
Method Article
Nano-Differential-Scanning-Fluorimetrie für das Screening in der fragmentbasierten Lead-Discovery
In diesem Artikel
Zusammenfassung
Die Überwachung von Änderungen der Schmelztemperatur eines Zielproteins(z. B.Thermal Shift Assay, TSA) ist eine effiziente Methode zum Screening von Fragmentbibliotheken von einigen hundert Verbindungen. Wir präsentieren ein TSA-Protokoll, das eine robotergestützte Nano-Differential Scanning Fluorimetry (Nano-DSF) zur Überwachung der intrinsischen Tryptophanfluoreszenz und der Lichtrückstreuung für das Fragmentscreening implementiert.
Zusammenfassung
Thermal Shift Assays (TSAs) untersuchen, wie sich die Schmelztemperatur (Tm)eines Zielproteins als Reaktion auf Veränderungen in seiner Umgebung(z. B.Pufferzusammensetzung) ändert. Der Nutzen von TSA und insbesondere der Nano-Differential Scanning Fluorimetry (Nano-DSF) wurde im Laufe der Jahre sowohl für die Suche nach Bedingungen, die zur Stabilisierung eines bestimmten Proteins beitragen, als auch für die Betrachtung der Ligandenbindung durch Überwachung von Veränderungen im scheinbaren Tmnachgewiesen. Dieser Artikel präsentiert ein effizientes Screening der Diamond-SGC-iNEXT Poised (DSi-Poised) Fragmentbibliothek (768 Verbindungen) unter Verwendung von Nano-DSF, wobei Tm überwacht wird, um eine potenzielle Fragmentbindung zu identifizieren. Die Voraussetzungen für die Proteinqualität und -konzentration für die Durchführung von Nano-DSF-Experimenten werden kurz skizziert, gefolgt von einem Schritt-für-Schritt-Protokoll, das einen Nano-Liter-Roboterspender verwendet, der üblicherweise in strukturbiologischen Labors zur Vorbereitung der erforderlichen Proben in 96-Well-Platten verwendet wird. Das Protokoll beschreibt, wie die Reagenzgemische auf die für Nano-DSF-Messungen benötigten Kapillaren übertragen werden. Darüber hinaus enthält dieses Papier Protokolle zur Messung der thermischen Denaturierung (Überwachung der intrinsischen Tryptophanfluoreszenz) und aggregation (Überwachung der Lichtrückstreuung) und der nachfolgenden Schritte für die Datenübertragung und -analyse. Abschließend werden Screening-Experimente mit drei verschiedenen Proteinzielen diskutiert, um den Einsatz dieses Verfahrens im Rahmen von Lead-Discovery-Kampagnen zu veranschaulichen. Das Gesamtprinzip der beschriebenen Methode lässt sich leicht auf andere Fragmentbibliotheken übertragen oder auf andere Instrumente anpassen.
Einleitung
Arzneimittelforschungsprogramme beginnen oft damit, chemische Verbindungen auf ihre Fähigkeit zu untersuchen, mit Arzneimittelzielen, meistens Proteinen, zu interagieren und / oder die Funktion zu modifizieren. Die sogenannten "Hits", die in solchen Screens zu finden sind, legen die Grundlage für die Entdeckung neuer Leads und Entwicklungskandidaten und für die meisten neuen Medikamente, die heutzutage zugelassen werden. Die Verfügbarkeit von Hochdurchsatzmethoden ist daher unerlässlich, um eine enorme Anzahl verfügbarer Targets mit einer Vielzahl unterschiedlicher Verbindungen zu screenen, um schnell ihre enge Bindung oder ihre Fähigkeit, eine bestimmte Funktion des Targets zu modulieren, zu identifizieren. Nachdem die Treffer identifiziert wurden, werden die vielversprechendsten Hit-Target-Kombinationen in eine umfangreiche Medikamentenentwicklungspipeline mit anderen, oft teuren und zeitaufwändigen Technologien geschoben, um "Struktur-Aktivitäts-Beziehungen" (SAR) zu verstehen.
Strukturbiologische Ansätze, wie sie die EU-geförderten Zugangsprogramme "Infrastructure for NMR, EM, and X-rays for Translational Research" (iNEXT) und ihr aktueller Nachfolger iNEXT-Discovery anbieten, werden häufig eingesetzt, um die Wechselwirkungen zahlreicher Verbindungen extrem detailliert zu untersuchen und gleichzeitig die Affinität und pharmakologischen Eigenschaften der ersten Treffer durch typischerweise mehrere Runden synthetischer Chemie zu verbessern1. Lead-Compounds, die aus diesen "from hit to lead"-Kampagnen hervorgehen, werden zu Entwicklungskandidaten und gehen in präklinische Studien. Die gut entwickelte molekulare Screening-Methodik lässt sich grob in zwei Ansätze einteilen, nämlich die ligandenbasierte Lead-Discovery (LBLD) und die fragmentbasierte Lead-Discovery (FBLD). In LBLD-Kampagnen werden Proteinrezeptoren entweder mit einigen tausend handverlesenen Liganden (basierend auf der Struktur natürlicher Liganden oder der Struktur des Ziels) oder mit vielen zehntausend Verbindungen in medikamentenähnlichen Ligandenbibliotheken gescreent, die einen großen Teil des chemischen Raums abdecken.
Normalerweise werden die Verbindungen in einem Aktivitätstest auf ihre hemmende Aktivität getestet, wobei typischerweise eine enzymatische Funktion überwacht wird. In den FBLD-Kampagnen2,3,4,5werden jedoch einige hundert Verbindungen, die typischerweise kleiner als Medikamente sind (100-200 Dalton), auf ihre Fähigkeit getestet, das Ziel direkt zu binden, ohne dass ein Aktivitätsassay verwendet wird. Diese Bindung könnte die Zielaktivität stören und kann durch viele biophysikalische Methoden gemessen werden, die direkt über die Fähigkeit von Fragmenten berichten, sich an das Ziel zu binden, oder durch strukturelle Methoden wie Röntgenkristallographie6 und Kernspinresonanzspektroskopie7und in jüngerer Zeit auch Kryo-Elektronenmikroskopie. Wenn Fragmente an verschiedenen Stellen binden, die nahe beieinander auf dem Protein liegen, können die verschiedenen, normalerweise niedrig affinen Bindungsfragmente chemisch rational kombiniert werden, um einen kleinen Satz von Leitungen zu erzeugen, die genauer untersucht werden können. Dies führt häufig zu höher affinen, stärkeren Verbindungen, und diese Methodik hat begonnen, wichtige Moleküle mit klinischem Potenzial zu liefern. Die Wahl einer "idealen" Fragmentbibliothek, die chemische Gruppen effizient nutzt, ist seit vielen Jahren ein aktives Forschungsgebiet8,9,10.
Während der schwerpunktige Schwerpunkt zunächst auf der Abdeckung des gesamten chemischen Raums lag, konzentrierte sich die nachfolgende Aufmerksamkeit darauf, die nachgeschaltete chemische Kombination von Fragmenttreffern zur Herstellung von Bleiverbindungen zu ermöglichen. Solche Recherchen haben zu den sogenannten "poised" Bibliotheken geführt. Diese enthalten Fragmente mit mindestens einer funktionellen Gruppe, die eine schnelle, kostengünstige Nachverfolgung der synthetischen Chemie für effiziente Fortschritte bei der Untersuchung von SAR ermöglichen. Eine der von iNEXT katalysierten Aktivitäten war die Aktualisierung der Von Forschern des Diamond Light Source and Structural Genomics Consortium entwickelten Bibliothek. Aus dieser gemeinsamen Anstrengung entstand die DSi-Poised Library11, die auch in iNEXT12validiert wurde. Später wurde diese Bibliothek auf die Verfügbarkeit von Verbindungen in der REAL Database von Enamine Ltd. ausgerichtet, einer chemischen Forschungsorganisation und Hersteller großer Sammlungen von Bausteinen und Verbindungsbibliotheken für das Screening. DSi-Poised ist jetzt für jedermann erhältlich, aber auch in vielen iNEXT-Discovery-Partnerlaboren für unterstützte Fragment-Screening-Projekte.
Die High-End-Röntgenkristallographie und die NMR-Strukturbiologie-Technologien haben beide ihre Vor- und Nachteile für FBLD. Beide erfordern isolierte Zielproben und liefern die hochauflösenden atomaren Details, die für FBLD erforderlich sind. Kristalle sind jedoch für die Röntgenkristallographie notwendig, und die Fragmente binden an Hohlräume in den wohlgeordneten Proteinregionen, die nicht am Aufbau des dreidimensionalen Kristallgitters beteiligt sind. Lösungs-NMR liefert oft unterschiedliche Treffer aus der Röntgenkristallographie, da sie von der Kristallumgebung nicht beeinflusst wird und die Bindung auch in teilweise geordneten Proteinregionen gut nachweisen kann. Obwohl ligandenbasierte NMR-Experimente relativ schnell sind, benötigen sie immer noch viel Zeit und Material und können routinemäßig nur für relativ kleine Proteinziele oder -domänen durchgeführt werden. Zur Priorisierung von Verbindungen für kristallographische oder NMR-Experimente wurden biophysikalische Ansätze verwendet13,14,15.
Da neuere Instrumente und Computerprotokolle ein effizientes kristallographisches Screening für FBLD ermöglichen, indem Strukturen bestimmt und ~ 1.000 Fragmente sehr effizient analysiert werden, ist diese Priorisierung in der röntgenbasierten Forschung weniger wichtig geworden. Für NMR bleibt es jedoch wünschenswert, billigere und schnellere Experimente zu verwenden, um das Bibliotheksscreening zu priorisieren und Instrumentenzeit auf Geräten zu sparen. Gleichzeitig kann die Verwendung einer Kombination von im Wesentlichen unterschiedlichen Technologien eine unabhängige Bestätigung von Bindungsereignissen oder sogar zusätzlichen Treffern liefern, die nicht nur mit der Kristallographie oder NMR-Methode aufgenommen werden. Kristallographische und NMR-Techniken erfordern beide sehr teure Ausrüstung und können oft nur in speziellen externen Einrichtungen mit Hilfe von lokalen, hochqualifizierten Experten durchgeführt werden. Darüber hinaus erfordert die richtige Analyse der Ergebnisse auch eine hohe Expertise. Während Programme wie iNEXT und iNEXT-Discovery den Zugang zu solchen Einrichtungen demokratisieren16, wurde erkannt, dass billiges, schnelles und durchsatzreiches FBLD-Screening mit anderen Methoden Drogen-Screening-Programme in einer viel breiteren Palette von Labors fördern kann. Solche Ergebnisse können dann als Hinweis verwendet werden, um Kooperationen mit medizinischen Chemikern aufzubauen und die teuersten Screening-Experimente den vielversprechendsten Verbindungen zu priorisieren, wenn NMR- und Kristallographieeinrichtungen die Anzahl der Verbindungen, die gescreent werden können, einschränkt.
Die TSA bildet eine schnelle, effiziente und relativ kostengünstige und zugängliche biophysikalische Methode17, die für das FBLD-Screening verwendet werden kann. Es wurde in mehreren Umgebungen verwendet, von der Unterstützung bei der Suche nach stabilen Proteinbedingungen für Kristallisationsversuche18bis hin zur Suche nach Verbindungen, die an bestimmte Ziele in Zellen binden19. TSAs wurden auch zur Messung der Dissoziationskonstanten für Liganden verwendet, die Zielproteine binden, da die Ligandenbindung häufig zu Veränderungen der thermischen Stabilität führt. In allen TSAs wird die Änderung der Denaturierungstemperatur eines Proteins (seine Stabilität) als Funktion eines langsamen Temperaturanstiegs gemessen. Eine effiziente Möglichkeit, die Proteindenaturierung beim Erhitzen zu verfolgen, ist DSF oder Thermofluor, das das Fluoreszenzabschrecken eines hydrophoben Farbstoffs (typischerweise Sypro Orange) bei Wechselwirkung mit exponierten hydrophoben Proteinregionen quantifiziert, die sich aufgrund des Temperaturanstiegs entfalten.
Nano-DSF bezieht sich typischerweise auf die Messung der thermischen Stabilität von Protein in Abwesenheit von externen Farbstoffen. Eines der ersten Instrumente, das diese Möglichkeit bot, war das OPTIM1000, das ein breites Spektrum der Lichtintensität sowie die Lichtstreuung einer Probe misst. Diese Maschine ermöglichte die gleichzeitige Messung der Proteinentfaltung (typischerweise nach Tryptophanfluoreszenz) und der Proteinaggregation (da gebildete Nanopartikel zu einer Erhöhung der Lichtstreuung bei ~ 400 nm führen). Später führte der Prometheus die Verwendung von Rückreflexion zur Messung der Aggregation und empfindlichen Detektion des Fluoreszenzsignals ein, was das Screening niedriger Proteinkonzentrationen mit guter Empfindlichkeit ermöglichte20. Im folgenden Abschnitt wird beschrieben, wie Prometheus verwendet wurde, um ein Fragment-Screening-Protokoll zum Nachweis von Treffern für verschiedene Proteinziele zu demonstrieren. Auf eine kurze Einführung in die erwartete Proteinqualität und -quantität folgt ein Schritt-für-Schritt-Protokoll zur Vorbereitung, Durchführung und Analyse der Fragmentscreening-Experimente. Screening-Ergebnisse für drei Proteine wurden als Beispieldaten gezeigt, die im Rahmen von iNEXT-Discovery-Kooperationen gewonnen wurden.
Protokoll
HINWEIS: Die in Nano-DSF-Experimenten verwendeten Proteine sollten rein (>95%) und homogen sein, gemessen an der Natriumdodecylsulfat-Polyacrylamid-Gelelektrophorese. Vor der Durchführung des Fragmentscreenings sollte die Stabilität der Proteine unter verschiedenen Pufferbedingungen bestimmt werden. Ein Puffer mit geringer Ionenstärke und niedrigem Salzgehalt, der das Protein minimal stört, sollte verwendet werden, um seine direkte Wechselwirkung mit den Fragmenten nicht zu beeinträchtigen. Die Puffer, die typischerweise in diesem Protokoll zur Überprüfung der Stabilität verwendet werden, sind in der Ergänzenden Tabelle S1 aufgeführt. Die Konzentration des Proteins, das für dieses Experiment als Stammlösung verwendet werden muss, beträgt typischerweise 0,2 mg ml-1. Für das Screening der gesamten DSi-Poised-Bibliothek (768 Verbindungen) werden insgesamt ~12 ml Protein dieser Konzentration, insgesamt ~2,5 mg, benötigt. Die in diesen Experimenten verwendete DSi-Poised-Bibliothek wurde im 96-Well-Format geliefert (Abbildung 1). Die Konzentration der Fragmente wurde auf 100 mM in 20% v/v Dimethylsulfoxid (DMSO) eingestellt. Es ist zu beachten, dass das hier beschriebene Mischprotokoll zu niedrigen DMSO-Endkonzentrationen von 0,4% v/v führt; Obwohl es sehr unwahrscheinlich ist, dass dies die Stabilität des Proteins beeinflusst, sollte die Wirkung von DMSO für jedes neue Protein überprüft werden.

Abbildung 1: Die für diese Experimente verwendeten Plattentypen. (A) U-Bodenplatte. (B) 96-Well-Platte. (C) Nahaufnahme der 96-Well-Platte, die die Unterwell 1 zeigt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
1. Plattenvorbereitung
- Nehmen Sie eine Fragmentplatte aus dem Gefrierschrank -20 °C/-80 °C heraus und lassen Sie sie bei Raumtemperatur auftauen, wobei Sie auf einem Tischschüttler sanft schütteln. Zentrifugieren Sie die Platte bei 500 × g für 30 s, um alle Tropfen zu sammeln, die an der Seite der Vertiefungen haften.
HINWEIS: Da die Fragmentbibliothek gelöst in DMSO-d6 (Schmelzpunkt 19 °C) verfügbar ist, ist es wichtig sicherzustellen, dass jede Verbindung vollständig aufgetaut und gelöst ist. - Nehmen Sie eine MRC-2-Well-Kristallisationsplatte (Abbildung 1A) und pipettieren Sie 14,7 μL der Proteinstocklösung in jede Sub-Vertiefung. Um dies zeiteffizient zu tun, halten Sie das Protein in einem Reagenzreservoir (Materialtabelle) und verwenden Sie eine Mehrkanalpipette zum Dosieren (Abbildung 2A, B).
HINWEIS: Für die DSi-Poised-Bibliothek enthalten je nach Format nicht alle Vertiefungen der 96-Well-Platte Verbindungen. In der Regel werden die Zeilen A, H und die Spalten 1, 12 mit DMSO gefüllt. Daher dürfen die Zeilen A, H und die Spalten 1, 12 nicht mit Protein gefüllt werden, da diese Vertiefungen am Ende des nächsten Schritts keine Fragmente enthalten; nur DMSO wird vom Roboter dorthin übertragen. Bitte beachten Sie, dass sich die leeren Zeilen und Spalten in einigen Platten unterscheiden.

Abbildung 2: Überblick über das Fragmentscreening-Verfahren. (A) Verwendung einer Mehrkanalpipette und eines Reagenzreservoirs zur Dosierung des Proteins. (B) Dosieren des Proteins in der 96-Well-Platte. (C) Fragmentdosierung durch den Spenderroboter. (D) Laden des Proteins in die Kapillaren. (E) Schublade mit dem Kapillarhalter. F) Nahaufnahme des Kapillarhalters. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Überblick über die in diesen Experimenten verwendeten Geräte. (A) Nanodispenser-Roboter, der für die Fragmentabgabe verwendet wird. Plattenpositionen sind angegeben. (B) Programmschnittstelle zum Definieren einer neuen Platte. (C) Schnittstelle des Dosierprogramms. (D) Das Dosierprogramm, mit dem die Fragmente dosiert werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
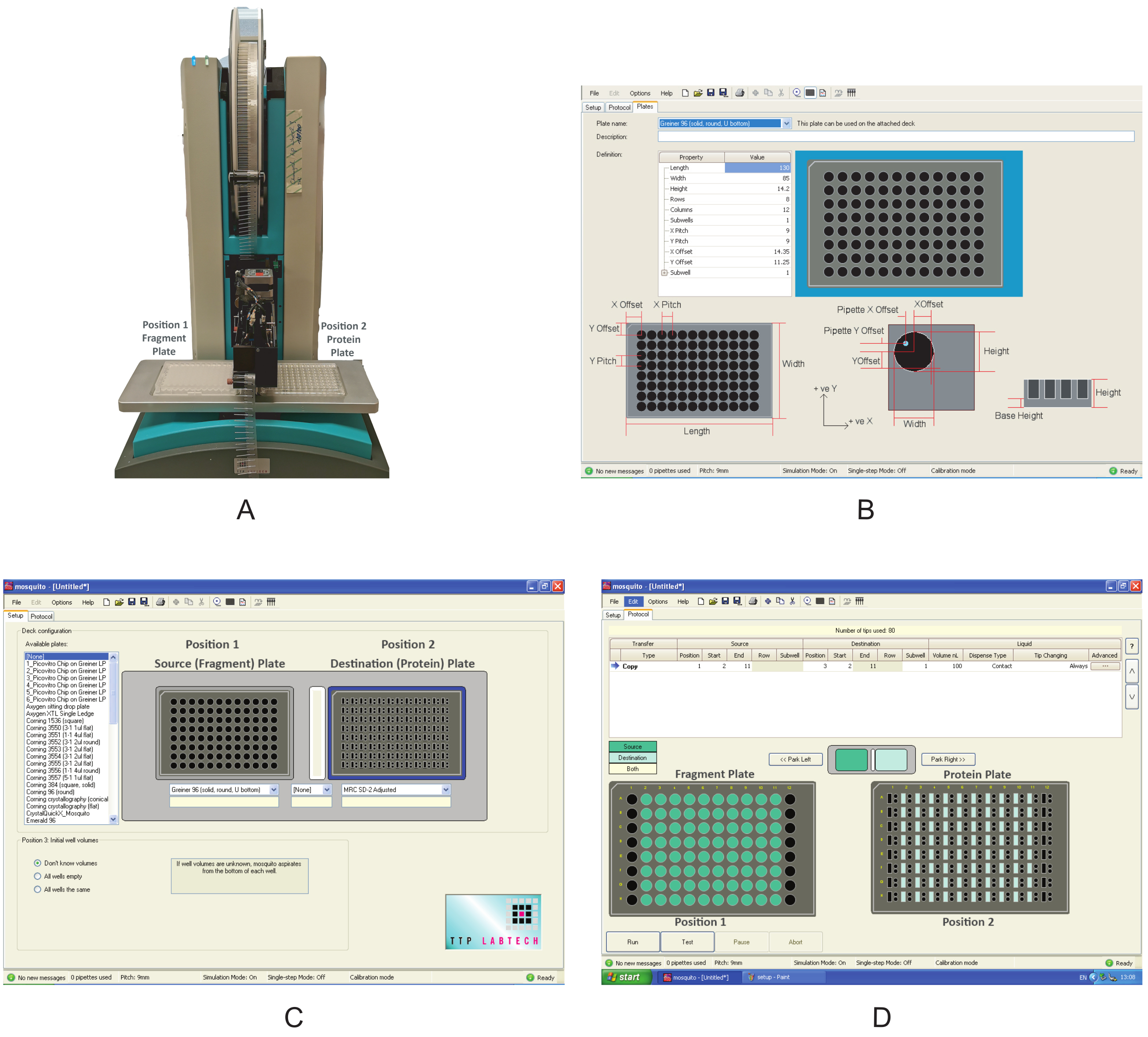{kind=link}
2. Fragment-Nano-Dispensing durch den Mosquito-Roboter
- Überprüfen Sie die Art der Platte, in der die Fragmente geliefert werden (Materialtabelle).
- Überprüfen Sie die Fragment- und Proteinplattendefinitionen auf der Mosquito.
- Schalten Sie das Nanodosierer ein (Abbildung 3A). Stellen Sie sicher, dass sich keine Hindernisse um die beweglichen Komponenten herum befinden.
- Öffnen Sie die grafische Benutzeroberfläche, klicken Sie auf die Registerkarte Setup und überprüfen Sie unter Deckkonfiguration,ob die Art der Platte, in die die Verbindungen geliefert werden, und diejenige, in die das Protein in Abschnitt 1 übertragen wurde, bereits in der Liste der verfügbaren Plattenvorhanden sind. Wenn nicht, klicken Sie auf Optionen| Platten und erstellen Sie eine neue Plattendefinition, indem Sie die richtigen Werte für den Eigenschaftstyp eingeben (Abbildung 3B).
HINWEIS: Diese Werte für die MRC-2-Well-Platte und die 96-Well-Platte, die in diesem Experiment verwendet werden, sind in der Ergänzenden Tabelle S2 bzw. der Ergänzenden Tabelle S3aufgeführt.
- Das Dosierprogramm
- Geben Sie auf der Registerkarte Setup die Plattenpositionen unter Deckkonfiguration an.
HINWEIS: Die in diesem Experiment verwendete Mosquito hat zwei Plattenpositionen auf dem Deck. Position 1 ist definiert als quellplatteund Position 2 ist die Zielplatte. - Wählen Sie im Dropdown-Menü Greiner U Bodenplatte für Position 1 und MRC 2-Well Plate für Position 2 (Abbildung 2C). Speichern Sie das Protokoll.
- Geben Sie auf der Registerkarte Setup die Plattenpositionen unter Deckkonfiguration an.
- Fragmentdosierung
- Platzieren Sie die Fragmentplatte an Position 1 und die Proteinplatte an Position 2 des Decks (Abbildung 3A).
- Klicken Sie auf der Registerkarte Protokoll (Abbildung 3D) auf Datei und wählen Sie das in Abschnitt 2.3 gespeicherte Protokoll zum Verteilen der Fragmente aus. Definieren Sie das Volumen der zu verteilenden Fragmente; verwenden Sie 0,3 μL. Definieren Sie für eine typische Platte, bei der die Spalten 1 und 12 keine Fragmente enthalten, die Startposition in Spalte 2 und die Endposition in Spalte 11. Wenn in einigen Platten die Spalten 2 oder 11 leer sind, verwenden Sie die Spalten 3 und 10 als Start- bzw. Endwerte.
HINWEIS: Aufgrund des Mosquito-Setups können nur Spalten übersprungen werden, keine Zeilen. Für die Reihen wird die Mücke DMSO pipetten. Stellen Sie sicher, dass die Option Tippänderung als Immerausgewählt ist, um die Fragmentbibliothek nicht zu kreuzen. - Klicken Sie auf Ausführen, um das Programm zu starten. Nach dem Dosieren, das ~2 min dauert, entfernen Sie das Protein und die Fragmentplatten aus dem Roboter und verschließen Sie sie mit einer Klebefolie. Zentrifugieren Sie kurz die Proteinplatte (500 × g,30 s), um alle Tropfen zu sammeln, die an den Seiten der Vertiefungen haften, bevor Sie mit dem nächsten Schritt fortfahren.
3. Messung von Nano-DSF
HINWEIS: Eine detaillierte Beschreibung zur Durchführung von TSAs mit dem Prometheus NT.48 wurde zuvor veröffentlicht20. Wichtige Punkte im Rahmen des Fragmentscreenings werden hier genannt.
- Platteninspektion vor der Messung
- Untersuchen Sie die Vertiefungen der Proteinplatte visuell auf Ausfällungen, die aufgrund der Zugabe der Fragmente aufgetreten sein könnten. Wenn in vielen Brunnen Niederschlag beobachtet wird, reduzieren Sie die Konzentration der Fragmente und wiederholen Sie das Experiment.
HINWEIS: Es wird empfohlen, die Proteinplatte bei Raumtemperatur zu halten; die Fragmente neigen dazu, bei niedrigeren Temperaturen unlöslich zu werden.
- Untersuchen Sie die Vertiefungen der Proteinplatte visuell auf Ausfällungen, die aufgrund der Zugabe der Fragmente aufgetreten sein könnten. Wenn in vielen Brunnen Niederschlag beobachtet wird, reduzieren Sie die Konzentration der Fragmente und wiederholen Sie das Experiment.
- Zubereitung des Prometheus
- Schalten Sie das Prometheus-Instrument ein. Drücken Sie auf dem Touchscreen open Drawer, um auf das Kapillarlademodul des Instruments zuzugreifen. Entfernen Sie den Magnetstreifen vom Lademodul und reinigen Sie den Spiegel mit Ethanol, um Staubpartikel zu entfernen.
- Übertragung von der Proteinfragmentplatte auf die Kapillaren
HINWEIS: Dieser Schritt beinhaltet die Übertragung der gemischten Protein- / Fragmentprobe von jeder Vertiefung in der Proteinplatte auf die Kapillaren zur Verwendung mit dem Prometheus. Obwohl hierfür typischerweise der Standardkapillartyp verwendet wird, ist es möglich, die High Sensitivity-Kapillaren für sehr niedrige Proteinkonzentrationen zu verwenden.- Platzieren Sie die Proteinplatte und die Kapillaren neben dem Instrument, um einfachen Zugang zum Kapillarlademodul zu haben.
- Nehmen Sie eine Kapillare, halten Sie sie an einem Ende und berühren Sie die Lösung in der Proteinplatte mit dem anderen Ende der Kapillare, um die Probe durch Kapillarwirkung zu übertragen. Tragen Sie immer Handschuhe und achten Sie darauf, die Kapillare in der Mitte nicht zu berühren, da Verunreinigungen(z. B.Staubpartikel) der Handschuhe die Messung beeinträchtigen würden.
- Platzieren Sie die Kapillare in der vorgesehenen Position des Halters und stellen Sie sicher, dass sie richtig ausgerichtet und zentriert ist.
- Wiederholen Sie die Schritte 3.3.2 und 3.3.3 und füllen Sie damit alle Positionen im Lademodul aus. Laden Sie alle Kapillaren auf, die Sie für die Messung benötigen.
HINWEIS: Für einen einzelnen Durchlauf können maximal 48 Kapillaren geladen werden. - Legen Sie am Ende den Magnetstreifen auf die Kapillaren, um sie an Ort und Stelle zu halten, und drücken Sie Close Drawer, um das Experiment zu starten.
- Führen Sie das Nano-DSF-Experiment durch
- Fluoreszenz-Scan
- Öffnen Sie die Prometheus-Anwendung PR. ThermControl, und erstellen Sie ein neues Projekt, indem Sie auf Neue Sitzung starten klicken, indem Sie den Namen ProteinName_ScreenName_PlateNumberverwenden. Führen Sie zunächst einen Discovery Scan durch, um die Fluoreszenz der Proben zu erkennen.
HINWEIS: Die Fluoreszenzwerte sollten idealerweise über 3.000 Zählungen für jede Probe liegen. Proben mit einer Fluoreszenz über der Sättigungsgrenze des Instruments (20.000 Zählungen) werden nicht gemessen. - Ändern Sie das Fluoreszenzsignal der Proben, indem Sie die Anregungsleistung des Lasers anpassen (Abbildung 4).
- Öffnen Sie die Prometheus-Anwendung PR. ThermControl, und erstellen Sie ein neues Projekt, indem Sie auf Neue Sitzung starten klicken, indem Sie den Namen ProteinName_ScreenName_PlateNumberverwenden. Führen Sie zunächst einen Discovery Scan durch, um die Fluoreszenz der Proben zu erkennen.
- Thermische Denaturierung
- Klicken Sie auf die Registerkarte Melting Scan. Um das Experiment durchzuführen, stellen Sie die Starttemperatur auf 20 °C, die Endtemperatur auf 95 °C und die Temperaturneigung auf 1 °C min-1ein. Klicken Sie auf Messung starten.
- Kommentieren des Experiments
- Klicken Sie nach Beginn der Experimente auf die Registerkarte Anmerkungen und Ergebnisse. Kommentieren Sie jede Kapillare, indem Sie ihr eine eindeutige Platten- und Brunnennummer geben.
HINWEIS: Zum Beispiel würde Kapillar 1B2 Platte 1 und Vertiefung B2 entsprechen. Auf dieser Registerkarte können zusätzliche Spalten für das Experiment hinzugefügt werden, z. B.Proteinname, Puffer, Proteinkonzentration. Nach Abschluss des Experiments werden die Ergebnisse ebenfalls auf dieser Registerkarte angezeigt. Dies ist eine gute Zeit, um am Ende des Tages innezuhalten; die Experimente finden über Nacht statt, die Ergebnisse können am nächsten Tag gesammelt werden.
- Klicken Sie nach Beginn der Experimente auf die Registerkarte Anmerkungen und Ergebnisse. Kommentieren Sie jede Kapillare, indem Sie ihr eine eindeutige Platten- und Brunnennummer geben.
- Datenvisualisierung und -export
- Klicken Sie nach Abschluss des Experiments auf die Registerkarte Schmelzscan, um die Schmelz- und Streukurven für die Proben anzuzeigen. Um die Ergebnisse in eine Tabelle zu exportieren, klicken Sie auf die Registerkarte Melting Scan, klicken Sie auf Exportierenund wählen Sie Verarbeitete Daten exportieren aus dem Dropdown-Menü.
- Fluoreszenz-Scan

Abbildung 4: Einstellung der Anregungsleistung und des Fluoreszenzsignals. (A) Bei 80% Anregungsleistung liegt das Fluoreszenzsignal für die meisten Proben über der Sättigungsgrenze. (B) Das Fluoreszenzsignal wird durch Verringern der Erregerleistung auf 60% auf messbare Werte reduziert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
4. Iteration
- Wiederholen Sie die Schritte 1-3, um 16 Durchläufe mit der gesamten Enamine-Bibliothek mit 768 Verbindungen durchzuführen (16 × 48 = 768).
- Da die Messung jeder Platte ~1,5 h dauert, vervollständigen Sie die Anmerkung (3.4.3) und bereiten Sie die nächste Proteinfragmentplatte vor, indem Sie die Schritte in den Abschnitten 1 und 2 wiederholen.
HINWEIS: An einem typischen Arbeitstag können je nach Erfahrung 4-6 Platten gemessen werden. Das Screening der gesamten DSi-Bibliothek kann in insgesamt 3-4 Tagen abgeschlossen werden.
5. Datenanalyse
- Untersuchen der generierten Daten
- Sobald jeder Durchlauf abgeschlossen ist, klicken Sie auf die Registerkarte Anmerkungen und Ergebnisse, um die Ergebnisse anzuzeigen. Konzentrieren Sie sich für jede Probe auf zwei berechnete Werte, die in dieser Übersicht am wichtigsten sind: 1) Die Streubeginntemperatur (Onset #1 für Streuung), die die Temperatur zu Beginn erhöhter Probenstreuereignisse angibt und für die Aggregation charakteristisch ist; 2) der Tm (Inflection Point #1 for Ratio)Wert, extrahiert aus dem Verhältnis zwischen Fluoreszenzereignissen bei 330 und 350 nm-Werten, die typischerweise den maximalen Veränderungen der Tryptophanfluoreszenz bei Veränderungen in ihrer Umgebung entsprechen.
- Überprüfen Sie die Streuung und die Schmelzkurven für jede Kapillare auf der Registerkarte Schmelzscan, um sicherzustellen, dass diese Werte zuverlässig sind.
- Exportieren und Prüfen in eine Tabelle
- Exportieren Sie die Daten aus allen verschiedenen Läufen zur Inspektion in eine Tabellenkalkulationssoftware, wie in 3.4.4 beschrieben, und erstellen Sie Übersichtstabellen und Diagramme. Klicken Sie auf die verschiedenen Blätter in der Tabellenkalkulationsdatei, um die Informationen in jedem Blatt zu notieren.
- Beachten Sie im Blatt Übersicht, dass jede Zeile einem Experiment entspricht. Beachten Sie entlang einer Übersicht der Parameter(z. B.Start- und Endtemperatur) die berechneten Werte für das Tm und den Streubeginn (siehe 5.1 oben für Namen und Erläuterungen und ergänzende Dateien 1 und 2 für ein Beispiel). Wenn die Software für einige Proben zwei oder mehr Tm-Werte (Wendepunkt #1 und #2 für Verhältnis) berechnet, schauen Sie sich die Schmelzübergangskurve an, um festzustellen, welcher der Tm-Werte korrekt ist.
- Beachten Sie im Blatt Verhältnis, dass jede Spalte einem Beispiel und jede Zeile den Daten entspricht, die bei jedem Temperaturschritt gelesen werden. Beobachten Sie die Fluoreszenzzahlwerte, die jedem Temperaturschritt entsprechen, und verwenden Sie sie, um die Schmelzkurven für bestimmte Proben zu zeichnen, wobei die Temperatursäule gegen die Fluoreszenzzahlsäule dargestellt wird. Beachten Sie, dass die Daten im Verhältnis (erste Ableitung), 330 nm, 330 nm (erste Ableitung), 350 nm, 350 nm (erste Ableitung) verwendet werden, um das Verhältnis und seine erste Ableitung (die ein Maximum bei der maximalen Änderungsrate hat) in einem ähnlichen Format zu berechnen.
- Beachten Sie ähnliche Daten für die Streuung im Streublatt. Generieren Sie die Streukurve für jede Probe, indem Sie die Temperatursäule gegen die Streusäule auftreiben. Suchen Sie nach dem Blatt für die erste Streuableitung.
- Exportieren Sie die Daten aus allen verschiedenen Läufen zur Inspektion in eine Tabellenkalkulationssoftware, wie in 3.4.4 beschrieben, und erstellen Sie Übersichtstabellen und Diagramme. Klicken Sie auf die verschiedenen Blätter in der Tabellenkalkulationsdatei, um die Informationen in jedem Blatt zu notieren.
- Erstellen und Validieren einer globalen Übersicht für alle Fragmente
- Nachdem Sie die korrekten Tm-Werte für die Fragmente überprüft haben, kombinieren Sie die Spalten Sample ID und Wendepunkt von 330/350 nm Ratio (Tm)aus allen Läufen in einer einzigen neuen Ergebnisdatei und kopieren Sie diese Spalten aus jedem Durchlauf.
- Verwenden Sie den durchschnittlichen Tm-Wert des nativen Proteins (typischerweise über zehn Durchläufe berechnet) und subtrahieren Sie ihn von den Tm-Werten jeder Probe, um das ΔTmzu erhalten. Sortieren Sie die Ergebnisse über ΔTm in absteigender Reihenfolge, um die Proben zu identifizieren, die zur größten Verschiebung führen.
- Teilen Sie die Fragmente in Abhängigkeit von der ΔTm-Verschiebung in Behälter auf und generieren Sie eine Häufigkeitstabelle für die gesamte Bibliothek, indem Sie die ΔTm für jeden Behälter gegen die Anzahl der Fragmente auftakten (Abbildung 5). Zeigen Sie der Einfachheit halber die Achse an, die die Anzahl der Fragmente in der Protokollskala darstellt. Bin die Probe mit ΔTm ±1 und passe die anderen Behälter an jeden Durchlauf an, um die Anzahl der Ausreißer empirisch anzupassen.
Ergebnisse
Ein Vollbild der DSi-Poised-Bibliothek (768 Fragmente) wurde an drei Proteinen von medizinischem Interesse durchgeführt, nämlich dem äußeren Kinetochor Highly Expressed in Cancer 1 Protein (Hec1 oder Ndc80), der regulatorischen Tetraricopeptid-Wiederholungsdomäne (TPR) der monopolaren Spindelkinase 1 (Mps1) und der SARS-CoV-2 3C-ähnlichen Protease Nsp5, die den C-Terminus des Replikatpolyproteins an 11 Stellen abspaltet. Die für jedes Protein gewählten Pufferbedingungen sowie die Proteinkonzentration und Tm der Proteine sind in der Ergänzenden Tabelle S4 aufgeführt.

Abbildung 5: Häufigkeitsverteilung der Verschiebung der Schmelztemperatur (ΔTm)für die drei Proteine Hec1, Mps1 und Nsp5, die in dieser Studie als repräsentative Ergebnisse dargestellt werden. Abkürzungen: Hec1 = Highly Expressed in Cancer 1 protein; Mps1 = monopolare Spindelkinase 1; Nsp5 = SARS-CoV-2 3C-ähnliche Protease. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
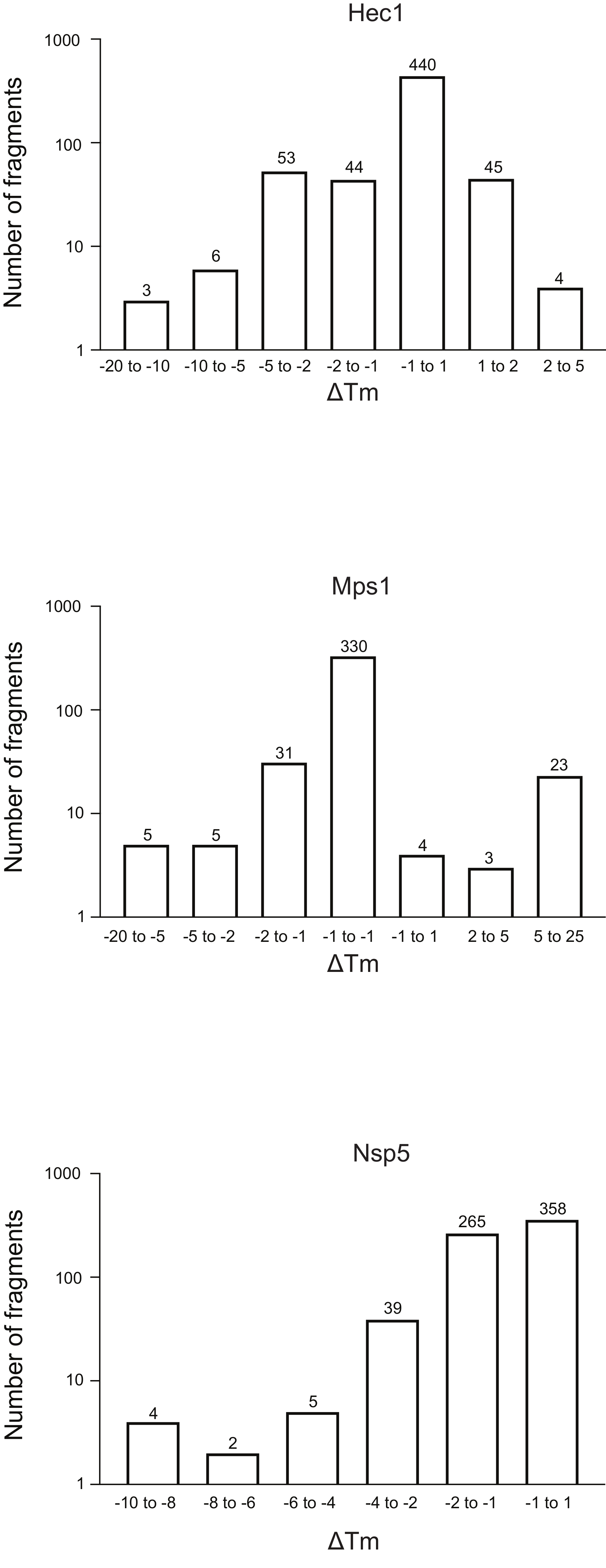{kind=link}
Die Ergebnisse der drei Screenings mit dem oben beschriebenen Protokoll, dargestellt als Häufigkeitsverteilung der Änderung in Tm gegenüber der Anzahl der Fragmente, sind in Abbildung 5 dargestellt. Diese Diagramme wurden wie in Abschnitt 5.3 des Protokolls beschrieben generiert, wobei die Häufigkeit der beobachteten Änderung in Tm dargestellt wird. Die Bedeutung der Verschiebung muss für jedes einzelne Projekt subjektiv definiert werden, wie im folgenden Diskussionsabschnitt beschrieben. Negative Werte zeigen eine Abnahme der Schmelztemperatur in Gegenwart eines Fragments, ein positiver Wert eine Erhöhung in Tman. Aus solchen Diagrammen ist leicht zu beobachten, dass für Nsp5 alle Fragmente eine destabilisierende Wirkung haben, während für Hec1 und Mps1 sowohl stabilisierende als auch destabilisierende Treffer beobachtet werden. Dies ist zu erwarten und wird diskutiert werden.
Diskussion
Dieses Protokoll beschreibt eine Methode mit mittlerem bis hohem Durchsatz zum Screening von Fragmentbibliotheken mit einigen gängigen Robotik- und Messgeräten. Screens wie die in diesem Protokoll beschriebenen können von der NKI Protein Facility in Amsterdam routinemäßig durchgeführt werden, zum Beispiel als iNEXT-Discovery-Service, oft sogar kostenlos für Benutzer nach Antragstellung und Peer-Review. In solchen Fällen kann die DSi-Poised-Bibliothek von der Einrichtung bereitgestellt werden, aber die Verwendung anderer Bibliotheken kann auch im Rahmen der jeweiligen Benutzeranwendung und Servicevereinbarung diskutiert werden. Die Wahl der Instrumente in diesem Protokoll stellt für viele Labore praktische Lösungen dar, sollte aber nicht als Goldstandard betrachtet werden. Für die Messung der thermischen Stabilität des Zielproteins für das Fragmentscreening werden markierungsfreie Methoden empfohlen, anstatt Methoden, die umweltempfindliche Markierungen zum Nachweis der Entfaltung in einem Thermocycler der Reverse-Transkriptionspolymerase-Kettenreaktion verwenden.
Markierungsfreie Methoden, wie die hier vorgestellte mit dem Prometheus-Instrument, haben einige Vorteile: Sie verwenden geringe Mengen an Protein, oft ein paar Größenordnungen weniger; sie können verwendet werden, um gleichzeitig die Streuung der Probe und damit die Aggregation zu messen; und die Markierungen, die zur Erkennung von Entfaltung in anderen Ansätzen verwendet werden, können mit jedem Fragment unterschiedlich interagieren, was zu Messartefakten führt. Dieses Protokoll wurde im Zusammenhang mit dem Mosquito-Roboter beschrieben, der das Pipettieren eines sehr kleinen Probenvolumens (0,3 μL) ermöglicht, das nicht manuell durchgeführt werden kann. Der Mosquito ist ein beliebter Roboter, der in vielen Labors präsent ist, die an Strukturbiologie- und Wirkstoffforschungsprojekten arbeiten. Das Protokoll kann jedoch eindeutig alternative Ansätze für das Pipettieren mit geringem Volumen verwenden.
Fragmentbibliotheken enthalten in DMSO gelöste Verbindungen. Eine der ersten Herausforderungen besteht darin, die optimale DMSO-Konzentration zu finden, bei der das Protein stabil bleibt und die Verbindungen löslich bleiben. Dabei werden die Messungen bei verschiedenen DMSO-Konzentrationen durchgeführt, um die optimalen Bedingungen für das Screening zu bestimmen. Die hier verwendete Protein-zu-Fragment-Verdünnung ergibt DMSO-Konzentrationen von 0,2%; Die meisten Proteine sind unter diesen Bedingungen ziemlich stabil. Die für die Durchführung des Screenings für die 768-Verbindungsbibliothek erforderliche Proteinmenge beträgt insgesamt ~2-3 mg, da die Messungen typischerweise bei niedrigen Proteinkonzentrationen (0,2 mg ml-1)durchgeführt werden. Die Arbeit mit solchen relativ niedrigen Proteinkonzentrationen reduziert nicht nur die Proteinproduktionskosten, sondern verringert auch die Wahrscheinlichkeit einer Proteinfällung. Die niedrige Proteinkonzentration hat keinen Einfluss auf den Nachweis der Fragmentbindung, da die Konzentration der Fragmente im Experiment ~ 2 mM beträgt, so dass auch schwache Bindemittel identifiziert werden können.
Da die Schmelzübergangsdetektion in diesen Experimenten auf der Fluoreszenzintensität basiert, ist ein kritischer Aspekt die Bestimmung der Anregungsleistung des Lasers, bei dem die Messungen durchgeführt werden sollen. Die Wechselwirkung der Verbindungen mit dem Protein kann (i) keinen Einfluss auf seine intrinsische Fluoreszenz haben, (ii) zu einem Abschrecken führen oder (iii) seine intrinsische Fluoreszenz erhöhen. Darüber hinaus bedeutet die Arbeit mit niedrigen Proteinkonzentrationen, dass die Fluoreszenzzahl für das native Protein niedrig wäre. Die Erregerleistung muss daher so eingestellt werden, dass die meisten Proben gemessen werden können. Das Streuprofil jedes Durchlaufs liefert wichtige Informationen über die Aggregationseffekte, die durch das Hinzufügen eines beliebigen Fragments ausgelöst werden können. Darüber hinaus ist der Einfluss der Temperatur auf die Löslichkeit der Verbindung auch auf das Streuprofil zu sehen.
Unerwarteterweise wurde bei vielen Verbindungen beobachtet, dass die Streuung mit steigender Temperatur tatsächlich abnahm (Abbildung 6). Es ist daher wichtig, sowohl die Schmelzübergangskurve als auch das zugehörige Streuprofil zu betrachten, um über die Zuverlässigkeit jedes Experiments zu entscheiden, insbesondere für diejenigen Fragmente, die als Kandidaten für anspruchsvollere Messungen mittels Röntgenkristallographie oder NMR-Spektroskopie oder sogar als Treffer für die Folgechemie gelten. Eine spezifische Einschränkung der Methode für Fragmentscreening-Zwecke besteht darin, dass viele Fragmente in der DSi-Poised-Bibliothek eine signifikante intrinsische Fluoreszenz aufweisen, manchmal sogar über die Sättigungsgrenze des Detektors hinaus, und daher können diese selbst bei geringer Anregungsleistung nicht richtig auf Zielbindung untersucht werden. Ein weiterer Punkt, der bei dieser Methode zu beachten ist, ist, dass sie nur mit Proteinen verwendet werden kann, die Tryptophanrückstände enthalten.

Abbildung 6: Einfluss der Temperatur auf die Löslichkeit der Verbindung. Schmelzübergangskurve und Streuprofil von Hec1 mit zwei verschiedenen Verbindungen. (A) Das Streuprofil zeigt, dass bei dieser Probe die Löslichkeit nicht durch die Temperatur beeinflusst wird. (B) Das Streuprofil zeigt, dass die Löslichkeit dieser Probe mit steigender Temperatur zunimmt. Die Schmelzübergangskurve ist in diesem Fall daher nicht zuverlässig. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Eine offene Frage ist, was als signifikante Veränderung von Tmbetrachtet werden sollte, nicht aus mathematischer Sicht, sondern aus praktischer Sicht: Welche Änderung in Tm ist wichtig, um als Hinweis auf die Bindung eines Liganden an ein Protein zu betrachten? In diesen Beispielen sind Verschiebungen von weniger als 1 °C für 74% der Fragmente für die Bindung von Hec1, 66% für Mps1 und 53% für Nsp5 zu sehen. In den Übersichtsgrafiken (Abbildung 5) wurden Behälter mit 1, 2, 5 oder mehr als 5 Grad Tm Verschiebung, entweder positiv oder negativ, berücksichtigt. Dies erfordert eine Anpassung an den jeweiligen Einzelfall, um einen guten Überblick zu geben und eine fundierte Entscheidungsfindung zu ermöglichen, die den nächsten Schritt bestimmt. Insbesondere bei einigen Proteinen wurden in Abhängigkeit von den betrachteten Fragmenten sowohl eine Stabilisierung als auch eine Destabilisierung des Ziels beobachtet. Beide Ereignisse sind interessant, da beide das Ergebnis einer Fragmentbindung sein können und beide zu einem guten Folgemolekül führen können, um das Proteinverhalten zu manipulieren.
Eine letzte Frage bleibt, nämlich "Was definiert einen nützlichen Treffer?". Tatsächlich hängt die Antwort von der spezifischen Situation ab. Zum Beispiel wurden für Hec1 alle Fragmente, die das Protein um mehr als 2 Grad stabilisieren oder um mehr als 5 Grad destabilisieren, unseren Chemie-Mitarbeitern mitgeteilt, die neue Moleküle basierend auf diesen Treffern entwickelten. Für Nsp5 wurden jedoch die destabilisierendsten Treffer an unsere NMR-Mitarbeiter kommuniziert, um die von nanoDSF abgeleiteten Treffer mit NMR-Experimenten zu bestätigen. Mit anderen Worten, die aus diesem Protokoll erhaltenen Screening-Ergebnisse sollten mit Vorsicht und kontextabhängig analysiert werden, wobei fundierte Entscheidungen auf der Grundlage der spezifischen Frage und der umgebenden Methodik getroffen werden sollten. In jedem Fall ist die hier beschriebene Methode ein komplementärer Ansatz zu bestehenden Methoden wie Röntgen- und NMR-basiertem Screening, die darauf abzielen können, neue Ideen für Chemiekampagnen zu bestätigen, zu priorisieren oder zu geben.
Ergänzende Tabelle S1: Liste der Puffer, die für das Screening von Proteinen verwendet werden. Abkürzungen: HEPES = 4-(2-hydroxyethyl)-1-piperazinethansulfonsäure; DTT = Dithiothreitol; MOBS = 4-(N-Morpholino)butansulfonsäure. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Ergänzende Tabelle S2: Eigenschaften für MRC 2-Well-Platte zur Verwendung mit Nanodosiererroboter. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Zusatztabelle S3: Eigenschaften für 96-Well-V-Bodenplatte zur Verwendung mit Nanodosiererrobotern. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Ergänzende Tabelle S4: Puffer, Proteinkonzentration und Tm der Proteine, die in repräsentativen Ergebnissen diskutiert werden. Abkürzungen: DTT = dithiothreitol; Hec1 = Hoch exprimiert in Krebs-1-Protein; Mps1 = monopolare Spindelkinase 1; Nsp5 = SARS-CoV-2 3C-ähnliche Protease. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Zusatzdatei 1: Übersichtsparameter-Beispieldaten. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Zusatzdatei 2: Tm und ΔTm Werte für 406 Fragmente - Beispieldaten. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Offenlegungen
Es gibt keine Offenlegungen.
Danksagungen
"Diese Arbeit profitierte vom Zugang zur NKI Protein Facility, einem Instruct-ERIC-Zentrum. Finanzielle Unterstützung wurde von iNEXT, Projektnummer 653706, und iNEXT-Discovery, Projektnummer 871037, finanziert durch das Horizon 2020-Programm der Europäischen Kommission" bereitgestellt.
Materialien
| Name | Company | Catalog Number | Comments |
| ClearVue Sheets | Molecular Dimensions | adhesive sealing film for protein plate | |
| CORNING 6570 Aluminium Sealing Tape | CORNING | adhesive sealing film for fragment plate | |
| DSi poised library | Enamine | Fragment library containing 768 compounds used in this study | |
| Elisa Reagent Reservior | ThermoFisher Scientific | 15075 | Reagent reservior used for pipetting the protein |
| Greiner round (U) bottom plates | Cat. No. 650201 | Fragments supplied in these plates | |
| Mosquito type X1 | sptlabtech | Part nr- 3019-0003 | Nanolitre dispenser |
| MRC 2-well crystallization plate | MRC96T-PS | ||
| Pierce ELISA Reagent Reservoirs | Pierce | ||
| Prometheus High Sensitivity capillaries | Catalog PR-C006 | ||
| Prometheus NT.48 nanoDSF | Nanotemper | Catalog nr PR001 (+ Aggregation Detection Optics, catalog nr PR-AGO) | nanoDSF and light back scattering |
| Prometheus Standard capillary type | Catalog PR-C002 | ||
| TX-1000 | Thermoscientific | Centrifuge for plates |
Referenzen
- Hoffer, L., Muller, C., Roche, P., Morelli, X. Chemistry-driven hit-to-lead optimization guided by structure-based approaches. Molecular Informatics. 37 (9-10), 1800059(2018).
- Lamoree, B., Hubbard, R. E. Current perspectives in fragment-based lead discovery (FBLD). Essays in Biochemistry. 61 (5), 453-464 (2017).
- Ress, D. C., Congreve, M., Murray, C. W., Carr, R. Fragment-based lead discovery. Nature Reviews Drug Discovery. 3 (8), 660-672 (2004).
- Carr, R. A. E., Congreve, M., Murray, C. W., Rees, D. C. Fragment-based lead discovery: Leads by design. Drug Discovery Today. 10 (14), 987-992 (2005).
- Bradley, A. R., et al. The SGC beyond structural genomics: Redefining the role of 3D structures by coupling genomic stratification with fragment-based discovery. Essays in Biochemistry. 61 (5), 495-503 (2017).
- Davies, T. G., Tickle, I. J. Fragment screening using X-ray crystallography. Topics in Current Chemistry. 317, 33-59 (2012).
- Ma, R., Wang, P., Wu, J., Ruan, K. Process of fragment-based lead discovery - A perspective from NMR. Molecules. 21 (7), 854(2016).
- Troelsen, N. S., Clausen, M. H. Library design strategies to accelerate fragment-based drug discovery. Chemistry. 26 (50), 11391-11403 (2020).
- Shi, Y., von Itzstein, M. How size matters: Diversity for fragment library design. Molecules. 24 (15), 2838(2019).
- Taylor, A., Doak, B. C., Scanlon, M. J. Design of a fragment-screening library. Methods in Enzymology. 610, 97-115 (2018).
- Cox, O. B., et al. A poised fragment library enables rapid synthetic expansion yielding the first reported inhibitors of PHIP(2), an atypical bromodomain. Chemical Science. 7, 2322-2330 (2016).
- Sreeramulu, S., et al. NMR quality control of fragment libraries for screening. Journal of Biomolecular NMR. 74 (10-11), 555-563 (2020).
- Pfaff, S. J., Chimenti, M. S., Kelly, M. J. S., Arkin, M. R. Biophysical methods for identifying fragment-based inhibitors of protein-protein interactions. Methods in Molecular Biology. 1278, 587-613 (2015).
- Fattori, D., Squarcia, A., Bartoli, S. Fragment-based approach to drug lead discovery: Overview and advances in various techniques. Drugs in R & D. 9 (4), 217-227 (2008).
- Winter, A., et al. Biophysical and computational fragment-based approaches to targeting protein-protein interactions: Applications in structure-guided drug discovery. Quarterly Reviews of Biophysics. 45 (4), 383-426 (2012).
- Boelens, R., et al. iNEXT: a European facility network to stimulate translational structural biology. FEBS Letters. 592 (12), 1909-1917 (2018).
- Zhang, R., Monsma, F. Fluorescence-based thermal shift assays. Current Opinion in Drug Discovery and Development. 13 (4), 389-402 (2010).
- Boivin, S., Kozak, S., Meijers, R. Optimization of protein purification and characterization using Thermofluor screens. Protein Expression and Purification. 91 (2), 192-206 (2013).
- Martinez Molina, D., Nordlund, P. The cellular thermal shift assay: a novel biophysical assay for in situ drug target engagement and mechanistic biomarker studies. Annual Review of Pharmacology and Toxicology. 56, 141-161 (2016).
- Bruce, D., Cardew, E., Freitag-Pohl, S., Pohl, E. How to stabilize protein: stability screens for thermal shift assays and nano differential scanning fluorimetry in the Virus-X Project. Journal of Visualized Experiments JoVE. (144), e58666(2019).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten