Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Hochauflösende Respirometrie zur Beurteilung der Bioenergetik in Zellen und Geweben mittels kammer- und plattenbasierter Respirometer
In diesem Artikel
Zusammenfassung
Die Beurteilung der oxidativen Phosphorylierung mit hochauflösenden Respirometern ist zu einem integralen Bestandteil der Funktionsanalyse von Mitochondrien und zellulärem Energiestoffwechsel geworden. Hier stellen wir Protokolle für die Analyse des zellulären Energiestoffwechsels mit kammer- und mikroplattenbasierten hochauflösenden Respirometern vor und diskutieren die wichtigsten Vorteile jedes Geräts.
Zusammenfassung
Die hochauflösende Respirometrie (HRR) ermöglicht die Überwachung der oxidativen Phosphorylierung in Echtzeit zur Analyse einzelner zellulärer Energiezustände und zur Beurteilung von Atmungskomplexen unter Verwendung diversifizierter Substrat-Entkoppler-Inhibitor-Titrationsprotokolle (SUIT). Hier wird die Verwendung von zwei hochauflösenden Atemschutzgeräten demonstriert und eine grundlegende Sammlung von Protokollen vorgestellt, die für die Analyse von kultivierten Zellen, Skelett- und Herzmuskelfasern sowie Weichteilen wie Gehirn und Leber anwendbar sind. Protokolle für kultivierte Zellen und Gewebe werden für ein kammerbasiertes Respirometer und kultivierte Zellen für ein mikroplattenbasiertes Respirometer bereitgestellt, die beide Standard-Atmungsprotokolle umfassen. Zu Vergleichszwecken werden CRISPR-entwickelte HEK293-Zellen, die einen Mangel an mitochondrialer Translation aufweisen und zu einem Mangel an multiplen Atemwegen führen, mit beiden Geräten verwendet, um zelluläre Defekte in der Atmung nachzuweisen. Beide Respirometer ermöglichen eine umfassende Messung der Zellatmung mit ihren jeweiligen technischen Vorzügen und ihrer Eignung in Abhängigkeit von der untersuchten Forschungsfrage und dem untersuchten Modell.
Einleitung
Mitochondrien erfüllen die Schlüsselenergieversorgung und sind eine unterteilte Organelle, die zu essentiellen zellulären bioenergetischen und metabolischen Prozessen wie Anabolismus von Nukleotiden, Lipiden und Aminosäuren, Eisen-Schwefel-Cluster-Biogenese beiträgt und an der Signalgebung beteiligt ist, wie z.B. kontrollierter Zelltod 1,2,3 . Mitochondriale Bioenergetik durch oxidative Phosphorylierung trägt zu fast allen zellulären Prozessen innerhalb der Zelle bei, und folglich sind mitochondriale Dysfunktionen primären oder sekundären Ursprungs mit einem breiten Spektrum von Krankheitszuständen verbunden 4,5. Mitochondriale Dysfunktion beinhaltet nicht nur Veränderungen in der Struktur oder der mitochondrialen Dichte, sondern auch in der Qualität und Regulierung des Atmungssystems6. Dieses qualitative Element umfasst Substratkontrolle, Kopplungseigenschaften, posttranslationale Modifikationen, Cristae-Dynamik und respiratorische Superkomplexe 7,8. Daher ist eine genaue Analyse der mitochondrialen Bioenergetik für experimentelle und diagnostische Ansätze zur Beurteilung des Energiestoffwechsels der Zelle wichtig für Gesundheit und Krankheit.
Die mitochondriale oxidative Phosphorylierung (OXPHOS) ist eine Abfolge von Reaktionen innerhalb des Atmungssystems oder Elektronentransfersystems (ETS) zur Erzeugung von Zellenergie durch Adenosintriphosphat (ATP)9. Der multienzymatische Schritt zur Nutzung der Energie vom Elektronenfluss durch die Komplexe I und II bis zum Komplex IV erzeugt einen elektrochemischen Protonengradienten über die innere mitochondriale Membran, der anschließend für die Phosphorylierung von Adenosindiphosphat (ADP) zu ATP über den Komplex V (F1FO ATP-Synthase) verwendet wird (Abbildung 1A).
Zunächst werden während des Tricarbonsäurezyklus (TCA), der Glykolyse und der Pyruvatoxidation Zwei-Elektronen-Träger erzeugt: Nicotinamidadenindinukleotid (NADH) und Dihydroflavinadenindinukleotid (FADH2). NADH wird am Komplex I (NADH-Dehydrogenase) oxidiert, bei dem zwei Elektronen auf Coenzym Q übertragen werden (Chinon wird zu Chinol reduziert), während Protonen in den Intermembranraum (IMS) gepumpt werden. Zweitens oxidiert Komplex II (Succinat-Dehydrogenase) FADH2 und speist die Elektronen dem Coenzym Q zu, ohne Protonen zu pumpen. Drittens werden im Komplex III (Cytochrom-c-Oxidoreduktase) Elektronen aus Coenzym Q auf Cytochrom c übertragen, während Protonen in das IMS gepumpt werden. Viertens überträgt Cytochrom c die Elektronen auf den Komplex IV (Cytochrom-c-Oxidase), den letzten Komplex, um Protonen zu pumpen, und wo Sauerstoff als Elektronenakzeptor fungiert, um Protonen zu assimilieren und schließlich Wasser zu bilden. Es ist dieser Sauerstoff, den die Mitochondrien verbrauchen, der mit einem Oxygraphen gemessen werden kann. Schließlich werden die Protonen, die aus Komplex I, Komplex III und Komplex IV erzeugt werden, verwendet, um Komplex V zu rotieren, wodurch ATP9 erzeugt wird.
Wichtig ist, dass der Elektronentransfer nicht nur linear erfolgt, sondern auch als Elektronentransportkette bezeichnet wird. Stattdessen können Elektronen über mehrere Atmungswege in den Coenzym-Q-Pool übertragen werden und erleichtern den konvergenten Elektronenfluss. NADH-Substrate und Succinat können beispielsweise über Komplex I bzw. Komplex II eintreten. Elektronen aus der Fettsäureoxidation können über den elektronenübertragenden Flavoproteinkomplex gespendet werden. Tatsächlich erfordert eine umfassende Analyse von OXPHOS einen ganzheitlichen Ansatz mit geeigneten Kraftstoffsubstraten (Abbildung 1A).

Abbildung 1: Mitochondriale oxidative Phosphorylierung und spezifische Substrat- und Inhibitorprotokolle. (A) Mitochondrium und Schema des Elektronentransfersystems (CI-CIV) und der mitochondrialen F1F0 ATP-Synthase (CV). Alle Strukturen sind von PDB. Die Abbildungen zeigen nur Substrate und Inhibitoren, die in dieser Studie beschrieben werden). (B) Probenspur des Sauerstoffflusses in intakten HEK293-Zellen unter Verwendung des Standardprotokolls in einem mHRR-Gerät. (C) Probenspur des Sauerstoffflusses in intakten HEK293-Zellen unter Verwendung des Standardprotokolls in einem cHRR-Gerät. (D) Probenspur des Sauerstoffflusses in permeabilisierten menschlichen Fibroblasten von einem gesunden Spender mit entsprechendem SUIT-Protokoll. Abkürzungen: 1 = Routinemäßige Atmung intakter Zellen; 2 = Zustand 2; 3 = Zustand 3(I); 4 = Zustand 3(I) mit cytC; 5 = Zustand 3 (I+II); 6 = Leck (OM); 7 = ETS-Kapazität; 8 = S(ROT); 9 = ROX; 10 = TMPD; 11 = Az. ROT = Rotenon, AM = Antimycin, ATP = Adenosintriphosphat, Az = Azid, OM = Oligomycin, FCCP = Carbonylcyanid p-trifluor-methoxyphenyl-hydrazon; Asc = Ascorbat, TMPD = N,N,N′,N′-Tetramethyl-p-phenylendiamin, Succ = Succinat, M = Malat, P = Pyruvat, ADP = Adenosindiphosphat, NAD = Nicotinamidadenindinukleotid, IMS = Intermembranraum, FAD = Flavinadenindinukleotid. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Die Analyse der mitochondrialen OXPHOS-Kapazität unter Verwendung von HRR ist zu einer instrumentellen biochemischen Methode von diagnostischem Wert geworden, nicht nur für primäre mitochondriale Defekte 10,11, sondern auch für alle anderen Bereiche der Biologie wie Krebs und Alterung 12. HRR ermöglicht die Bestimmung der Zellatmung durch die Analyse der mitochondrialen OXPHOS-Kapazität, die den individuellen oder kombinierten Mangel an mitochondrialen Atemwegskomplexen direkt widerspiegelt und indirekt mit zellulärer Dysfunktion und verändertem Energiestoffwechsel assoziiert ist9. Methodisch werden Atmungsmessungen mit Zellen, Gewebe oder isolierten Mitochondrien 11,13,14 durchgeführt, wobei gefrorenes Material nur teilweise geeignet ist 15,16. Es wird gezeigt, dass gefrorenes Gewebe ein intaktes ETS mit beibehaltener superkomplexer Stabilitätaufweist 15. So werden im Gegensatz zu herkömmlichen TCA-Zwischenprodukten entsprechende Substrate direkt in das ETS eingespeist. Die Kopplung zwischen der ETS- und ATP-Synthese geht jedoch verloren, da die Membranintegrität durch Frostschäden (Eiskristallbildung) beeinträchtigt wird.
Respirationsexperimente finden normalerweise bei einer physiologischen Temperatur von 37 °C für Endotherme in nicht permeabilisierten oder permeabilisierten Zellen oder Geweben statt. Während erstere den zytosolischen Stoffwechselkontext betrachtet, liefert letztere den energetischen Beitrag einzelner OXPHOS-Komplexe und der ATPase durch die Zugabe spezifischer Substrate (und Inhibitoren). Die Sequenz und Variation von Substraten und Inhibitoren hat zur Entwicklung einer Vielzahl von SUIT-Protokollen17 und Assays18 geführt, um verschiedene wissenschaftliche Fragen der OXPHOS-Funktion zu beantworten (überprüft unter12). Das Basisprotokoll der Zellatmung bewertet vier verschiedene Zustände: i) Routineatmung - die Atmung in einem jeweiligen Atmungsmedium ohne Zugabe von Substraten oder Inhibitoren, die aber endogene Substrate verbrauchen. Dieser Zustand kann allgemeine OXPHOS- oder sekundär induzierte Atmungsdefekte aufdecken, die beispielsweise durch veränderte Metabolitenprofile verursacht werden. Als nächstes zeigt die Zugabe des ATPase-Inhibitors Oligomycin die Permeabilität der inneren mitochondrialen Membran für Protonen, definiert als ii) Leckrespiration. Die anschließende Titration eines Protonophors wie des Entkopplers Carbonylcyanid p-trifluor-methoxyphenyl-hydrazon (FCCP) ermöglicht es, den Zustand zu bestimmen, in dem die ETS-Kapazität in einem offenen Transmembran-Protonenschaltungsmodus, definiert als iii) entkoppelte Atmung, maximal ist. Wichtig ist, dass ein entkoppelter Zustand auch durch experimentelle Eingriffe durch übermäßige mechanische Schädigung der mitochondrialen Membranen auftreten kann. Umgekehrt bezieht sich der nicht gekoppelte Zustand auf die respiratorische Entkopplung durch einen intrinsischen Mechanismus, der physiologisch kontrolliert wird. Schließlich bestimmt die vollständige Hemmung des ETS durch Zugabe des Komplex-III-Inhibitors Antimycin und des Komplex-I-Inhibitors Rotenon den Restsauerstoffverbrauch (ROX) aus nicht-mitochondrialen sauerstoffverbrauchenden Prozessen (Abbildung 1A-C).
Die mitochondriale Bioenergetik besteht aus fünf verschiedenen Atmungszuständen19,20. Zustand 1 Atmung ist ohne zusätzliche Substrate oder ADP, außer für das, was endogen verfügbar ist. Nach der Zugabe von ADP, aber immer noch keine Substrate, wird Zustand 2 Atmung erreicht. Wenn Substrate hinzugefügt werden, die Elektronentransfer und ATP-Synthese ermöglichen, wird die Atmung im Zustand 3 erreicht. In diesem Zustand kann die OXPHOS-Kapazität bei gesättigten Konzentrationen von ADP, anorganischem Phosphat, Sauerstoff, NADH- und Succinat-verknüpften Substraten definiert werden. Zustands-4-Atmung oder LEAK-Atmung kann als ein Zustand ohne ADP oder chemisch gehemmte ATP-Synthasen bei ausreichenden Substraten definiert werden. Schließlich, wenn der gesamte Sauerstoff in einer geschlossenen Kammer erschöpft (anoxisch) ist, wird die Atmung von Zustand 5 beobachtet.
Es gibt mehrere Methoden, um zelluläre Energiezustände14 zu bewerten, wobei zwei Geräte die aktuelle Echtzeitbewertung von OXPHOS durch Analyse des Sauerstoffverbrauchs dominieren, gemessen als Funktion der Abnahme des Sauerstoffs im Laufe der Zeit in einem geschlossenen Kammersystem mit unterschiedlicher Anwendbarkeit, abhängig vom experimentellen Modell und der Forschungsfrage: dem hochauflösenden Respirometer Oroboros 2k und dem extrazellulären Flussanalysator Seahorse XF. Beide Geräte erfassen die Sauerstoffverbrauchsraten als Abnahme der Picomolen (pmol) von Sauerstoff (O2) pro Sekunde als absoluten Wert innerhalb der Kammer oder Mikrotiterplattenvertiefung. Der spezifische Sauerstoffverbrauch pro Masse wird durch Normalisierung des jeweiligen Sauerstoffverbrauchs in einem bestimmten Pufferrezept pro Anzahl von Zellen (Millionen), Gewebegewicht (mg) oder Proteinmenge erhalten.
Das O2k (Oroboros Instruments) ist ein geschlossenes Zweikammersystem, das mit einem polarographischen Sauerstoffsensor (abgekürzt als kammerbasiertes hochauflösendes Respirometer: cHRR) ausgestattet ist. Jede Versuchskammer fasst 2 ml Flüssigkeit, die durch Magnetrührer homogen gehalten wird. Der polarographische Sauerstoffsensor verwendet einen amperometrischen Ansatz, um den Sauerstoff zu messen: Er enthält eine Goldkathode, eine Silber/Silberchlorid-Anode und dazwischen eine KCI-Lösung, die eine elektrochemische Zelle erzeugt, an die eine Spannung (0,8 V) angelegt wird. Sauerstoff aus dem Assay-Medium diffundiert durch eine 25 μm fluorierte Ethylen-Propylen-Membran (O2-permeable) und wird an der Kathode reduziert, wodurch Wasserstoffperoxid entsteht. An der Anode wird Silber durch Wasserstoffperoxid oxidiert und erzeugt einen elektrischen Strom. Dieser elektrische Strom (Ampere) steht in einem linearen Verhältnis zum Sauerstoffpartialdruck. Der Partialdruck von Sauerstoff und der Sauerstofflöslichkeitsfaktor des Assaymediums werden verwendet, um die Sauerstoffkonzentration zu berechnen. Da der Sauerstoffpartialdruck von der Versuchstemperatur abhängig ist und polarographische Messungen temperaturempfindlich sind, müssen Temperaturschwankungen durch einen Peltier-Heizblock präzise (±0,002 °C) geregelt werden. Die Temperatur kann in einem Bereich von 4 °C und 47 °C geregelt werden.
Der Seahorse XF extracellular flux analyzer (Agilent) ist ein plattenbasiertes System mit 24- oder 96-Well-Mikroplattenformat, bei dem drei Fluoreszenzelektroden den Sauerstoffverbrauch im Laufe der Zeit in jedem Well messen (abgekürzt als microplate-based high-resolution respirometer: mHRR). Für die automatisierte Injektion während des Assays stehen maximal vier Ports in der Assay-Kartusche zur Verfügung. Ein Assay enthält mehrere Zyklen mit jeweils drei Phasen: 1) Mischen, 2) Warten und 3) Messung. Während der Messphase werden Sensorsonden in die Mikroplatte abgesenkt, wodurch eine vorübergehend geschlossene Kammer mit 7-10 μL Volumen zur Messung des emittierten Lichts entsteht. Dieses Licht wird von polymereingebetteten Fluorophoren an der Spitze der Sensorsonden emittiert, dieO2 basierend auf Phosphoreszenzlöschung erfassen. Die Intensität des Fluoreszenzsignals ist proportional zuO2 und wird durch die Temperatur des Sensors und des Assay-Mediums beeinflusst. Daher erfordert eine genaue Sauerstoffschätzung einen relativen Ansatz mit einer Hintergrundbohrung ohne Probe. Die Wiederherstellung der Sauerstoffkonzentration erfolgt während der Mischphase, wenn sich der Sensor nach oben und unten bewegt, um das Volumen über der temporären Kammer zu mischen. Jeder Zyklus berechnet eine Sauerstoffverbrauchsrate. Die Temperatur kann in einem Bereich von 16 °C und 42 °C geregelt werden.
HRR ist der Goldstandard zur Beurteilung der zellulären Bioenergetik bei primären und Mitochondrien-assoziierten Erkrankungen und des allgemeinen Zellstoffwechsels. In dieser Studie werden grundlegende Protokolle für HRR bereitgestellt, um die OXPHOS-Funktion in Zellen und Geweben zu beurteilen.

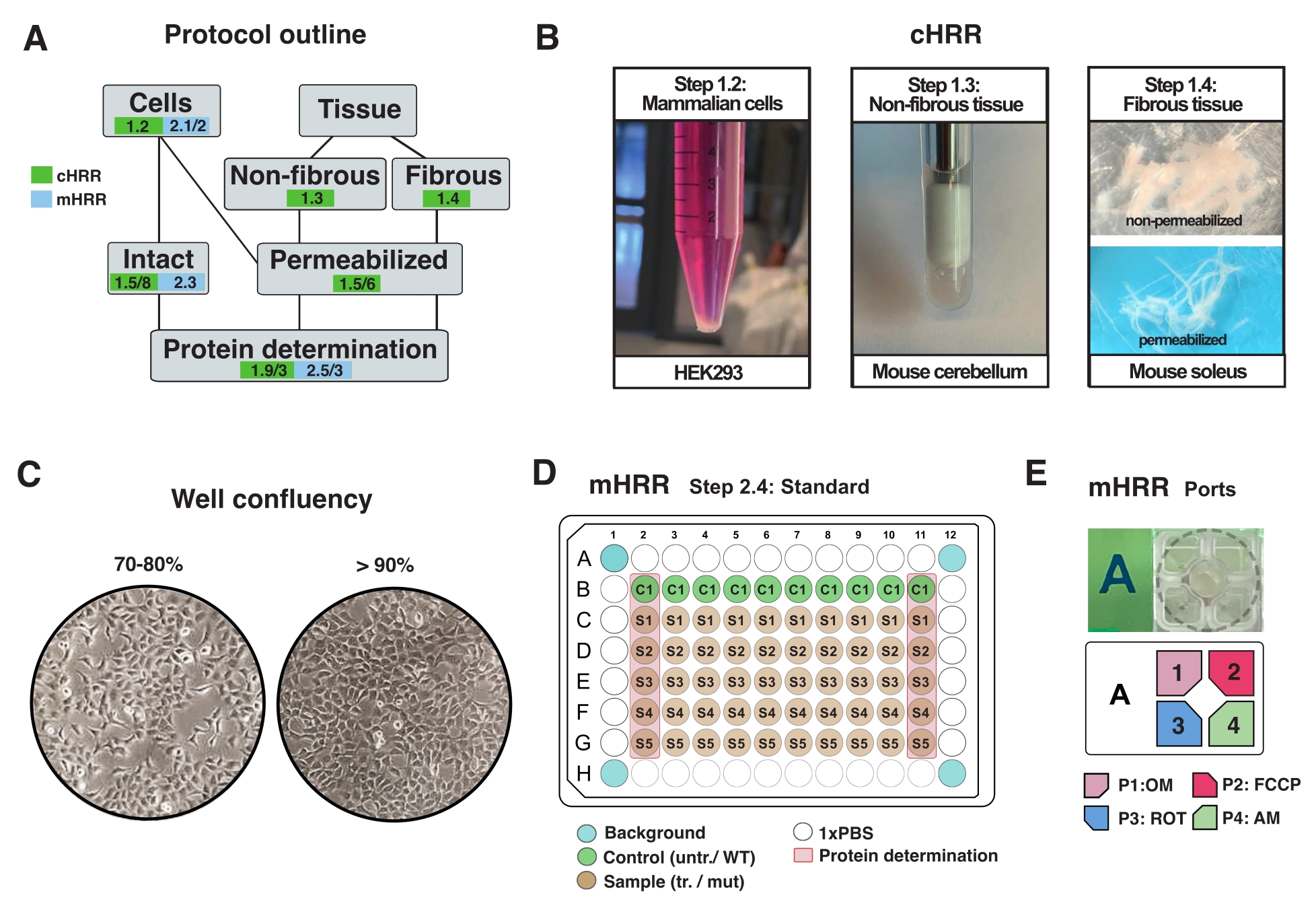
Abbildung 2: Arbeitsablauf für Zell- und Gewebepräparate für cHRR und Zellpräparation für die mHRR-Respirometrie. (A) Überblick über die bereitgestellten Protokolle. (B) Säugetierzellen (Schritt 1.2): HEK293-Pellet entspricht 3 x 106 Zellen (linkes Feld). Nicht faseriges Gewebe (Schritt 1.3): Herstellung von murinem Kleinhirnlysat in 2 ml Teflon-Töpfer (mittlere Tafel). Saponin-induzierte Permeabilisierung der Skelettmuskulatur (Schritt 1.4) rechtes Panel) für die cHRR-Respirometrie. (C) Standard-Mikrotiterplatten-Seeding-Layout (Schritt 2.4) und Konfluenzprüfung für die Analyse eukaryotischer Zellen (HEK293) für die mHRR-Respirometrie. (D, E) Schema der Injektionsöffnungsbeladung für die mHRR-Respirometrie (Schritt 2.4). Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Protokoll
Alle Tierversuche werden in Übereinstimmung mit dem National Animal Experiment Review Board und der regionalen staatlichen Verwaltungsagentur für Südfinnland durchgeführt. In dieser Studie wurden männliche C57BL/6JOlaHsd-Mäuse (4-6 Monate alt) verwendet. Die Zustimmung zur Verwendung menschlicher Zelllinien wurde von der institutionellen Ethikkommission der Universität Helsinki eingeholt.
1. Hochauflösende Respirometrie: Kammerbasiertes Respirometer (cHRR)
HINWEIS: Die Experimente in diesem Abschnitt des Protokolls wurden mit dem Oroboros O2k-Core durchgeführt: Oxygraph-2k (Tabelle der Materialien)
- Kalibrierung von Sauerstoffsensoren
- Respirometer vor dem Lauf bei 37 °C in 2,1 ml mitochondrialem Atmungsmedium (MiR05, Tabelle 1, Löslichkeitsfaktor: 0,92) für >45 min und Sauerstoffkalibrierung wie beschrieben21. Fahren Sie fort, wenn die Baseline-Variation innerhalb von ± 4 pmol/s liegt.
HINWEIS: Große Schwankungen des Hintergrundsignals können auf die erforderliche Wartung der Sensormembran oder auf Spuren von Inhibitoren hinweisen, die aus früheren Experimenten in der Kammer verbleiben. Eine instrumentelle Hintergrundkorrektur des Sauerstoffflusses wird vor einer Reihe von Experimentenempfohlen 25. - Zeichnen Sie Sauerstoffkalibrierwerte auf, um die Leistung der Sensormembran im Laufe der Zeit zu überwachen.
HINWEIS: Dies zeigt die Sensorfunktion, die Signal-Rausch-Stabilität und wann eine Wartung der Sensormembran erforderlich ist. Je nach Umgebungsdruck werden in MiR05 zwischen 180-200 μmol Sauerstoff gelöst. - Entfernen Sie die gesamte Flüssigkeit in der Kammer, bevor Sie eine Probe in das Atmungsmedium geben.
HINWEIS: Bewerten Sie das Volumen der Atemkammern regelmäßig auf genau 2 ml.
- Respirometer vor dem Lauf bei 37 °C in 2,1 ml mitochondrialem Atmungsmedium (MiR05, Tabelle 1, Löslichkeitsfaktor: 0,92) für >45 min und Sauerstoffkalibrierung wie beschrieben21. Fahren Sie fort, wenn die Baseline-Variation innerhalb von ± 4 pmol/s liegt.
- Vorbereitung der Zellen für die hochauflösende Respirometrie
- Kultur HEK293-Zellen in Schalen mit einem Durchmesser von 10 cm 2 in Dulbeccos Modified Eagle's Medium (DMEM) mit hoher Glukose, ergänzt mit 10% hitzeinaktiviertem fetalem Rinderserum (FBS), GlutaMax, nicht-essentiellen Aminosäuren und Na-Pyruvat22 und Uridin23, um den OXPHOS-defekten Stoffwechsel in einem Inkubator bei 37 °C bei 5% CO2 zu unterstützen.
HINWEIS: Jede Art von eukaryotischen Zellen kann kultiviert werden. Bei den meisten Zelltypen führt die Kultivierung einer 10 cm2 Schale zu ausreichend Zellen (normalerweise >3 x 106 Zellen). Überprüfen Sie routinemäßig auf Mykoplasmeninfektionen, um Auswirkungen auf den Zellstoffwechsel und die Atmung zu vermeiden. - Züchten Sie Zellen, ohne 90% Konfluenz zu überschreiten (Abbildung 2C).
HINWEIS: Zellen mit >90% Konfluenz können wachstumsabhängige hemmende Wirkungen auf die Atmung zeigen (wenn sie nicht synchronisiert oder postmitotisch sind). - Waschen Sie die Zellen mit 1x PBS, lösen Sie sie mit 1 ml warmem 0,25% Trypsin, deaktivieren Sie Trypsin durch Zugabe von warmem DMEM (5 ml / 10 cm2 Platte) und zählen Sie die Zellen mit einem Hämozytometer.
- Zentrifugieren Sie vorsichtig die Zelllösung gleich 2,5 x 10 6 Zellen bei 300 x g für 5 min, entfernen Sie den Überstand vollständig und resuspendieren Sie in 2,5 ml warmem MiR05 (1 x 106 Zellen/ml) (Abbildung 2A).
- Für Suspensionszellen ist eine Lösung in Höhe von 2,5 x 106 Zellen zu zählen und zu entfernen, zu pelletieren und wie in Schritt 1.2.4 beschrieben fortzufahren.
- Ausführen des SUIT-Protokolls zur Optimierung der Permeabilisierung (Schritt 1.6), permeabilisierter Zellen oder Gewebe (Schritt 1.5) oder intakter Zellen (Schritt 1.7)
HINWEIS: Für konsistente Ergebnisse wird empfohlen, die Zellkonzentration konstant zu halten (z. B. 1 x 106 Zellen / ml). Obwohl die Atmung unabhängig von der Zelldichte im Respirometer24 ist, sind Substrate und Inhibitoren während der gesamten Experimente in vergleichbarer Konzentration, wenn die Zellzahlen konstant gehalten werden.
- Kultur HEK293-Zellen in Schalen mit einem Durchmesser von 10 cm 2 in Dulbeccos Modified Eagle's Medium (DMEM) mit hoher Glukose, ergänzt mit 10% hitzeinaktiviertem fetalem Rinderserum (FBS), GlutaMax, nicht-essentiellen Aminosäuren und Na-Pyruvat22 und Uridin23, um den OXPHOS-defekten Stoffwechsel in einem Inkubator bei 37 °C bei 5% CO2 zu unterstützen.
- Aufbereitung von nicht-fibrösem Gewebe (z.B. Gehirn, Leber) für die hochauflösende Respirometrie
- Schneiden Sie ein homogenes Stück Gewebe mit einem Gewicht von 30-40 mg aus oder verwenden Sie das gesamte Organ (in diesem Fall das Kleinhirn der Maus).
HINWEIS: Wenn Gewebe nicht sofort verwendet wird, bewahren Sie 2 ml eiskaltes MiR05 auf, so dass die Konservierung für die meisten Gewebe bis zu 2 Stunden möglich ist. Einzelne Gewebelagerzeiten müssen in Zeitreihen bewertet werden. - Befeuchten Sie das Gewebe trocken mit einem Whatman-Filterpapier (Vorsicht: Weichteilsubstanz neigt dazu, zu haften).
- Legen Sie das 30-40 mg Gewebestück in einen eisgekühlten 2-ml-Polytetrafluorethylen-Töpfer-Elvehjem-Homogenisator.
- Fügen Sie eine angemessene Menge MiR05 hinzu, um 20 mg / ml zu erhalten, um das Verhältnis von Gewebe zu Puffer aufrechtzuerhalten. Halten Sie die Gesamtmenge >1,5 ml und <2 ml, um eine unzureichende oder übermäßige Flüssigkeit für eine geeignete mechanische Permeabilisierung zu vermeiden.
- Setzen Sie den Stößel ein, lysieren Sie das Gewebe langsam, indem Sie den Stößel vorsichtig zurückziehen, während Sie die Erzeugung eines Vakuums vermeiden, das übermäßige Gewebeschäden verursacht.
- Führen Sie insgesamt 7 Striche (1x definiert als ein Auf- und Abwärtsstrich) durch, bis sie lysiert sind (erkennbar als trübe Flüssigkeit ohne größere Ablagerungen) (Abbildung 2B).
HINWEIS: Die Anzahl der Schlaganfälle für eine geeignete Lyse muss für jedes Gewebe getestet werden, indem die Integrität der äußeren mitochondrialen Membran über die Cytochrom-C-Reaktion bewertet wird (Schritt 1.5.11). Schwer zu lysierende Bindegewebe- oder Gefäßteile können verbleiben. - Dekantieren Sie das lysierte Gewebe in ein 15 ml Zentrifugenröhrchen.
- Waschen Sie das Innere des Töpfers mit einer gleichen Menge MiR05, die im Lysing-Schritt verwendet wird (z. B. 1,5 ml) und fügen Sie dem 15-ml-Röhrchen, das jetzt 3-4 ml MiR05 bei 10 mg / ml Gewebelysat enthält, hinzu.
- Fügen Sie 2 ml einfache MiR05 pro Kammer hinzu, um sie auf 37 °C zu erwärmen.
- Schwenken Sie das Rohr für eine gleichmäßige Verteilung, bevor Sie 500 μL (entspricht 5 mg) jedes Lysats pro Kammer langsam pipettieren, um die Spannung von Kälte auf 37 ° C zu minimieren.
- Warten Sie >3 Minuten, bis sich der Kammerinhalt auf 37 °C erwärmt hat, bevor Sie die Kammer schließen. Entfernen Sie überschüssige Flüssigkeit oben auf dem Stopfen (Menge pro Kammer nach dem Schließen: 4 mg).
- Führen Sie das SUIT-Protokoll für standardpermeabilisiert aus (Schritt 1.5).
- Schneiden Sie ein homogenes Stück Gewebe mit einem Gewicht von 30-40 mg aus oder verwenden Sie das gesamte Organ (in diesem Fall das Kleinhirn der Maus).
- Aufbereitung von fibrösem Gewebe (Skelettmuskulatur, Herzmuskel) für die hochauflösende Respirometrie
- Extrahieren Sie das harte Gewebe, entfernen Sie das Bindegewebe und Fett aus den Muskeln mit einer scharfen Pinzette in 2 ml eiskaltem BIOPS (Tabelle 2) unter einem Dissektionsmikroskop.
- Trennen Sie die Faserbündel (~ 4 mg) entlang der Längsachse mit einer scharfen Pinzette. Herauskitzeln Sie die Fasern ausreichend heraus, um eine netzartige Struktur zu erhalten (Abbildung 2B).
HINWEIS: Eine ordnungsgemäße mechanische Fasertrennung und Permeabilisierung wird durch den Verlust des roten Pigments Myoglobin und erhöhte Transluzenz angezeigt. - Waschen und permeabilisieren Sie das Faserbündel in Saponin (50 μg/ml in BIOPS, frisch zubereitet) für 20 min bei 4 °C (Fasern werden durchscheinend, was auf eine vollständige Permeabilisierung hinweist, Abbildung 2B).
- Waschen Sie die Fasern zweimal in MiR05 für 5 min pro Wäsche bei 4 °C.
- Mit Filterpapier trocken tupfen und wiegen, bevor Sie sie in die mit 2,1 mL MiR05 gefüllte Kammer geben.
- Führen Sie Stopfen ein, ohne sie vollständig zu schließen, dann die Kammern mit 2 ml reinemO2 mit einer 20-ml-Spritze mit Sauerstoff zu versorgen und die Kammern zu schließen, indem Sie die Stopfen in einer rotierenden Bewegung drehen. Halten Sie dieO2-Konzentration während des Experiments zwischen 300 und 500 μM, um eine Einschränkung der Sauerstoffdiffusion zu vermeiden.
- Protokoll zur Beurteilung der routinemäßigen Atmung in Zellen oder Geweben
- Probe in die Kammer geben, wie in den Schritten 1.5.2-1.5.3 beschrieben.
- Zugabe von 2,3 ml warmer MiR05-Zellsuspension (Standardeingang: 1 x 106 Zellen/ml wie in Schritt 1.2 oder 2 mg Gewebe/ml wie in Schritt 1.3)
- Skelett- und Herzmuskel (Schritt 1.4): Fügen Sie ~ 4 mg saponinpermeabilisierte Fasern zu vorgewärmten 2,3 ml warmem MiR05 hinzu, wobei die Schritte 1.4.4-1.4.6 berücksichtigt werden
- Laufkammern bei 37 °C und einer Rührgeschwindigkeit von 700 U/min. Warten Sie >3 Minuten, damit die Medien die Kammern entgasen und schließen können, indem Sie den Stopfen in einer rotierenden Bewegung drehen. Die Peltierblockstabilisierung zeigt das Erreichen der eingestellten Temperatur an.
- (OPTIONAL) Ändern Sie die Rührerdrehzahl auf 300 U/min, damit die verbleibenden Blasen durch die Kapillare des Stoppers entweichen können.
- Saugen Sie überschüssige Flüssigkeit auf den Stopfen. Warten Sie 10 Minuten, bis ein stabiles Sauerstoffflusssignal mit einem beliebigen Probentyp erreicht ist, um die Routine-/Zustands-1-Atmung aufzuzeichnen ( Abbildung 1B).
- Für Atemmessungen in permeabilisierten Zellen und Geweben fahren Sie mit Schritt 1.6 fort. Für intakte Zellen mit Schritt 1.8.
- Protokoll für die OXPHOS-Analyse in permeabilisierten Zellen oder Geweben
- Verwenden Sie lysierte (permeabilisierte) Gewebeproben oder permeabilisierende Zellen, indem Sie 1 μL Digitonin (8,1 mM Digitonin-Stamm in Dimethylsulfoxid (DMSO)) für eine Endkonzentration von 5 μg/ml hinzufügen, um Zellen zu permeabilisieren. Der Fluss sinkt und sollte sich bei >5 min stabilisieren.
VORSICHT: Digitonin ist akut toxisch für die Atemwege, bei Kontakt mit der Haut oder beim Verschlucken.
HINWEIS: Die Injektion aller Chemikalien erfolgt mit Präzisionsglasspritzen. Verwenden Sie Spritzen nur für angegebene Chemikalien, um Kreuzkontaminationen zu vermeiden, und waschen Sie sie nach Gebrauch gründlich in Wasser und EtOH. Blockierte Spritzen können Ultraschall in warmem ddH2O oder einem Reinigungsdraht erfordern, um chemische Verstopfungen zu entfernen. Ziehen Sie immer einen Überschuss der jeweiligen Stammlösung in die Spritze zurück, um zu vermeiden, dass Luft in die Kammern eindringt. Untersuchen Sie das Innere der Kammern auf das Einbringen von Luft nach jeder Injektion. Zeichnen Sie jeden Schritt auf, bis die Flussplateaus erreicht sind. - In schneller Folge hinzufügen: 5 μL 0,4 M Malat (M) für eine Endkonzentration von 1 mM, 5 μL 2,0 M Pyruvat (P; frisch zubereitet), für eine Endkonzentration von 5 mM, 4 μL 2,5 M Glutamat (G) für eine Endkonzentration von 5 mM.
- Nach vorherigem Flussplateau fügen Sie 5 μL (10 μL für Muskelgewebe) 0,5 M Adenosindiphosphat (ADP, Aliquote, die bei -80 °C gelagert werden) für eine Endkonzentration von 1,25 mM hinzu.
HINWEIS: Gewebe wie Muskeln benötigen möglicherweise eine andere Konzentration, um eine Sättigung zu erreichen. - Fügen Sie 5 μL 4 mM Cytochrom C (CytC) für eine Endkonzentration von 10 μM hinzu.
HINWEIS: Optional für Zellen zur Beurteilung der Qualität der Permeabilisierung. - 16 μL 1,25 M Succinat (S) für eine Endkonzentration von 10 mM zugeben. (OPTIONAL) Fügen Sie 3 μL 0,5 M ADP für eine Endkonzentration von 2 mM hinzu, um die Sättigung der ADP-Konzentration zu kontrollieren.
- Für Zellen und nichtfaseriges Gewebe 2 μL 1 mg/ml Oligomycin (OM) für eine Endkonzentration von 1 μg/ml zugeben.
ACHTUNG: Alle verwendeten ETS-Hemmer sind hochgiftig.
HINWEIS: Oligomycin kann Titration für eine optimale Konzentration erfordern, da es die ETS-Kapazität unterdrücken kann und für Muskelgewebe weggelassen wird. Reoxygenieren Sie hier, wenn Muskelgewebe untersucht wird und wennO2 unter 300 μM liegt. - Titrieren Sie FCCP aus einem 2-mM-Vorrat, fügen Sie 0,6 μL mit nachfolgenden 0,2 μL-Schritten hinzu, bis kein Anstieg der Atmung und der Atmung maximal entkoppelt ist (theoretisch: nicht gekoppelt).
- 1 μL 1 mM Rotenon (ROT) für eine Endkonzentration von 0,5 μM hinzufügen. 2 μL 1 mg/ml Antimycin (AM)-Vorrat für eine Endkonzentration von 1 μg/ml hinzufügen.
- Reoxygenieren Sie die Kammern, um einen ähnlichen Sauerstoffgehalt (~ 150 μM) in allen Kammern zu erreichen, indem Sie den Kolben langsam in Drehbewegung anheben.
- Fügen Sie 5 μL 0,8 m Ascorbat für eine Endkonzentration von 2 mM hinzu, unmittelbar gefolgt von 5 μL 0,2 M N,N,N′,N′-Tetramethyl-p-phenylendiamin (TMPD) für eine Endkonzentration von 0,5 mM, um die komplexe IV-Aktivität zu beurteilen (optional).
- Fügen Sie 5 μL 4 M Azid für eine Endkonzentration von 10 mM sofort hinzu, wenn der maximaleO2-Fluss mit TMPD erreicht ist. Setzen Sie den Lauf für >5 Minuten fort, um die Autooxidation von TMPD für die komplexe IV-Basisberechnung zu untersuchen.
- Zählen Sie die Zellen auf, um die Zellenanzahl vor dem Lauf zu bestätigen, und fahren Sie mit Schritt 1.9 fort.
HINWEIS: Die Digitonin-Permeabilisierung (nur für Zellen) muss in Versuchsexperimenten titriert werden, um den maximalen Fluss zu erreichen und die Integrität der mitochondrialen Membran nicht zu beeinträchtigen (siehe Schritt 1.7). Permeabilisierte Proben (insbesondere Muskelgewebe) mit einem Anstieg der Atemfrequenz um >10% nach Zugabe von Cytochrom c sollten aufgrund einer Schädigung der äußeren mitochondrialen Membran von der weiteren Analyse ausgeschlossen werden. Ein kurzzeitiger Rückgang des Flusses nach der Zugabe von EtOH-gelösten Chemikalien wird erwartet.
- Verwenden Sie lysierte (permeabilisierte) Gewebeproben oder permeabilisierende Zellen, indem Sie 1 μL Digitonin (8,1 mM Digitonin-Stamm in Dimethylsulfoxid (DMSO)) für eine Endkonzentration von 5 μg/ml hinzufügen, um Zellen zu permeabilisieren. Der Fluss sinkt und sollte sich bei >5 min stabilisieren.
- Protokoll zur Bestimmung optimaler Permeabilisierungsbedingungen für Zellen
- Fügen Sie Zellen hinzu, wie in den Schritten 1.2 und 1.5.2 beschrieben.
- Nehmen Sie 10 μL 10 mg/ml Digitonin-Vorrat und fügen Sie 10 μL DMSO hinzu, um sie auf 5 mg/ml zu verdünnen.
- 1 μL Rotenon (1 mM Stamm) hinzufügen. 10 μL Succinat (2 mM Stamm) und 5 μL ADP (0,5 Mio. Bestand) hinzufügen.
- Titrieren Sie 1 μL Digitonin (2,5 mg pro Schritt) wiederholt, bis die Atmung nicht weiter ansteigt und maximal ist.
HINWEIS: Eine Abnahme der Atmung weist auf eine übermäßige Konzentration von Digitonin hin.
- Protokoll zur OXPHOS-Analyse in intakten Zellen
- Nach der routinemäßigen Atmung (Schritt 1.6.1-1.6.6) werden 2 μL 0,01 mM Oligomycin für eine Endkonzentration von 10 nM zugegeben.
- Titrieren Sie FCCP aus 2 mM Bestand, fügen Sie 0,6 μL mit anschließenden 0,2 μL Schritten hinzu, bis keine weitere Zunahme der Atmung und Atmung maximal entkoppelt ist (theoretisch: nicht gekoppelt)
- 1 μL 1 mM Rotenon für eine Endkonzentration von 0,5 μM hinzufügen. 2 μL 1 mg/ml Antimycin-Vorrat für eine Endkonzentration von 1 μg/ml hinzufügen.
- Reoxygenieren Sie die Kammer auf den gleichen Sauerstoffgehalt (~ 150 μM), indem Sie den Kolben langsam in Drehbewegung anheben.
- Zugabe von 5 μL 0,8 M Ascorbat für eine Endkonzentration von 2 mM. Sofort 5 μL 0,2 M TMPD für eine Endkonzentration von 0,5 mM hinzufügen, um die komplexe IV-Aktivität zu beurteilen.
HINWEIS: Bereiten Sie vor größeren Experimenten eine neue Charge vor, da TMPD anfällig für Autooxidation ist. Die Aktivität kann im Laufe der Zeit abnehmen, wenn sie bei -20 °C gelagert wird. - Fügen Sie 5 μL 4 M Azid für eine Endkonzentration von 10 mM sofort hinzu, wenn der maximaleO2-Fluss mit TMPD erreicht ist. Fahren Sie >5 Minuten lang fort, um die Autooxidation von TMPD für die komplexe Berechnung des IV-Basisniveaus zu untersuchen.
- Zählen Sie die Zellen neu, um die Zellenanzahl vor dem Durchlauf zu bestätigen, und fahren Sie mit Schritt 1.9 fort.
- Beispielsammlung nach der Ausführung
- Sammeln Sie genau 1 mL MiR05-Suspension aus jeder Kammer (mit eingeschalteten Rührern) in ein 1,5 mL Rohr 1,5 ml Rohr.
- Zentrifuge bei 1000 x g für permeabilisierte Zellen oder bei 20.000 x g für Gewebelysat. Entfernen Sie den Überstand und frieren Sie das Pellet bei -80 °C zur Weiterverarbeitung ein (Abschnitt 3).
- Analyse von SUIT-Protokollen
- Analysieren Sie den Sauerstofffluss (pmol/s, normalisiert auf Input) an jedem Plateau nach Zugabe eines Substrats oder Inhibitors (Abbildung 1C und Abbildung 3A). Exportieren Sie die Werte in eine Tabelle.
- Subtrahieren Sie den Restsauerstoffverbrauch (ROX, Abbildung 1C und Abbildung 3C) von allen Werten jedes Versuchslaufs. Subtrahieren Sie die Azidrestatmung von TMPD, um eine komplexe IV-Atmung zu erhalten.
- Zeichnen Sie die für Zelle (Abbildung 3A, B) oder Gewebeeintrag (Abbildung 5A,B) normalisierten absoluten Werte auf. Berechnen Sie die Flusskontrollverhältnisse (Schritt 1.11) oder normalisieren Sie sie auf Proteineintrag (Abbildung 3C).
- Berechnung des Flussregelverhältnisses
- Erfassen Sie einen Index der Atmungsfunktion und der Kopplungskontrolle unter Verwendung von Flusskontrollverhältnissen (FCR)9,26.
HINWEIS: Dies ermöglicht die Beurteilung der intrinsischen mitochondrialen Qualität, unabhängig von der mitochondrialen Menge. Darüber hinaus sind die Flusskontrollverhältnisse (FCR) innerhalb derselben Zelllinien vergleichbar, was eine Qualitätskontrolle des Reagenzes ermöglicht (die jeweiligen FCRs werden durch die angegebenen nummerierten Referenzwerte in Abbildung 1B-D und Abbildung 3C ermittelt). - Berechnen Sie das Atemkontrollverhältnis für die Kopplung von OXPHOS an LEAK unter Verwendung von Gleichung 1.
Gleichung 1: FCRADP = 5/6 = Zustand 3 / Zustand 4 - Berechnen Sie die FCR zur Beurteilung der NADH-abhängigen Atmung mit Gleichung 2
Gleichung 2: FCR-Zustand 3 (I) = 3/5 = Zustand 3 (I) /Zustand 3 ( I+II) - Berechnen Sie die FCR, um die Succinat-abhängige Atmung mit Gleichung 3 zu beurteilen.
Gleichung 3:FCR-Zustand 3 (II) = 8/7 =S-Fäulnis /ETS-Kapazität - Berechnen Sie die FCR, um gekoppelt mit entkoppelt mit Gleichung 4 zu bewerten.
Gleichung 4: FCR gekoppelt/entkoppelt = 5/7 = Zustand 3 (I+II) / ETS-Kapazität - Um die Integrität der mitochondrialen äußeren Membran zu testen, verwenden Sie Gleichung 5.
Gleichung 5: % mitochondriale äußere Membranschädigung = 3/4 = Zustand 3 (I) / Zustand 3 (I) mit cyt c
- Erfassen Sie einen Index der Atmungsfunktion und der Kopplungskontrolle unter Verwendung von Flusskontrollverhältnissen (FCR)9,26.
2. Hochauflösende Respirometrie: Mikroplatten-basiertes Respirometer (mHRR)
HINWEIS: Die Experimente in diesem Abschnitt des Protokolls wurden mit dem Seahorse XFe96 Extracellular Flux Analyzer (Table of Materials) durchgeführt.
- Zellkultur
- Kultivieren Sie jede Art von Zelle. Anhänger (z. B. Kollagen, Laminin) können verwendet werden, um die Zellbefestigung zu erleichtern. Hier werden HEK293-Zellen wie bisher kultiviert (Schritt 1.3).
- Trennen Sie die Zellen am Tag vor dem Experiment und übertragen Sie sie in eine bestimmte mHRR 96-Well-Mikroplatte, um am Tag des Experiments eine ideale Konfluenz (80% -100%) zu erhalten (Abbildung 2C).
HINWEIS: Für mHRR sind Mikroplattenzelldichten kritisch. Individuelle Wachstumseigenschaften von Zelllinien oder Behandlungen, die das Wachstum beeinflussen, müssen berücksichtigt werden, um eine vergleichbare Konfluenz am Tag des Experiments zu gewährleisten.
- Vorbereitung der Zellen für die hochauflösende Respirometrie
- Die Zellen vor der Aussaat ausreichend ernten und resuspendieren
HINWEIS: Es wird empfohlen, Zellen aus derselben Verdünnung für Replikate zu säen. - Samen Sie die Zellen entsprechend den Wachstumsraten einzelner Zelllinien oder den Wachstumseigenschaften in Behandlung.
HINWEIS: Optimieren Sie auf einer Standard-96-Well-Mikrotiterplatte und extrapolieren Sie die Zelldichte auf eine 96-Well-Assay-spezifische Microplatte. In diesem Setup wurden 7 x 104 HEK293 WT-Zellen pro Bohrloch einer 96-Bohrung gesät. Die erste und letzte Säule der 96-Well-Platte werden für die Proteinbestimmung verwendet (Abbildung 2C). Die vier Eckvertiefungen sollten keine Zellen enthalten und dienen der experimentellen Hintergrundkorrektur. Idealerweise sind Vertiefungen in der Nähe der Ränder leer, um den Kanteneffekt zu minimieren (z. B. zeigen Zellen veränderte Wachstumsraten, die durch Temperatureffekte verursacht werden) (Abbildung 2C, D).
- Die Zellen vor der Aussaat ausreichend ernten und resuspendieren
- Herstellung von Sensorplatten, Beladung von Inhibitoren
- Ergänzen Sie am Tag des Assays 38,8 ml Medium mit 0,4 ml 1 M Glukose, 0,4 ml 200 mM Glutamin und 0,4 ml 100 mM Na-Pyruvat.
HINWEIS: Die mHRR-Atmung erfordert ein spezielles, nicht gepuffertes DMEM-Medium bei pH 7,4. Im Allgemeinen sollten 40 ml für ein Experiment mit einer 96-Well-Mikrotiterplatte ausreichen. - Erwärmen Sie das Atmungsmedium auf 37 °C und tauschen Sie das Zellkulturmedium gegen das Atmungsmedium aus, indem Sie es zweimal mit 80 μL pro Vertiefung waschen.
- Stellen Sie die Platte mit den Zellen in einem 37 °C Inkubator ohne CO2 für 60 Minuten vor dem Assay ein.
HINWEIS: Dieser Schritt ist wichtig, um die Platte zu entgasen, da CO 2 die Atmungsergebnisse beeinflussen kann und Serum im Medium während des AssaysBlasen erzeugen kann. - Vorwärmte Inhibitoraliquoten für OM, FCCP, ROT und AM bis 37 °C und entnehmen die Sensorplatte aus dem Inkubator.
- Verdünnen Sie OM, FCCP, ROT und AM in 3 ml Assay-Medium bis zu einer endgültigen Bohrlochkonzentration von 1,5 μM, 1,125 μM bzw. 1 μM. Füllen Sie die einzelnen Ports aus, wie in Abbildung 2E dargestellt.
HINWEIS: Zum Befüllen der Sensorpatrone wird eine Mehrkanalpipette empfohlen. Da Druckluft zum Einspritzen von Verbindungen verwendet wird, müssen alle Anschlüsse mit einer gleichen Menge an Flüssigkeitsvolumen gefüllt werden, wenn ein Port mit einer Verbindung gefüllt wird. ROT und AM können in einem Port kombiniert werden. Inhibitoren können in EtOH oder DMSO gelöst werden. - Prüfen Sie die Einspritzanschlüsse und überprüfen Sie ein gleichmäßiges Ladevolumen für jeden Anschluss.
HINWEIS: Alle Anschlüsse enthalten ein Loch an der Unterseite für die Injektion. Beim Bewegen der Sensorplatte ist Vorsicht geboten. Luftblasen können mit einer Nadel entfernt werden.
- Ergänzen Sie am Tag des Assays 38,8 ml Medium mit 0,4 ml 1 M Glukose, 0,4 ml 200 mM Glutamin und 0,4 ml 100 mM Na-Pyruvat.
- Protokoll zur Sauerstoffbewertung in intakten Zellen
- Führen Sie am Tag vor dem Assay die Schritte 2.4.2-2.4.7 aus.
- Aliquot 20 ml der Kalibrantenlösung in ein 50 mL konisches Rohr.
- Öffnen Sie das Extracellular Flux Assay Kit und entfernen Sie den Inhalt.
- Legen Sie die Sensorkassette invertiert neben die Utility-Platte. Pipette 200 μL Kalibrantlösung in jede Vertiefung der Gebrauchsplatte.
- Befestigen Sie die Sensorpatrone auf der Betriebsanzeige und achten Sie darauf, dass alle Sensoren untergetaucht sind.
- Stellen Sie die Platte über Nacht oder mindestens 12 h in einen 37 °C Inkubator ohne CO2 ein. Stellen Sie sicher, dass die Luftfeuchtigkeit im Inkubator ausreicht, um eine Verdunstung des Kalibers zu verhindern.
- Schalten Sie das mikrotiterplattenbasierte System und den Computer ein, um am nächsten Tag einsatzbereit zu sein (die Maschine benötigt mindestens 3 Stunden, um sich vor der Durchführung eines Assays auf 37 °C auszugleichen).
HINWEIS: Für die Signalstabilität erhöhen Sie die Messpunkte auf 6 statt 3 Messzyklen pro Atemzustand. Jeder Zyklus besteht aus 3 Minuten Mischen und 3 Minuten Messen. - Führen Sie am Tag des XF-Assays die Schritte 2.4.9-2.4.20 aus.
- Überprüfen Sie die Konfluenz der Zellkulturplatte, die Morphologie der Zellen und dass die Hintergrundvertiefungen leer sind.
- Die Zellen werden mit dem vorbereiteten Atmungsmedium gewaschen, wie in den Schritten 2.4.11-2.4.12 beschrieben.
- Entfernen Sie alle bis auf 20 μL des Kulturmediums aus jedem Bohrloch. Entfernen Sie 55 μL, wenn das Kulturmedium 80 μL aufgrund der Verdampfung über Nacht (ca. 5 μL) betrug.
- Waschen Sie die Zellen zweimal mit 90 μL Assay-Medium. Zum Schluss 100 μL Assay-Medium hinzufügen. Das Endvolumen sollte 120 μL betragen.
HINWEIS: Für diesen Schritt wird eine Mehrkanalpipette empfohlen, um sicherzustellen, dass bei jedem Versuchszustand das gleiche Waschverfahren angewendet wurde (abhängig vom Plattenaufbau). Neigen Sie die Platte beim Ansaugen in einen Winkel von 45° und platzieren Sie die Pipettenspitzen in der Ecke der Vertiefungen zum Absaugen und Einspritzen von Flüssigkeiten. Es ist unerlässlich, während des Waschens vorsichtig zu sein, da sich bestimmte Zellen leicht vom Boden der Zellkulturplatte lösen können. - Stellen Sie die Platte vor dem Assay 60 min lang in einem 37 °C Inkubator ohne CO2 ein.
- Holen Sie sich die hydratisierte Sensorkartuschenplatte aus dem CO2-freien Inkubator.
- Entsorgen Sie die alte Kalibrantlösung und ersetzen Sie sie durch frische Kalibrantlösung, die auf 37 °C vorgewärmt ist.
- Bereiten Sie Inhibitoren und Assay-Medium vor (3 ml pro Inhibitor für insgesamt 12 mL Assay-Medium) und verwenden Sie ein Pipettenreservoir für die Inhibitorladung in Ports.
- Öffnen Sie die Software und führen Sie eine vorgefertigte oder neue Vorlage aus. Füllen Sie die Plattenkarte, passen Sie die Titrationen und Messzyklen an und drücken Sie dann Start , um die Kalibrierung der optischen Sensoren zu starten.
- Entfernen Sie den Deckel von der eingelegten Kassette und legen Sie ihn in den Steckplatz, der automatisch aus dem Gerät gleitet, und überprüfen Sie, ob die Markierungen in der unteren rechten Ecke der Platte mit dem Dreieck in der unteren rechten Ecke des Steckplatzes ausgerichtet sind.
- Klicken Sie auf Weiter , um eine automatische Kalibrierung durchzuführen, die ca. 20 Minuten dauert.
- Entfernen Sie nach der Kalibrierung das Gebrauchskennzeichen, das das Kaliber enthält.
- Entfernen Sie den Deckel von der Mikrotiterplatte, die die Zellen enthält, und legen Sie die Platte in den Schlitz, wenn Sie von der Maschine dazu aufgefordert werden. Klicken Sie auf Weiter , um den Lauf zu starten.
- Beispielsammlung nach der Ausführung
- Nehmen Sie die Platte aus der Maschine, entfernen Sie vorsichtig die verbleibenden Assay-Medien, ohne die Zellen zu stören, und frieren Sie die gesamte Platte bei -80 °C zur weiteren Verarbeitung ein (Abschnitt 3).
3. Bestimmung des Proteins mit dem Bicinchoninsäure-Assay (BCA-Assay)
- Herstellung von verdünntem Rinderserumalbumin (BSA) in einem Puffer, der für die Proteinextraktion verwendet wird und mit BCA kompatibel ist: 2 mg/ml, 1,5 mg/ml, 1 mg/ml, 0,5 mg/ml, 0,25 mg/ml und 0 mg/ml für die Standardkurve in Duplikaten.
- Extrahieren Sie Proteine durch Resuspension in einem geeigneten Lysepuffer (z. B. RIPA) mit 20 μL pro Vertiefung für mHRR oder 100 μL pro Pellet, die in einem 1,5 ml-Röhr für cHRR enthalten sind.
- Inkubieren Sie die mHRR-Platte oder den 1,5 ml Schlauch mit Proteinlysaten für 30 min auf Eis.
- Das 1,5-ml-Röhrchen mit dem Proteinlysat bei 4 °C bei 20.000 x g für 20 min zentrifugieren und den resultierenden Überstand in ein neues sauberes 1,5-ml-Röhrchen überführen.
- Verwenden Sie 10 μL pro Probe in Duplikaten und Standards in einer Mikrotiterplatte. 200 μL BCA-Arbeitsreagenz zugeben und >15 min inkubieren.
- Lesen Sie die Platte in einem Standard-Spektralphotometer bei einer Wellenlänge von 562 nm ab und berechnen Sie die Proteinkonzentrationen mit einer BSA-Standardkurve.
- Normalisieren Sie die Atmungsergebnisse auf die Proteinkonzentration.
HINWEIS: Die Normalisierung auf die Proteinmenge ermöglicht es, die Zellaussaatdichten oder den Nassgewichtseintrag zu bestätigen. Die extrahierten Proteine eignen sich beispielsweise für ein nachträgliches Immunoblotting gegen Untereinheiten des ETS, bilden die native Probe jedoch nicht vollständig ab (z. B. Verlust von Phosphorylierungsstellen).
Ergebnisse
Hier stellen wir Protokolle zur Bestimmung der mitochondrialen Bioenergetik in eukaryotischen Zellen, nicht-fibrösem Gewebe (z. B. Kleinhirn) und fibrösem Gewebe (z. B. Skelettmuskulatur) zur Verfügung. Für eukaryotische Zellen wurden HEK293 mit CRISPR-manipuliertem Knockout von zwei verschiedenen Proteinen, die mit einer mitochondrialen Translation assoziiert sind, was zu einem multiplen (CRISPR KO1) und schweren/vollständigen OXPHOS-Mangel (CRISPRKO2) führte, entweder mit cHRR (Abbildung 3A-C) oder mHR...
Diskussion
Traditionell wurde die mitochondriale Bioenergetik mit Clark-artigen Sauerstoffelektroden untersucht. Ein Mangel an Auflösung und Durchsatz rechtfertigte jedoch den technologischen Fortschritt. Bis heute sind der O2k (als cHRR bezeichnet) und der Seahorse XF96 Flux Analyzer (als mHRR bezeichnet) im Bereich der zellulären Bioenergetik weit verbreitet. Hier präsentieren wir eine verständliche Sammlung von Protokollen für die Analyse des zellulären Energiestoffwechsels durch Bewertung der mitochondrialen Atmung mit cH...
Offenlegungen
Kein Interessenkonflikt offenzulegen.
Danksagungen
Diese Arbeit wurde durch Mittel der Akademie von Finnland (C.B.J), der Magnus Ehrnroot Foundation (C.B.J.) und eines Doktorandenstipendiums der Integrated Life Sciences Graduate School (R.A.) unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| 2 mL Potter-Elvehjem Glass/PTFE Tissue Grinder/Homogenizer | Omni International | 07-358029 | |
| 95% O2, 5% CO2 medical gas mixture | Potter for tissue grinding | ||
| ADP | Sigma | A 4386 | |
| Antimycin A | Sigma | A 8674 | Chemical |
| Ascorbate | Merck | PHR1279-1G | Chemical, dissolve in ethanol |
| BSA (fatty accid free) | Sigma | A 6003 | Chemical |
| CaCO3 | Sigma | C 4830 | Chemical |
| Cytochrome c | Sigma | C 7752 | Chemical |
| Digitonin | Sigma | D 5628 | Chemical |
| Dithiothreitol | Sigma | D 0632 | Chemical, dissolve in DMSO |
| D-Sucrose | Roth | 4621.1 | Chemical |
| Dulbecco’s modified Eagle’s medium (High glucose) | Fisher Scientific | 41965-039 | Chemical |
| Dulbecco’s modified Eagle’s medium (No Glucose) | Fisher Scientific | A14430-01 | |
| EGTA | Sigma | E 4378 | |
| Etomoxir | Sigma | E1905 | Chemical |
| Falcon 15 ml Conical Centrifuge Tubes | Fisher Scientific | AM12500 | Chemical |
| Falcon 50 ml Conical Centrifuge Tubes | Fisher Scientific | AM12501 | |
| FCCP | Sigma | C 2920 | |
| Glucose | Sigma | G7021 | Chemical, dissolve in ethanol |
| Glutamate | Sigma | G 1626 | Chemical |
| GlutaMax (100x) (200 nM L-alanyl-L-glutamine dipeptide) | Fisher Scientific | 35050061 | Chemical |
| HEK293 cells | ATTC | CRL-1573 | |
| Hemocytometer | Fisher Scientific | 0267151B | Instrument for cell counting |
| Hepes | Sigma | H 7523 | Chemical |
| Imidazole | Fluka | 56750 | Chemical |
| KCl | Merck | 1.04936 | Chemical |
| L-carnitine | Sigma | C0283 | Chemical |
| Malate | Sigma | M 1000 | Chemical |
| MES hydrate | Sigma | M8250 | Chemical |
| MgCl2 | Sigma | M 9272 | Chemical |
| Na2ATP | Sigma | A 2383 | Chemical |
| Na2Phosphocreatine | Sigma | P 7936 | Chemical |
| Na-pyruvate (100 mM) (100x) | Fisher Scientific | 11360070 | |
| NEAA (Non-essential amino acids) 100x | Fisher Scientific | 11140035 | |
| Normal FBS (10x) | Fisher Scientific | 10500064 | |
| O2k-Core: Oxygraph-2k | Oroboros Instruments | 10000-02 | High-resolution respirometry instrument |
| O2k-Titration Set | Oroboros Instruments | 20820-03 | Hamilton syringes for chemical injections |
| Oligomycin | Sigma | O 4876 | Chemical, dissolve in ethanol |
| Palmitoylcarnitine | Sigma | P 4509 | Chemical |
| Penicillin-Streptomycin | Fisher Scientific | 15140122 | |
| Pierce BCA Protein Assay Kit | Fisher Scientific | 23227 | |
| Pyruvate | Sigma | P 2256 | Chemical |
| RIPA-Buffer | Fisher Scientific | 89900 | Chemical |
| Rotenone | Sigma | R 8875 | Chemical, dissolve in ethanol |
| Saponin | Sigma | S7900 | Chemical |
Seahorse XF DMEM assay medium pack, pH 7.4 | Agilent, Santa Clara, CA | 103680-100 | |
| Seahorse XFe96 Extracellular Flux Analyzer | Agilent, Santa Clara, CA | High-throughput respirometry instrument | |
| Seahorse XFe96 FluxPak | Agilent, Santa Clara, CA | Includes assay plates, cartridges, loading guides for transferring compounds to the assay cartridge, and calibrant solution. | |
| Small scissors | Fisher Scientific | 08-951-20 | |
| Sodium azide | Sigma | S2002 | Chemical |
| Succinate | Sigma | S 2378 | Chemical |
| Taurine | Sigma | T 8691 | Chemical |
| TMPD | Sigma | T 3134 | Chemical |
| Trypan Blue solution | Merck | 72-57-1 | Chemical |
| Trypsin 0.25% EDTA | Fisher Scientific | 25200056 | |
| Two thin-edged forceps | Fisher Scientific | 12-000-122 | |
| Uridine stock (500x) | Sigma | U3750 | Chemical |
Referenzen
- McBride, H. M., Neuspiel, M., Wasiak, S. Mitochondria: More than just a powerhouse. Current Biology. 16 (14), 551-560 (2006).
- Mehta, M. M., Weinberg, S. E., Chandel, N. S. Mitochondrial control of immunity. Beyond ATP. Nature Reviews Immunology. 17 (10), 608-620 (2017).
- Spinelli, J. B., Haigis, M. C. The multifaceted contributions of mitochondria to cellular metabolism. Nature Cell Biology. 20 (7), 745-754 (2018).
- Gnaiger, E. Capacity of oxidative phosphorylation in human skeletal muscle. New perspectives of mitochondrial physiology. International Journal of Biochemistry and Cell Biology. 41 (10), 1837-1845 (2009).
- Gorman, G. S., et al. Mitochondrial diseases. Nature Reviews Disease Primers. 2, 1-23 (2016).
- Boushel, R., Gnaiger, E., Schjerling, P., Skovbro, M., Kraunsøe, R., Dela, F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia. 50 (4), 790-796 (2007).
- Cogliati, S., et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 155 (1), 160-171 (2013).
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biology. 13, 1-11 (2015).
- Gnaiger, E. Mitochondrial pathways and Respiratory control. An introduction to OXPHOS analysis. Bioenergetics communications. 5th ed. , (2020).
- Jackson, C. B., et al. Mutations in SDHD lead to autosomal recessive encephalomyopathy and isolated mitochondrial complex II deficiency. Journal of Medical Genetics. 51 (3), 170-175 (2014).
- Pesta, D., Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods in Molecular Biology. 810, 25-58 (2012).
- Horan, M. P., Pichaud, N., Ballard, J. W. O. Review: Quantifying mitochondrial dysfunction in complex diseases of aging. Journals of Gerontology - Series A Biological Sciences and Medical Sciences. 67 (10), 1022-1035 (2012).
- Doerrier, C., Garcia-Souza, L. F., Krumschnabel, G., Wohlfarter, Y., Mészáros, A. T., Gnaiger, E. High-resolution fluorespirometry and oxphos protocols for human cells, permeabilized fibers from small biopsies of muscle, and isolated mitochondria. Methods in Molecular Biology. 1782, 31-70 (2018).
- Zhang, J., et al. Measuring energy metabolism in cultured cells, including human pluripotent stem cells and differentiated cells. Nature Protocols. 7 (6), 1068-1085 (2012).
- García-Roche, M., Casal, A., Carriquiry, M., Radi, R., Quijano, C., Cassina, A. Respiratory analysis of coupled mitochondria in cryopreserved liver biopsies. Redox Biology. 17, 207-212 (2018).
- Acin-Perez, R., et al. A novel approach to measure mitochondrial respiration in frozen biological samples. The EMBO Journal. 39 (13), 1-18 (2020).
- Cell metabolism assay kits. Seahorse assay kits and media Available from: https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-assay-lits-reagents-cell-assay-media (2021)
- Chance, B., Williams, G. R. A method for the localization of sites for oxidative phosphorylation. Nature. 176 (4475), 250-254 (1955).
- Gnaiger, E., et al. Mitochondrial respiratory states and rates. MitoFit Preprint Arch. , (2019).
- Gnaiger, E. O2k-procedures: SOP O2k quality control 1: Polarographic oxygen sensors and accuracy of calibration Section Page. Oroboros. 03 (18), 1-21 (2020).
- Robinson, B. H., Petrova-Benedict, R., Buncic, J. R., Wallace, D. C. Nonviability of cells with oxidative defects in galactose medium: A screening test for affected patient fibroblasts. Biochemical Medicine and Metabolic Biology. 48 (2), 122-126 (1992).
- King, M. P., Attardi, G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science. 246 (4929), 500-503 (1989).
- Makrecka-Kuka, M., Krumschnabel, G., Gnaiger, E. High-resolution respirometry for simultaneous measurement of oxygen and hydrogen peroxide fluxes in permeabilized cells, tissue homogenate and isolated mitochondria. Biomolecules. 5 (3), 1319-1338 (2015).
- Fasching, M., Gnaiger, E. O2k quality control 2: Instrumental oxygen background correction and accuracy of oxygen flux. Mitochondrial Physiology Network. 14 (06), 1-14 (2016).
- Gnaiger, E., Lassnig, B., Kuznetsov, A., Rieger, G., Margreiter, R. Excess capacity of cytochrome c oxidase. Journal of Experimental Biology. 1139, 1129-1139 (1998).
- Gnaiger, E., et al. Mitochondria in the Cold. Life in the Cold. , 431-442 (2000).
- Fontana-Ayoub, M., Fasching, E., Gnaiger, Selected media and chemicals for respirometry with mitochondrial preparations. Mitochondrial Physiology Network. 02 (17), 1-9 (2014).
- Gerencser, A. A., et al. Quantitative microplate-based respirometry with correction for oxygen diffusion. Analytical Chemistry. 81 (16), 6868-6878 (2009).
- Krumschnabel, G., Eigentler, A., Fasching, M., Gnaiger, E. Use of safranin for the assessment of mitochondrial membrane potential by high-resolution respirometry and fluorometry. Methods in Enzymology. 542, 163-181 (2014).
- Nászai, A., Terhes, E., Kaszaki, J., Boros, M., Juhász, L. Ca(2+)N it be measured? Detection of extramitochondrial calcium movement with high-resolution fluorespirometry. Scientific Reports. 9 (1), 1-13 (2019).
- Pajak, B., et al. 2-Deoxy-d-Glucose and its analogs: From diagnostic to therapeutic agents. International Journal of Molecular Sciences. 21 (1), 234 (2019).
- Mercier-Letondal, P., Marton, C., Godet, Y., Galaine, J. Validation of a method evaluating T cell metabolic potential in compliance with ICH Q2 (R1). Journal of Translational Medicine. 19 (1), 1-15 (2021).
- Sauerbeck, A., et al. Analysis of regional brain mitochondrial bioenergetics and susceptibility to mitochondrial inhibition utilizing a microplate based system. Journal of Neuroscience Methods. 198 (1), 36-43 (2011).
- Jackman, M. R., Willis, W. T. Characteristics of mitochondria isolated from type I and type IIb skeletal muscle. American Journal of Physiology - Cell Physiology. 270 (2), 673-678 (1996).
- Ponsot, E., et al. Mitochondrial tissue specificity of substrates utilization in rat cardiac and skeletal muscles. Journal of Cellular Physiology. 203 (3), 479-486 (2005).
- Schönfeld, P., Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy-Reflections on disadvantages of the use of free fatty acids as fuel for brain. Journal of Cerebral Blood Flow and Metabolism. 33 (10), 1493-1499 (2013).
- Calderon-Dominguez, M., Mir, J. F., Fucho, R., Weber, M., Serra, D., Herrero, L. Fatty acid metabolism and the basis of brown adipose tissue function. Adipocyte. 5 (2), 98-118 (2016).
- Divakaruni, A. S., Rogers, G. W., Murphy, A. N. Measuring mitochondrial function in permeabilized cells using the seahorse XF analyzer or a clark-type oxygen electrode. Current Protocols in Toxicology. 2014, 1-16 (2014).
- Iuso, A., Repp, B., Biagosch, C., Terrile, C., Prokisch, H. Assessing mitochondrial bioenergetics in isolated mitochondria from various mouse tissues using Seahorse XF96 analyzer. Methods in Molecular Biology. 1567, 217-230 (2017).
- Rogers, G. W., et al. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS ONE. 6 (7), 21746 (2011).
- Jordá, A., Zaragozá, R., Portolés, M., Báguena-Cervellera, R., Renau-Piqueras, J. Long-term high-protein diet induces biochemical and ultrastructural changes in rat liver mitochondria. Archives of Biochemistry and Biophysics. 265 (2), 241-248 (1988).
- Jackson, C. B., Gallati, S., Schaller, A. QPCR-based mitochondrial DNA quantification: Influence of template DNA fragmentation on accuracy. Biochemical and Biophysical Research Communications. 423 (3), 441-447 (2012).
- Hirsch, H. M. Tissue autoxidation inhibitors: II. The presence of inhibitor in intact cells; Assay of liver and hepatoma effect on radio-oxidations. Cancer Research. 16 (11), 1076-1082 (1956).
- Picard, M., et al. Mitochondrial structure and function are disrupted by standard Isolation methods. PLoS ONE. 6 (3), 18317 (2011).
- Tanumihardja, E., Slaats, R. H., Van Der Meer, A. D., Passier, R., Olthuis, W., Van Den Berg, A. Measuring both pH and O2 with a single On-Chip sensor in cultures of human pluripotent stem cell-derived cardiomyocytes to track induced changes in cellular metabolism. ACS Sensors. 6 (1), 267-274 (2021).
- Harms, F., Stolker, R. J., Mik, E. Cutaneous respirometry as novel technique to monitor mitochondrial function: A feasibility study in healthy volunteers. PLoS ONE. 11 (7), 159544 (2016).
- Levitsky, Y., et al. Micro-respirometry of whole cells and isolated mitochondria. RSC Advances. 9 (57), 33257-33267 (2019).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten