
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
Time-resolved Förster Resonance Energy Transfer Assays for Measurement of Endogenous Phosphorylated STAT Proteins in Human Cells
In This Article
Summary
Time-resolved Förster resonance energy transfer cell-based assay protocols are described for the simple, specific, sensitive, and robust quantification of endogenous phosphorylated signal transducer and activator of transcription (STAT) 1/3/4/5/6 proteins in cell lysates in a 384-well format.
Abstract
The Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling pathway plays a crucial role in mediating cellular responses to cytokines and growth factors. STAT proteins are activated by tyrosine phosphorylation mediated mainly by JAKs. The abnormal activation of STAT signaling pathways is implicated in many human diseases, especially cancer and immune-related conditions. Therefore, the ability to monitor STAT protein phosphorylation within the native cell signaling environment is important for both academic and drug discovery research. The traditional assay formats available to quantify phosphorylated STAT proteins include western blotting and the enzyme-linked immunosorbent assay (ELISA). These heterogeneous methods are labor-intensive, low-throughput, and often not reliable (specific) in the case of western blotting. Homogeneous (no-wash) methods are available but remain expensive.
Here, detailed protocols are provided for the sensitive, robust, and cost-effective measurement in a 384-well format of endogenous levels of phosphorylated STAT1 (Y701), STAT3 (Y705), STAT4 (Y693), STAT5 (Y694/Y699), and STAT6 (Y641) in cell lysates from adherent or suspension cells using the novel THUNDER time-resolved Förster resonance energy transfer (TR-FRET) platform. The workflow for the cellular assay is simple, fast, and designed for high-throughput screening (HTS). The assay protocol is flexible, uses a low-volume sample (15 µL), requires only one reagent addition step, and can be adapted to low-throughput and high-throughput applications. Each phospho-STAT sandwich immunoassay is validated under optimized conditions with known agonists and inhibitors and generates the expected pharmacology and Z'-factor values. As TR-FRET assays are ratiometric and require no washing steps, they provide much better reproducibility than traditional approaches. Together, this suite of assays provides new cost-effective tools for a more comprehensive analysis of specific phosphorylated STAT proteins following cell treatment and the screening and characterization of specific and selective modulators of the JAK/STAT signaling pathway.
Introduction
The JAK/STAT signaling pathway plays a key role in mediating cellular responses to diverse cytokines, interferons, growth factors, and related molecules1,2. The binding of these ligands to specific cell-surface receptors results in the activation of JAKs, which in turn activate STAT proteins by phosphorylation of specific tyrosine residues. STAT phosphorylation results in their dimerization and translocation into the nucleus, where they exert their effect on the transcription of regulated target genes. The STAT family consists of seven members: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. The members play a complex and essential role in the regulation of physiologic cell processes, including proliferation, differentiation, apoptosis, angiogenesis, and immune system regulation. The abnormal activation of STAT signaling pathways is implicated in many human diseases, especially cancer and immune-related conditions3,4. Therefore, the ability to assess STAT protein phosphorylation within the native cell signaling environment is important for both academic and drug discovery research.
To date, the conventional methods used to measure intracellular phosphorylated protein levels, including STATs, are antibody-based and include western blotting, ELISA, and phosphoflow cytometry. These heterogeneous methods are labor-intensive, time-consuming, error-prone, low-throughput, and often unreliable (e.g., specificity issues) in the case of western blotting5. In contrast, homogeneous assays require fewer experimental steps, use smaller sample volumes, and are amenable to HTS. There are five homogeneous cell-based immunoassay platforms commercially available that can be used to quantitatively monitor JAK-dependent phosphorylation of STATs in cell lysates: SureFire, HTRF, LANCE, LanthaScreen, and Lumit. Each of these platforms has its advantages and disadvantages.
SureFire is based on luminescent oxygen channeling technology, which utilizes donor and acceptor beads coated to specifically capture a pair of antibodies, one of which is biotinylated. In the presence of phosphorylated protein, the two antibodies bring the donor and acceptor beads into close proximity, enabling the generation of a chemiluminescent signal6. While versatile and sensitive, this technology is expensive, is affected by biotin in the culture medium, is very sensitive to ambient temperature and light, and requires a special reader for detection. HTRF and LANCE are both based on TR-FRET technology that utilizes long-lifetime luminescent lanthanide ion complexes (Europium or Terbium chelates, or Europium cryptate) as the donor molecules and far-red fluorophores as the acceptor molecules7. When two protein-specific antibodies labeled with either donor or acceptor molecules are brought into close proximity, FRET takes place, causing an increase in acceptor fluorescence and a decrease in donor fluorescence. These long-lived fluorescent signals can be measured in a time-resolved and ratiometric manner to reduce assay interference and increase data quality. Other advantages of TR-FRET are that it is not light-sensitive, allows repeated readings, and exhibits long signal stability. While TR-FRET is widely implemented in HTS due to its versatility, sensitivity, and high robustness, all commercial TR-FRET-based assay platforms are expensive, thereby precluding its wide adoption in academic and small industrial laboratories. The LanthaScreen assay also uses a TR-FRET based-readout but is reliant on an engineered U2OS cell line that stably expresses green fluorescent protein (GFP)-STAT1 fusion protein combined with a terbium-labeled phospho-specific STAT1 antibody8. In addition to being limited in terms of choice of signaling proteins, this method requires purchasing expensive transfected cell lines, reducing its applicability and increasing the possibility of experimental artifacts. Lumit is a generic bioluminescent immunoassay platform that utilizes secondary antibodies (anti-mouse and anti-rabbit) chemically labeled with the small and large NanoBit subunits of NanoLuc Luciferase9. The binding of two primary antibodies to the target protein brings the secondary antibodies into proximity to form an active enzyme that generates a luminescence signal. While luminescence is generally a sensitive and robust readout, the requirement for primary antibodies raised in two different species limits the choices for assay design. In addition, the use of secondary antibodies in complex sample matrixes may be prone to assay interference.
Thus, a need still exists for a reliable, rapid, yet affordable cell-based assay platform for measuring individual phosphorylated and total STAT proteins in a manner compatible with HTS. To address this need, a new high-throughput cell-based immunoassay platform was developed based on an enhanced TR-FRET technology (THUNDER) and designed to enable simple, sensitive, robust, and cost-effective measurement of endogenously expressed intracellular proteins (phosphorylated or total) in cell lysates. The advantages of this technology stem from the combination of a donor/acceptor FRET pair exhibiting exceptional spectral compatibility and TR-FRET signal, rigorously validated antibodies, and optimized lysis buffers. These assays are formatted as sandwich immunoassays and use a straightforward, three-step workflow (Figure 1). Cells are first treated to modulate protein phosphorylation and then lysed with the specific lysis buffer provided in the kit. The target phosphorylated or total STAT protein in the cell lysate is detected in a single reagent addition and incubation step with a pair of fluorophore-labeled antibodies that recognize distinct epitopes on the target protein (Figure 2). One antibody is labeled with a Europium chelate donor (Eu-Ab1), while the second antibody is labeled with a far-red acceptor fluorophore (FR-Ab2). The two labeled antibodies bind to the protein in solution, bringing the two labels into close proximity. Excitation of the donor Europium chelate at 320 or 340 nm triggers a FRET to the acceptor, which emits a long-lived TR-FRET signal at 665 nm proportional to the concentration of target protein (phosphorylated or total) in the cell lysate.

Figure 1: TR-FRET assay workflow. The workflow consists of three steps: cell treatment, cell lysis, and protein detection using TR-FRET. In the two-plate assay protocol, lysates are transferred to a white 384-well detection plate, whereas in the one-plate protocol, all steps are conducted in the same white 384-well detection plate (all-in-one-well protocol). Regardless of the assay protocol used, protein detection is performed in the same total volume (20 µL per well). Abbreviation: TR-FRET = time-resolved Förster resonance energy transfer. Please click here to view a larger version of this figure.
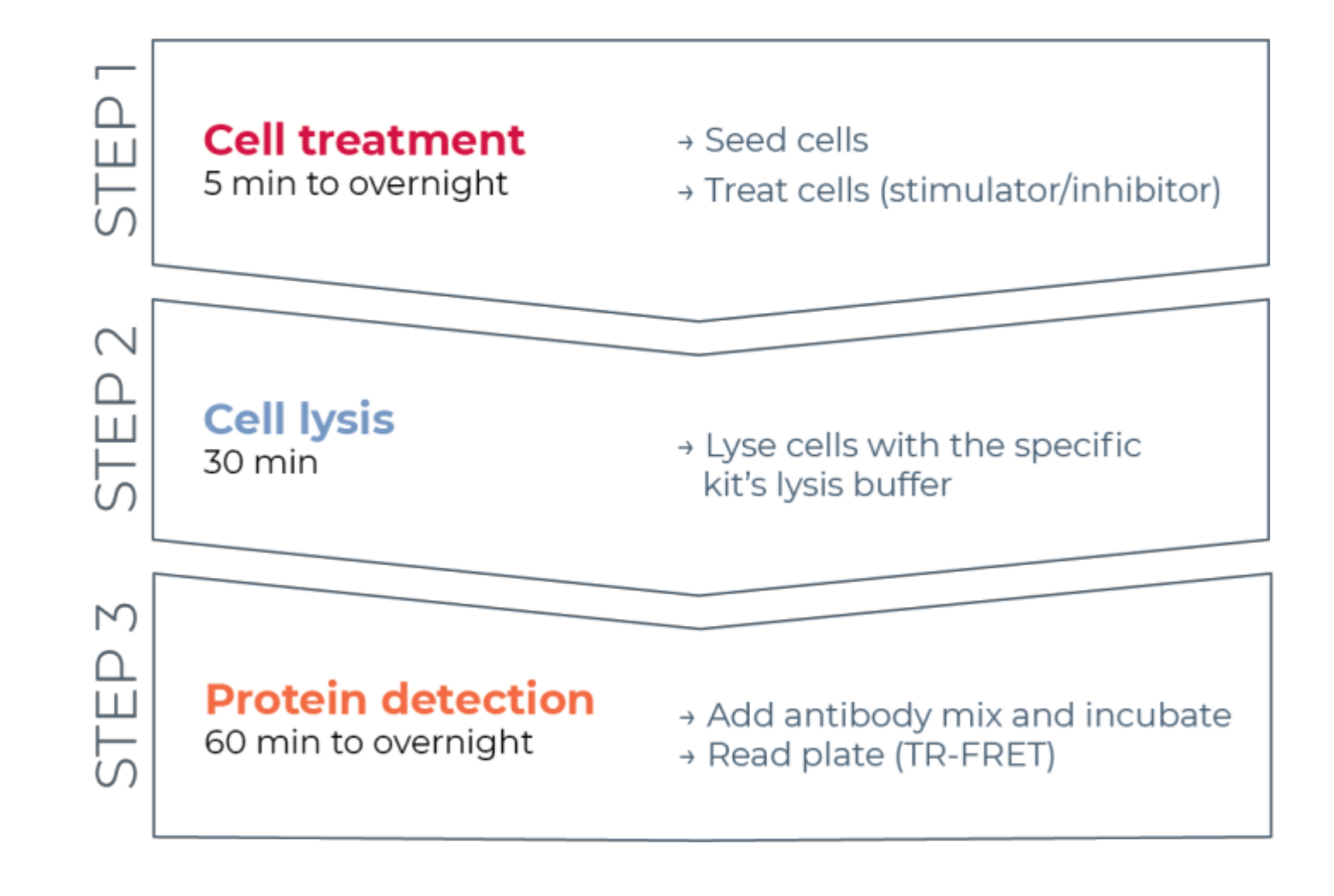{kind=link}

Figure 2: TR-FRET sandwich immunoassay principle. One antibody is labeled with the Europium chelate donor (Eu-Ab1) and the second with the far-red small fluorophore acceptor (FR-Ab2). The two labeled antibodies bind specifically to distinct epitopes on the target protein (phosphorylated or total) in the cell lysate, bringing the two fluorophores into close proximity. Excitation of the donor Europium chelate at 320 or 340 nm triggers a FRET from the donor to the acceptor molecules, which in turn emit a signal at 665 nm. This signal is proportional to the concentration of protein in the cell lysate. In the absence of the specific target protein, the donor and acceptor fluorophores are too distant from each other for FRET to occur. Abbreviations: FRET = Förster resonance energy transfer; TR-FRET = time-resolved FRET; Ab = antibody; FR = far-red; Eu - Europium chelate; P = phosphorylation. Please click here to view a larger version of this figure.
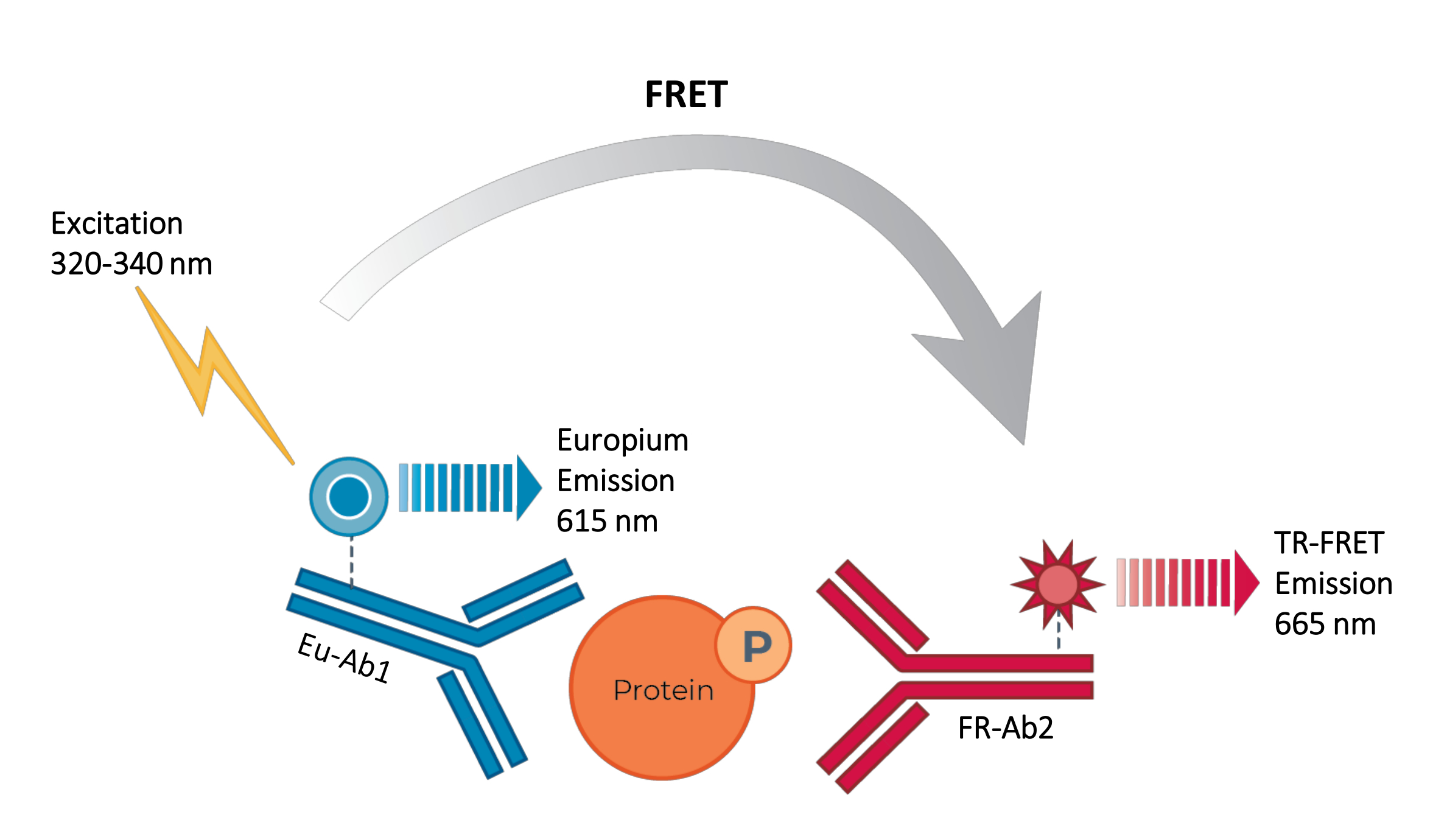{kind=link}
Here, detailed protocols are provided for measuring, in a 384-well format, the intracellular levels of phosphorylated STAT1 (Y701), STAT3 (Y705), STAT4 (Y693), STAT5 (Y694/Y699), and STAT6 (Y641), together with total STAT1, STAT3, STAT5, and STAT6, in cell lysates from adherent or suspension cells using the THUNDER TR-FRET platform. These protocols define steps for cell treatment, lysis, and TR-FRET-based target protein detection using either a two-plate transfer protocol or a one-plate all-in-one-well protocol. These cell-based assays are applied for determining the pharmacological profile of known activators and inhibitors of the JAK/STAT pathway. The robustness and suitability of selected assays for HTS are demonstrated. Lastly, key experiments for assay optimization are discussed, along with recommendations for assay troubleshooting.
Protocol
1. Cell culture
- Maintain cells in a humidified 37 °C/5% CO2 incubator and culture with either DMEM supplemented with 10% fetal bovine serum (FBS) (HeLa and A431 cells) or RPMI supplemented with 15% FBS (U266B1 cells). Culture the cells until they reach 70-80% confluence, then trypsinize them and passage or use them for the assays.
NOTE: Culture media contained phenol red. No serum starvation was conducted for any cell line prior to conducting the assays.
2. Stimulator or inhibitor titration using the two-plate assay protocol with adherent cells
NOTE: This procedure describes how to determine stimulator or inhibitor potencies by generating a concentration-response curve from a dilution series of the test compound.
- Cell seeding
- Dispense 50 µL of cells at the pre-optimized density (40,000 HeLa cells/well for both STAT3 and STAT6; 75,000 A431 cells/well for STAT5) into a 96-well tissue culture-treated plate in the appropriate culture medium. Incubate overnight at 37 °C/5% CO2.
NOTE: Optimal cell density and culture incubation time need to be determined.
- Dispense 50 µL of cells at the pre-optimized density (40,000 HeLa cells/well for both STAT3 and STAT6; 75,000 A431 cells/well for STAT5) into a 96-well tissue culture-treated plate in the appropriate culture medium. Incubate overnight at 37 °C/5% CO2.
- Dilutions of test compounds
- Prepare intermediate 2x dilution series of test compound(s) by serially diluting compound(s) (half-log interval dilutions) across 12 wells of a polypropylene 96-well plate into serum-free medium.
NOTE: It is recommended to conduct a 12-point, half-log interval concentration-response curve in at least duplicate for an accurate estimation of the EC50 or IC50. - Alternatively, for hydrophobic, dimethylsulfoxide (DMSO)-soluble test compounds, perform the initial dilutions in 100% DMSO, and then dilute the compound dilution series into serum-free medium.
NOTE: The assay tolerance to DMSO must be established before conducting a test compound titration in DMSO vehicle. It is important to keep equal solvent concentrations between treated and untreated cells. In addition, when testing serial dilutions of compounds, the solvent concentrations should always remain constant across the dilution series.
- Prepare intermediate 2x dilution series of test compound(s) by serially diluting compound(s) (half-log interval dilutions) across 12 wells of a polypropylene 96-well plate into serum-free medium.
- Cell treatment
- For cell stimulation, add 50 µL of serum-free medium alone (untreated cells) or containing the stimulator (2x).
- Incubate for the pre-optimized time at either room temperature (RT) or 37 °C (interferon (IFN) α2b/20 min at RT for STAT3; epidermal growth factor (EGF)/10 min at RT for STAT5; interleukin (IL)-4/20 min at RT for STAT6). Proceed then to section 2.4.
NOTE: Optimal incubation temperature needs to be determined. - For cell inhibition, add 25 µL of serum-free medium alone (untreated cells) or containing the inhibitor (4x).
- Incubate for the pre-optimized time at either RT or 37 °C (JAK Inhibitor 1/30 min at RT for STAT3 and STAT6; Erlotinib/15 min at RT for STAT5).
- Add 25 µL of serum-free medium alone (untreated cells) or containing the stimulator (4x) at its EC80.
- Incubate for the pre-optimized time at either RT or 37 °C (same conditions as for step 2.3.2).
- Cell lysis
- Prepare the kit's specific 1x Supplemented Lysis Buffer as indicated by the manufacturer.
NOTE: It is mandatory to supplement the 1x Lysis Buffer with the 100x Phosphatase Inhibitor Cocktail diluted to a final concentration of 1x. The 1x Supplemented Lysis Buffer contains 1 mM sodium fluoride, 2 mM sodium orthovanadate, and 2 mM beta-glycerophosphate. Other phosphatase inhibitors are not required and should be avoided. Lysis buffers and phosphatase inhibitors other than those included in the kit are not recommended as they might contain ingredients that could interfere with the measurement. - Carefully remove and discard the cell culture medium by aspirating the supernatant.
- Immediately add 50 µL of 1x Supplemented Lysis Buffer.
NOTE: Lysis Buffer volume (25-50 µL) may be optimized. - Incubate for 30 min at RT under shaking (orbital plate shaker set at 400 rpm; moderate agitation).
NOTE: Lysis incubation time (30-60 min) may be optimized. Lysates can be used immediately for target protein detection or frozen at -80 °C.
- Prepare the kit's specific 1x Supplemented Lysis Buffer as indicated by the manufacturer.
- TR-FRET detection
- Prepare the 4x Antibody Detection Mix in 1x Detection Buffer as indicated by the manufacturer.
- In this transfer step, carefully pipette 15 µL of cell lysate from the 96-well culture plate to a well of a white, low-volume 384-well microplate.
- Add 15 µL of the Positive Control Lysate and 15 µL of 1x Lysis Buffer (negative control) to separate assay wells.
- Add 5 μL of 4x Antibody Detection Mix (either Eu-Ab1/FR-Ab2 for detection of the phospho-protein or Eu-Ab3/FR-Ab4 for detection of the total protein) to each of the assay wells.
- Cover the plate with a plate sealer and incubate for 1 h up to overnight at RT, depending on the assay (see the corresponding Technical Data Sheet).
NOTE: Optimal reading time needs to be optimized for each assay and cell line. The plate can be read several times without a negative effect on the assay performance. - Remove the adhesive plate sealer and read the plate on a TR-FRET compatible microplate reader.
NOTE: Filter-based fluorometers are recommended, though some monochromator instruments can be used. Verify that the appropriate optic module (filters and mirror) for TR-FRET is installed. Use an excitation wavelength of 320 or 340 nm to excite the Europium chelate. Read assays at both 615 nm (or 620 nm) and 665 nm to detect both the emission from the donor Europium and the acceptor fluorophore, respectively. The instrument settings will depend on the particular reader. Data presented here were obtained using lamp-based excitation, 90 µs delay, 300 µs integration time, and 100 flashes per well. The phospho-STAT4 assay, however, was read using laser excitation to generate higher signal-to-background (S/B) ratios.
3. Stimulator or inhibitor titration using the two-plate assay protocol with suspension cells
- Dilution of test compounds
- Prepare intermediate 2x dilution series of test compound(s) as described in steps 2.2.1 and 2.2.2.
- Cell seeding and treatment
- Dispense 20 µL of cells at the pre-optimized density (200,000 U266B1 cells/well for STAT1; 400,000 U266B1 cells/well for STAT4) into a 96-well tissue culture-treated plate in the appropriate culture medium. Directly proceed to cell treatment or incubate 2-4 h at 37 °C, 5% CO2.
NOTE: This step needs to be optimized for different cell types. - For cell stimulation, add 20 µL of serum-free medium alone (untreated cells) or containing the stimulator (2x).
- Incubate for the pre-optimized time at either RT or 37 °C (IFNα2b/15 min at RT for STAT1; IFNα2b/25 min at 37 °C for STAT4). Proceed then to section 3.3.
NOTE: Optimal incubation temperature needs to be determined. - For cell inhibition, add 10 µL of serum-free medium alone (untreated cells) or containing the inhibitor (4x).
- Incubate for the pre-optimized time at either RT or 37 °C (JAK Inhibitor 1/30 min at RT for STAT1 and STAT4).
- Add 10 µL of serum-free medium alone (untreated cells) or containing the stimulator (4x) at its EC80.
- Incubate for the pre-optimized time at either RT or 37 °C (same conditions as for step 3.2.3).
- Dispense 20 µL of cells at the pre-optimized density (200,000 U266B1 cells/well for STAT1; 400,000 U266B1 cells/well for STAT4) into a 96-well tissue culture-treated plate in the appropriate culture medium. Directly proceed to cell treatment or incubate 2-4 h at 37 °C, 5% CO2.
- Cell lysis
- Prepare the kit's specific 5x Supplemented Lysis Buffer as indicated by the manufacturer.
NOTE: It is mandatory to supplement the 5x Lysis Buffer with the 100x Phosphatase Inhibitor Cocktail diluted to a final concentration of 5x. The 5x Supplemented Lysis Buffer contains 5 mM sodium fluoride, 10 mM sodium orthovanadate, and 10 mM beta-glycerophosphate. Other phosphatase inhibitors are not required and should be avoided. - Add 10 µL of 5x Supplemented Lysis Buffer.
- Incubate for 30 min at RT under shaking (orbital plate shaker set at 400 rpm; moderate agitation).
NOTE: Lysis incubation time (30-60 min) may be optimized. Lysates can be used immediately for target protein detection or frozen at -80 °C.
- Prepare the kit's specific 5x Supplemented Lysis Buffer as indicated by the manufacturer.
- TR-FRET detection
- Following cell lysis, conduct the TR-FRET detection step as described in section 2.5 for the 2-plate assay protocol for adherent cells.
4. Stimulator titration using the one-plate assay protocol with adherent or suspension cells
- Dilution of test compounds
- Prepare intermediate dilution series of test compound(s) at 3x by serially diluting compound(s) (half-log interval dilutions) across 12 wells of a polypropylene 96-well plate into serum-free medium.
- Cell seeding and treatment
- Dispense 8 µL of cells at the pre-optimized density (160,000 U266B1 cells/well for STAT4; 80,000 HeLa cells/well for STAT6), in the appropriate serum-free culture medium, into a white, low-volume 384well assay plate. Directly proceed to cell treatment or incubate 2-4 h at 37 °C, 5% CO2.
NOTE: The requirement for a cell culture incubation period before treatment needs to be determined for different cell types. - For cell stimulation, add 4 µL of serum-free medium alone (untreated cells) or containing the stimulator (3x).
- Incubate for the pre-optimized time at either room temperature or 37 °C (IFNα2b/25 min at 37°C for STAT4; IL-4/20 min at 37°C for STAT6). Proceed then to section 4.3.
- Dispense 8 µL of cells at the pre-optimized density (160,000 U266B1 cells/well for STAT4; 80,000 HeLa cells/well for STAT6), in the appropriate serum-free culture medium, into a white, low-volume 384well assay plate. Directly proceed to cell treatment or incubate 2-4 h at 37 °C, 5% CO2.
- Cell lysis
- Prepare the kit's specific 5x Supplemented Lysis Buffer as indicated by the manufacturer.
NOTE: It is mandatory to supplement the 5x Lysis Buffer with the 100x Phosphatase Inhibitor Cocktail diluted to a final concentration of 5x. - Add 3 µL of 5x Supplemented Lysis Buffer.
- Incubate for 30 min at RT under shaking (orbital plate shaker at 400 rpm).
NOTE: Lysis incubation time (30-60 min) may be optimized. Lysates can be used immediately or frozen at 80 °C.
- Prepare the kit's specific 5x Supplemented Lysis Buffer as indicated by the manufacturer.
- TR-FRET detection
- Add 15 µL of Positive Control Lysate (undiluted) and 15 µL of 1x Supplemented Lysis Buffer (negative control) to separate assay wells.
- Add 5 µL of 4x Antibody Detection Mix (either Eu-Ab1/FR-Ab2 for the phospho-protein or Eu-Ab3-/FR-Ab4 for the total protein) prepared in 1x Detection Buffer to each of the assay wells.
- Cover the plate with a plate sealer and incubate for 1 h up to overnight at RT, depending on the assay.
NOTE: Optimal reading time needs to be optimized for each assay and cell line. The plate can be read several times without a negative effect on the assay performance. - Remove the adhesive plate sealer. Read the plate on a TR-FRET-compatible microplate reader.
5. Data analysis
- Calculate the TR-FRET ratio for each well using the following formula (1):
 (1)
(1)
NOTE: Because the TR-FRET signal is read in a time-resolved mode, background subtraction is usually not necessary. If background subtraction is conducted, use the cell-free wells containing the 1x Supplemented Lysis Buffer (negative control) for background subtraction. Determine the average TR-FRET ratio from the cell-free wells and then subtract this value from the TR-FRET ratio of each well. - For concentration-response curves, analyze the data according to a nonlinear regression using the 4 parameter logistic equation (sigmoidal dose-response curve with variable slope) and a 1/Y2 data weighting to generate EC50 or IC50 values.
- For the Z' factor experiment, analyze the data according to the following formula (2)10
 (2)
(2)
Where µ and σ are the mean values and standard deviations for the positive control (p; stimulated cells) and negative control (n; untreated cells), respectively.
Results
Each THUNDER TR-FRET assay was pharmacologically validated by treating adherent (HeLa or A431) or suspension cells (U266B1) with JAK/STAT pathway-specific activators or inhibitors and then measuring the levels of specific phosphorylated and total STATs, when applicable. Assays were conducted in 384-well format using the two-plate transfer protocol and pre-optimized assay conditions. Figure 3, Figure 4, Figure 5,
Discussion
Compared to conventional methods for phosphoprotein analysis such as western blotting and ELISA-based methods, the workflow for a THUNDER TR-FRET cellular assay is simple and fast, uses a low-volume sample (15 µL), is designed for HTS in a 384-well format, and is highly amenable to automation. The assay protocol is flexible and can readily be adapted to both medium- and high-throughput applications. Assays can be run using either a two-plate transfer protocol or a one-384-well plate protocol. In the two-plate transf...
Disclosures
Competing Interests: Jaime Padros, Mireille Caron, and Geneviève Chatel are employees of BioAuxilium Research, which manufactures the THUNDER TR-FRET assay kits used in this study. In addition, Jaime Padros and Mireille Caron are shareholders of BioAuxilium Research. This does not alter the authors' adherence to all JoVE policies on sharing data and materials.
Acknowledgements
None.
Materials
| Name | Company | Catalog Number | Comments |
| 96-well microplate, clear, flat bottom, polystyrene, tissue culture-treated, sterile | Corning | 3595 | This is the plate for culturing cells when using the two-plate assay protocol. Other cell culture 96-well plates can be used |
| 384-well microplate, white, low-volume | PerkinElmer | 6007290 | This is the plate for TR-FRET detection when using the two- or one-plate assay protocols. Other low-volume, white 384-well plates can be used |
| A431 cell line | ATCC | CRL-1555 | |
| Adhesive microplate seal | PerkinElmer | 6050185 | |
| DMSO | Fisher | D159-4 | |
| Dulbecco’s modified Eagle medium (DMEM) | Wisent | 319-005-CL | THUNDER TR-FRET is compatible with culture medium containing phenol red |
| EnVision Xcite Multilabel plate reader | PerkinElmer | 2104-0020A | The assays can be performed on a variety of plate readers equipped with the TR-FRET option |
| Erlotinib hydrochloride | Sigma | CDS022564 | |
| Falcon tissue culture treated flasks | Fisher | 13-680-65 | |
| Fetal bovine serum (FBS) | Wisent | 098-150 | |
| HeLa cell line | ATCC | CCL-2 | |
| JAK Inhibitor 1 | Cayman Chemical | 15146 | |
| Orbital plate shaker | Many options available | Not applicable | |
| Recombinant human EGF | PeproTech | AF-100-15 | |
| Recombinant human IFNα2b | ProSpec | CYT-460 | |
| Recombinant human IL-4 | R&D Systems | 204-IL | |
| Roswell Park Memorial Institute 1640 medium (RPMI) | Wisent | 350-007-CL | THUNDER TR-FRET is compatible with culture medium containing phenol red |
| THUNDER Phospho-STAT1 (Y701) + Total STAT1 TR-FRET Cell Signaling Assay Kit | BioAuxilium Research | KIT-STAT1PT-500 | |
| THUNDER Phospho-STAT3 (Y705) + Total STAT3 Cell Signaling Assay Kit | BioAuxilium Research | KIT-STAT3PT-500 | |
| THUNDER Phospho-STAT4 (Y693) TR-FRET Cell Signaling Assay Kit | BioAuxilium Research | KIT-STAT4P-500 | |
| THUNDER Phospho-STAT5 (Y694/Y699) + Total STAT5 TR-FRET Cell Signaling Assay Kit | BioAuxilium Research | KIT-STAT5PT-500 | |
| THUNDER Phospho-STAT6 (Y641) + Total STAT6 TR-FRET Cell Signaling Assay Kit | BioAuxilium Research | KIT-STAT6PT-500 | |
| Trypsin/EDTA 0.05% | Wisent | 325-542-CL | |
| U266B1 cell line | ATCC | TIB-196 | |
| Ultrapure water | NA | NA | Use Milli-Q grade water (18 MΩ.cm) to dilute Lysis Buffer and Detection Buffer |
References
- Villarino, A., Kanno, Y., O'Shea, J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nature Immunology. 18 (4), 374-384 (2017).
- Hammarén, H. M., Virtanen, A. T., Raivola, J., Silvennoinen, O. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine. 118, 48-63 (2019).
- O'Shea, J. J., et al. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annual Review of Medicine. 66, 311-328 (2015).
- Verhoeven, Y., et al. et al. potential and controversy of targeting STAT family members in cancer. Seminars in Cancer Biology. 60 (2), 41-56 (2020).
- Gilda, J. E., et al. et al. blotting inaccuracies with unverified antibodies: need for a Western blotting minimal reporting standard (WBMRS). PLoS One. 10 (8), 0135392 (2015).
- Binder, C., et al. et al. and utilization of the SureFire phospho-STAT5 assay for a cell-based screening campaign. Assay and Drug Development Technologies. 6 (1), 27-37 (2008).
- Ayoub, M. A., et al. Homogeneous time-resolved fluorescence-based assay to monitor extracellular signal-regulated kinase signaling in a high-throughput format. Frontiers in Endocrinology. 5, 94 (2014).
- Robers, M. B., Machleidt, T., Carlson, C. B., Bi, K. Cellular LanthaScreen and beta-lactamase reporter assays for high-throughput screening of JAK2 inhibitors. Assay and Drug Development Technologies. 6 (4), 519-529 (2008).
- Hwang, B., Engel, L., Goueli, S. A., Zegzouti, H. A. Homogeneous bioluminescent immunoassay to probe cellular signaling pathway regulation. Communications Biology. 3 (8), 1-12 (2020).
- Zhang, J. H., Chung, T. D., Oldenburg, K. R. A simple statistical parameter for use in evaluation and validation of high-throughput screening assays. Journal of Biomolecular Screening. 4 (2), 67-73 (1999).
- Osmond, R. I. W., Das, S., Crouch, M. F. Development of cell-based assays for cytokine receptor signaling, using an AlphaScreen SureFire assay format. Analytical Biochemistry. 403 (1-2), 94-101 (2010).
- Haan, C., et al. Jak1 has a dominant role over Jak3 in signal transduction through γc-containing cytokine receptors. Chemistry & Biology. 18 (3), 314-323 (2011).
- Kim, Y., Apetri, M., Luo, B., Settleman, J. E., Anderson, K. S. Differential effects of tyrosine kinase inhibitors on normal and oncogenic EGFR signaling and downstream effectors. Molecular Cancer Research. 13 (4), 765-774 (2015).
- Qian, J., et al. Comparison of two homogeneous cell-based kinase assays for JAK2 V617F: SureFire pSTAT5 and GeneBLAzer fluorescence resonance energy transfer assays. ASSAY and Drug Development Technologies. 10 (2), 212-217 (2012).
- Iversen, P. W., et al., Markossian, S., et al. HTS assay validation. Assay Guidance Manual. , (2012).
- Muelas, M. W., Ortega, F., Breitling, R., Bendtsen, C., Westerhoff, H. V. Rational cell culture optimization enhances experimental reproducibility in cancer cells. Scientific Reports. 8, 3029 (2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved