillary in the Melting Scan tab to make sure that these values are reliable.
- Export and inspection to a spreadsheet
- Export the data from all different runs for inspection to a spreadsheet software, as described in 3.4.4, and to create overview tables and plots. Click on the various Sheets in the spreadsheet file to note the information in each sheet.
- In the Overview sheet, note that each row corresponds to one experiment. Along an overview of parameters (e.g., start and end temperature), observe the calculated values for the Tm and the scattering onset (see 5.1 above for names and explanation, and Supplemental files 1 and 2 for an example). As the software calculates two or more Tm values (Inflection Point #1 and #2 for Ratio) for some samples, look at the melting transition curve to determine which of the Tm values is correct.
- In the Ratio sheet, note that each column corresponds to a sample, and each row to the data read at each temperature step. Observe the fluorescence count values corresponding to each temperature step, and use them to plot the melting curves for specific samples, plotting the temperature column against the fluorescence count column. Note that the data in the Ratio (first derivative), 330 nm, 330 nm (first derivative), 350 nm, 350 nm (first derivative) are used to calculate the ratio and its first derivative (which has a maximum at the maximum rate of change) in a similar format.
- Observe similar data for scattering in the Scattering sheet. Generate the scattering curve for each sample by plotting the temperature column against the scattering column. Look for the Sheet for the scattering first derivative.
- Export the data from all different runs for inspection to a spreadsheet software, as described in 3.4.4, and to create overview tables and plots. Click on the various Sheets in the spreadsheet file to note the information in each sheet.
- Creating and validating a global overview for all fragments
- After verifying the correct Tm values for the fragments, combine the columns Sample ID and Inflection point of 330/350 nm ratio (Tm) from all the runs in a single new result file, copying these columns from each run.
- Use the average Tm value of the native protein (typically calculated over ten runs) and subtract it from the Tm values of each sample, to get the ΔTm. Sort the results over ΔTm in descending order to identify the samples that result in the largest shift.
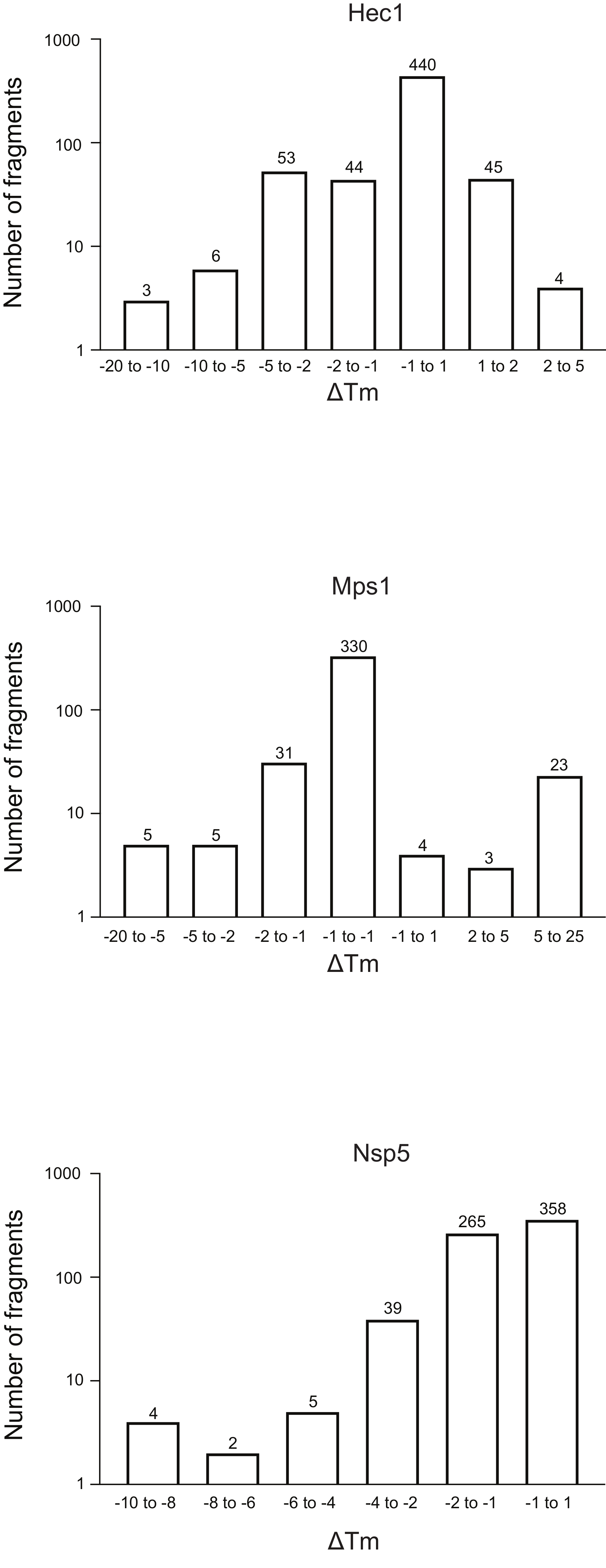
- Divide the fragments into bins depending on the ΔTm shift, and generate a frequency table for the whole library by plotting the ΔTm for each bin against the number of fragments (Figure 5). For convenience, show the axis representing the number of fragments in the log scale. Bin the sample with ΔTm ±1, and adapt the other bins to each run to adjust the number of outliers empirically.
Results
A full screen of the DSi-Poised library (768 fragments) was performed on three proteins of medical interest, namely, the outer kinetochore Highly Expressed in Cancer 1 protein (Hec1, or Ndc80), the regulatory tetraricopeptide repeat (TPR) domain of the monopolar spindle kinase 1 (Mps1), and the SARS-CoV-2 3C-like protease, Nsp5, which cleaves off the C-terminus of the replicase polyprotein at 11 sites. The buffer conditions chosen for each protein, as well as the protein concentration and Tm of the proteins, are shown in Supplemental Table S4.

Figure 5: Frequency distribution of the shift in melting temperature (ΔTm) for the three proteins, Hec1, Mps1, and Nsp5, presented in this study as representative results. Abbreviations: Hec1 = Highly Expressed in Cancer 1 protein; Mps1 = monopolar spindle kinase 1; Nsp5 = SARS-CoV-2 3C-like protease. Please click here to view a larger version of this figure.
{kind=link}
The results of the three screenings using the protocol described above, displayed as the frequency distribution of the change in Tm versus the number of fragments, are shown in Figure 5. These plots have been generated as described in section 5.3 of the protocol, plotting the frequency of the observed change in Tm. The significance of the shift needs to be defined in a subjective manner for every different project, as described in the discussion section below. Negative values indicate a reduction in the melting temperature in the presence of a fragment, a positive value an increase in Tm. From such plots, it is easy to observe that for Nsp5, all the fragments have a destabilizing effect, whereas for Hec1 and Mps1, both stabilizing and destabilizing hits are observed. This can be expected and will be discussed.
Discussion
This protocol describes a medium-to-high throughput method for screening fragment libraries using some common robotics and measurement instruments. Screens like those described in this protocol can be routinely performed by the NKI Protein Facility in Amsterdam, for instance as an iNEXT-Discovery service, often even for free for users after proposal application and peer review. In such cases, the DSi-Poised library can be provided by the facility, but the use of other libraries can also be discussed in the context of each different user application and service agreement. The choice of instruments in this protocol represents practical solutions for many laboratories, but should not be considered as a gold standard. Label-free methods are recommended for measuring the thermal stability of the target protein for fragment screening, rather than methods that use environmentally sensitive labels for detecting unfolding in a reverse-transcription polymerase chain reaction thermocycler.
Label-free methods, such as the one presented here using the Prometheus instrument, have some advantages: they use low amounts of protein, often a couple of orders of magnitude less; they can be used to simultaneously measure scattering of the sample and thus aggregation; and the labels used for detecting unfolding in other approaches can interact differently with each fragment, resulting in measurement artifacts. This protocol has been described in the context of the Mosquito robot, which allows the pipetting of a very small volume of sample (0.3 µL) that cannot be done manually. The Mosquito is a popular robot, present in many laboratories working on structural biology and drug discovery projects; however, the protocol can clearly use alternative approaches for low-volume pipetting.
Fragment libraries contain compounds dissolved in DMSO. One of the initial challenges is to find the optimal DMSO concentration at which the protein remains stable, and the compounds remain soluble. This involves performing the measurements at various DMSO concentrations to determine the optimal conditions for screening. The protein to fragment dilution used here results in DMSO concentrations of 0.2%; most proteins are fairly stable in these conditions. The amount of protein required for carrying out the screening for the 768-compound library is ~2-3 mg in total, as the measurements are typically carried out at low protein concentrations (0.2 mg mL-1). Working with such relatively low protein concentrations not only reduces protein production costs, but also reduces the chances of protein precipitation. The low protein concentration does not affect detection of fragment binding, as the concentration of the fragments in the experiment is ~2 mM, allowing also weak binders to be identified.
As the melting transition detection in these experiments is based on fluorescence intensity, a critical aspect is to determine the excitation power of the laser at which to carry out the measurements. The interaction of the compounds with the protein can (i) have no effect on its intrinsic fluorescence, (ii) result in quenching, or (iii) increase its intrinsic fluorescence. In addition to this, working with low protein concentrations means that the fluorescence count for the native protein would be low. The excitation power therefore must be adjusted in such a manner that most of the samples can be measured. The scattering profile of every run provides important information about the aggregation effects that might be triggered by the addition of any fragment. In addition, the effect of temperature on compound solubility can also be seen on the scattering profile.
Unexpectedly, for many compounds it was observed that the scattering actually decreased with increasing temperature (Figure 6). It is therefore important to look at both the melting transition curve and the accompanying scattering profile to decide about the reliability of each experiment, especially for those fragments that are considered candidates for more demanding measurements by X-ray crystallography or NMR spectroscopy, or even considered as hits for follow-up chemistry. One specific limitation of the method for fragment screening purposes is that many fragments in the DSi-Poised library have significant intrinsic fluorescence, sometimes even beyond the saturation limit of the detector, and therefore these cannot be properly screened for target binding even at low excitation power. Another point to note for this method is that it can only be used with proteins containing tryptophan residues.

Figure 6: Effect of temperature on compound solubility. Melting transition curve and scattering profile of Hec1 with two different compounds. (A) The scattering profile shows that for this sample, the solubility is not affected by temperature. (B) The scattering profile shows that the solubility of this sample increases with increasing temperature. The melting transition curve in this case is therefore not reliable. Please click here to view a larger version of this figure.
{kind=link}
An open question is what should be considered as a significant change in Tm, not from a mathematical perspective, but from the practical point of view: what change in Tm is important to consider as indicative of binding of a ligand to a protein? In these examples, shifts of less than 1 °C are seen for 74% of the fragments for binding Hec1, 66% for Mps1, and 53% for Nsp5. Considering 1 °C as 'significant change' would hardly provide hits that are worthy to pursue by follow-up chemistry. In the overview graphs (Figure 5), bins of 1, 2, 5, or more than 5 degrees of Tm shift, either positive or negative, were considered. This requires modification according to each specific case to give a good overview and to allow informed decision-making, determining the next step. Notably, for some proteins, both stabilization and de-stabilization of the target were observed depending on the fragments considered. Both events are interesting, as both can be the result of fragment binding, and both can lead to a good follow-up molecule to manipulate protein behavior.
A final question remains, namely, "what defines a useful hit?". In fact, the answer depends on the specific situation. For example, for Hec1, all fragments that stabilize the protein by more than 2 degrees or destabilize it by more than 5 were communicated to our chemistry collaborators, who designed new molecules based on these hits. For Nsp5, however, the most de-stabilizing hits were communicated to our NMR collaborators to confirm the nanoDSF-derived hits with NMR experiments. In other words, the screening results obtained from this protocol should be analyzed with caution and in a context-dependent manner, making informed decisions based on the specific question and surrounding methodology. In any case, the method described here is a complementary approach to existing methodologies such as X-ray and NMR-based screening, which can aim to confirm, prioritize, or give new ideas for chemistry campaigns.
Supplemental Table S1: List of buffers used for screening of proteins. Abbreviations: HEPES = 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; DTT = dithiothreitol; MOBS = 4-(N-morpholino)butanesulfonic acid. Please click here to download this Table.
Supplemental Table S2: Properties for MRC 2-well plate for use with nanodispenser robot. Please click here to download this Table.
Supplemental Table S3: Properties for 96-well V-bottom plate for use with nanodispenser robot. Please click here to download this Table.
Supplemental Table S4: The buffer, protein concentration, and Tm of the proteins discussed in representative results. Abbreviations: DTT = dithiothreitol; Hec1 = Highly Expressed in Cancer 1 protein; Mps1 = monopolar spindle kinase 1; Nsp5 = SARS-CoV-2 3C-like protease. Please click here to download this Table.
Supplemental File 1: Overview parameters-example data. Please click here to download this File.
Supplemental File 2: Tm and ΔTm values for 406 fragments-example data. Please click here to download this File.
Disclosures
There are no disclosures.
Acknowledgements
"This work benefited from access to the NKI Protein Facility, an Instruct-ERIC centre. Financial support has been provided by iNEXT, project number 653706, and iNEXT-Discovery, project number 871037, funded by the Horizon 2020 program of the European Commission".
Materials
| Name | Company | Catalog Number | Comments |
| ClearVue Sheets | Molecular Dimensions | adhesive sealing film for protein plate | |
| CORNING 6570 Aluminium Sealing Tape | CORNING | adhesive sealing film for fragment plate | |
| DSi poised library | Enamine | Fragment library containing 768 compounds used in this study | |
| Elisa Reagent Reservior | ThermoFisher Scientific | 15075 | Reagent reservior used for pipetting the protein |
| Greiner round (U) bottom plates | Cat. No. 650201 | Fragments supplied in these plates | |
| Mosquito type X1 | sptlabtech | Part nr- 3019-0003 | Nanolitre dispenser |
| MRC 2-well crystallization plate | MRC96T-PS | ||
| Pierce ELISA Reagent Reservoirs | Pierce | ||
| Prometheus High Sensitivity capillaries | Catalog PR-C006 | ||
| Prometheus NT.48 nanoDSF | Nanotemper | Catalog nr PR001 (+ Aggregation Detection Optics, catalog nr PR-AGO) | nanoDSF and light back scattering |
| Prometheus Standard capillary type | Catalog PR-C002 | ||
| TX-1000 | Thermoscientific | Centrifuge for plates |
References
- Hoffer, L., Muller, C., Roche, P., Morelli, X. Chemistry-driven hit-to-lead optimization guided by structure-based approaches. Molecular Informatics. 37 (9-10), 1800059(2018).
- Lamoree, B., Hubbard, R. E. Current perspectives in fragment-based lead discovery (FBLD). Essays in Biochemistry. 61 (5), 453-464 (2017).
- Ress, D. C., Congreve, M., Murray, C. W., Carr, R. Fragment-based lead discovery. Nature Reviews Drug Discovery. 3 (8), 660-672 (2004).
- Carr, R. A. E., Congreve, M., Murray, C. W., Rees, D. C. Fragment-based lead discovery: Leads by design. Drug Discovery Today. 10 (14), 987-992 (2005).
- Bradley, A. R., et al. The SGC beyond structural genomics: Redefining the role of 3D structures by coupling genomic stratification with fragment-based discovery. Essays in Biochemistry. 61 (5), 495-503 (2017).
- Davies, T. G., Tickle, I. J. Fragment screening using X-ray crystallography. Topics in Current Chemistry. 317, 33-59 (2012).
- Ma, R., Wang, P., Wu, J., Ruan, K. Process of fragment-based lead discovery - A perspective from NMR. Molecules. 21 (7), 854(2016).
- Troelsen, N. S., Clausen, M. H. Library design strategies to accelerate fragment-based drug discovery. Chemistry. 26 (50), 11391-11403 (2020).
- Shi, Y., von Itzstein, M. How size matters: Diversity for fragment library design. Molecules. 24 (15), 2838(2019).
- Taylor, A., Doak, B. C., Scanlon, M. J. Design of a fragment-screening library. Methods in Enzymology. 610, 97-115 (2018).
- Cox, O. B., et al. A poised fragment library enables rapid synthetic expansion yielding the first reported inhibitors of PHIP(2), an atypical bromodomain. Chemical Science. 7, 2322-2330 (2016).
- Sreeramulu, S., et al. NMR quality control of fragment libraries for screening. Journal of Biomolecular NMR. 74 (10-11), 555-563 (2020).
- Pfaff, S. J., Chimenti, M. S., Kelly, M. J. S., Arkin, M. R. Biophysical methods for identifying fragment-based inhibitors of protein-protein interactions. Methods in Molecular Biology. 1278, 587-613 (2015).
- Fattori, D., Squarcia, A., Bartoli, S. Fragment-based approach to drug lead discovery: Overview and advances in various techniques. Drugs in R & D. 9 (4), 217-227 (2008).
- Winter, A., et al. Biophysical and computational fragment-based approaches to targeting protein-protein interactions: Applications in structure-guided drug discovery. Quarterly Reviews of Biophysics. 45 (4), 383-426 (2012).
- Boelens, R., et al. iNEXT: a European facility network to stimulate translational structural biology. FEBS Letters. 592 (12), 1909-1917 (2018).
- Zhang, R., Monsma, F. Fluorescence-based thermal shift assays. Current Opinion in Drug Discovery and Development. 13 (4), 389-402 (2010).
- Boivin, S., Kozak, S., Meijers, R. Optimization of protein purification and characterization using Thermofluor screens. Protein Expression and Purification. 91 (2), 192-206 (2013).
- Martinez Molina, D., Nordlund, P. The cellular thermal shift assay: a novel biophysical assay for in situ drug target engagement and mechanistic biomarker studies. Annual Review of Pharmacology and Toxicology. 56, 141-161 (2016).
- Bruce, D., Cardew, E., Freitag-Pohl, S., Pohl, E. How to stabilize protein: stability screens for thermal shift assays and nano differential scanning fluorimetry in the Virus-X Project. Journal of Visualized Experiments JoVE. (144), e58666(2019).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved