
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
La respirométrie à haute résolution pour évaluer la bioénergétique dans les cellules et les tissus à l’aide de respiromètres à chambre et à plaques
Dans cet article
Résumé
L’évaluation de la phosphorylation oxydative à l’aide de respiromètres à haute résolution est devenue une partie intégrante de l’analyse fonctionnelle des mitochondries et du métabolisme énergétique cellulaire. Ici, nous présentons des protocoles pour l’analyse du métabolisme énergétique cellulaire à l’aide de respiromètres haute résolution à chambre et à microplaques et discutons des principaux avantages de chaque appareil.
Résumé
La respirométrie à haute résolution (HRR) permet de surveiller la phosphorylation oxydative en temps réel pour l’analyse des états énergétiques cellulaires individuels et l’évaluation des complexes respiratoires à l’aide de protocoles diversifiés de titrage substrat-découpleur-inhibiteur (SUIT). Ici, l’utilisation de deux appareils de respirométrie à haute résolution est démontrée, et une collection de base de protocoles applicables à l’analyse des cellules cultivées, des fibres musculaires squelettiques et cardiaques et des tissus mous tels que le cerveau et le foie sont présentés. Des protocoles pour les cellules et tissus cultivés sont fournis pour un respiromètre à chambre et des cellules cultivées pour un respiromètre à microplaques, tous deux englobant les protocoles de respiration standard. À des fins de comparaison, des cellules HEK293 conçues par CRISPR et déficientes en traduction mitochondriale provoquant une déficience multiple du système respiratoire sont utilisées avec les deux appareils pour démontrer des défauts cellulaires dans la respiration. Les deux respiromètres permettent une mesure complète de la respiration cellulaire avec leurs mérites techniques respectifs et leur adéquation en fonction de la question de recherche et du modèle à l’étude.
Introduction
Les mitochondries remplissent la fourniture clé d’énergie et sont un organite compartimenté contribuant aux processus bioénergétiques et métaboliques cellulaires essentiels tels que l’anabolisme des nucléotides, des lipides et des acides aminés, la biogenèse des amas fer-soufre et sont impliquées dans la signalisation telle que la mort cellulaire contrôlée 1,2,3 . La bioénergétique mitochondriale par phosphorylation oxydative contribue à presque tous les processus cellulaires au sein de la cellule et, par conséquent, les dysfonctionnements mitochondriaux d’origine primaire ou secondaire sont associés à un large éventail de maladies 4,5. Le dysfonctionnement mitochondrial implique non seulement des altérations de la structure ou de la densité mitochondriale, mais aussi de la qualité et de la régulation du système respiratoire6. Cet élément qualitatif englobe le contrôle du substrat, les caractéristiques de couplage, les modifications post-traductionnelles, la dynamique des cristae et les supercomplexes respiratoires 7,8. Par conséquent, une analyse précise de la bioénergétique mitochondriale pour des approches expérimentales et diagnostiques visant à évaluer le métabolisme énergétique de la cellule est importante pour la santé et la maladie.
La phosphorylation oxydative mitochondriale (OXPHOS) est une séquence de réactions au sein du système respiratoire ou du système de transfert d’électrons (ETS) pour la génération d’énergie cellulaire par l’adénosine triphosphate (ATP)9. L’étape multi-enzymatique visant à exploiter l’énergie du flux d’électrons à travers les complexes I et II vers le complexe IV génère un gradient électrochimique de protons à travers la membrane mitochondriale interne, utilisé par la suite pour la phosphorylation de l’adénosine diphosphate (ADP) en ATP via le complexe V (F1FO ATP synthase) (Figure 1A).
Tout d’abord, des transporteurs à deux électrons sont générés au cours du cycle tricarboxylique (TCA), de la glycolyse et de l’oxydation du pyruvate : nicotinamide adénine dinucléotide (NADH) et dihydroflavine adénine dinucléotide (FADH2). Le NADH est oxydé au niveau du complexe I (NADH déshydrogénase), au cours duquel deux électrons sont transférés à la coenzyme Q (la quinone est réduite en quinol), tandis que les protons sont pompés dans l’espace intermembranaire (IMS). Deuxièmement, le complexe II (Succinate déshydrogénase) oxyde le FADH2 et alimente les électrons en coenzyme Q sans pomper de protons. Troisièmement, au complexe III (cytochrome c oxydoréductase), les électrons de la coenzyme Q sont transférés au cytochrome c tandis que les protons sont pompés dans l’IMS. Quatrièmement, le cytochrome c transfère les électrons au complexe IV (Cytochrome c oxydase), le complexe final pour pomper les protons, et où l’oxygène fonctionne comme un accepteur d’électrons pour assimiler les protons, formant finalement de l’eau. C’est cet oxygène que les mitochondries consomment qui peut être mesuré par un oxygraphe. Enfin, les protons générés à partir des complexes I, III et IV sont utilisés pour faire tourner le complexe V, générant ainsi de l’ATP9.
Il est important de noter que le transfert d’électrons ne se produit pas seulement de manière linéaire, autrement désigné comme la chaîne de transport d’électrons. Au lieu de cela, les électrons peuvent être transférés au pool de coenzyme Q par de multiples voies respiratoires et faciliter le flux d’électrons convergents. Les substrats de NADH et le succinate, par exemple, peuvent entrer par les complexes I et II, respectivement. Les électrons issus de l’oxydation des acides gras peuvent être donnés via le complexe flavoprotéique de transfert d’électrons. En effet, une analyse complète d’OXPHOS nécessite une approche holistique avec des substrats de combustible appropriés (Figure 1A).

Figure 1 : Phosphorylation oxydative mitochondriale et protocoles spécifiques de substrat et d’inhibiteur. (A) Mitochondrie et schéma du système de transfert d’électrons (CI-CIV) et de la F1F0 ATP synthase mitochondriale (CV). Toutes les structures sont de PDB. Les figures ne représentent que les substrats et les inhibiteurs décrits dans cette étude). (B) Échantillon de trace de flux d’oxygène dans des cellules HEK293 intactes utilisant un protocole standard dans un dispositif mHRR. (C) Échantillon de trace de flux d’oxygène dans des cellules HEK293 intactes utilisant un protocole standard dans un dispositif cHRR. (D) Échantillon de trace de flux d’oxygène dans des fibroblastes humains perméabilisés provenant d’un donneur sain avec le protocole SUIT respectif. Abréviations : 1 = Respiration de routine des cellules intactes; 2 = État 2; 3 = État 3(I); 4 = État 3(I) avec cytC; 5 = État 3 (I+II); 6 = Fuite (OM); 7 = capacité du SEQE; 8 = S(ROT); 9 = ROX; 10 = TMPD; 11 = Az. ROT = Roténone, AM = Antimycine, ATP = Adénosine triphosphate, Az = Azide, OM = Oligomycine, FCCP = Cyanure de carbonyle p-trifluoro-méthoxyphényl-hydrazone; Asc = Ascorbate, TMPD = N,N,N′,N′-tétraméthyl-p-phénylènediamine, Succ = Succinate, M = Malate, P = Pyruvate, ADP = Adénosine diphosphate, NAD = Nicotinamide adénine dinucléotide, IMS = Espace intermembranaire, FAD = Flavine adénine dinucléotide. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
L’analyse de la capacité OXPHOS mitochondriale à l’aide de HRR est devenue une méthode biochimique instrumentale de valeur diagnostique non seulement pour les défauts mitochondriaux primaires10,11, mais s’étendant à tous les autres domaines de la biologie tels que le cancer et le vieillissement12. HRR permet la détermination de la respiration cellulaire par l’analyse de la capacité mitochondriale OXPHOS, qui reflète directement une déficience individuelle ou combinée du complexe respiratoire mitochondrial, et est indirectement associée à un dysfonctionnement cellulaire et à une altération du métabolisme énergétique9. Sur le plan méthodologique, les mesures de la respiration sont effectuées à l’aide de cellules, detissus ou de mitochondries isolées 11,13,14, le matériel congelé ne convenant que partiellement15,16. Il est démontré que les tissus congelés ont un ETS intact avec une stabilité supercomplexe maintenue15. Ainsi, contrairement aux intermédiaires TCA traditionnels, les substrats respectifs sont directement introduits dans l’ETS. Cependant, le couplage entre la synthèse de l’ETS et de l’ATP est perdu car l’intégrité de la membrane est compromise par les dommages causés par le gel (formation de cristaux de glace).
Les expériences de respiration ont normalement lieu à une température physiologique de 37 °C pour les endothermes dans des cellules ou des tissus non perméabilisés ou perméabilisés. Alors que le premier considère le contexte métabolique cytosolique, le second fournit la contribution énergétique des complexes OXPHOS individuels et de l’ATPase par l’ajout de substrats spécifiques (et d’inhibiteurs). La séquence et la variation des substrats et des inhibiteurs ont conduit au développement d’un large éventail de protocoles SUIT17 etde tests 18 pour répondre à diverses questions scientifiques de la fonction OXPHOS (examinés sous12). Le protocole de base de la respiration cellulaire évalue quatre états différents: i) la respiration de routine - la respiration dans un milieu respiratoire respectif sans aucun ajout de substrats ou d’inhibiteurs consommant mais endogènes. Cet état peut révéler des OXPHOS généraux ou des défauts respiratoires induits secondaires causés, par exemple, par des profils de métabolites modifiés. Ensuite, l’ajout de l’inhibiteur de l’ATPase oligomycine révèle la perméabilité de la membrane mitochondriale interne aux protons, définie comme ii) la respiration des fuites. Le titrage ultérieur d’un protonophore tel que le décupleur cyanure de carbonyle p-trifluoro-méthoxyphényl-hydrazone (FCCP) permet de déterminer l’état auquel la capacité ETS est maximale dans un mode de circuit proton transmembranaire ouvert, défini comme iii) respiration non couplée. Il est important de noter qu’un état non couplé peut également se produire par des interventions expérimentales par des dommages mécaniques excessifs aux membranes mitochondriales. Inversement, l’état non couplé fait référence au découplage respiratoire par un mécanisme intrinsèque qui est physiologiquement contrôlé. Enfin, l’inhibition complète de l’ETS par addition de l’inhibiteur du complexe III antimycine et de l’inhibiteur du complexe I roténone détermine la consommation résiduelle d’oxygène (ROX) des processus non mitochondriaux consommant de l’oxygène (Figure 1A-C).
La bioénergétique mitochondriale se compose de cinq états respiratoires distincts19,20. La respiration de l’état 1 est sans substrats supplémentaires ni ADP, à l’exception de ce qui est disponible de manière endogène. Après l’ajout d’ADP, mais toujours, pas de substrats, la respiration de l’état 2 est atteinte. Lorsque des substrats sont ajoutés, permettant le transfert d’électrons et la synthèse de l’ATP, la respiration de l’état 3 est atteinte. Dans cet état, la capacité OXPHOS peut être définie à des concentrations saturantes d’ADP, de phosphate inorganique, d’oxygène, de substrats liés au NADH et au succinate. La respiration d’état 4 ou respiration LEAK peut être définie comme un état sans ADP ou ATP synthases inhibées chimiquement tout en ayant suffisamment de substrats. Enfin, lorsque tout l’oxygène est épuisé (anoxique) dans un cadre à chambre fermée, la respiration de l’état 5 est observée.
Plusieurs méthodes existent pour évaluer les états d’énergie cellulaire14 avec deux dispositifs dominant l’évaluation actuelle en temps réel d’OXPHOS par l’analyse de la consommation d’oxygène, mesurée comme la fonction de la diminution de l’oxygène au fil du temps dans un système à chambre fermée avec une applicabilité différente en fonction du modèle expérimental et de la question de recherche: le respiromètre haute résolution Oroboros 2k et l’analyseur de flux extracellulaire Seahorse XF. Les deux appareils enregistrent les taux de consommation d’oxygène sous forme de diminution des picomoles (pmol) d’oxygène (O2) par seconde en tant que valeur absolue dans la chambre ou le puits de microplaque. La consommation spécifique d’oxygène par masse est obtenue en normalisant la consommation d’oxygène respective dans une recette tampon spécifique par nombre de cellules (millions), poids tissulaire (mg) ou quantité de protéines.
L’O2k (Oroboros Instruments) est un système fermé à deux chambres équipé d’un capteur d’oxygène polarographique (abrégé en respiromètre haute résolution à chambre: cHRR). Chaque chambre expérimentale contient 2 mL de liquide qui est maintenu homogène par des agitateurs magnétiques. Le capteur d’oxygène polarographique utilise une approche ampérométrique pour mesurer l’oxygène: il contient une cathode d’or, une anode argent/chlorure d’argent et entre les deux une solution KCI créant une cellule électrochimique sur laquelle une tension (0,8 V) est appliquée. L’oxygène du milieu d’essai diffuse à travers une membrane d’éthylène propylène fluoré de 25 μm (perméable àL’O2) et subit une réduction à la cathode, produisant du peroxyde d’hydrogène. À l’anode, l’argent est oxydé par le peroxyde d’hydrogène, générant un courant électrique. Ce courant électrique (ampère) est linéairement lié à la pression partielle d’oxygène. La pression partielle de l’oxygène et le facteur de solubilité en oxygène du milieu d’essai sont utilisés pour calculer la concentration en oxygène. Étant donné que la pression partielle d’oxygène dépend de la température expérimentale et que les mesures polarographiques sont sensibles à la température, les fluctuations de température nécessitent une régulation précise (±0,002 °C) par un bloc chauffant Peltier. La température peut être contrôlée dans une plage de 4 °C et 47 °C.
L’analyseur de flux extracellulaire Seahorse XF (Agilent) est un système à base de plaques au format microplaque à 24 ou 96 puits dans lequel trois électrodes de fluorescence mesurent la consommation d’oxygène au fil du temps dans chaque puits (abrégé en respiromètre haute résolution à base de microplaques: mHRR). Un maximum de quatre ports dans la cartouche de dosage sont disponibles pour l’injection automatisée pendant le test. Un essai contient plusieurs cycles, chacun avec trois phases: 1) mélange, 2) attente et 3) mesure. Pendant la phase de mesure, les sondes de capteur sont abaissées dans la microplaque, créant une chambre temporairement fermée contenant un volume de 7 à 10 μL pour mesurer la lumière émise. Cette lumière est émise par des fluorophores incorporés dans des polymères à l’extrémité des sondes du capteur, qui détectentO2 en fonction de la trempe par phosphorescence. L’intensité du signal de fluorescence est proportionnelle àO2 et influencée par la température du capteur et du milieu d’essai. Par conséquent, une estimation précise de l’oxygène nécessite une approche relative avec un puits de fond sans aucun échantillon. La restauration de la concentration en oxygène se produit pendant la phase de mélange lorsque le capteur se déplace de haut en bas pour mélanger le volume au-dessus de la chambre temporaire. Chaque cycle calcule un taux de consommation d’oxygène. La température peut être contrôlée dans une plage de 16 °C et 42 °C.
HRR est l’étalon-or pour évaluer la bioénergétique cellulaire dans les maladies primaires et associées aux mitochondries et le métabolisme cellulaire général. Dans cette étude, des protocoles de base pour hrr sont fournis pour évaluer la fonction OXPHOS dans les cellules et les tissus.

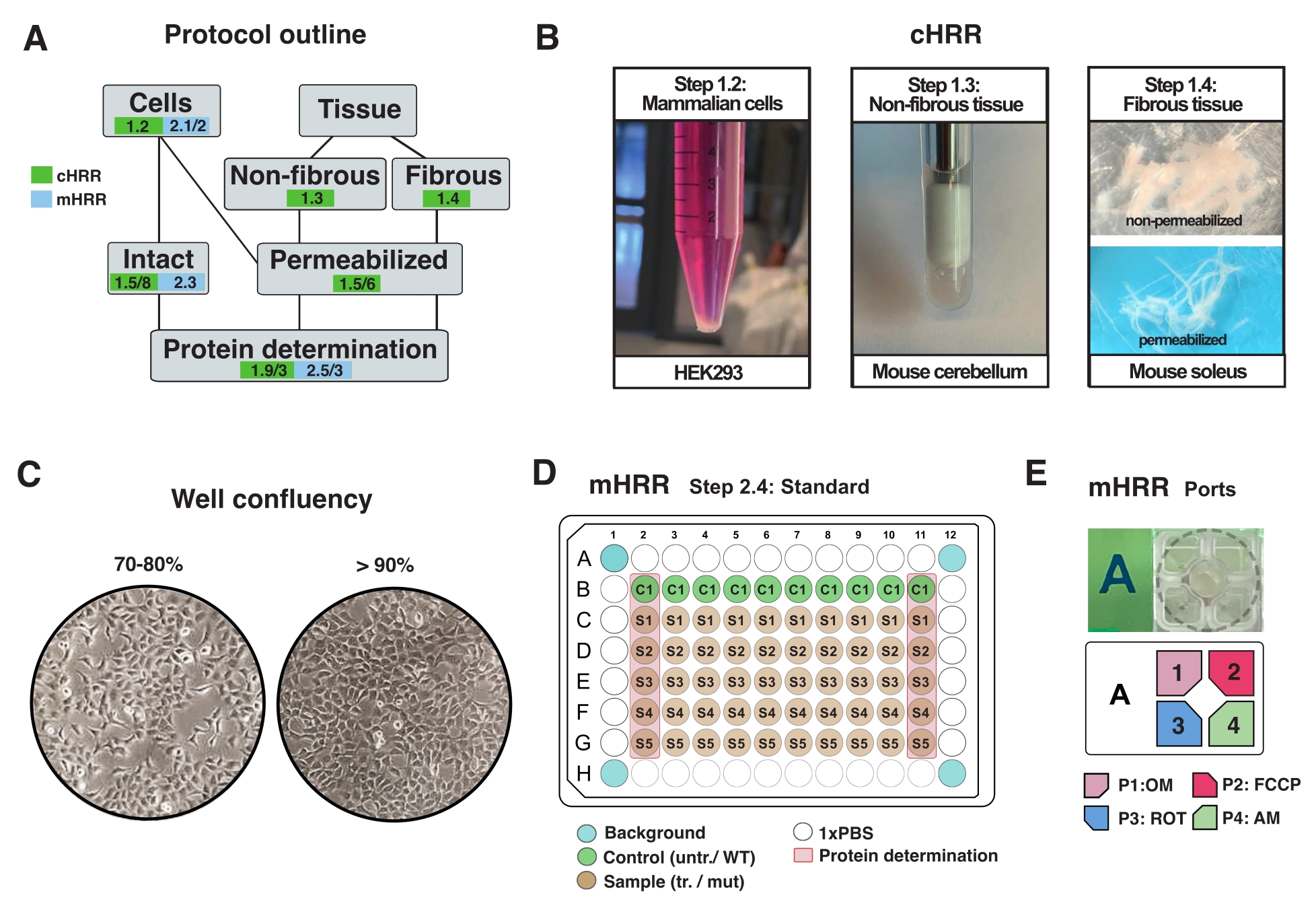
Figure 2 : Flux de travail pour les préparations cellulaires et tissulaires pour la RDH et préparation cellulaire pour la respirométrie mHRR. (A) Aperçu des protocoles fournis. (B) Cellules de mammifères (étape 1.2) : pastille HEK293 égale à 3 x 106 cellules (panneau de gauche). Tissu non fibreux (étape 1.3) : Préparation du lysat de cervelet murin dans 2 mL de potier en téflon (panneau central). Perméabilisation musculaire squelettique induite par la saponine (étape 1.4) panneau de droite) pour la respirométrie cHRR. (C) Schéma standard d’ensemencement des microplaques (étape 2.4) et contrôle de la confluence pour l’analyse des cellules eucaryotes (HEK293) pour la respirométrie mHRR. (D, E) Schéma de chargement de l’orifice d’injection pour la respirométrie mHRR (étape 2.4). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Protocole
Tous les expérimentations animales sont effectués conformément au Conseil national d’examen de l’expérimentation animale et à l’Agence administrative régionale de l’État pour le sud de la Finlande. Des souris mâles C57BL/6JOlaHsd (âgées de 4 à 6 mois) ont été utilisées dans cette étude. Le consentement pour l’utilisation de lignées cellulaires humaines a été obtenu auprès du comité d’éthique institutionnelle de l’Université d’Helsinki.
1. Respirométrie à haute résolution : respiromètre à chambre (cHRR)
REMARQUE: Les expériences de cette section du protocole ont été réalisées à l’aide de l’Oroboros O2k-Core: Oxygraph-2k (Table des matériaux)
- Étalonnage des capteurs d’oxygène
- Respiromètres pré-exécutés à 37 °C dans 2,1 mL de milieu respiratoire mitochondrial (MiR05, tableau 1, facteur de solubilité : 0,92) pendant >45 min et effectuer l’étalonnage de l’oxygène comme décrit21. Procéder si la variation de base se situe dans ± 4 pmol/s.
REMARQUE: De grandes fluctuations du signal de fond pourraient indiquer un entretien nécessaire de la membrane du capteur ou des traces d’inhibiteurs restant dans la chambre lors d’expériences antérieures. Une correction instrumentale du flux d’oxygène de fond est recommandée avant un lot d’expériences25. - Enregistrez les valeurs d’étalonnage de l’oxygène pour surveiller les performances de la membrane du capteur au fil du temps.
REMARQUE: Cela révèle la fonction du capteur, la stabilité signal-bruit et quand l’entretien de la membrane du capteur est nécessaire. En fonction de la pression ambiante, entre 180 et 200 μmol d’oxygène sont solubilisés dans MiR05. - Retirer tout le liquide dans la chambre avant l’ajout de tout échantillon dans le milieu respiratoire.
REMARQUE: Évaluez le volume des chambres de respiration à exactement 2 mL régulièrement.
- Respiromètres pré-exécutés à 37 °C dans 2,1 mL de milieu respiratoire mitochondrial (MiR05, tableau 1, facteur de solubilité : 0,92) pendant >45 min et effectuer l’étalonnage de l’oxygène comme décrit21. Procéder si la variation de base se situe dans ± 4 pmol/s.
- Préparation de cellules pour la respirométrie à haute résolution
- Cultiver des cellules HEK293 dans des boîtes de 10 cmet 2 diamètres dans le milieu de l’Aigle modifié (DMEM) de Dulbecco avec un taux élevé de glucose complété par du sérum fœtal bovin (FBS) inactivé par la chaleur à 10%, du GlutaMax, des acides aminés non essentiels et du Na-Pyruvate22 et de l’uridine23 pour soutenir le métabolisme défectueux d’OXPHOS dans un incubateur à 37 ° C à 5% de CO2.
REMARQUE: Tout type de cellule eucaryote peut être cultivé. Pour la plupart des types de cellules, la culture d’un plat de 10 cm2 conduit à suffisamment de cellules (généralement >3 x 106 cellules). Vérifiez régulièrement l’infection à mycoplasmes pour éviter les effets sur le métabolisme cellulaire et la respiration. - Cultiver des cellules sans dépasser 90 % de confluence (Figure 2C).
REMARQUE: Les cellules ayant une confluence >90% peuvent présenter des effets inhibiteurs dépendants de la croissance sur la respiration (si elles ne sont pas synchronisées ou post-mitotiques). - Lavez les cellules avec 1x PBS, détachez avec 1 mL de trypsine chaude à 0,25%, désactivez la trypsine en ajoutant du DMEM chaud (5 mL / 10 cm2 plaque) et comptez les cellules avec un hémocytomètre.
- Centrifugez doucement la solution cellulaire égale à 2,5 x 106 cellules à 300 x g pendant 5 min, retirez complètement le surnageant et remettez en suspension dans 2,5 mL de MiR05 chaud (1 x 106 cellules/mL) (Figure 2A).
- Pour les cellules de suspension, compter et retirer la solution égale à 2,5 x 106 cellules, pastille et continuer comme mentionné à l’étape 1.2.4.
- Exécutez le protocole SUIT pour l’optimisation de la perméabilisation (étape 1.6), la cellule ou le tissu perméabilisé (étape 1.5) ou les cellules intactes (étape 1.7)
REMARQUE: Pour des résultats cohérents, il est recommandé de maintenir la concentration cellulaire constante (par exemple, 1 x 106 cellules / mL). Bien que la respiration soit indépendante de la densité cellulaire dans le respiromètre24, les substrats et les inhibiteurs sont à une concentration comparable tout au long des expériences si le nombre de cellules est maintenu constant.
- Cultiver des cellules HEK293 dans des boîtes de 10 cmet 2 diamètres dans le milieu de l’Aigle modifié (DMEM) de Dulbecco avec un taux élevé de glucose complété par du sérum fœtal bovin (FBS) inactivé par la chaleur à 10%, du GlutaMax, des acides aminés non essentiels et du Na-Pyruvate22 et de l’uridine23 pour soutenir le métabolisme défectueux d’OXPHOS dans un incubateur à 37 ° C à 5% de CO2.
- Préparation de tissus non fibreux (p. ex. cerveau, foie) pour la respirométrie à haute résolution
- Excisez un morceau de tissu homogène, de 30 à 40 mg de poids, ou utilisez l’organe entier (cervelet de souris dans ce cas).
REMARQUE: Si le tissu n’est pas immédiatement utilisé, conserver dans 2 mL de MiR05 glacé permettant une conservation jusqu’à 2 h pour la plupart des tissus. Les temps de stockage individuels des tissus doivent être évalués dans des séries chronologiques. - Épongez le tissu avec un papier filtre Whatman (attention: la matière des tissus mous a tendance à coller).
- Placez le morceau de tissu de 30 à 40 mg dans un homogénéisateur Elvehjem de potier en polytétrafluoroéthylène refroidi à la glace de 2 mL.
- Ajouter une quantité appropriée de MiR05 pour obtenir 20 mg/mL afin de maintenir le rapport tissu-tampon. Conserver la quantité totale >1,5 mL et <2 mL pour éviter un liquide insuffisant ou excessif pour une perméabilisation mécanique appropriée.
- Insérez le pilon, lysez le tissu lentement en rétractant soigneusement le pilon tout en évitant la génération d’un vide causant des dommages excessifs aux tissus.
- Effectuer 7 coups au total (1x défini comme un coup vers le haut et vers le bas) jusqu’à ce qu’il soit lysé (apparent comme un liquide trouble sans débris majeurs) (Figure 2B).
REMARQUE: Le nombre d’AVC pour une lyse appropriée doit être testé pour chaque tissu en évaluant l’intégrité de la membrane mitochondriale externe via la réponse du cytochrome C (étape 1.5.11). Des parties de tissu conjonctif ou de vaisseau difficiles à lyser peuvent rester. - Décantez le tissu lysé dans un tube de centrifugeuse de 15 mL.
- Lavez l’intérieur du potier avec une quantité égale de MiR05 utilisé dans l’étape de lysage (par exemple, 1,5 mL) et ajoutez au tube de 15 mL contenant maintenant 3-4 mL de MiR05 à 10 mg / mL de lysat de tissu.
- Ajouter 2 mL de MiR05 ordinaire par chambre pour chauffer à 37 °C.
- Faire tourbillonner le tube pour une distribution égale avant de pipeter lentement 500 μL (soit 5 mg) de chaque lysat par chambre pour minimiser la contrainte du froid à 37 °C.
- Attendez >3 min pour que le contenu de la chambre se réchauffe à 37 °C avant de fermer la chambre. Retirer l’excès de liquide sur le dessus du bouchon (quantité par chambre après fermeture: 4 mg).
- Exécutez le protocole SUIT pour la perméabilisation standard (étape 1.5).
- Excisez un morceau de tissu homogène, de 30 à 40 mg de poids, ou utilisez l’organe entier (cervelet de souris dans ce cas).
- Préparation de tissu fibreux (muscle squelettique, muscle cardiaque) pour la respirométrie à haute résolution
- Extraire le tissu dur, retirer le tissu conjonctif et la graisse des muscles à l’aide de pinces pointues dans 2 mL de BIOPS glacé (tableau 2) sous un microscope à dissection.
- Séparez les faisceaux de fibres (~4 mg) le long de l’axe longitudinal avec des pinces pointues. Extraire suffisamment les fibres pour obtenir une structure en forme de maille (Figure 2B).
REMARQUE: Une séparation mécanique et une perméabilisation appropriées des fibres sont indiquées par la perte de la myoglobine pigmentaire rouge et une translucidité accrue. - Laver et perméabiliser le faisceau de fibres dans de la saponine (50 μg/mL dans BIOPS, préparé frais) pendant 20 min à 4 °C (les fibres deviennent translucides, indiquant une perméabilisation complète, Figure 2B).
- Lavez les fibres deux fois dans MiR05 pendant 5 min par lavage à 4 °C.
- Sécher avec du papier filtre et peser avant d’ajouter à la chambre remplie de 2,1 mL de MiR05.
- Introduisez les bouchons sans fermer complètement, puis oxygénez les chambres avec 2 mLd’O2 pur à l’aide d’une seringue de 20 mL et fermez les chambres en tordant les bouchons dans un mouvement de rotation. Maintenirla concentration d’O2 entre 300 et 500 μM pendant l’expérience pour éviter la limitation de la diffusion de l’oxygène.
- Protocole d’évaluation de la respiration de routine dans les cellules ou les tissus
- Ajouter l’échantillon à la chambre comme indiqué aux étapes 1.5.2-1.5.3.
- Ajouter 2,3 mL de suspension chaude de cellules MiR05 (entrée standard : 1 x 106 cellules/mL comme à l’étape 1,2 ou 2 mg de tissu/mL comme à l’étape 1.3)
- Muscle squelettique et cardiaque (étape 1.4): Ajouter ~ 4 mg de fibres perméabilisées à la saponine à 2,3 mL de MiR05 chaud préavertissé en tenant compte des étapes 1.4.4-1.4.6
- Faites fonctionner les chambres à 37 °C et à une vitesse d’agitation de 700 tr/min. Attendez >3 min pour permettre au média de dégazer et de fermer les chambres en tordant le bouchon dans un mouvement de rotation. La stabilisation du bloc Peltier indique l’atteinte de la température réglée.
- (FACULTATIF) Modifiez la vitesse de l’agitateur à 300 tr/min pour permettre aux bulles restantes de s’échapper par le capillaire du bouchon.
- Aspirer tout excès de liquide sur le dessus du bouchon. Attendez 10 minutes jusqu’à ce qu’un signal de flux d’oxygène stable soit obtenu avec n’importe quel type d’échantillon pour enregistrer la respiration de routine/état 1, Figure 1B).
- Pour les mesures de la respiration dans les cellules et les tissus perméabilisés, passez à l’étape 1.6. Pour les cellules intactes avec l’étape 1.8.
- Protocole pour l’analyse OXPHOS dans les cellules ou tissus perméabilisés
- Utiliser un échantillon de tissu lysé (perméabilisé) ou perméabiliser des cellules en ajoutant 1 μL de digitonine (8,1 mM de digitonine dans du diméthylsulfoxyde (DMSO)) pour une concentration finale de 5 μg/mL pour perméabiliser les cellules. Le flux va baisser et devrait se stabiliser à >5 min.
ATTENTION : La digitonine est extrêmement toxique pour les voies respiratoires, au contact de la peau ou lorsqu’elle est avalée.
REMARQUE: L’injection de tous les produits chimiques est effectuée avec des seringues en verre de précision. Utilisez des seringues uniquement pour les produits chimiques indiqués afin d’éviter la contamination croisée et lavez soigneusement à l’eau et à l’EtOH après utilisation. Les seringues bloquées peuvent nécessiter des ultrasons dans du ddH2O chaud ou un fil de nettoyage pour déloger les bouchons chimiques. Rétractez toujours un surplus de la solution mère respective dans la seringue pour éviter d’introduire de l’air dans les chambres. Inspectez l’intérieur des chambres pour l’introduction d’air après chaque injection. Enregistrez chaque étape jusqu’à ce que le flux plafonne. - Ajouter successivement : 5 μL de 0,4 M de malate (M) pour une concentration finale de 1 mM, 5 μL de 2,0 M de pyruvate (P; préparé fraîchement), pour une concentration finale de 5 mM, 4 μL de glutamate (G) de 2,5 M pour une concentration finale de 5 mM.
- Après le plateau du flux précédent, ajouter 5 μL (10 μL pour le tissu musculaire) de 0,5 M d’adénosine diphosphate (ADP, aliquotes stockées à -80 °C) pour une concentration finale de 1,25 mM.
REMARQUE: Les tissus tels que les muscles peuvent avoir besoin d’une concentration différente pour atteindre la saturation. - Ajouter 5 μL de cytochrome C (cytC) de 4 mM pour une concentration finale de 10 μM.
REMARQUE : Facultatif pour les cellules afin d’évaluer la qualité de la perméabilisation. - Ajouter 16 μL de succinate (S) de 1,25 M pour une concentration finale de 10 mM. (FACULTATIF) Ajouter 3 μL de 0,5 M ADP pour une concentration finale de 2 mM afin de contrôler la saturation de la concentration d’ADP.
- Pour les cellules et les tissus non fibreux, ajouter 2 μL de 1 mg/mL d’oligomycine (OM) pour une concentration finale de 1 μg/mL.
ATTENTION : Tous les inhibiteurs de l’ETS utilisés sont très toxiques.
REMARQUE: L’oligomycine peut nécessiter un titrage pour une concentration optimale car elle peut réduire la capacité ETS et est omise pour le tissu musculaire. Réoxygénez ici lorsque le tissu musculaire est dosé et siO2 est inférieur à 300 μM. - Titrer le FCCP à partir d’un stock de 2 mM, ajouter 0,6 μL avec les étapes suivantes de 0,2 μL jusqu’à ce qu’aucune augmentation de la respiration et la respiration ne soient découplées au maximum (théorique: non couplé).
- Ajouter 1 μL de roténone (ROT) de 1 mM pour une concentration finale de 0,5 μM. Ajouter 2 μL de 1 mg/mL d’antimycine (AM) pour une concentration finale de 1 μg/mL.
- Réoxygénez les chambres pour atteindre un niveau d’oxygène similaire (~150 μM) dans toutes les chambres en soulevant lentement le piston en mouvement de torsion.
- Ajouter 5 μL d’ascorbate de 0,8 M pour une concentration finale de 2 mM immédiatement suivie de 5 μL de 0,2 M de N,N,N′,N′-tétraméthyl-p-phénylènediamine (TMPD) pour une concentration finale de 0,5 mM afin d’évaluer l’activité du complexe IV (facultatif).
- Ajouter 5 μL d’azoture de 4 M pour une concentration finale de 10 mM immédiatement lorsque le flux maximald’O2 est atteint avec le TMPD. Poursuivez la course pendant >5 min pour tester l’auto-oxydation du TMPD pour le calcul complexe du niveau de base IV.
- Recomptez les cellules pour confirmer le nombre de cellules avant l’exécution et passez à l’étape 1.9.
REMARQUE: La perméabilisation de la digitonine (pour les cellules uniquement) doit être titrée dans les expériences d’essai pour atteindre le flux maximal et ne pas affecter l’intégrité de la membrane mitochondriale (voir l’étape 1.7). Les échantillons perméabilisés (en particulier le tissu musculaire) présentant une augmentation >10% de la fréquence respiratoire après l’ajout du cytochrome c doivent être exclus d’une analyse plus approfondie en raison de lésions de la membrane mitochondriale externe. Une baisse de flux de courte durée après l’ajout de produits chimiques dissous dans l’EtOH est attendue.
- Utiliser un échantillon de tissu lysé (perméabilisé) ou perméabiliser des cellules en ajoutant 1 μL de digitonine (8,1 mM de digitonine dans du diméthylsulfoxyde (DMSO)) pour une concentration finale de 5 μg/mL pour perméabiliser les cellules. Le flux va baisser et devrait se stabiliser à >5 min.
- Protocole pour déterminer les conditions optimales de perméabilisation des cellules
- Ajoutez des cellules comme décrit aux étapes 1.2 et 1.5.2.
- Prendre 10 μL de 10 mg/mL de digitonine et ajouter 10 μL de DMSO pour diluer à 5 mg/mL.
- Ajouter 1 μL de roténone (1 mM de bouillon). Ajouter 10 μL de succinate (2 mM de bouillon) et 5 μL d’ADP (0,5 M de bouillon).
- Titrer 1 μL de digitonine (2,5 mg par étape) à plusieurs reprises jusqu’à ce que la respiration n’augmente plus et soit maximale.
REMARQUE: Une diminution de la respiration indique une concentration excessive de digitonine.
- Protocole pour l’analyse OXPHOS dans des cellules intactes
- Après la respiration de routine (étape 1.6.1-1.6.6), ajouter 2 μL d’oligomycine de 0,01 mM pour une concentration finale de 10 nM.
- Titrer le FCCP à partir d’un stock de 2 mM, ajouter 0,6 μL avec les étapes suivantes de 0,2 μL jusqu’à ce qu’aucune autre augmentation de la respiration et la respiration ne soit découplée au maximum (théorique: non couplée)
- Ajouter 1 μL de roténone de 1 mM pour une concentration finale de 0,5 μM. Ajouter 2 μL de 1 mg/mL de stock d’antimycine pour une concentration finale de 1 μg/mL.
- Réoxygénez la chambre au même niveau d’oxygène (~150 μM) en soulevant lentement le piston en mouvement de torsion.
- Ajouter 5 μL d’ascorbate de 0,8 M pour une concentration finale de 2 mM. Ajouter immédiatement 5 μL de 0,2 M TMPD pour une concentration finale de 0,5 mM afin d’évaluer l’activité du complexe IV.
REMARQUE: Préparez un nouveau lot avant tout ensemble d’expériences plus important, car le TMPD est sujet à l’auto-oxydation. L’activité peut diminuer avec le temps lorsqu’elle est stockée à -20 °C. - Ajouter 5 μL d’azoture de 4 M pour une concentration finale de 10 mM immédiatement lorsque le flux maximald’O2 est atteint avec le TMPD. Continuer l’exécution pendant >5 minutes pour tester l’auto-oxydation du TMPD pour le calcul complexe du niveau de base IV.
- Recomptez les cellules pour confirmer le nombre de cellules avant l’exécution et passez à l’étape 1.9.
- Collecte d’échantillons post-exécution
- Collectez exactement 1 mL de suspension MiR05 de chaque chambre (avec agitateurs allumés) dans un tube de 1,5 mL de 1,5 mL.
- Centrifuger à 1000 x g pour les cellules perméabilisées ou à 20 000 x g pour le lysat tissulaire. Retirer le surnageant et congeler la pastille à -80 °C pour un traitement ultérieur (section 3).
- Analyse des protocoles SUIT
- Analyser le flux d’oxygène (pmol/s, normalisé à l’entrée) à chaque plateau après l’ajout d’un substrat ou d’un inhibiteur (Figure 1C et Figure 3A). Exportez les valeurs dans une feuille de calcul.
- Soustrayez la valeur de la consommation résiduelle d’oxygène (ROX, Figure 1C et Figure 3C) de toutes les valeurs de chaque essai expérimental. Soustrayez la respiration résiduelle d’azoture de la TMPD pour obtenir une respiration IV complexe.
- Tracer les valeurs absolues normalisées pour l’entrée de cellules (Figure 3A, B) ou de tissu (Figure 5A,B). Calculez les rapports de contrôle du flux (étape 1.11) ou normalisez-les en fonction de l’apport en protéines (Figure 3C).
- Calcul du rapport de contrôle de flux
- Acquérir un indice de la fonction respiratoire et du contrôle du couplage à l’aide des rapports de contrôle de flux (FCR)9,26.
REMARQUE: Cela permet d’évaluer la qualité mitochondriale intrinsèque, indépendamment de la quantité mitochondriale. De plus, les rapports de contrôle du flux (FCR) sont comparables au sein des mêmes lignées cellulaires, ce qui permet le contrôle de la qualité des réactifs (les FCR respectifs sont obtenus à l’aide des valeurs de référence numérotées indiquées dans les figures 1B-D et 3C). - Calculer le rapport de contrôle respiratoire pour le couplage d’OXPHOS à LEAK à l’aide de l’équation 1.
Équation 1 : FCRADP = 5/6 = État 3 / État 4 - Calculer le FCR pour évaluer la respiration dépendante du NADH à l’aide de l’équation 2
Équation 2 : État FCR3 (I) = 3/5 = État 3 (I) / État 3 (I+II) - Calculez le FCR pour évaluer la respiration dépendante du succinate à l’aide de l’équation 3.
Équation 3 : État FCR3 (II) = 8/7 =pourriture S /capacité ETS - Calculez le FCR pour évaluer couplé à découplé à l’aide de l’équation 4.
Équation 4 : FCRcouplé/découplé = 5/7 = État 3 (I+II) /Capacité ETS - Pour tester l’intégrité de la membrane externe mitochondriale, utilisez l’équation 5.
Équation 5 : % de lésions de la membrane externe mitochondriale = 3/4 = État 3 (I) / État 3 (I) avec cyt c
- Acquérir un indice de la fonction respiratoire et du contrôle du couplage à l’aide des rapports de contrôle de flux (FCR)9,26.
2. Respirométrie haute résolution : respiromètre à microplaques (mHRR)
REMARQUE: Les expériences de cette section du protocole ont été effectuées à l’aide de l’analyseur de flux extracellulaire Seahorse XFe96 (table des matériaux)
- Culture cellulaire
- Cultivez n’importe quel type de cellule. Les adhérents (p. ex. collagène, laminine) peuvent être utilisés pour faciliter l’attachement cellulaire. Ici, les cellules HEK293 sont cultivées comme avant (étape 1.3).
- La veille de l’expérience, détachez les cellules et transférez-les dans une microplaque mHRR 96 puits désignée pour obtenir une confluence idéale le jour de l’expérience (80%-100%) (Figure 2C).
REMARQUE: Pour mHRR, les densités de cellules de microplaques sont critiques. Les propriétés de croissance individuelles des lignées cellulaires ou des traitements affectant la croissance doivent être prises en compte pour assurer une confluence comparable le jour de l’expérience.
- Préparation de cellules pour la respirométrie à haute résolution
- Récolter et remettre en suspension les cellules suffisamment avant l’ensemencement
REMARQUE: Il est recommandé d’ensemencer des cellules de la même dilution pour les répliques. - Ensemencez les cellules en fonction des taux de croissance des lignées cellulaires individuelles ou des propriétés de croissance sous traitement.
REMARQUE: Optimisez sur une microplaque standard de 96 puits et extrapolez la densité cellulaire à une microplaque spécifique au test de 96 puits. Dans cette configuration, 7 x 104 cellules HEK293 WT ont été ensemencées par puits d’un puits de 96. Les première et dernière colonnes de la plaque de 96 puits sont utilisées pour la détermination des protéines (figure 2C). Les quatre puits d’angle ne doivent contenir aucune cellule et sont utilisés pour la correction expérimentale du fond. Idéalement, les puits près des bords sont vides pour minimiser l’effet de bord (p. ex., les cellules présentent des taux de croissance modifiés causés par les effets de la température) (figure 2C, D).
- Récolter et remettre en suspension les cellules suffisamment avant l’ensemencement
- Préparation des plaques de capteur, chargement des inhibiteurs
- Le jour du dosage, compléter 38,8 mL de milieu avec 0,4 mL de 1 M glucose, 0,4 mL de glutamine 200 mM et 0,4 mL de Na-Pyruvate 100 mM.
REMARQUE: la respiration mHRR nécessite un milieu DMEM spécialisé non tamponné à pH 7,4. En général, 40 mL devraient suffire pour une expérience avec une microplaque de 96 puits. - Réchauffer le milieu d’essai respiratoire à 37 °C et échanger le milieu de culture cellulaire contre le milieu d’essai respiratoire en lavant deux fois avec 80 μL par puits.
- Placez la plaque avec les cellules dans un incubateur à 37 °C sans CO2 pendant 60 min avant le test.
REMARQUE: Cette étape est essentielle pour dégazer la plaque car le CO2 peut affecter les résultats respiratoires et le sérum dans le milieu peut produire des bulles pendant le test. - Les aliquotes inhibitrices préwarm pour OM, FCCP, ROT et AM à 37 °C et sortent la plaque du capteur de l’incubateur.
- Diluer OM, FCCP, ROT et AM dans 3 mL de milieu d’essai jusqu’à une concentration finale du puits de 1,5 μM, 1,125 μM et 1 μM, respectivement. Remplissez des ports séparés comme indiqué à la figure 2E.
REMARQUE: Une pipette multicanal est recommandée pour remplir la cartouche du capteur. Étant donné que l’air sous pression est utilisé pour injecter des composés, tous les orifices doivent être remplis avec une quantité égale de volume de liquide chaque fois qu’un orifice est rempli d’un composé. ROT et AM peuvent être combinés en un seul port. Les inhibiteurs peuvent être dissous dans l’EtOH ou le DMSO. - Inspectez les ports d’injection et vérifiez un volume de chargement uniforme pour chaque port.
REMARQUE: Tous les ports contiennent un trou en bas pour l’injection. Des précautions doivent être prises lors du déplacement de la plaque du capteur. Les bulles d’air peuvent être enlevées à l’aide d’une aiguille.
- Le jour du dosage, compléter 38,8 mL de milieu avec 0,4 mL de 1 M glucose, 0,4 mL de glutamine 200 mM et 0,4 mL de Na-Pyruvate 100 mM.
- Protocole pour l’évaluation de l’oxygène dans les cellules intactes
- La veille du test, effectuez les étapes 2.4.2 à 2.4.7.
- Aliquote 20 mL de la solution d’étrier dans un tube conique de 50 mL.
- Ouvrez le kit de test de flux extracellulaire et retirez le contenu.
- Placez la cartouche du capteur inversée à côté de la plaque utilitaire. Pipette 200 μL de solution d’étriquant dans chaque puits de la plaque utilitaire.
- Fixez la cartouche du capteur sur la plaque d’utilité en veillant à ce que tous les capteurs soient immergés.
- Placer la plaque dans un incubateur à 37 °C sans CO2 pendant la nuit ou un minimum de 12 h. Vérifiez que l’humidité à l’intérieur de l’incubateur est suffisante pour empêcher l’évaporation de l’étrier.
- Allumez le système à base de microplaques et l’ordinateur pour qu’ils soient prêts à être utilisés le lendemain (la machine nécessite un minimum de 3 h pour s’équilibrer à 37 °C avant de procéder à un essai).
REMARQUE: Pour la stabilité du signal, augmentez les points de mesure à 6 au lieu de 3 cycles de mesure par état respiratoire. Chaque cycle consiste en 3 min de mélange et 3 min de mesure. - Le jour du test XF, effectuez les étapes 2.4.9 à 2.4.20.
- Vérifiez la confluence de la plaque de culture cellulaire, la morphologie des cellules et que les puits de fond sont vides.
- Laver les cellules avec le milieu respiratoire préparé comme mentionné aux étapes 2.4.11-2.4.12.
- Retirer la totalité du milieu de culture, sauf 20 μL, de chaque puits. Retirer 55 μL si le milieu de culture était de 80 μL en raison de l’évaporation pendant la nuit (environ 5 μL).
- Lavez les cellules deux fois avec 90 μL de milieu d’essai. Enfin, ajouter 100 μL de milieu d’essai. Le volume final doit être de 120 μL.
REMARQUE: Une pipette multicanal est recommandée pour cette étape afin de s’assurer que la même procédure de lavage a été appliquée à chaque condition expérimentale (dépend de la configuration de la plaque). Lors de l’aspiration, inclinez la plaque à un angle de 45 ° et placez les pointes de la pipette dans le coin des puits pour l’aspiration et l’injection de liquides. Il est impératif de faire attention pendant le lavage car certaines cellules peuvent facilement se détacher du fond de la plaque de culture cellulaire. - Placer la plaque dans un incubateur à 37 °C sans CO2 pendant 60 min avant le test.
- Récupérez la plaque de cartouche de capteur hydratée de l’incubateur sans CO2.
- Jeter l’ancienne solution d’étriquant et la remplacer par une solution d’étriquant frais, préavertissée à 37 °C.
- Préparer les inhibiteurs et le milieu d’essai (3 mL par inhibiteur pour un total de 12 mL de milieu d’essai) et utiliser un réservoir de pipette pour le chargement de l’inhibiteur dans les orifices.
- Ouvrez le logiciel et exécutez un modèle prédéfini ou nouveau. Remplissez la carte des plaques, ajustez les titrages et les cycles de mesure, puis appuyez sur Démarrer pour lancer l’étalonnage des capteurs optiques.
- Retirez le couvercle de la cartouche chargée et placez-le dans la fente qui glisse automatiquement hors de la machine, en vérifiant que les marques dans le coin inférieur droit de la plaque s’alignent avec le triangle dans le coin inférieur droit de la fente.
- Cliquez sur Continuer pour effectuer un étalonnage automatique, d’une durée d’environ 20 min.
- Après l’étalonnage, retirez la plaque d’utilité contenant l’étrier.
- Retirez le couvercle de la microplaque contenant les cellules et placez la plaque dans la fente lorsque la machine y invite. Cliquez sur Continuer pour démarrer la course.
- Collecte d’échantillons post-exécution
- Sortez la plaque de la machine, retirez soigneusement le support d’essai restant sans perturber les cellules et congelez la plaque entière à -80 °C pour un traitement ultérieur (section 3).
3. Détermination des protéines à l’aide du dosage de l’acide bicinchoninique (dosage BCA)
- Préparer l’albumine sérique bovine diluée (BSA) dans un tampon utilisé pour l’extraction des protéines et compatible avec l’ACA : 2 mg/mL, 1,5 mg/mL, 1 mg/mL, 0,5 mg/mL, 0,25 mg/mL et 0 mg/mL pour la courbe standard en double.
- Extraire les protéines en les réutilisant dans un tampon de lyse approprié (p. ex., RIPA) avec 20 μL par puits pour le mHRR ou 100 μL par pastille contenu dans un tube de 1,5 mL pour le cHRR.
- Incuber la plaque mHRR ou le tube de 1,5 mL contenant des lysats de protéines pendant 30 min sur de la glace.
- Centrifuger le tube de 1,5 mL contenant le lysat de protéine à 4 °C à 20 000 x g pendant 20 min et transférer le surnageant résultant dans un nouveau tube propre de 1,5 mL.
- Utiliser 10 μL par échantillon en doublons et étalons dans une plaque de microtitrage. Ajouter 200 μL de réactif de travail BCA et incuber pendant 15 min >.
- Lisez la plaque dans un spectrophotomètre standard à une longueur d’onde de 562 nm et calculez les concentrations de protéines à l’aide d’une courbe standard BSA.
- Normaliser les résultats de la respiration à la concentration en protéines.
REMARQUE: La normalisation en quantité de protéines permet de corroborer les densités d’ensemencement cellulaire ou l’apport de poids humide. Les protéines extraites conviennent à l’immunobuvardage ultérieur contre les sous-unités de l’ETS par exemple, mais ne représentent pas entièrement l’échantillon natif (par exemple, perte de sites de phosphorylation).
Résultats
Ici, nous fournissons des protocoles pour déterminer la bioénergétique mitochondriale dans les cellules eucaryotes, les tissus non fibreux (par exemple, le cervelet) et les tissus fibreux (par exemple, le muscle squelettique). Pour les cellules eucaryotes, HEK293 avec knockout conçu par CRISPR de deux protéines différentes associées à la traduction mitochondriale entraînant un déficit multiple (CRISPRKO1) et sévère/complet en OXPHOS (CRISPRKO2) a été mesuré avec cHRR (
Discussion
Traditionnellement, la bioénergétique mitochondriale a été étudiée avec des électrodes d’oxygène de type Clark. Un manque de résolution et de débit, cependant, justifiait le progrès technologique. À ce jour, l’O2k (appelé cHRR) et l’analyseur de flux Seahorse XF96 (appelé mHRR) ont été largement adoptés dans le domaine de la bioénergétique cellulaire. Ici, nous présentons une collection compréhensible de protocoles pour l’analyse du métabolisme énergétique cellulaire via l’évaluation de...
Déclarations de divulgation
Aucun conflit d’intérêts à divulguer.
Remerciements
Ce travail a été soutenu par un financement de l’Académie de Finlande (C.B.J), de la Fondation Magnus Ehrnroot (C.B.J) et d’une bourse de doctorat de l’Integrated Life Sciences Graduate School (R.A.).
matériels
| Name | Company | Catalog Number | Comments |
| 2 mL Potter-Elvehjem Glass/PTFE Tissue Grinder/Homogenizer | Omni International | 07-358029 | |
| 95% O2, 5% CO2 medical gas mixture | Potter for tissue grinding | ||
| ADP | Sigma | A 4386 | |
| Antimycin A | Sigma | A 8674 | Chemical |
| Ascorbate | Merck | PHR1279-1G | Chemical, dissolve in ethanol |
| BSA (fatty accid free) | Sigma | A 6003 | Chemical |
| CaCO3 | Sigma | C 4830 | Chemical |
| Cytochrome c | Sigma | C 7752 | Chemical |
| Digitonin | Sigma | D 5628 | Chemical |
| Dithiothreitol | Sigma | D 0632 | Chemical, dissolve in DMSO |
| D-Sucrose | Roth | 4621.1 | Chemical |
| Dulbecco’s modified Eagle’s medium (High glucose) | Fisher Scientific | 41965-039 | Chemical |
| Dulbecco’s modified Eagle’s medium (No Glucose) | Fisher Scientific | A14430-01 | |
| EGTA | Sigma | E 4378 | |
| Etomoxir | Sigma | E1905 | Chemical |
| Falcon 15 ml Conical Centrifuge Tubes | Fisher Scientific | AM12500 | Chemical |
| Falcon 50 ml Conical Centrifuge Tubes | Fisher Scientific | AM12501 | |
| FCCP | Sigma | C 2920 | |
| Glucose | Sigma | G7021 | Chemical, dissolve in ethanol |
| Glutamate | Sigma | G 1626 | Chemical |
| GlutaMax (100x) (200 nM L-alanyl-L-glutamine dipeptide) | Fisher Scientific | 35050061 | Chemical |
| HEK293 cells | ATTC | CRL-1573 | |
| Hemocytometer | Fisher Scientific | 0267151B | Instrument for cell counting |
| Hepes | Sigma | H 7523 | Chemical |
| Imidazole | Fluka | 56750 | Chemical |
| KCl | Merck | 1.04936 | Chemical |
| L-carnitine | Sigma | C0283 | Chemical |
| Malate | Sigma | M 1000 | Chemical |
| MES hydrate | Sigma | M8250 | Chemical |
| MgCl2 | Sigma | M 9272 | Chemical |
| Na2ATP | Sigma | A 2383 | Chemical |
| Na2Phosphocreatine | Sigma | P 7936 | Chemical |
| Na-pyruvate (100 mM) (100x) | Fisher Scientific | 11360070 | |
| NEAA (Non-essential amino acids) 100x | Fisher Scientific | 11140035 | |
| Normal FBS (10x) | Fisher Scientific | 10500064 | |
| O2k-Core: Oxygraph-2k | Oroboros Instruments | 10000-02 | High-resolution respirometry instrument |
| O2k-Titration Set | Oroboros Instruments | 20820-03 | Hamilton syringes for chemical injections |
| Oligomycin | Sigma | O 4876 | Chemical, dissolve in ethanol |
| Palmitoylcarnitine | Sigma | P 4509 | Chemical |
| Penicillin-Streptomycin | Fisher Scientific | 15140122 | |
| Pierce BCA Protein Assay Kit | Fisher Scientific | 23227 | |
| Pyruvate | Sigma | P 2256 | Chemical |
| RIPA-Buffer | Fisher Scientific | 89900 | Chemical |
| Rotenone | Sigma | R 8875 | Chemical, dissolve in ethanol |
| Saponin | Sigma | S7900 | Chemical |
Seahorse XF DMEM assay medium pack, pH 7.4 | Agilent, Santa Clara, CA | 103680-100 | |
| Seahorse XFe96 Extracellular Flux Analyzer | Agilent, Santa Clara, CA | High-throughput respirometry instrument | |
| Seahorse XFe96 FluxPak | Agilent, Santa Clara, CA | Includes assay plates, cartridges, loading guides for transferring compounds to the assay cartridge, and calibrant solution. | |
| Small scissors | Fisher Scientific | 08-951-20 | |
| Sodium azide | Sigma | S2002 | Chemical |
| Succinate | Sigma | S 2378 | Chemical |
| Taurine | Sigma | T 8691 | Chemical |
| TMPD | Sigma | T 3134 | Chemical |
| Trypan Blue solution | Merck | 72-57-1 | Chemical |
| Trypsin 0.25% EDTA | Fisher Scientific | 25200056 | |
| Two thin-edged forceps | Fisher Scientific | 12-000-122 | |
| Uridine stock (500x) | Sigma | U3750 | Chemical |
Références
- McBride, H. M., Neuspiel, M., Wasiak, S. Mitochondria: More than just a powerhouse. Current Biology. 16 (14), 551-560 (2006).
- Mehta, M. M., Weinberg, S. E., Chandel, N. S. Mitochondrial control of immunity. Beyond ATP. Nature Reviews Immunology. 17 (10), 608-620 (2017).
- Spinelli, J. B., Haigis, M. C. The multifaceted contributions of mitochondria to cellular metabolism. Nature Cell Biology. 20 (7), 745-754 (2018).
- Gnaiger, E. Capacity of oxidative phosphorylation in human skeletal muscle. New perspectives of mitochondrial physiology. International Journal of Biochemistry and Cell Biology. 41 (10), 1837-1845 (2009).
- Gorman, G. S., et al. Mitochondrial diseases. Nature Reviews Disease Primers. 2, 1-23 (2016).
- Boushel, R., Gnaiger, E., Schjerling, P., Skovbro, M., Kraunsøe, R., Dela, F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia. 50 (4), 790-796 (2007).
- Cogliati, S., et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 155 (1), 160-171 (2013).
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biology. 13, 1-11 (2015).
- Gnaiger, E. Mitochondrial pathways and Respiratory control. An introduction to OXPHOS analysis. Bioenergetics communications. 5th ed. , (2020).
- Jackson, C. B., et al. Mutations in SDHD lead to autosomal recessive encephalomyopathy and isolated mitochondrial complex II deficiency. Journal of Medical Genetics. 51 (3), 170-175 (2014).
- Pesta, D., Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods in Molecular Biology. 810, 25-58 (2012).
- Horan, M. P., Pichaud, N., Ballard, J. W. O. Review: Quantifying mitochondrial dysfunction in complex diseases of aging. Journals of Gerontology - Series A Biological Sciences and Medical Sciences. 67 (10), 1022-1035 (2012).
- Doerrier, C., Garcia-Souza, L. F., Krumschnabel, G., Wohlfarter, Y., Mészáros, A. T., Gnaiger, E. High-resolution fluorespirometry and oxphos protocols for human cells, permeabilized fibers from small biopsies of muscle, and isolated mitochondria. Methods in Molecular Biology. 1782, 31-70 (2018).
- Zhang, J., et al. Measuring energy metabolism in cultured cells, including human pluripotent stem cells and differentiated cells. Nature Protocols. 7 (6), 1068-1085 (2012).
- García-Roche, M., Casal, A., Carriquiry, M., Radi, R., Quijano, C., Cassina, A. Respiratory analysis of coupled mitochondria in cryopreserved liver biopsies. Redox Biology. 17, 207-212 (2018).
- Acin-Perez, R., et al. A novel approach to measure mitochondrial respiration in frozen biological samples. The EMBO Journal. 39 (13), 1-18 (2020).
- Cell metabolism assay kits. Seahorse assay kits and media Available from: https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-assay-lits-reagents-cell-assay-media (2021)
- Chance, B., Williams, G. R. A method for the localization of sites for oxidative phosphorylation. Nature. 176 (4475), 250-254 (1955).
- Gnaiger, E., et al. Mitochondrial respiratory states and rates. MitoFit Preprint Arch. , (2019).
- Gnaiger, E. O2k-procedures: SOP O2k quality control 1: Polarographic oxygen sensors and accuracy of calibration Section Page. Oroboros. 03 (18), 1-21 (2020).
- Robinson, B. H., Petrova-Benedict, R., Buncic, J. R., Wallace, D. C. Nonviability of cells with oxidative defects in galactose medium: A screening test for affected patient fibroblasts. Biochemical Medicine and Metabolic Biology. 48 (2), 122-126 (1992).
- King, M. P., Attardi, G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science. 246 (4929), 500-503 (1989).
- Makrecka-Kuka, M., Krumschnabel, G., Gnaiger, E. High-resolution respirometry for simultaneous measurement of oxygen and hydrogen peroxide fluxes in permeabilized cells, tissue homogenate and isolated mitochondria. Biomolecules. 5 (3), 1319-1338 (2015).
- Fasching, M., Gnaiger, E. O2k quality control 2: Instrumental oxygen background correction and accuracy of oxygen flux. Mitochondrial Physiology Network. 14 (06), 1-14 (2016).
- Gnaiger, E., Lassnig, B., Kuznetsov, A., Rieger, G., Margreiter, R. Excess capacity of cytochrome c oxidase. Journal of Experimental Biology. 1139, 1129-1139 (1998).
- Gnaiger, E., et al. Mitochondria in the Cold. Life in the Cold. , 431-442 (2000).
- Fontana-Ayoub, M., Fasching, E., Gnaiger, Selected media and chemicals for respirometry with mitochondrial preparations. Mitochondrial Physiology Network. 02 (17), 1-9 (2014).
- Gerencser, A. A., et al. Quantitative microplate-based respirometry with correction for oxygen diffusion. Analytical Chemistry. 81 (16), 6868-6878 (2009).
- Krumschnabel, G., Eigentler, A., Fasching, M., Gnaiger, E. Use of safranin for the assessment of mitochondrial membrane potential by high-resolution respirometry and fluorometry. Methods in Enzymology. 542, 163-181 (2014).
- Nászai, A., Terhes, E., Kaszaki, J., Boros, M., Juhász, L. Ca(2+)N it be measured? Detection of extramitochondrial calcium movement with high-resolution fluorespirometry. Scientific Reports. 9 (1), 1-13 (2019).
- Pajak, B., et al. 2-Deoxy-d-Glucose and its analogs: From diagnostic to therapeutic agents. International Journal of Molecular Sciences. 21 (1), 234 (2019).
- Mercier-Letondal, P., Marton, C., Godet, Y., Galaine, J. Validation of a method evaluating T cell metabolic potential in compliance with ICH Q2 (R1). Journal of Translational Medicine. 19 (1), 1-15 (2021).
- Sauerbeck, A., et al. Analysis of regional brain mitochondrial bioenergetics and susceptibility to mitochondrial inhibition utilizing a microplate based system. Journal of Neuroscience Methods. 198 (1), 36-43 (2011).
- Jackman, M. R., Willis, W. T. Characteristics of mitochondria isolated from type I and type IIb skeletal muscle. American Journal of Physiology - Cell Physiology. 270 (2), 673-678 (1996).
- Ponsot, E., et al. Mitochondrial tissue specificity of substrates utilization in rat cardiac and skeletal muscles. Journal of Cellular Physiology. 203 (3), 479-486 (2005).
- Schönfeld, P., Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy-Reflections on disadvantages of the use of free fatty acids as fuel for brain. Journal of Cerebral Blood Flow and Metabolism. 33 (10), 1493-1499 (2013).
- Calderon-Dominguez, M., Mir, J. F., Fucho, R., Weber, M., Serra, D., Herrero, L. Fatty acid metabolism and the basis of brown adipose tissue function. Adipocyte. 5 (2), 98-118 (2016).
- Divakaruni, A. S., Rogers, G. W., Murphy, A. N. Measuring mitochondrial function in permeabilized cells using the seahorse XF analyzer or a clark-type oxygen electrode. Current Protocols in Toxicology. 2014, 1-16 (2014).
- Iuso, A., Repp, B., Biagosch, C., Terrile, C., Prokisch, H. Assessing mitochondrial bioenergetics in isolated mitochondria from various mouse tissues using Seahorse XF96 analyzer. Methods in Molecular Biology. 1567, 217-230 (2017).
- Rogers, G. W., et al. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS ONE. 6 (7), 21746 (2011).
- Jordá, A., Zaragozá, R., Portolés, M., Báguena-Cervellera, R., Renau-Piqueras, J. Long-term high-protein diet induces biochemical and ultrastructural changes in rat liver mitochondria. Archives of Biochemistry and Biophysics. 265 (2), 241-248 (1988).
- Jackson, C. B., Gallati, S., Schaller, A. QPCR-based mitochondrial DNA quantification: Influence of template DNA fragmentation on accuracy. Biochemical and Biophysical Research Communications. 423 (3), 441-447 (2012).
- Hirsch, H. M. Tissue autoxidation inhibitors: II. The presence of inhibitor in intact cells; Assay of liver and hepatoma effect on radio-oxidations. Cancer Research. 16 (11), 1076-1082 (1956).
- Picard, M., et al. Mitochondrial structure and function are disrupted by standard Isolation methods. PLoS ONE. 6 (3), 18317 (2011).
- Tanumihardja, E., Slaats, R. H., Van Der Meer, A. D., Passier, R., Olthuis, W., Van Den Berg, A. Measuring both pH and O2 with a single On-Chip sensor in cultures of human pluripotent stem cell-derived cardiomyocytes to track induced changes in cellular metabolism. ACS Sensors. 6 (1), 267-274 (2021).
- Harms, F., Stolker, R. J., Mik, E. Cutaneous respirometry as novel technique to monitor mitochondrial function: A feasibility study in healthy volunteers. PLoS ONE. 11 (7), 159544 (2016).
- Levitsky, Y., et al. Micro-respirometry of whole cells and isolated mitochondria. RSC Advances. 9 (57), 33257-33267 (2019).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.