A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
The Fabrication and Operation of a Continuous Flow, Micro-Electroporation System with Permeabilization Detection
In This Article
Summary
This protocol describes the microfabrication techniques required to build a lab-on-a-chip, microfluidic electroporation device. The experimental setup performs controlled, single-cell-level transfections in a continuous flow and can be extended to higher throughputs with population-based control. An analysis is provided showcasing the ability to electrically monitor the degree of cell membrane permeabilization in real-time.
Abstract
Current therapeutic innovations, such as CAR-T cell therapy, are heavily reliant on viral-mediated gene delivery. Although efficient, this technique is accompanied by high manufacturing costs, which has brought about an interest in using alternative methods for gene delivery. Electroporation is an electro-physical, non-viral approach for the intracellular delivery of genes and other exogenous materials. Upon the application of an electric field, the cell membrane temporarily allows molecular delivery into the cell. Typically, electroporation is performed on the macroscale to process large numbers of cells. However, this approach requires extensive empirical protocol development, which is costly when working with primary and difficult-to-transfect cell types. Lengthy protocol development, coupled with the requirement of large voltages to achieve sufficient electric-field strengths to permeabilize the cells, has led to the development of micro-scale electroporation devices. These micro-electroporation devices are manufactured using common microfabrication techniques and allow for greater experimental control with the potential to maintain high throughput capabilities. This work builds off a microfluidic-electroporation technology capable of detecting the level of cell membrane permeabilization at a single-cell level under continuous flow. However, this technology was limited to 4 cells processed per second, and thus a new approach for increasing the system throughput is proposed and presented here. This new technique, denoted as cell-population-based feedback control, considers the cell permeabilization response to a variety of electroporation pulsing conditions and determines the best-suited electroporation pulse conditions for the cell type under test. A higher-throughput mode is then used, where this 'optimal' pulse is applied to the cell suspension in transit. The steps for fabricating the device, setting up and running the microfluidic experiments, and analyzing the results are presented in detail. Finally, this micro-electroporation technology is demonstrated by delivering a DNA plasmid encoding for green fluorescent protein (GFP) into HEK293 cells.
Introduction
Current therapeutic innovations in biomedical research, such as CAR-T (Chimeric Antigen Receptor Engineered T cell) cell therapy and genetic editing using CRISPR (clustered regularly interspaced short palindromic repeat DNA sequences)/Cas9, heavily rely on the ability to deliver exogenous material both successfully and efficiently into the intracellular space1. In CAR-T therapy, the gold standard to perform the gene delivery step in cell therapy manufacturing is using viral vectors2. Though viral-mediated gene delivery is an efficient delivery modality, it also has several drawbacks. These include manufacturing costs, cytotoxicity, immunogenicity, mutagenesis/tumorigenesis potential, and size limitations on the gene(s) to be delivered3. These limitations have led to the research and development of alternative, non-viral delivery technologies.
Electroporation, an alternative to viral-mediated gene delivery, relies on the application of an optimal electrical pulse waveform to perform DNA, RNA, and protein transfections of cells. Following the application of an external electric field, the cell membrane is briefly compromised, making the cell susceptible to the intracellular delivery of otherwise impermeable exogenous materials4. Compared to viral-mediated delivery, electroporation is advantageous as it is generally safe, easy to operate, and has low operating costs. Electroporation can deliver both small and large molecular cargo and can be efficient in transfecting cells regardless of lineage5. To achieve desirable outcomes following electroporation, i.e., good viability and good electro-transfection efficiency, a variety of experimental parameters need to be co-optimized. These include cell type6, cell density, molecule concentration7, electroporation buffer properties (e.g., molecular composition, conductivity, and osmolarity)8, electrode size/geometry9, and electrical pulse waveform (shape, polarity, number of pulses)10 (refer to Figure 1 for an illustration). Although each of these parameters can have a significant effect on the outcomes of electroporation experiments, pulse waveform has been especially studied in great detail, as the electrical energy of the applied pulse(s) is the root of the intrinsic trade-off between the resulting cell viability and electro-transfection efficiency8.
Typically, electroporation experiments are performed on the macro-scale, where cells are suspended in 100s of microliters of buffer between a set of large, parallel-plate electrodes within an electroporation cuvette. The electrodes are commonly manufactured out of aluminum with an electrode distance of 1-4 mm. Once the cells are manually loaded via pipette, the cuvette is electrically connected to a bulky, electrical pulse generator where the user can set and apply the pulse waveform parameters to electroporate the cell suspension. Although macro-scale or bulk electroporation can process cell densities >106 cells/mL, this feature can be wasteful when optimizing the electrical pulse waveform settings. This is particularly of concern when electroporating primary cell types where the cell population numbers can be limited. Additionally, due to the large distance between the electrodes, the pulse generator must be able to supply large voltages to achieve electric field strengths >1kV/cm11. These high voltages cause resistive power dissipation through the electrolyte buffer resulting in Joule heating, which can be detrimental to the resulting cell viability12. Lastly, performing electroporation on a dense suspension of cells will consistently be burdened with an innate variability in the resulting electro-transfection efficiency and cell viability. Each cell in suspension could experience a different electric field strength due to the surrounding cells. Depending on whether the experienced electric field strength is either increased or decreased, the resulting cell viability or electro-transfection efficiency may each be negatively impacted11. These downsides to macro-scale electroporation have led to the pursuit and development of alternative technologies that operate on the micro-scale and allow for better control at the single-cell level.
The field of BioMEMS, or biomedical micro-electro-mechanical systems, stems from the technological advancements made in the microelectronics industry. Specifically, utilizing microfabrication processes to develop micro-devices for the advancement of biomedical research. These advancements include the development of micro-electrode arrays for in vivo electrical monitoring13, capacitive micro-electrodes for in situ electroporation14, miniaturized organ-on-a-chip devices15, microfluidic point-of-care diagnostics16, biosensors17, and drug delivery systems18, including nano- and micro-electroporation devices19,20,21. Due to the ability to design and manufacture devices at the same size scale as biological cells, nano- and micro-electroporation technologies are advantageous when compared to their macro-scale counterpart22,23. These electroporation devices eliminate the requirement of high voltage pulse applications, as electrode sets with spacings of 10s to 100s of micrometers are typically integrated. This feature drastically reduces the current through the electrolyte, which in turn reduces the accumulation of toxic electrolysis products and the effects of Joule heating in these systems. The micro-scale channels also ensure that a much more uniform electric field is reliably applied to the cells during pulse application, resulting in more consistent outcomes24. In addition, it is also commonplace for micro-electroporation devices to be integrated into a microfluidic platform which lends itself for future integration into a fully automated technology, a highly desirable capability in cell therapy manufacturing25. Lastly, micro-scale electroporation allows for the electrical interrogation of electroporation events. For example, the degree of cell membrane permeabilization can be monitored in real-time at a single cell level26,27. The purpose of this method is to describe the microfabrication, system operation, and analysis of a microfluidic, single-cell micro-electroporation device capable of measuring the degree of cell membrane permeabilization for optimizing electroporation protocols, yet increasing throughput over the previous state-of-the-art.
Performing single-cell level electroporation is no longer a novel technique, as it was first demonstrated by Rubinsky et al. in 2001 with the development of a static cell electroporation technology28. Their micro-device was innovative as they were the first to demonstrate the ability to electrically monitor the event of electroporation. This has further led to the development of static, single-cell electroporation technologies capable of electrically detecting the degree of cell membrane permeabilization in a parallelized manner to increase the throughputs of the devices. However, even with parallelization and batch processing, these devices severely lack the total number of cells they can process per unit time29,30. This limitation has led to the development of flow-through devices capable of performing single-cell level micro-electroporation at much greater throughputs31. This device transition, from static to flow-through environment, limits the capability of electrically monitoring the degree of cell membrane permeabilization following the application of the electroporation pulse. The method described in this work bridges the gap between these two technologies, a micro-electroporation technology capable of electrically detecting, pulsing, and monitoring the degree of cell membrane permeabilization of individual cells, in a continuous-flow, serial fashion.
This technology was recently described in Zheng et al. In that work, the capabilities of this technology were introduced with the completion of a parametric study, where both the amplitude and duration of the electroporation pulse were varied, and the ensuing electrical signal, indicative of cell membrane permeabilization, was explored32. The results showed that an increase in the intensity of the electroporation pulse (i.e., increase in applied electric field or increase in pulse duration) caused an increase in the measured cell membrane permeabilization. To further validate the system, a common fluorescent indicator of successful electroporation, propidium iodide33, was added to the cell suspension, and a fluorescence image was captured immediately following the application of the electrical pulse. The optical signal, i.e., the fluorescence intensity of propidium iodide inside the cell, was strongly correlated with the electrical measurement of the degree of cell membrane permeabilization, verifying the reliability of this electrical measurement. However, this work only considered the delivery of the small molecule propidium iodide, which has little to no translatable significance.
In this work, a new application of this technology is introduced to improve upon the throughput of the system while delivering a biologically active plasmid DNA (pDNA) vector and assessing the electro-transfection efficiency of cells replated and cultured following electroporation. Though the previous work outperforms existing micro-electroporation technologies that are capable of electrically measuring the event of electroporation, the current state of the device still requires long cell transit times between the electrode set (~250 ms) to perform the cell detection, pulse application, and the cell membrane permeabilization measurement. With a single channel, this limits the throughput to 4 cells/s. To combat this limitation, a new concept of cell-population-based feedback-controlled electroporation is introduced to perform pDNA electro-transfection. By using a hypo-physiologic conductivity electroporation buffer, this system allows for the electrical interrogation of single cells across a multitude of electroporation pulse applications. Based on the electrical response, an 'optimal' electroporation pulse is then determined. A 'high-throughput' mode is then implemented where the cell membrane permeabilization determination is nullified, the flow rate is increased, and the electroporation pulse duty cycle is matched to the cell transit time to ensure one pulse per cell in transit between the electrodes. This work will provide extensive details into the microfabrication steps for the manufacturing of the micro-device, the material/equipment and their setup required to perform the experimentation, and the operation/analysis of the device and its electro-transfection efficiency (eTE).

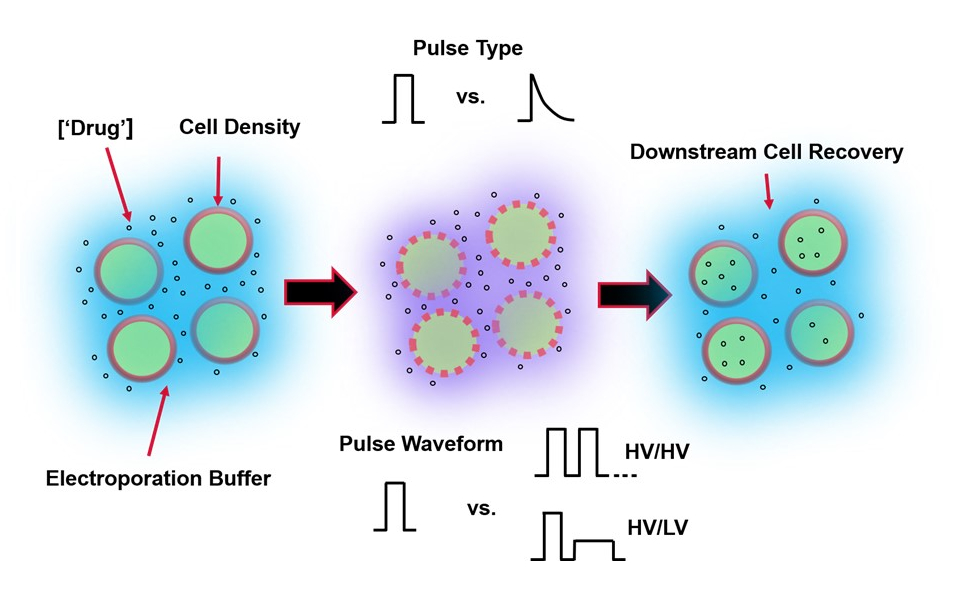
Figure 1: Experimental factors affecting electroporation outcomes. (Left) Cell Suspension-Important factors to consider prior to the onset of electroporation include: Payload (in this case, pDNA), concentration, cell density, and electroporation buffer properties. Electroporation buffer properties to consider are conductivity, osmolarity, and the exact molecular composition contributing to these values. (Middle) Pulse Application-The exact pulse-type (square wave vs. exponential decay) and pulse waveform (single pulse vs. pulse train) must be optimized to maximize both the resulting cell viability and electro-transfection efficiency. Common pulse trains implemented in electroporation processes are typically composed of a series of High Voltage (HV) pulses or series of pulses rotating between HV and Low Voltage (LV) pulse magnitudes. (Right) Cell Recovery-Down-stream processing steps, in particular, the recovery cell culture media that cells are transferred to, should be optimized. Not featured (Far Left), additional upstream cell processing steps can be implemented for overall electroporation process optimization. Please click here to view a larger version of this figure.
{kind=link}
Protocol
NOTE: Users should review all MSDS for the materials and supplies used in this protocol. Appropriate PPE should be worn at each step and sterile technique used during experimentation. Sections 1-7 discuss the device fabrication.
1. Device fabrication- Mask design
NOTE: Refer to Figure 2 for an illustration of the microfabrication process. The microfabrication steps are to be carried out in a cleanroom environment. Additional PPE is necessary (hair net, facial hair net, mask, cleanroom suit, shoe covers).
- Install a CAD software of choice, design a 2-Dimensional 'mask' of both the microfluidic channel and electrodes and save the design in desired file format (i.e., .dxf, .dwg).
NOTE: Refer to Supplementary Figure 1 for an example of a 2-Dimensional mask schematic. - Send to a supplier of choice to be printed. Ensure the dimensions of the designs are within the resolution capabilities of the supplier.
2. Device fabrication- Photolithography
NOTE: The provided microfabrication recipes are adopted from the photoresists' manufacturer's recommendations and should only be used as a starting point34. Exact values for baking times, exposure times, etc., need to be optimized for each fabrication protocol. It is recommended to use wafer tweezers for handling both silicon wafers and glass slides.
- Microfluidic channel fabrication
- Silicon wafer and soda-lime glass slide cleaning: Follow steps 2.1.2-2.1.3 to perform silicon wafer and 1" × 3" soda-lime glass slide cleaning (both referred to as 'substrate').
- Submerge the substrates in an acetone bath, an isopropanol (IPA) bath and a deionized water bath for 10 min each. Perform this 3-step wash serially at room temperature.
- Remove and dry the surface using a pressurized nitrogen or filtered air gas source. Place the substrates into a 150 °C oven for a minimum of 30 min to allow evaporation of the remaining moisture.
- SU-8 photolithography on silicon wafer: Perform photolithography on the silicon wafer following steps 2.1.5-2.1.14.
NOTE: To achieve a microfluidic channel height of 20 µm, SU-8 2000 series negative photoresist was used. Exact spin rates will vary depending on the formulation of SU-8 (i.e., 2010, 2015, etc.); however, the following conditions are for the SU-8 2010 formulation35. - Remove the silicon wafer from the 150 °C oven and allow it to cool to room temperature (RT).
- Secure the wafer to the chuck of the wafer spin coater using the spin coater's vacuum system. Program the spinner. Step 1 - 500 rpm for 10 s at an acceleration of 100 rpm/s, Step 2 - 1000 rpm for 30 s at an acceleration of 300 rpm/s.
- Dispense 4 mL of SU-8 2010 photoresist onto the center of the silicon wafer. Run the program. Once the system comes to a halt, turn off the vacuum.
- Using tweezers, transfer the SU-8 coated silicon wafer on a hot plate at 95 °C for 4-5 min for soft bake. Then remove the wafer from the hot plate and allow it to cool to RT.
NOTE: Follow the proper start up procedure for the lab-specific photolithographic mask aligner. - Secure the photomask with the 2D microfluidic channel designs onto the mask holder. Insert the silicon wafer, with the SU-8 coating facing upwards, onto the wafer chuck.
- Set the exposure settings for 150 mJ/cm2 and run the machine.
CAUTION: Do not look directly at the UV light source to avoid potential eye damage. - Place the SU-8 coated silicon wafer on a hot plate at 95 °C for 4-5 min for post-exposure bake.
- Submerge the silicon wafer in the SU-8 developer solution (see Table of Materials) for 3-4 min. Apply gentle agitation. Remove the wafer from the solution and rinse the surface with IPA.
- Dry the surface using a pressurized nitrogen or filtered air gas source. Inspect the features under a microscope using a UV filter and ensure no obvious defects in the microfluidic channels.
- Place the silicon wafer into a 150 °C oven for a minimum of 30 min for a hard bake.
- Allow to cool down to RT and use stylus profilometry to measure the exact height and slope of the channel sidewalls.
- Photolithography on Glass Slides
NOTE: Hexamethyldisilazane (HMDS) is used as an adhesion promoter for the S1818 positive photoresist36.- Remove the glass slide from the 150 °C oven and allow it to cool to RT.
- Secure the glass slide to the chuck of the spinner using vacuum and program the spinner. Step 1 - 500 rpm for 10 s at an acceleration of 100 rpm/s. Step 2 - 3000 rpm for 30 sat an acceleration of 300 rpm/s.
- Dispense 3-4 droplets of HMDS across the surface of the glass slide. Run the program.
NOTE: To achieve a surface coating of ~3 µm, S1800 positive photoresist series should be used. Exact spin rates will vary depending on the formulation; the recommendations below are for the S1818 formulation34. - Dispense 1 mL of photoresist onto the surface of the glass slide. Ensure enough to cover the surface area.
- Run the program. Once the system comes to a halt, turn off the vacuum, and remove the glass slide.
- Place the S1818 coated glass slide on a hot plate at 120 °C for 4 min for a soft bake. Remove and allow to come to RT.
- Secure the photomask with the 2D electrode designs onto the mask holder.
- Insert and align the glass slide, with the S1818 coating facing upwards, onto the wafer chuck. Set the exposure settings for 250 mJ/cm2 and run the machine.
NOTE: Different contact aligner models may be more or less accommodating to non-circular, varying thickness substrates. - Submerge the glass slide in MF-319 developer solution for 2 min. Apply gentle agitation. Rinse the surface of the glass slide with deionized water.
- Dry the surface using a pressurized nitrogen or filtered air gas source and observe the features under a microscope using a UV filter. Make sure there are no obvious defects in the lithographic patterns.
- Place glass slide into the 150 °C oven, ensuring substrate surface of interest is facing up, for a minimum of 30 min for a hard bake. Remove from the oven and keep protected from light.
3. Device fabrication: Hydrofluoric acid (HF) etch
CAUTION: This step involves the handling and disposal of hydrofluoric acid (HF), which can cause deep, painful chemical burns. Additional PPE should be used to protect the handler (face shield, elbow-length chemically resistant gloves, chemically resistant apron with sleeves). Calcium gluconate acid neutralizer and skin gel should be kept in proximity of the lab bench. This step should not be performed alone. HF should never be stored in or dispensed into glass containers as the container will be etched by the acid.
NOTE: The HF uniformly etches the exposed glass (i.e., the electrode design) to form a recess in the glass, allowing for better edge resolution of the electrode pattern after metal deposition (section 4).
- Submerge the glass slide in 10:1 buffered HF solution for 1 min in a polytetrafluoroethylene container. Transfer and wash the glass slides in deionized water. Repeat the wash step 3 times.
- Dry the surface using a pressurized nitrogen or filtered air gas source. Place glass substrates in a 65 °C oven overnight to remove any remaining moisture. Cover the substrates from light.
4. Device fabrication: Physical vapor deposition
NOTE: This step involves the metal deposition onto the glass slide substrates to define the electrode patterns. Commonly used metal electrodes are chromium/gold and titanium/platinum. Gold and platinum do not adhere to the glass substrate, so a seed adhesion layer of chromium or titanium, respectively, is required to promote adhesion37.
- Follow the cleanroom-specific protocol to operate the in-house PVD system. This work uses a DC sputtering system and sputter with 100 SCCM Argon gas at a pressure of ~8 mTorr and 200 W power.
- Sputter titanium for 8 min at a rate of ~100 Å/min. Sputter platinum for 10 min at a rate of ~200 Å/min. Remove the substrates from the PVD chamber.
5. Device fabrication: Photoresist lift-off
NOTE: This step involves dissolving the photoresist layer in an acetone bath, leaving the adhered platinum electrodes patterned on the glass slides.
- Submerge the metal-coated glass slides in an acetone bath for ~10 min.
- Sonicate the bath to introduce agitation to break up the unadhered metal film. Use an acetone-soaked wipe to remove any residues if necessary.
- Once all photoresist/metal is removed, wash the electrode patterns with deionized water, and place them in a 65 °C oven overnight to remove any remaining surface moisture.
- Use stylus profilometry to measure the profile of the patterned electrodes.
6. Device fabrication: Soft lithography
NOTE: This step involves replica molding the microfluidic channel onto the SU-8 master relief structure using an elastomer, polydimethylsiloxane (PDMS).
- Silicon wafer silanization
NOTE: This is an optional step; however, it will increase the lifetime of the SU-8 relief structure that was fabricated in subsection 2.1. This step should be performed in a chemical fume hood.- Secure the wafer to the bottom of a Petri dish and place the Petri dish into a desiccator.
- Surround the perimeter of the silicon wafer with approximately 50 µL of Trichloro(1H,1H,2H,2H-perfluorooctyl) silane. Connect vacuum (vacuum pump or house vacuum line) and run for 20 min.
- PDMS replica molding
- In a disposable container (e.g., weigh boat, plastic cup), mix PDMS elastomer base to hardener at a 10:1 weight ratio on top of an electronic balance. Pour the PDMS solution over the silicon wafer and place the mixture under a vacuum to remove all air bubbles.
- Cure at 65 °C for a minimum of 4 h allowing the PDMS to solidify. Using the tip of a razor blade, cut out the molded PDMS and peel from the silicon wafer.
- Using a sharpened biopsy punch, remove PDMS from the inlet/outlets of the device. For this device, 0.75 mm and 3 mm biopsy punches were used for the inlets and the outlets, respectively.
NOTE: The biopsy punch used should have a slightly smaller diameter than the outer diameter of the interconnecting tubing to ensure a tight seal of tubing in the reservoirs.
- Sonication cleaning of PDMS
- Submerge the PDMS devices in IPA and place them into a sonicator for 30-45 min to remove any PDMS debris from the inlet/outlets. PDMS may swell in the IPA solution.
- Rinse with deionized water and place in a 65 °C oven overnight to allow the PDMS to de-swell back to the normal size.
NOTE: Any leftover debris can clog the device during experimentation. Large pieces of debris can be removed from the PDMS surface using a piece of scotch tape prior to sonication.
7. Device fabrication: PDMS bonding/wire attachment
NOTE: This step involves treating the surface of the PDMS and glass substrate with an oxygen plasma to form an irreversible bond between the PDMS and glass38. The recipe provided may need to be adapted to the exact system used in the laboratory.
- Cut the devices to size and ensure the surface of the PDMS device is clean. If not reclean, follow the steps in subsection 6.3.
- Program the plasma generator. Set Power to 70 W, Time to 35 s, Pressure to 325 mTorr, Flow Rate of oxygen gas to 60 SCCM. Place PDMS and electrode glass slide into the system with the features facing up and run the program.
- Remove the devices and quickly align channel features to the electrodes using a stereoscope. Firmly apply pressure from the center of the PDMS towards the sides to remove any unwanted air bubbles at the bonding interface.
- Place on a hot place at 95 °C for at least 2 min to finalize the bonding procedure and let the device cool down at RT.
- Cut 2 pieces of 22-G solid wire at ~6" lengths and strip the insulator from both ends.
- Bond the wires to electrode pads using silver conductive epoxy. Place completed devices in a 65 °C oven overnight.

Figure 2: Microdevice fabrication. (A) Microfluidic Channel Fabrication-Key Steps: Silicon Wafer Cleaning (steps 2.1.1-2.1.3), Photoresist Coating and Soft Bake (steps 2.1.7-2.1.8), UV Exposure (step 2.1.10), Development (step 2.1.12), and PDMS Pouring (subsection 6.2). (B) Electrode Fabrication-Key Steps: Glass Slide Cleaning (steps 2.1.1-2.1.3), HMDS Coating and Photoresist Coating (steps 2.2.3-2.2.4), UV Exposure (step 2.2.8), Development (step 2.2.9), HF Etch (section 3), Physical Vapor Deposition (section 4), and Photoresist Lift-off (section 5). (C) Device Finalization-Key Steps: Inlet/Outlet Access and Sonication (step 6.2.3 and section 6.3), PDMS Bonding, and Wire Attachment (section 7). Please click here to view a larger version of this figure.
{kind=link}
8. Cell culture and harvest
NOTE: Standard cell culture and sterile handling procedures should be utilized. Follow cell-type-specific protocol for cell culture.
- Cell culture
- Cell passage: Culture and passage the cells following steps 8.1.2-8.1.5.
- Culture HEK293 cells in complete DMEM solution (88% DMEM, 10% heat-inactivated fetal bovine serum, 1% L-glutamine, 1% penicillin-streptomycin) in a T25 flask in an incubator at 37 °C, 95% O2, 5% CO2. Passage cells on schedule when reaching ~80% confluency.
- Aspirate the media using either a pipette or vacuum system and incubate the cells in 0.25% trypsin-EDTA (2 mL-T25 flask) for 2 min at 37 °C. Neutralize trypsin with twice the volume of culture media.
- Transfer the cell suspension into a 15 mL conical tube and centrifuge HEK293 cells at 770 x g for 2 min. Aspirate the supernatant using either a pipette or vacuum system
- Resuspend HEK293 cells in 1 mL of pre-warmed DMEM.
- Cell plating: Plate the cells following steps 8.1.7-8.1.8
- Plate the cells at a 10:1 to 20:1 dilution in a T25 flask (5 mL of DMEM) to continue the culture.
- Plate the cells at a 5:1 to 20:1 dilution in a 6-well plate (2 mL of DMEM per well) to be harvested for electroporation experiments.
NOTE: HEK293 cells plated 24 h prior to electroporation experiments to achieve ~70% confluency at cell harvest (subsection 8.3). An inconsistent harvest schedule can lead to variability in electroporation results.
- Electroporation buffer
- Prepare electroporation buffer
NOTE: Refer to Sherba et al. for specifics on the electroporation buffer preparation8. The buffer composition was 285 mM Sucrose, 0.7 mM MgCl2, 1 mM KCl, 10 mM HEPES, 3 mM NaOH (pH: 7.4; osmolality: 310 mOsm, conductivity: 500 µS/cm). Electroporation buffer should be formulated in a sterile fashion and stored at 4 °C for a shelf life of ~1 month. Electroporation buffer formulation should be optimized on a per cell type basis.
- Prepare electroporation buffer
- Cell harvest and pDNA addition
- Follow the same steps as cell passage (8.1.2-8.1.4).
- Wash the cells in sterile 1x PBS, transfer-cell suspension into a 15 mL conical tube, and centrifuge cells at 770 x g for 2 min.
- Wash HEK293 cell pellet in the electroporation buffer and centrifuge at 770 x g for 2 min. Resuspend the cells in the electroporation buffer at ~5 million cells/mL.
NOTE: Cell density should be optimized per cell type. - Add pDNA encoding for green fluorescent protein (GFP) to a final concentration of 20 µg/mL. Gently mix the pDNA/cell suspension and transfer the suspension into a 1 cc syringe for experimentation.
9. Hardware/experimental setup
NOTE: Prior to harvesting cells for experimentation, ensure the experimental setup is completed to minimize the amount of time the cells are suspended in the electroporation buffer. Turn on electronics 20-30 min prior to experiments to warm up. Refer to Figure 3 for a schematic of the experimental setup for the operation of the single-cell detection module.
NOTE: A custom-built PA90 Op-Amp circuit was developed to accommodate both the sensitivity required for single-cell level detection using the lock-in amplifier and the high voltages required to apply sufficiently strong electroporation pulses. Refer to PA90 datasheet for specifications on recommended circuitry39.
- Initialize the lock-in amplifier with current pre-amplifier settings and set via the algorithm. Refer to Zheng et al. for specifics on the lock-in settings32.
- Power supplies, function generator, and amplifier
- Power Supply 1: Set to -15 V to power the negative end of the circuit.
- Power Supply 2 (Function Generator): Set to output DC signal and set the amplitude to 2 V. Connect to 50x amplifier input.
- Program Electroporation Pulse Generator for the square wave: Set the desired pulse width (duty cycle) and desired pulse amplitude (Volts).
- Set output to trigger mode (1 pulse). Connect the output to the input of the 50x amplifier.
NOTE: Remember the 50x gain when programming the pulse amplitude. I.e., to achieve an electric field strength of 1 kV/cm, a total of 30 V is required, 30 V/300 µm (distance between electrodes), therefore the function generator output should be set to 30/50, or 600 mV. - Verify the outputs of the 50x Amplifier using an oscilloscope. Output 1-100 V from Power Supply 2 (9.2.2). Output 2-Variable amplitude for the electroporation pulse (9.2.4).
- Connect a 10x probe to an oscilloscope channel and to the completed micro-device (device under test, DUT) in step 7.6 where the electroporation pulse is going to be applied. Monitor the system during experimentation to ensure pulses are being applied.
- Ensure lock-in USB is connected and registered. Double-check all lock-in settings in the algorithm code (most importantly, lock-in output frequency).
- Microscope/CCD camera
- Place the micro-device onto the stage of the microscope via a slide holder. Turn on the CCD camera and bring the microfluidic channel into focus. Use a 4x or 10x objective.

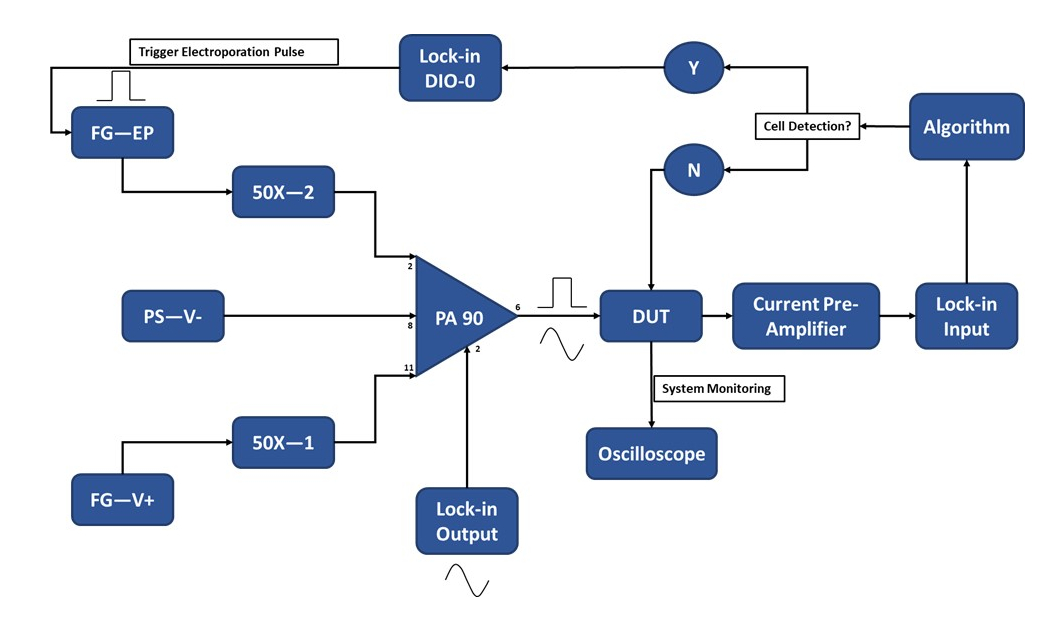
Figure 3: Experimental setup schematic-Single cell detection. The high-power op-amp (PA-90) allows for the superposition of the high voltage electroporation pulse onto the lock-in Output AC signal that is required for the single-cell detection. This excitation signal passes through the micro-electroporation device (Device Under Test, DUT) where the current is then amplified by the current pre-amplifier and fed into the algorithm. The system continuously monitors for the cell detection event. Upon cell entry, a digital signal is generated by the lock-in amplifier to trigger the application of the electroporation pulse to the cell(s) in transit. Legend: PA-90 (high power op amp), DUT (device under test), DIO (digital input/output), FG-EP (function generator / electroporation pulse), 50X (50X amplifier), PS-V- (power supply / negative voltage for PA 90), FG-V+ (Function Generator, positive voltage for PA 90). Please click here to view a larger version of this figure.
{kind=link}
10. Experimental operation
- Microfluidic channel priming
- Remove all air bubbles from the cell-loaded syringe. Attach a 30 G needle to the cell-loaded syringe.
- Using tweezers, slide tygon tubing down the length of the needle. Pre-fill the outlet reservoir with recovery media (same as step 8.1.2 without the antibiotics), ~40-50 µL.
- Using the thumb, gently apply pressure to the plunger such that the fluid slowly reaches the end of the tubing line.
- Secure the syringe to the syringe pump. Turn the syringe pump on and ensure it is set to forward perfusion.
- Program the pump for the proper diameter of the syringe to ensure flow rates are accurate. Refer to pump manual for specifics on syringe diameters.
NOTE: To prevent cells from settling in the syringe, secure the syringe pump on a clamp stand such that it can operate in a vertical position with the syringe end facing downwards. - Set syringe pump flow rate, ~10-20 µL/min, and allow the pump to run until the fluid reaches the end of the tubing line. Secure tubing to the microfluidic device.
- Lower the syringe pump flow rate, ~5-10 µL/min, and allow the pump to run until all air is expelled from the microfluidic device and cells are traversing to the device outlet.
- Remove the cells from the outlet via pipette aspiration. Re-fill the outlet reservoir with recovery media (same as step 8.1.2 without the antibiotics), ~40-50 µL.
- Single-cell electroporation-cell membrane permeabilization mapping
NOTE: Refer to Figure 4 and Figure 5 for a better understanding of the electrical data indicative of cell membrane permeabilization and the cell membrane permeabilization mapping, respectively.- Set the syringe pump flow rate to ~0.1-0.3 µL/min to ensure a flow of single cells through the electrode set. The cell transit time between the electrodes should be ~250 ms.
- Start the computer program by clicking on Run. Ensure the system is saving the electrical data.
- Ensure the system is reliably detecting cells to trigger the computer-controlled pulse applications. Adjust Detection Threshold accordingly.
- Set the pulse parameters for the initial, lowest electrical energy electroporation pulse. Refer to Table 1 for electroporation pulsing parameters in this study.
- Turn on the Output channel for the electroporation pulse generator (step 9.2.3.).
- Follow a pre-determined number of cell detection/pulse applications (n =100). At the end of each tested condition, aspirate cells from the microdevice outlet and replenish the outlet with recovery media.
- Iterate to the next electroporation pulse condition. Repeat until all electroporation pulse conditions are tested.
- Determine the degree of cell membrane permeabilization for each pulse application tested. (Post-process validation is described in subsection 11.1). Generate the cell membrane permeabilization map (Figure 5).
- Determine the electroporation pulse parameters for high-throughput, population-based feedback.
- Turn off the syringe pump, remove cells from the outlet reservoir, and replenish the outlet with recovery media.
- Population-based feedback-controlled electroporation-high throughput
NOTE: Refer to Figure 6 for a schematic illustrating the population-based feedback process.- Set the syringe pump flow rate to ~1-3 µL/min to ensure a flow of single cells through the electrode set. The cell transit time between the electrodes should be ~25 ms.
- Set the pulse amplitude to the 'optimized' condition (10.2.9), turn off trigger mode, and set the pulse width to match the cell transit time.
- Set the duty cycle such that pulse ON time matches the 'optimized' condition. Refer to Table 1.
- Set the Output channel function generator to ON, turn on the syringe pump, and allow the system to run until the desired number of cells have been electroporated.
- When done, turn off both the syringe pump and the function generator.
- Transfer the cells from the outlet reservoir into the appropriately sized cell culture flask/plate filled with pre-warmed recovery media and transfer culture flask/plate into the incubator.
11. Analysis
- Single-cell level membrane permeabilization detection
NOTE: To ensure the 'optimal' pulse was used during the high throughput module, a post-experiment analysis should be performed to verify the electrical data exported from subsection 10.2. Please refer to Figure 4 for a graphical representation of the electrical signal representative of membrane permeabilization due to electroporation.- Load data into an analysis software (MATLAB, Python, etc.). Generate a plot of Current versus Time for each pulsing condition.
- Manually determine the degree of cell membrane permeabilization (ΔIP/ΔIC). Refer to Figure 4. Generate the Cell Membrane Permeabilization Map (ΔIP / ΔIC versus Electrical Energy, Figure 5) over all tested pulse conditions. Verify 'optimal' pulsing condition.
- electro-Transfection Efficiency (eTE)
- Following the 24-h incubation period, remove the electroporated cells from the incubator.
- Perform a live cell stain. Dilute DRAQ5 1:1000 to a final concentration of 5 µM in the cell culture vessel. Gently mix the cells/staining solution and incubate at 37 °C for 5-30 min.
NOTE: A different stain can be implemented in this step. Ensure that the fluorescent properties do not overlap with the fluorescent marker indicating successful electro-transfection (i.e., GFP is in the green wavelength and DRAQ5 is the far-red). - Turn on an epifluorescent microscope, lamp, and cameras (see Table of Materials).
- Remove the cells from the incubator and bring them into focus on the microscope.
- Capture a phase-contrast image (brightfield) of the selected field.
- Capture epifluorescent images of the same field using FITC (GFP) and Far-Red (DRAQ5) filters. Analyze the image sets manually or via an algorithm.
NOTE: Refer to Figure 7 for representative images. - Count the total number of GFP-positive cells in all the images. Count the total number of DRAQ5 stained cells in all the images. Calculate eTE (ratio of GFP positive cells to DRAQ5 stained cells).
Results
Figure 4 highlights the operating principles behind the single-cell-level membrane permeabilization detection for a single pulse amplitude. Following the initiation of the electroporation experiment, the cell detection algorithm determines an optimal threshold for cell detection via a point-by-point, slope-based detection method. The system then continuously monitors (1) for a significant negative change in the measured electrical current, which is indicative of the entry of a cell. This is ...
Discussion
The methodology presented within this protocol primarily focuses on the microfabrication of a microfluidic device that is then integrated into a specialized electroporation experimental setup. The term 'recipe', which is often used when describing the specifics of the microfabrication process, hints at the importance of following/optimizing each step to successfully fabricate a functioning device. However, certain critical steps within the process, when not optimized, such as UV exposure time/energy, PVD sputteri...
Disclosures
The authors have nothing to disclose.
Acknowledgements
The authors would like to acknowledge financial support by the National Science Foundation (NSF CBET 0967598, DBI IDBR 1353918) and the U.S. Department of Education's Graduate Training in Emerging Areas of Precision and Personalized Medicine (P200A150131) for funding graduate student J.J.S. on fellowship.
Materials
| Name | Company | Catalog Number | Comments |
| 150-mm diameter petri dishes | VWR | 25384-326 | step 6.1.1 to secure wafer |
| 24-well tissue culture plates | VWR | 10062-896 | step 10.3.6 to plate electroporated cells |
| 33220A Waveform/Function generator | Agilent | step 9.2.3 electroporation pulse generator | |
| 4'' Si-wafers | University Wafer | subsection 2.1 for microfluidic channel fabrication | |
| 6-well tissue culture plates | VWR | 10062-892 | step 8.1.8 to plate cells |
| Acetone | Fisher Scientific | A18-4 | step 2.1.2 for cleaning and step 5.1 photoresist lift-off |
| Allegra X-22R Centrifuge | Beckman Coulter | steps 8.1.4 , 8.3.2. and 8.3.3. to spin down cells | |
| AutoCAD 2018 | Autodesk | subsection 1.1. to design transparency masks | |
| Buffered oxide etchant 10:1 | VWR | 901621-1L | subsection 3.1 for HF etching |
| CCD Monochrome microscope camera | Hamamatsu | Orca 285 C4742-96-12G04 | step 11.2.3. for imaging |
| CMOS camera- Sensicam QE 1.4MP | PCO | subsection 9.3 part of the experimental setup | |
| Conductive Epoxy | CircuitWorks | CW2400 | subsection 7.6. for wire attachement |
| Conical Centrifuge Tubes, 15 mL | Fisher Scientific | 14-959-70C | step 8.1.4. for cell centrifuging |
| Dektak 3ST Surface Profilometer | Veeco (Sloan/Dektak) | step 2.1.15 and 5.4 for surface profilometry | |
| Disposable biopsy punch, 0.75 mm | Robbins Instruments | RBP075 | step 6.2.3 for inlet access |
| Disposable biopsy punch, 3 mm | Robbins Instruments | RBP30P | step 6.2.3 for outlet access |
| DRAQ5 | abcam | ab108410 | step 11.2.2. for live cell staining |
| Dulbecco’s Modified Eagle’s Medium | ThermoFisher Scientific | 11885084 | step 8.1.2. part of media composition |
| E3631A Bipolar Triple DC power supply | Agilent | step 9.2.1.-9.2.2.part of the experimental setup | |
| Eclipse TE2000-U Inverted Microscope | Nikon | subsection 9.3. part of the experimental setup | |
| EVG620 UV Lithography System | EVG | step 2.1.9. and 2.2.7. for UV Exposure | |
| Fetal Bovine Serum | Neuromics | FBS001 | step 8.1.2. part of media composition |
| FS20 Ultrasonic Cleaner | Fisher Scientific | subsection 5.1. for photoresist lift-off | |
| Glass Media Bottle with Cap, 100mL | Fisher Scientific | FB800100 | step 8.2.1. for buffer storage |
| Glass Media Bottle with Cap, 500mL | Fisher Scientific | FB800500 | step 8.1.2.for media storage |
| HEK-293 cell line | ATCC | CRL-1573 | subsection 8.1 for cell culturing |
| HEPES buffer solution | Sigma Aldrich | 83264-100ML-F | step 8.2.1 part of electroporation buffer composition |
| Hexamethyldisilazane | Sigma Aldrich | 379212-25ML | step 2.2.3 adhesion promoter |
| HF2LI Lock-in Amplifier | Zurich Instruments | subsection 9.2 part of the experimental setup | |
| HF2TA Current amplifier | Zurich Instruments | subsection 9.2 part of the experimental setup | |
| Isopropyl Alcohol | Fisher Scientific | A459-1 | step 2.1.2 for cleaning, step 2.1.14 for rinsing wafer following SU-8 development, and step 6.3.1 for cleaning PDMS |
| IX81 fluorescence microscope | Olympus | step 11.2.3 for imaging | |
| L-Glutamine Solution | Sigma Aldrich | G7513-20ML | step 8.1.2. part of media composition |
| M16878/1BFA 22 gauge wire | AWC | B22-1 | subsection 7.5 for device fabrication |
| Magnesium chloride | Sigma Aldrich | 208337-100G | step 8.1.2 part of electroporation buffer composition |
| MF 319 Developer | Kayaku Advanced Materials | 10018042 | step 2.2.9. photoresist developer |
| Microposit S1818 photoresist | Kayaku Advanced Materials | 1136925 | step 2.2.4 positive photoresist for electrode patterning |
| Microscope slides, 75 x 25 mm | VWR | 16004-422 | step 2.2.1 electrode soda lime glass substrate |
| Model 2350 High voltage amplifier | TEGAM | 2350 | step 9.2.5. part of the experimental setup |
| National Instruments LabVIEW | National Instruments | data acquisition | |
| Needle, 30G x 1 in | BD Scientific | 305128 | step 10.1.1. part of the system priming |
| PA90 IC OPAMP Power circuit | Digi-key | 598-1330-ND | Part of the custom circuit |
| Penicillin-Streptomycin | Sigma Aldrich | P4458-20ML | step 8.1.2. part of media composition |
| Plasmid pMAX-GFP | Lonza | VCA-1003 | step 8.3.4. for intracellular delivery |
| Plastic tubing, 0.010'' x 0.030" | VWR | 89404-300 | step 10.1.2. for system priming |
| Platinum targets | Kurt J. Lesker | subsection 4.2. for physical vapor deposition | |
| Potassium chloride | Sigma Aldrich | P9333-500G | step 8.2.1. part of electroporation buffer composition |
| Pump 11 PicoPlus microfluidic syringe pump | Harvard Apparatus | MA1 70-2213 | step 10.1.4. for system priming |
| PVD75 Physical vapor deposition system | Kurt J. Lesker | subsection 4.1. for physical vapor deposition | |
| PWM32 Spinner System | Headway Research | steps 2.1.6 and 2.2.2. for substrate coating with photoresist | |
| PX-250 Plasma treatment system | March Instruments | subsection 7.2 for PDMS and glass substrate bonding | |
| SDG1025 Function/Waveform generator | Siglent | step 9.2.2. part of the experimental setup | |
| Sodium hydroxide | Sigma Aldrich | S8045-500G | step 8.2.1. part of electroporation buffer composition |
| SU-8 2010 negative photoresist | Kayaku Advanced Materials | Y111053 | step 2.1.7. for microfluidic channel patterning |
| SU-8 developer | Microchem | Y010200 | step 2.1.12. for photoresist developing |
| Sucrose | Sigma Aldrich | S7903-1KG | step 8.2.1. part of electroporation buffer composition |
| Sylgard 184 elastomer kit | Dow Corning | 3097358-1004 | step 6.2.1 10 : 1 mixture of PDMS polymer and hardening agent |
| Syringe, 1 ml | BD Scientific | 309628 | step 8.3.4. part of system priming |
| SZ61 Stereomicroscope System | Olympus | subsection 7.3. for channel and electrode alignment | |
| Tissue Culture Treated T25 Flasks | Falcon | 353108 | step 8.1.2 for cell culturing |
| Titanium targets | Kurt J. Lesker | subsection 4.2. for physical vapor deposition | |
| Transparency masks | CAD/ART Services | steps 2.1.9. and 2.2.7. for photolithography | |
| Trichloro(1H,1H,2H,2H-perfluorooctyl)silane | Sigma Aldrich | 448931-10G | step 6.1.2. for wafer silanization |
| Trypsin-EDTA solution | Sigma Aldrich | T4049-100ML | steps 8.1.3. and 8.3.1. for cell harvesting |
References
- Gao, Q. Q., et al. Therapeutic potential of CRISPR/Cas9 gene editing in engineered T-cell therapy. Cancer Medicine. 8 (9), 4254-4264 (2019).
- Aijaz, A., et al. Biomanufacturing for clinically advanced cell therapies. Nature Biomedical Engineering. 2 (6), 362-376 (2018).
- Milone, M. C., O'Doherty, U. Clinical use of lentiviral vectors. Leukemia. 32 (7), 1529-1541 (2018).
- Weaver, J. C., Chizmadzhev, Y. A. Theory of electroporation: A review. Bioelectrochemistry and Bioenergetics. 41 (2), 135-160 (1996).
- Kotnik, T., Rems, L., Tarek, M., Miklavcic, D. Membrane electroporation and electropermeabilization: mechanisms and models. Annual Review of Biophysics. 48, 63-91 (2019).
- Rosazza, C., Meglic, S. H., Zumbusch, A., Rols, M. P., Miklavcic, D. Gene electrotransfer: A mechanistic perspective. Current Gene Therapy. 16 (2), 98-129 (2016).
- Clauss, J., et al. Efficient non-viral T-cell engineering by sleeping beauty minicircles diminishing DNA toxicity and miRNAs silencing the endogenous T-cell receptors. Human Gene Therapy. 29 (5), 569-584 (2018).
- Sherba, J. J., et al. The effects of electroporation buffer composition on cell viability and electro-transfection efficiency. Scientific Reports. 10 (1), 3053 (2020).
- Lu, H., Schmidt, M. A., Jensen, K. F. A microfluidic electroporation device for cell lysis. Lab on a Chip. 5 (1), 23-29 (2005).
- Kar, S., et al. Single-cell electroporation: current trends, applications and future prospects. Journal of Micromechanics and Microengineering. 28 (12), (2018).
- Shi, J. F., et al. A review on electroporation-based intracellular delivery. Molecules. 23 (11), (2018).
- Wang, S. N., Zhang, X. L., Wang, W. X., Lee, L. J. Semicontinuous flow electroporation chip for high-throughput transfection on mammalian cells. Analytical Chemistry. 81 (11), 4414-4421 (2009).
- Wei, W. J., et al. An implantable microelectrode array for simultaneous L-glutamate and electrophysiological recordings in vivo. Microsystems & Nanoengineering. 1, (2015).
- Maschietto, M., Dal Maschio, M., Girardi, S., Vassanelli, S. In situ electroporation of mammalian cells through SiO2 thin film capacitive microelectrodes. Scientific Reports. 11 (1), (2021).
- Wu, Q. R., et al. Organ-on-a-chip: recent breakthroughs and future prospects. Biomedical Engineering Online. 19 (1), (2020).
- Pandey, C. M., et al. Microfluidics Based Point-of-Care Diagnostics. Biotechnology Journal. 13 (1), (2018).
- Vigneshvar, S., Sudhakumari, C. C., Senthilkumaran, B., Prakash, H. Recent advances in biosensor technology for potential applications - An overview. Frontiers in Bioengineering and Biotechnology. 4, (2016).
- Nuxoll, E. BioMEMS in drug delivery. Advanced Drug Delivery Reviews. 65 (11-12), 1611-1625 (2013).
- Kang, S., Kim, K. H., Kim, Y. C. A novel electroporation system for efficient molecular delivery into Chlamydomonas reinhardtii with a 3-dimensional microelectrode. Scientific Reports. 5, (2015).
- Zheng, M. D., Shan, J. W., Lin, H., Shreiber, D. I., Zahn, J. D. Hydrodynamically controlled cell rotation in an electroporation microchip to circumferentially deliver molecules into single cells. Microfluidics and Nanofluidics. 20 (1), (2016).
- Santra, T. S., Kar, S., Chang, H. Y., Tseng, F. G. Nano-localized single-cell nano-electroporation. Lab on a Chip. 20 (22), 4194-4204 (2020).
- Lee, W. G., Demirci, U., Khademhosseini, A. Microscale electroporation: challenges and perspectives for clinical applications. Integrative Biology. 1 (3), 242-251 (2009).
- Santra, T. S., Chang, H. Y., Wang, P. C., Tseng, F. G. Impact of pulse duration on localized single-cell nano-electroporation. Analyst. 139 (23), 6249-6258 (2014).
- Geng, T., Lu, C. Microfluidic electroporation for cellular analysis and delivery. Lab on a Chip. 13 (19), 3803-3821 (2013).
- Hsi, P., et al. Acoustophoretic rapid media exchange and continuous-flow electrotransfection of primary human T cells for applications in automated cellular therapy manufacturing. Lab on a Chip. 19 (18), 2978-2992 (2019).
- Khine, M., Ionescu-Zanetti, C., Blatz, A., Wang, L. P., Lee, L. P. Single-cell electroporation arrays with real-time monitoring and feedback control. Lab on a Chip. 7 (4), 457-462 (2007).
- Ye, Y. F., et al. Single-cell electroporation and real-time electrical monitoring on a microfluidic chip. 2020 33rd Ieee International Conference on Micro Electro Mechanical Systems (Mems 2020). , 1040-1043 (2020).
- Huang, Y., Rubinsky, B. Microfabricated electroporation chip for single cell membrane permeabilization. Sensors and Actuators a-Physical. 89 (3), 242-249 (2001).
- Guo, X. L., Zhu, R. Controllable in-situ cell electroporation with cell positioning and impedance monitoring using micro electrode array. Scientific Reports. 6, (2016).
- Punjiya, M., Nejad, H. R., Mathews, J., Levin, M., Sonkusale, S. A flow through device for simultaneous dielectrophoretic cell trapping and AC electroporation. Scientific Reports. 9, (2019).
- Wang, H. Y., Lu, C. Microfluidic electroporation for delivery of small molecules and genes into cells using a common DC power supply. Biotechnology and Bioengineering. 100 (3), 579-586 (2008).
- Zheng, M. D., et al. Continuous-flow, electrically-triggered, single cell-level electroporation. Technology. 5 (1), 31-41 (2017).
- Batista Napotnik, T., Miklavcic, D. In vitro electroporation detection methods - An overview. Bioelectrochemistry. 120, 166-182 (2018).
- MICROPOSIT™ S1800® G2 Series Photoresists. KAYAKU Available from: https://kayakuam.com/wp-content/uploads/2019/09/S1800-G2.pdf (2021)
- SU-8 2000 Permanent Negative Epoxy Photoresist. KAYAKU Available from: https://kayakuam.com/wp-content/uploads/2020/08/KAM-SU-8-2000-2000.5-2015-Datasheet-8.13.20-final.pdf (2001)
- Substrate Preparation. MicroChemicals Available from: https://www.microchemicals.com/technical_information/subtrate_cleaning_adhesion_photoresist.pdf (2021)
- Lisinenkova, M., Hahn, L., Schulz, J. . 4M 2006 - Second International Conference on Multi-Material Micro Manufacture. , 91-94 (2006).
- Beh, C. W., Zhou, W. Z., Wang, T. H. PDMS-glass bonding using grafted polymeric adhesive - alternative process flow for compatibility with patterned biological molecules. Lab on a Chip. 12 (20), 4120-4127 (2012).
- PA90 High Voltage Power Operational Amplifiers. APEX Available from: https://www.apexanalog.com/resources/products/pa90u.pdf (2021)
- Lissandrello, C. A., et al. High-throughput continuous-flow microfluidic electroporation of mRNA into primary human T cells for applications in cellular therapy manufacturing. Scientific Reports. 10 (1), 18045 (2020).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved