
Method Article
Chemical Triphosphorylation of Oligonucleotides
In This Article
Summary
Oligonucleotide 5′-triphosphates are ubiquitous components in essential biological pathways and have seen increasing use in biotechnology applications. Here, we describe techniques for the routine synthesis and purification of oligonucleotide 5′-triphosphates, starting from oligonucleotides prepared by standard automated synthesis techniques.
Abstract
The 5′-triphosphate is an essential nucleic acid modification found throughout all life and increasingly used as a functional modification of oligonucleotides in biotechnology and synthetic biology. Oligonucleotide 5′-triphosphates have historically been prepared in vitro by enzymatic methods. However, these methods are limited to natural RNA oligonucleotides, have strong sequence preferences, and tend to produce heterogeneous products. New methods of chemical triphosphorylation complement both the reduced cost of automated oligonucleotide synthesis by phosphoramidite chemistry and the diverse range of nucleotide modifications now available. Thus, the synthesis of oligonucleotide triphosphates of arbitrary sequence and length, and optionally containing various nonnatural modifications, is now accessible.
This paper presents the appropriate methods and techniques for chemical triphosphorylation of oligonucleotides using salicyl phosphorochloridite and pyrophosphate. This method uses commercially available reagents, is compatible with most oligonucleotides prepared by standard solid-phase synthesis methods, and can be completed in 2 h following oligonucleotide synthesis, before deprotection and purification. Two uses of chemically triphosphorylated oligonucleotides as substrates for catalytic RNA enzymes are demonstrated, including the synthesis of a mirror-image version of the hammerhead ribozyme from nonbiological L-RNA triphosphates.
Introduction
The 5′-triphosphorylated form of RNA is ubiquitous in biology as it is generated by RNA transcription in all domains of life and by RNA replication during the life cycle of many RNA viruses. These triphosphates serve as the substrate for the formation of 7-methylguanylate-capped mRNA in eukaryotes and, therefore, play an essential role in protein expression1. In contrast, the triphosphate is retained in bacteria and viruses; thus, RNA 5′-triphosphates are recognized by innate immunity response regulators in eukaryotes2,3,4,5,6,7. Outside biology, a host of RNA ligase ribozymes have been evolved to use the 5′-triphosphate in vitro8 and modified for use in diagnostic assays9,10,11,12,13,14,15. One such ribozyme can be used for template-dependent synthesis of L-RNA, the nonbiological "mirror-image" enantiomer of natural D-RNA, from small L-RNA oligonucleotide 5′-triphosphates16,17,18. The routine preparation of triphosphorylated oligonucleotides of varying sequence and backbone composition is essential to investigating these systems.
The most common and accessible method for preparing RNA 5′-triphosphates in the laboratory is by in vitro transcription. However, RNA produced by this method is restricted in sequence and size by promoter and substrate requirements of the RNA polymerase enzyme. T7 RNA polymerase and specialized derivatives are the most common polymerases used for this purpose19,20,21,22. In vitro transcribed RNA prepared with these enzymes must be initiated with a 5′-terminal purine and is strongly biased toward purines in the first 10 nucleotides23,24. Moreover, enzymatic incorporation of base- or backbone-modified nucleotides is at best inefficient and more often impossible with natural polymerases, limiting the opportunity to produce oligonucleotide 5′-triphosphates composed of anything but natural D-RNA. Another limiting factor is that RNA generated by in vitro transcription can contain substantial 5′- and 3′- heterogeneity and is produced as extremely heterogeneous products when shorter than 20 nt23,24,25,26,27.
In contrast, chemical triphosphorylation of oligonucleotides prepared by solid-phase phosphoramidite synthesis28,29,30,31,32,33,34,35 can be used to prepare oligonucleotide 5′-triphosphates 3-50 nt long, of any sequence. Additionally, a vast array of nucleic acid modifications accessible to phosphoramidite synthesis can be added to oligonucleotides prior to 5′-triphosphorylation14,15,16,17,18,29,36. Many of these methods use the phosphitylation reagent salicyl phosphorochloridite, which was developed by Ludwig and Eckstein for the solution-phase triphosphorylation of mononucleosides37. Triphosphorylation of oligonucleotides with this reagent is achieved on the solid phase by phosphitylation of the oligonucleotide 5′-hydroxyl, conversion to the triphosphate by reaction with pyrophosphate and oxidation, followed by standard procedures for cleavage of the oligonucleotide from the solid support, deprotection, and purification (Figure 1)28.

Figure 1: Scheme for triphosphorylation of synthetic oligonucleotides. In the first step, the oligonucleotide 5ʹ-hydroxyl is phosphitylated with SalPCl. In the next step, the 5ʹ-salicyl phosphite is reacted with TBAP to form the cyclic metaphosphite, then in the third step oxidized to generate the cyclic 5ʹ-trimetaphosphate in DNA/RNA synthesizer oxidation solution (0.1 M Iodine/pyridine/H2O/THF), which is rapidly hydrolyzed to yield the linear 5ʹ-triphosphate in the same solution28,33,37. Subsequent alkaline cleavage from the solid CPG support and deprotection of the oligonucleotide in aqueous MeNH2/ammonia will hydrolyze any residual cyclic trimetaphosphate to the linear form. Abbreviations: SalPCl = salicyl phosphorochloridite; TBAP = tributylammonium pyrophosphate; THF = tetrahydrofuran; CPG = controlled pore glass; MeNH2 = methylamine. Please click here to view a larger version of this figure.
{kind=link}
Although early published reports using this method often suffered from poor yields and undesired side products28,37,38, careful maintenance of anhydrous conditions is all that is necessary for routinely obtaining high yields. This can be achieved by careful preparation of reagents and the use of a simple reaction device assembled from standard plastic components. Here, we demonstrate the appropriate steps for chemical triphosphorylation of oligonucleotides, including preparation of reagents, assembly of the reaction chamber, the triphosphorylation reaction, and subsequent deprotection and purification of the triphosphorylated oligonucleotides. Also included is the representative use of 5′-triphosphorylated oligonucleotides as substrates for ligase ribozymes for the synthesis of larger nucleic acid products with natural D-RNA and abiotic L-RNA backbones.
Protocol
1. Automated solid-phase synthesis of 5′-hydroxyl oligonucleotides on a solid support
- Prepare the automated DNA/RNA synthesizer with reagents and phosphoramidites according to the target oligonucleotide composition and instrument instructions.
- Load a synthesis column containing solid-support onto the synthesizer and synthesize oligonucleotides according to the synthesizer instrument protocols.
NOTE: The triphosphorylation procedure has been optimized for oligonucleotides prepared at the 1 µmole scale. - Remove the 5′-dimethoxytrityl protecting group to yield the solid-supported oligonucleotide 5′-hydroxyl as part of the oligonucleotide synthesis in the previous step, or by performing a terminal detritylation step according to the synthesizer instrument protocols.
- Remove the column containing the 5′-hydroxyl oligonucleotide on solid support from the synthesizer, dry under a house vacuum for 10 min to remove residual solvent, and proceed to triphosphorylation (sections 3 and 4) once materials for triphosphorylation are prepared (section 2).
NOTE: If not used immediately, the dried column can be stored under a normal atmosphere in a sealed plastic container with desiccant at -20 °C. Further drying is not needed at this stage as the column is thoroughly dried prior to triphosphorylation in section 3.
2. Preparing materials for triphosphorylation
- Attach a dry argon source with adjustable pressure to a gas manifold with at least two lines and connect to a bubbler. Ensure that the lines terminate in 1 mL plastic syringes to facilitate connection to the reaction apparatus.
- Gather equipment to be used during triphosphorylation, including 1 mL plastic syringes, a three-way polypropylene stopcock, noncoring needles, 1.5 mL polypropylene tubes, and a small metal spatula. Store them in a sealed container or desiccator with desiccant at room temperature for at least 1 day prior to use.
- Prepare 30 mL each of anhydrous 1,4-dioxane, 3:1 dioxane:pyridine by volume, N,N-dimethylformamide (DMF), and acetonitrile (ACN) in dried 30 mL glass bottles with drying traps (4 Å molecular sieves in sealed membrane packets) at least 1 day prior to use. Seal with rubber septa, and store in a desiccator with desiccant.
- Store solid 2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one (salicyl phosphorochloridite, SalPCl) in its original container inside a sealed jar with desiccant at 4 °C. Always flush the container with argon between uses.
- Prepare a tributylammonium pyrophosphate (TBAP) solution at least 5 days in advance of the triphosphorylation reaction:
- Weigh 1-5 g of solid TBAP in a dried 30-mL glass bottle and dissolve in 1 mL of DMF and 0.5 mL of tributylamine per g of TBAP.
- Add three drying traps, seal the bottle with a rubber septum under argon, and bubble with argon for 30 min to degas.
- Store inside a sealed jar with desiccant at 4 °C for 5 days to allow the molecular sieves to absorb all trace water. Store the jar at -20 °C and prepare fresh after 6 months.
3. Assembling and using the triphosphorylation apparatus
- Allow the synthesis column to warm to room temperature if retrieving from storage at -20 °C.
- Assemble the reaction chamber shown in Figure 2:
- Prepare the antechamber: remove the plunger from a dry 1 mL syringe, cut off the top of the syringe using scissors or a razor blade, and attach the syringe to the synthesis column. Attach the three-way stopcock to the top of the syringe and attach the side inlet of the stopcock to the dry argon source with the bubbler, so that the top inlet of the stopcock is the reagent injection port.
- Secure this apparatus to a stand with clamps and seal all upstream joints with wax sealing film. Adjust the stopcock so that the injection port is closed, and the apparatus is open to the argon source. Close the bubbler, and allow argon at low pressure (<10 psi) to stream through the reaction chamber for 5 min.
NOTE: Multiple reaction chambers can be set in parallel to triphosphorylate 2-4 oligonucleotides. However, one line from the manifold should be reserved for supplying argon to the reagent bottles. - Reopen the bubbler and attach a syringe to the bottom of the synthesis column, which will be the waste syringe. Pull argon through the column repeatedly using the waste syringe; then, reattach the syringe with the plunger pushed fully in.
NOTE: Unless loading reagents, the stopcock should be set so that the injection port is closed and the apparatus open to the argon source, as shown in Figure 2A. Similarly, the waste syringe should be attached and the joint to the synthesis column sealed with wax sealing film unless actively removing reagents.
- To add a reagent or solvent:
- Attach a needle to the dry argon source and insert it into the reagent or solvent bottle septum, taking care not to immerse the needle in the bottle contents.
- Assemble a dry syringe with a needle and insert it into the reagent or solvent bottle septum, without immersing it into the bottle contents. Fill the syringe with argon, withdraw the needle from the septum, and expel the argon. Fill the syringe with argon and expel again; then, fill the syringe with the required volume of solvent or reagent under argon pressure.
- Adjust the stopcock on the apparatus so that the argon source is closed, and the injection port is open (Figure 2B). Quickly remove the filled syringe and needle from the source bottle, wipe away any solvent stuck to the side or tip of the needle, and insert the needle into the injection port. Expel the reagent into the antechamber of the apparatus, remove the needle, and close the injection port, reopening the apparatus to the argon source.
- Gently draw the liquid from the antechamber all the way through the synthesis column using the waste syringe so that all liquid is now held in the waste syringe. Now, slowly push the solution back up into the synthesis column, ensuring no gas bubbles are in the column. To mix or agitate, gently pull the solution up and down over the column with the waste syringe.
NOTE: Always move liquid slowly and gently through the reaction chamber to ensure that no seals are broken, which allows air to enter the apparatus.
- To remove a reagent or solvent from the column:
- Slowly pull the solution into the waste syringe. After the bulk of the solution has passed into the waste syringe, pull argon through to flush the remaining solvent from the column.
- Remove the wax sealing film around the waste syringe joint, then remove the syringe and discard the waste solution. Replace the waste syringe with a new, dry syringe, and reseal the joint with wax sealing film.

Figure 2: Triphosphorylation apparatus. During mixing or reactions, the device (A) is open to the argon source (i) and closed to the air by adjusting the three-way stopcock (ii). Reagents are drawn from the antechamber (iii) into the synthesis column (iv) by means of the waste syringe (v). Reagents are removed by drawing all liquid into the waste syringe (v) and discarding it. When loading reagents (B), the three-way stopcock (ii) is open to the atmosphere, and the reagent is loaded into the antechamber (iii) by means of a syringe and needle (vi). (C) A photograph of the assembled apparatus set as in (A) for reagent mixing and reacting. Please click here to view a larger version of this figure.
{kind=link}
4. On-column triphosphorylation of synthetic 5′-hydroxyl oligonucleotide
- Remove the SalPCl and TBAP solution from storage at -20 °C and allow them to warm to room temperature before use.
- Add 200 µL of pyridine/dioxane to the antechamber, according to steps 3.3.1-3.3.3. However, do not load solvent onto the synthesis column until step 4.4.
- Use a dry metal spatula to weigh 6-12 mg of SalPCl into a dry 1.5 mL microcentrifuge tube, and dissolve in 100 µL of dioxane by gently syringing the solvent up and down within the microcentrifuge tube.
- Add the dissolved SalPCl to the antechamber and load it onto the synthesis column, following step 3.3. Let it react for 15 min, agitating the solution every 5 min. Remove and discard the SalPCl solution according to step 3.4.
NOTE: SalPCl is added in large excess and will scavenge any water absorbed during preparation and loading into the reaction chamber. However, the introduction of any moisture during steps 4.5 and 4.6 will compromise the final oligonucleotide 5′-triphosphate yield. - Add 250 µL of TBAP solution to the antechamber and load it onto the synthesis column, following step 3.3. Let it react for 20 min, agitating every 5 min. Remove and discard the TBAP solution according to step 3.4.
- Wash the column with 0.5 mL of DMF, then 0.5 mL of ACN, removing the solvent after each addition according to steps 3.3 and 3.4.
- Add 250 µL of oxidizer solution (0.1 M iodine in tetrahydrofuran (THF)/pyridine/water, 88:10:2) to the antechamber and load it onto the synthesis column, following step 3.3. Let it react for 30 min, agitating every 10 min. Remove and discard the oxidizer solution according to step 3.4.
- Wash the column with 0.5 mL of ACN and remove, according to steps 3.3 and 3.4.
- Disassemble the reaction apparatus. Wash the synthesis column with 5 mL of ACN and dry the column.
5. Cleavage from solid support, deprotection, and purification
- Remove the dried solid support resin from the synthesis column and transfer it to a 1.5 mL polypropylene screw-cap sealable tube with a silicone o-ring.
- Suspend the resin in 1 mL of a 1:1 mixture of 28%-30% aqueous ammonia and 40% aqueous methylamine (AMA) and seal the tube tightly. Incubate at 65 °C for 10 min with intermittent mixing by gentle inversion39. Use a milder treatment at room temperature for 2 h for oligonucleotides longer than 40 nt.
CAUTION: Heating of the AMA solution will put the tube under high pressure. If the tube is not sealed tightly or does not use an ammonia-compatible (silicone) o-ring, the tube may vent gas or leak solvent, potentially compromising safety or the final product yield. Never open the tube while it is above ambient temperature, as hot AMA solution can violently evolve gas. - Chill the tube on ice and briefly centrifuge it (6,000-12,000 × g for 10 s). Remove the supernatant from the resin, filter through a syringe equipped with a 0.2 µm filter, and transfer to a new, sterile polypropylene tube. Evaporate the solution to dryness using a vacuum centrifuge fitted with an ammonia-neutralizing chemical trap.
NOTE: If the synthetic oligonucleotide does not contain any RNA nucleotides with 2′-silyl protecting groups, skip to step 5.8. - Remove silyl protecting groups by dissolving the dried material in 1 mL of 1 M tetrabutylammonium fluoride (TBAF) in THF, heating to 55 °C, shaking if necessary to fully dissolve the oligonucleotide, and incubating at room temperature for 16-24 h40,41.
- Quench the TBAF solution with 1 mL of 1 M Tris buffer, pH 7.5, and remove THF using a vacuum centrifuge.
- Remove TBAF salts using a disposable size exclusion column, following the manufacturer's instructions. For oligonucleotides shorter than 15 nt, confirm elution of the product by collecting the eluate in fractions, and identifying the main product fractions by absorbance at 260 nm on a UV-Vis spectrophotometer.
- Concentrate the deprotected RNA oligonucleotide by lyophilization or vacuum centrifuge if less than 15 nt, or by ethanol precipitation if greater than 15 nt.
- Prepare a preparative scale 10%-20% polyacrylamide/8 M urea/1x TBE gel using 19:1 mono:bis acrylamide stock, according to the appropriate protocols for oligonucleotide size, gel plate, and stand. Assemble the gel plate in the gel stand with 1x TBE in reservoirs and pre-run at 35 W (or as appropriate for gel plate format) for at least 30 min.
- Dissolve the solid oligonucleotide in urea gel loading buffer (8 M urea, 10% sucrose, 50 mM Tris, pH 8, 1 mM EDTA with bromophenol and xylene cyanol running dyes) and heat to 80 °C. Load on the polyacrylamide gel and run for 1-2 h at 25-35 W (or as appropriate for the gel plate format and oligonucleotide size).
NOTE: Bromophenol blue or xylene cyanol should be excluded from the gel loading buffer for oligonucleotides shorter than 15 nt if either co-migrates with the product, as it is difficult to remove these dyes from the eluted oligonucleotide without ethanol precipitation. - When gel electrophoresis is completed, remove the gel plate from the gel stand, disassemble the gel plate, and wrap the gel in polyvinylchloride film. Identify the product bands by back-shadowing with 254 nm UV light, and excise the major product band using a razor blade.
- Extract the oligonucleotide from the excised gel by the crush and soak method42.
- Crush the excised gel by extruding it through a plastic syringe or mechanically.
- For oligonucleotides longer than 15 nt:
- Elute in 3x volumes of crush and soak buffer (300 mM NaCl; 10 mM Tris, pH 8; 1 mM EDTA) for at least 12 h with agitation or shaking.
- Remove solid gel pieces by passing the solution through a syringe fitted with a 0.2 µm filter and concentrate the oligonucleotide by ethanol precipitation.
- For oligonucleotide shorter than 15 nt:
- Elute in 3x volumes of nuclease-free water for at least 12 h with agitation or shaking.
- Remove solid gel pieces by passing the solution through a syringe fitted with a 0.2 µm filter and concentrate by lyophilization.
- Remove residual salts and solutes using a disposable size exclusion column as in step 5.6. Concentrate by lyophilization.
- Store purified triphosphorylated oligonucleotides at -20 °C in TE buffer (10 mM Tris, pH 8; 1 mM EDTA) or in a similar oligonucleotide storage buffer.
- Determine the concentration of the oligonucleotide by measuring the absorbance at 260 nm using a UV-Vis spectrophotometer.
NOTE: The estimated extinction coefficient of the oligonucleotide should not be affected by its 5′-phosphorylation state, and can be calculated from its sequence using an oligonucleotide extinction coefficient calculator. - Verify triphosphorylation by mass spectrometry. Look for an expected mass of +239.94 Da relative to that of the 5′-hydroxyl oligonucleotide.
NOTE: Either matrix-assisted laser desorption/ionization or electrospray ionization mass spectrometry (MALDI-MS or ESI-MS, respectively) are suitable for identifying the triphosphorylation state of the oligonucleotide when using protocols optimized for nucleic acids. ESI-MS provides more consistent results, however, due to lower rates of ion fragmentation; a representative commercial service is provided in the Table of Materials.
- Determine the concentration of the oligonucleotide by measuring the absorbance at 260 nm using a UV-Vis spectrophotometer.
6. Triphosphorylated oligonucleotides as substrates for ribozyme self-replication
CAUTION: 32P is a radioactive isotope and the following steps should be performed using standard safety protocols for working with radioactive materials in a laboratory and by a researcher certified for use of radioactive materials by the relevant Environmental Health and Safety departments. As an alternative, self-replicating ribozyme substrate A can be prepared synthetically with a 5′-fluorescein label14 and imaged fluorescently, as in step 7.9.
- Prepare solution A as a mixture of self-replicator ribozyme E and 5′-32P-labeled RNA substrate A14 to concentrations of 0.30 µM and 30 µM, respectively. Prepare solutions B-transcribed and B-synthetic with 15 µM triphosphorylated RNA substrate B, prepared by in vitro transcription14 or chemical triphosphorylation as above, respectively, in 75 mM EPPS buffer, pH 8.5, and 37.5 MgCl2. Bring both solutions to 42 °C.
NOTE: All RNA components are listed in the Table of Materials. - To initiate self-replication, rapidly mix 5 µL of solution A with 10 µL of solution B-transcribed or B-synthetic to a final concentration of 0.1 µM E, 10 µM A, 10 µM B, 25 mM MgCl2, and 50 mM EPPS buffer, pH 8.5. Incubate the reaction mixture at 42 °C.
- At regular intervals, take 0.5 µL aliquots and quench in 9.5 µL of formamide gel loading buffer (95% formamide; 10 mM EDTA, pH 8).
- Prepare an analytical polyacrylamide/8 M urea/1x TBE gel, according to appropriate protocols for the gel plate and stand. Assemble the cast gel and plate in a gel stand, fill the reservoirs with 1x TBE, and prerun at 40 W (or as appropriate for different gel plates and stands) for 30 min.
- Heat the quenched reaction samples to 80 °C, load 5 µL of the sample in each well, and run the gel at 40 W for approximately 40 min (or as appropriate for different gel plates and stands).
- Remove the gel plate from the gel stand, disassemble, and wrap the gel in polyvinylchloride film. Cover the gel with a phosphor screen and expose for 1 h (or as appropriate for 32P cpm); scan the screen using a fluorescent/phosphorescent gel scanner.
- Quantify the reaction yield using gel image quantification software.
- Open the gel image using the Analysis Toolbox, choose Analysis | Shape Definition, select Areas | the rectangle shape, and draw rectangles of the same size around bands corresponding to unreacted A and product E for each time and both reactions.
- Choose Analysis | Background Subtraction, select Areas | the rectangle shape, and draw a rectangle of the same size in an empty portion of the gel image. Change the background subtraction method for all rectangles to Image Rectangle.
- Choose Window | 2 Area Window, then Edit | Export to Excel to export the quantified band pixel volumes to a spreadsheet file.
- Plot the concentration of product E versus time, and fit the data to the logistic growth Eq (1) using statistical data-fitting software:
[E] = (1)
(1)
Where a is reaction maximum extent, b is degree of sigmoidicity, and c is exponential growth rate.- In the exported spreadsheet, divide the product E volume by the sum of substrate A and product E volumes to determine the fractional yield of product for each time and both reactions. Multiply by the initial concentration of substrate A (10 µM) to determine the yield of product E as a function of time.
- In statistical data-fitting software, choose File | New | New Project File, select XY under New Table & Graph, and click Create. Enter the reaction times and product E yields for both reactions in adjacent columns, and label columns correspondingly (e.g., "time", "transcribed B", "synthetic B").
- Choose Insert | New Analysis, select XY analyses | Nonlinear regression (curve fit) then click OK. Choose Growth curves | Logistic growth and click OK; do not adjust any other parameters. Observe the fit parameters and confidence intervals under Results and the plot of data points and fitted curves under Graphs.
7. Cross-chiral copying of L-RNA
- Prepare 10 µL of RNA solution containing 20 µM D-RNA 27.3t cross-chiral polymerase, 2 µM 5′-fluorescein-labeled L-RNA primer, 4 µM biotinylated L-RNA hammerhead template, and 20 µM each of L-RNA pppCUG, pppAUG, pppAGG, and pppCGC, in 10 µL of 50 mM Tris, pH 8.3. Anneal the RNA by heating it to 90 °C for 1 min and cooling to 23 °C at 0.2 °C/s in a PCR thermocycler. See the Table of Materials for details about the polymerase, primer, and template.
- Incubate the RNA solution at 17 °C and start the reaction by adding 10 µL of 2x start buffer (400 mM MgCl2, 500 mM NaCl, and 50 mM Tris, pH 8.3). Ensure that the final concentrations of all components of the reaction are 10 µM polymerase, 1 µM primer, 2 µM template, 10 µM each trinucleotide 5′-triphosphate, 200 mM MgCl2, 250 mM NaCl, and 50 mM Tris, pH 8.3.
- As the reaction proceeds, take 10 µL aliquots and quench with 5 µL of 0.5 M EDTA, pH 8.
- To each quenched reaction sample, add 0.1 mg of streptavidin-coated magnetic beads (20 pmol biotin-oligonucleotide binding capacity) suspended in 10 µL of 1 M NaCl in TE buffer with 0.05% neutral detergent to capture trimer-extended primers via the biotinylated template and incubate for 30 min at room temperature with shaking.
- Capture the beads on a bead-capture magnet, remove and discard liquid, and add 50 µL of wash solution (250 mM NaCl in TE with 0.05% neutral detergent). Mix the beads, capture them again, and remove the wash solution. Repeat once more.
- To elute extended primers from beads, add 50 µL of elution solution (25 mM NaOH with 0.05% neutral detergent), and mix the beads. Recapture the beads, remove the elution solution, quench with 100 mM Tris (pH 7.5), and precipitate with ethanol.
- Prepare an analytical polyacrylamide/8 M urea/1x TBE gel, according to the appropriate protocols for the gel plate and stand. Assemble the cast gel and plate in a gel stand, fill the reservoirs with 1x TBE, and prerun at 40 W (or as appropriate for different gel plates and stands) for 30 min.
- Dissolve the precipitated RNA in 10 µL of formamide gel loading buffer, and prepare unreacted end-labeled primer at 0.5 µM in formamide gel loading buffer. Heat samples to 80 °C, load 5 µL of the sample in each well, and run the gel at 40 W for approximately 40 min (or as appropriate for different gel plates and stands).
- Remove the gel plate from the stand, and scan using a fluorescent/phosphorescent gel scanner to visualize cross-chiral L-RNA extension products.
Results
Oligonucleotides should be synthesized using standard protocols appropriate to the phosphoramidites and automated DNA/RNA synthesizer, leaving the product oligonucleotide uncleaved from the solid support in the original plastic synthesis column, with the 5ʹ-terminal dimethoxytrityl group removed to yield the free 5ʹ-hydroxyl (section 1). All oligonucleotides used in this demonstration were prepared using 1,000 Å controlled pore glass (CPG) resin as the solid support and conducted at the 0.2 or 1 µmole scale. Representative examples of synthesizer columns, resins, reagents, and phosphoramidites are provided in the Table of Materials. For larger-scale reactions, volumes and times used in subsequent steps may need to be adjusted.
The triphosphorylation reaction is conducted on-column in a custom-built reaction chamber (Figure 2, section 3) using standard, commercially available components listed in the Table of Materials and follows the scheme illustrated in Figure 1 (section 4)28. It is essential that conditions be kept strictly anhydrous during triphosphorylation, and that all solvents and reagents be prepared over molecular sieves in advance and allowed to fully dry before use (section 2). Triphosphorylation typically takes 2 h to occur, and afterward, the washed and dried column can be treated according to standard oligonucleotide deprotection and purification procedures (section 5).
After deprotection, oligonucleotide triphosphates are purified by denaturing polyacrylamide gel electrophoresis (PAGE), showing a single major product band by UV back-shadowing that can be excised and eluted from the gel. The 5′-triphosphate product is readily separated from reaction side products for short oligonucleotides, as shown for DNA trinucleotide 5ʹ-triphosphates, pppAAA and pppCCC, and L-RNA trinucleotide 5ʹ-triphosphate pppGAA in Figure 3A,B. Both the 5′-hydroxyl and 5′-triphosphate products for AAA and CCC DNA trimers were excised and identified by mass spectrometry and correspondingly labeled in Figure 3A. Additional bands, as visible for the AAA DNA trimer, generally do not contain enough material to recover and identify. The presence of these bands, however, correlates with additional product masses in the unpurified reaction products (Figure 3C), typically representing 5′-diphosphate, monophosphate, and H-phosphonate side products, as discussed below.
After PAGE purification, larger oligonucleotides can be eluted using the crush and soak method42 and subsequent ethanol precipitation. However, oligonucleotides less than 15 nt cannot be ethanol precipitated efficiently and, thus, require a modified procedure for gel elution (step 5.11.3). The disposable size exclusion column listed in the Table of Materials is rated only for use with oligonucleotides longer than 10 nt. However, we have found that oligonucleotides as short as trimers can be effectively desalted using the manufacturer's recommended protocol. Nevertheless, it is recommended when desalting short oligonucleotides (as in steps 5.6 and 5.11.3) that the column eluate be collected in fractions, and product fractions be identified by absorbance at 260 nm using a UV-Vis spectrophotometer. A size exclusion column optimized for shorter oligonucleotides is provided in the Table of Materials as an alternative choice. The final yield from 1 µmole scale oligonucleotide synthesis after purification is 50-300 nmol.
Triphosphorylation can be confirmed by mass spectrometry, where the triphosphorylated product has a mass +239.94 Da greater than the 5′-hydroxyl oligonucleotide, although the presence of materials corresponding to the 5′-di- and monophosphate (+159.96 and +79.98 Da, respectively) are often observed. A 5′-H-phosphonate side product with a mass +63.98 Da from the 5′-OH mass may also be observed, and high levels of this product indicate conditions during triphosphorylation were not sufficiently anhydrous. Prior to purification, deprotected oligonucleotides will typically show all these products (Figure 3C), while purified material will show a peak corresponding to the 5′-triphosphate product along with 5′-di- and monophosphates (Figure 3D,E).
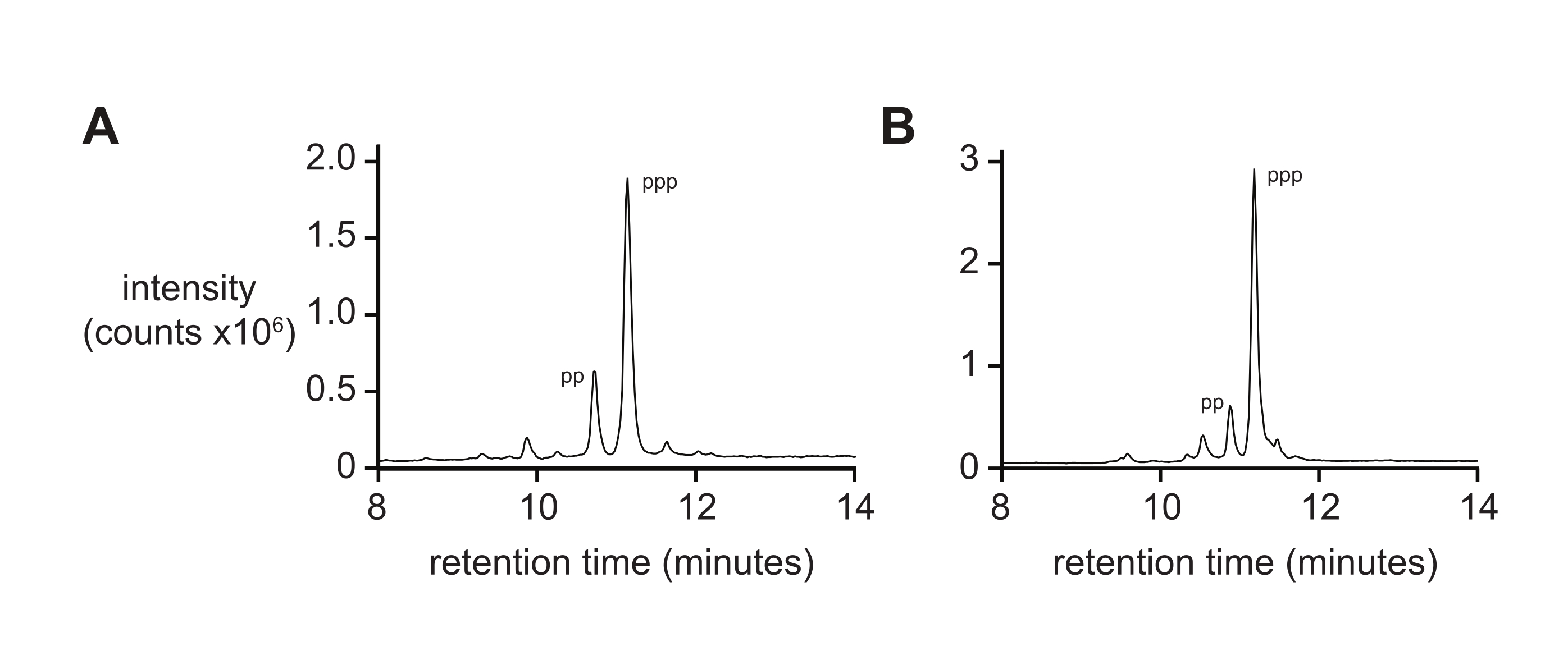
Mass spectrometry alone will typically not give a rigorous measure of 5′-triphosphate purity due to differential rates of ionization and fragmentation of the triphosphate during ionization. To measure final product purity, reverse-phase liquid chromatography and tandem ESI-MS (RP-LC/ESI-MS) are recommended, particularly for longer oligonucleotides. Analysis of D-RNA 5ʹ-triphosphates pppACGAGG and pppGAGACCGCAACUUA by RP-LC/ESI-MS (Figure 4A,B, respectively) show typical final product purity, containing 20% 5ʹ-diphosphate as these two species are difficult to separate when present on longer oligonucleotides.
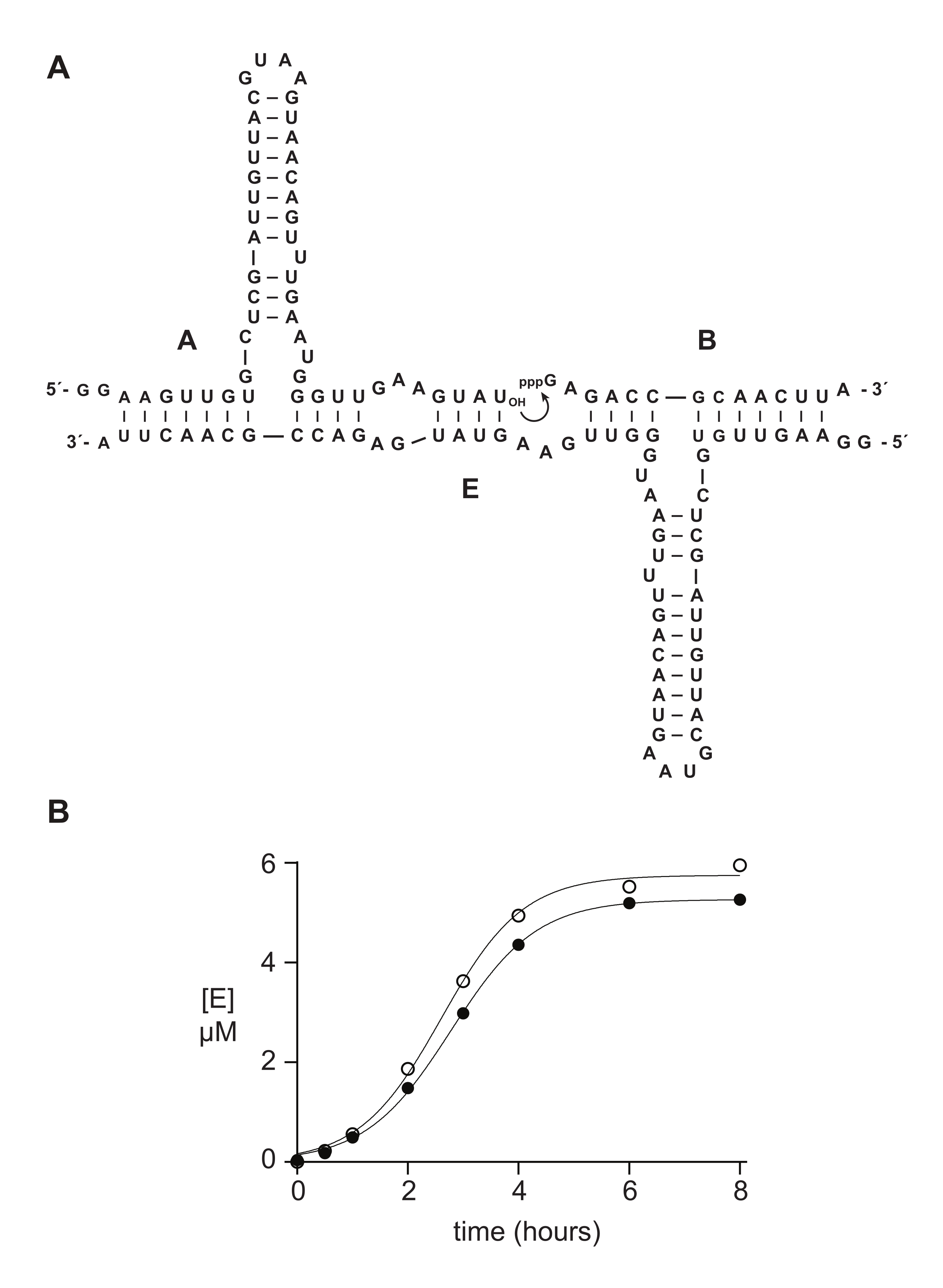
Synthetic 5′-triphosphate oligonucleotides typically function as well or better than materials prepared enzymatically in biochemical studies. In section 6, as an example, 5′-triphosphate 14 nt RNA substrates prepared either synthetically or by in vitro transcription were compared in an RNA-catalyzed self-replication reaction14,15,43,44,45. Ribozyme E catalyzes the joining of substrates A and B to yield a new copy of E in an autocatalytic reaction capable of exponential growth (Figure 5A). E and 32P-labeled A components were prepared by in vitro transcription, and triphosphorylated substrate B was prepared either synthetically, as described above, or by in vitro transcription14. Self-replication reaction progress was monitored by taking periodic samples that were analyzed by denaturing PAGE and quantified via a fluorescent/phosphorescent gel scanner. The resulting data, fit to a logistic growth function, revealed that either transcribed or synthetic B substrate supports exponential growth, but synthetic B gives a slightly greater amount of product (Figure 5B). This result may reflect compositional heterogeneity at the 5′-end of RNA prepared by in vitro transcription23,24.
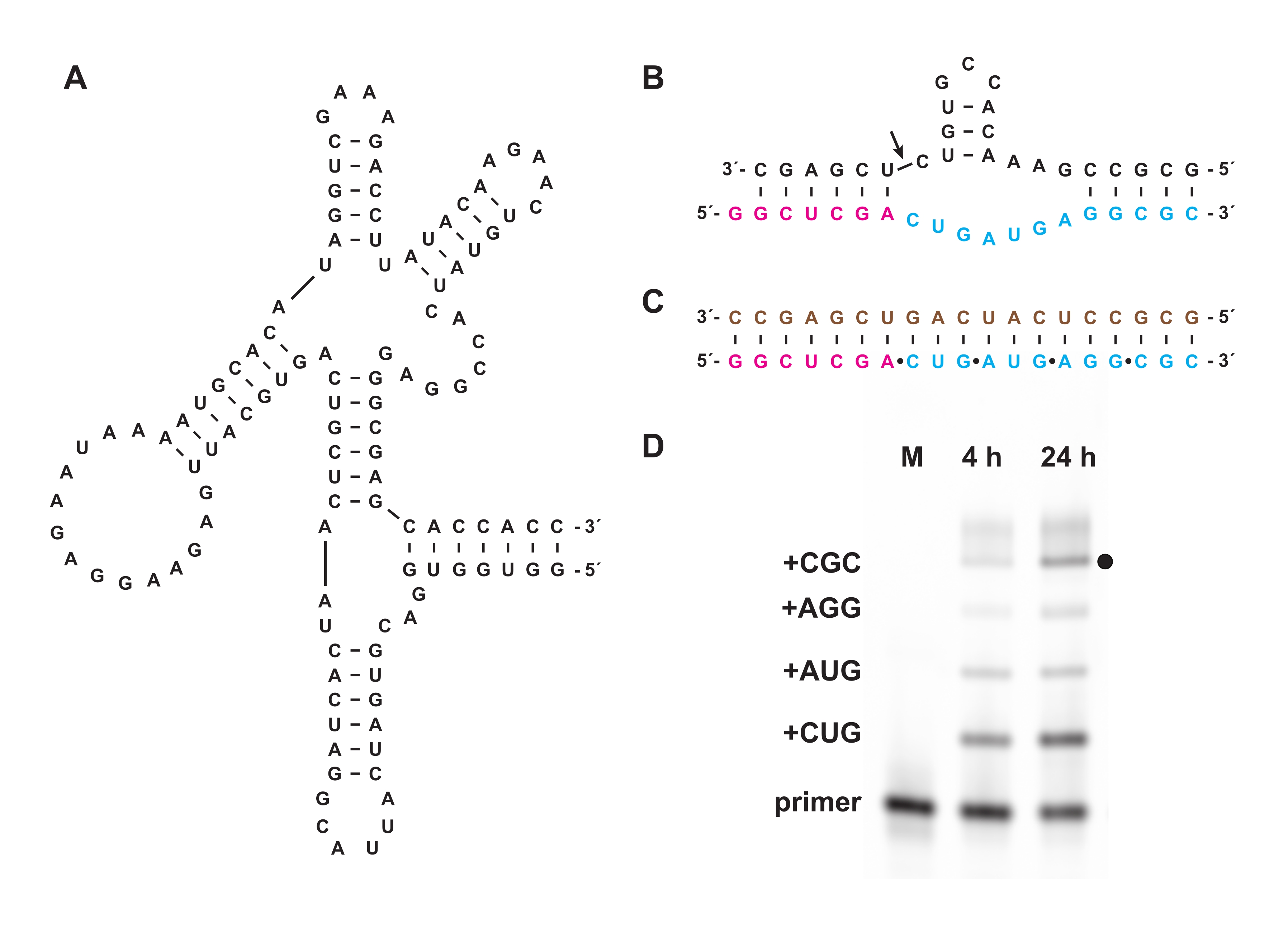
Chemical triphosphorylation also enables the synthesis of oligonucleotide triphosphates that cannot be prepared biologically, either in vitro or in cells. In section 7, nonbiological oligonucleotide triphosphates composed of L-RNA, the enantiomer of natural D-RNA, prepared as in sections 1-5, were used as substrates for the D-RNA "cross-chiral" polymerase ribozyme 27.3t (Figure 6A), which catalyzes the template-directed polymerization of a longer L-RNA product from short L-RNA oligonucleotide 5′-triphosphates in a sequence-general manner. As an example, the ribozyme can synthesize an L-RNA version of the hammerhead self-cleavage motif (Figure 6B)18. Purified L-RNA trinucleotide triphosphates were combined with a fluorescein-labeled L-RNA primer and L-RNA template (Figure 6C) and reacted with the cross-chiral ligase. Samples over the course of the reaction were analyzed by PAGE and imaged using a fluorescent/phosphorescent gel scanner to demonstrate synthesis of an L-RNA version of the hammerhead ribozyme encoded by the template (Figure 6D).

Figure 3: Purification of trinucleotide 5ʹ-triphosphates. (A) PAGE analysis (visualized by UV-back-shadowing) of triphosphorylation of DNA trinucleotides tri-deoxyadenosine (AAA, blue) and tri-deoxycytidine (CCC, red), intentionally overloaded to visualize minor side-products. Both the 5ʹ-triphosphate product (ppp) and 5ʹ-hydroxyl (OH) starting material were excised and identified by MALDI-MS. (B) Preparative PAGE of triphosphorylation of L-RNA trinucleotide GAA, with major product band excised and identified as the 5ʹ-triphosphate (ppp) by ESI-MS. (C) MALDI-MS of crude reaction products after deprotection and (D) purified products from (A). 5ʹ-triphosphate (ppp; pppAAA expected 1,119 Da, observed 1,118 Da; pppCCC expected 1,047 Da, observed 1,046); 5ʹ-diphosphate (pp), 5ʹ-monophosphate (p), 5ʹ-hydroxyl (OH), and 5ʹ-H-phosphonate (Hp) are labeled. (E) Deconvoluted mass spectrum from direct injection ESI-MS of isolated 5ʹ-triphosphate product from (B), with identified peaks labeled (expected 1,181.6 Da, observed 1,181.0 Da). 5ʹ-diphosphate (pp) products are also observed, as are sodium ion peaks for both the tri- and di-phosphate products (+22 Da). Common contaminant peaks are labeled with an asterisk. For ease of comparison, mass spectra were normalized to the highest intensity measured in each spectrum and are reported as a percentage relative to that value. Abbreviations: PAGE = polyacrylamide gel electrophoresis; MALDI-MS = matrix-assisted laser desorption/ionization; ESI-MS = electrospray ionization mass spectrometry. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Analytical RP-LC of 6 nt and 14 nt D-RNA oligonucleotide triphosphates. (A) 5ʹ-pppACGAGG-3ʹ and (B) 5ʹ-pppGAGACCGCAACUUA-3ʹ. Tandem ESI-MS identified the major peak of both (~70%) as the 5ʹ-triphosphate (ppp), with lesser amounts of the 5ʹ-diphosphate (pp). Abbreviations: RP-LC = reverse-phase liquid chromatography; nt = nucleotides; ESI-MS = electrospray ionization mass spectrometry. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Comparison of oligonucleotide 5ʹ-triphosphate substrates prepared by chemical synthesis or in vitro transcription. (A) The self-replicating ribozyme E ligates RNA A and 5′-triphosphorylated RNA B. (B) Comparison of self-replication reactions using 10 µM A and 10 µM B, either synthetic (open circles) or in vitro transcribed (filled circles). (B) Data were fit to the logistic growth equation: [E] = a / (1 + be-ct), where a is the final yield, b is the degree of sigmoidicity, and c is the exponential growth rate. Growth rates for the two reactions were identical, at 1.14 h-1, while the final extent was 10% higher for reactions with synthetic B. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Cross-chiral L-RNA polymerization with a ribozyme. (A) The D-RNA 27.3t polymerase ribozyme, which catalyzes template-dependent ligation of L-RNA. (B) The L-RNA product synthesized by 27.3t forms part of a hammerhead endonuclease motif. (C) L-RNA polymerization catalyzed by 27.3t using a biotinylated L-RNA template (brown), an end-labeled L-RNA primer (magenta), and four L-RNA trinucleotide triphosphates (cyan), prepared synthetically. (D) PAGE analysis of extension products of (B) at 4 h and 24 h, showing each trinucleotide incorporation up to full-length product (black dot). Unreacted L-RNA primer is included as a reference marker. Abbreviations: PAGE = polyacrylamide gel electrophoresis; M = reference marker. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The triphosphorylation procedure described here is broadly compatible with oligonucleotide synthesis using standard phosphoramidite chemistry. Nucleoside phosphoramidites should have base-labile protecting groups compatible with rapid deprotection in AMA39, including the standard β-cyanoethyl on the phosphite, and isobutyryl, dimethylformamidyl, acetyl, phenoxyacetyl, or 4-isopropylphenoxyacetyl groups on the exocyclic amines of the nucleobases. Ribose 2'-hydroxyl groups should be protected by silyl protecting groups, either t-butyldimethylsilyl (TBDMS) or tri-iso-propylsilyloxymethyl (TOM)40,41. The base-labile pivaloyloxymethyl (PivOM) group also has been reported to be compatible with chemical triphosphorylation30.
Multiple methods have been described for the chemical triphosphorylation of synthetic oligonucleotides28,29,30,31,32,33,34,35. We have found triphosphorylation using the Ludwig-Eckstein reagent37 to be one of the most accessible, requiring no specialized synthesis of reagents and no specialized equipment. Oligonucleotide 5′-triphosphates prepared by this method have been used routinely as substrates for RNA ligase ribozymes, including the use of enzymatically inaccessible L-RNA oligonucleotide triphosphates to achieve template-dependent synthesis and replication of this "mirror-image" nucleic acid14,16,17,18. The method is also suitable for the preparation of small 5′-triphosphorylated stem-loop RNAs that are potent activators of the innate immune response in vertebrates6,7.
The Ludwig-Eckstein reagent, salicyl phosphorochloridite37, is highly reactive to water, and effectively scavenges any water introduced when dissolving the reagent prior to loading onto the oligonucleotide column. After this point, however, the 5′-phosphitylated oligonucleotide will preferentially react with any water introduced into the system over pyrophosphate, forming a 5′-H-phosphonate side product after workup28,37,38. Careful preparation of triphosphorylation reagents and the triphosphorylation reaction chamber ensure this side product is not formed. For solvent drying, type 4 Å molecular sieves are sold prepackaged in Teflon bags compatible with most organic solvents under various brand names by most oligonucleotide synthesis reagent companies. Additional precautions, such as performing triphosphorylation in a glove box under an anhydrous atmosphere, are generally not necessary.
Reaction of the 5′-phosphitylated oligonucleotide with TBAP forms a cyclic 5′-trimetaphosphite intermediate, which is then oxidized to the cyclic 5′-trimetaphosphate using oligonucleotide synthesis oxidizer solution (iodine in water/pyridine/THF). It should be noted that commercial oxidizer solutions use varying amounts of iodine, and it is essential to use the high 0.1 M iodine concentration to ensure complete oxidation to the triphosphate. The cyclic product is hydrolyzed to the final linear 5′-triphosphate in the same solution37, and alternative, anhydrous oxidizer solutions must be used if linearization with nucleophiles other than water is desired (see below for applications)33. Any residual cyclic trimetaphosphate, however, will be linearized during subsequent alkaline deprotection of the oligonucleotide. Hydrolysis of the cyclic 5′-trimetaphosphate yields only the linear, rather than branched triphosphate37,46.
Oligonucleotide deprotection typically does not need to be modified to accommodate 5′-triphosphorylation, but a few precautions should be taken. The triphosphate is relatively stable to brief exposure to alkaline conditions, but care should be taken not to expose the triphosphate to AMA longer than necessary. Protecting groups that require more prolonged treatment in ammonia or AMA for more than 10 min at 65 °C should be avoided. More gentle treatments, such as 2 h in ammonia at room temperature are acceptable when compatible with other phosphoramidite protecting groups. A common, fast deprotection method for silyl-protected synthetic RNA oligonucleotides uses triethylamine trihydrofluoride and high temperature47; however, this should be avoided when preparing RNA 5′-triphosphates as the prolonged acidic conditions are found to accelerate triphosphate hydrolysis31,32.
Preparative PAGE has proven to be the simplest and most reliable method for postdeprotection purification of 5′-triphosphorylated oligonucleotides (Figure 3 and Figure 4). However, preparative reverse-phase HPLC can also be used to purify triphosphorylated products. The presence of 5′-diphosphate and, to a lesser extent, 5′-monophosphate products is routinely observed when verifying triphosphorylation by mass spectrometry. We have observed 5′-triphosphate fragmentation during mass spectrometry from highly pure material prepared by chemical synthesis or transcription, particularly if the instrument is not optimized for analysis of oligonucleotides. Nevertheless, RP-LC analysis often shows 10%-20% of the 5′-diphosphate side product is present in longer 5′-triphosphorylated oligonucleotides (Figure 4). Commercial preparations of tributylammonium pyrophosphate can be contaminated with as much as 20% monophosphate, which will yield 5′-diphosphate as a side product during triphosphorylation30,31. Careful preparation of this reagent in-house can yield much more pure TBAP stocks31. However, we have found oligonucleotides triphosphorylated using commercial sources of TBAP still show comparable or greater reactivity when used as substrates in enzymatic reactions (Figure 5B), compared to material prepared by in vitro transcription.
One notable further use of oligonucleotide triphosphorylation with the Ludwig-Eckstein reagent takes advantage of the cyclic trimetaphosphite intermediate33. If the subsequent oxidation step is conducted with 1 M t-butyl peroxide in hexanes, which is often used for oligonucleotide oxidation under anhydrous conditions, oxidation of the phosphite occurs without ring opening hydrolysis, yielding the cyclic trimetaphosphate. This intermediate can then be reacted with primary amine or alcohol nucleophiles to yield 5′-triphosphates with modifications at the γ-phosphate. These modifications include the addition of a lipophilic tag linked by a phosphoramidate bond, which facilitates rapid triphosphate-specific purification by RP-LC, followed by acidic hydrolysis of the tag from the triphosphate33. Fluorescent modifications at the γ-phosphate position can also be introduced for use as real-time fluorescent reporters for ribozyme-catalyzed ligation reactions15,33.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgements
The authors are grateful to Greg Springsteen, Natasha Paul, Charles Olea, Jr., Jonathan Sczepanski, and Katrina Tjhung for useful discussions on best practices for chemical triphosphorylation reactions and to Gerald Joyce for helpful comments. This work was supported by grant MCB 2114588 from the National Science Foundation.
Materials
| Name | Company | Catalog Number | Comments |
| 0.22 µm polyethersulfone syringe filter | MilliporeSigma | SLMP025SS | Syringe filter for removing crushed polyacrylamide gel particles (Section 5) |
| 0.22 µm PTFE syringe filter | MilliporeSigma | SLLG013SL | Syringe filter for removing CPG resin (Section 5) |
| 1 mL plastic syringes | ThermoFisher Scientific | 14-823-434 (BD 309659) | Components of triphosphorylation apparatus (sections 2–4) |
| 1,4-Dioxane, anhydrous | MilliporeSigma | 296309 | Triphosphorylation solvent (sections 2–4) |
| 2-Chloro-4H-1,3,2-benzodioxaphosphorin-4-one, Salicyl Phosphorochloridite (SalPCl) | MilliporeSigma | 324124 | Triphosphorylation reagent (sections 2–4) |
| 30 mL glass bottles | MilliporeSigma | 23232 | Bottles for preparing triphosphorylation solvents and TBAP solution (section 2) |
| 3-way Stopcock, polycarbonate/polypropylene | Bio-Rad Laboratories | 7328103 | Component of triphosphorylation apparatus (sections 2–4) |
| 40% acrylamide/bis-acrylamide solution, 19:1 | Bio-Rad Laboratories | 1610144 | For PAGE (sections 5–7) |
| Acetonitrile, anhydrous, 100 mL | Glen Research | 40-4050-50 | Triphosphorylation solvent (sections 2–4) |
| Ammonia-neutralizing Trap | ThermoFisher Scientific | ANT100 and ANS121 | For use with Speedvac DNA130 (section 5) |
| Ammonium persulfate (APS) | Bio-Rad Laboratories | 1610700 | For PAGE, catalyst for acrylamide polymerization (sections 5–7) |
| Aqueous ammonia, 28% | MilliporeSigma | 338818 | For preparing AMA deprotection reagent (section 5) |
| Aqueous methylamine, 40% | TCI America | TCI-M0137 | For preparing AMA deprotection reagent (section 5) |
| Automated DNA/RNA oligonucleotide synthesizer | PerSeptive Biosystems | Expedite 8909 DNA/RNA Synthesizer | any column-based synthesizer is acceptable (section 1) |
| Bead-capture magnet | ThermoFisher Scientific | 12320D | For streptavidin bead capture (section 7) |
| Bromophenol blue | Bio-Rad Laboratories | 1610404 | For PAGE urea loading buffer (section 5) |
| Deep vacuum oil pump | ThermoFisher Scientific | VLP200-115 | For use with lyophilizer (section 5) |
| Drierite dessicant, 10-20 mesh | MilliporeSigma | 737828 | Desiccant for storing triphosphorylation chemicals and equipment (sections 1–2) |
| D-RNA 27.3t cross-chiral polymerase | prepared in house18 | 5′-GGUGGUGGAC GUGAUCAUUA CGGAUCACUA ACUCGUCAGU GCAUUGAGAA GGAGAAUAAA AUGCACAUAG GUCGAAAGAC CUUAUACAAG AACUGUAUCA CCGGAGGGCG AGCACCACC-3′ | For cross-chiral ribozyme reactions (section 7) |
| D-RNA CPG solid supports, 1,000Å, prepackaged 1 µmole synthesis columns | Glen Research | 20-3404-41E, 20-3415-41E, 20-3424-41E, 20-3430-41E | representative, for D-RNA oligonucleotide synthesis (section 1) |
| D-RNA TOM-protected phosphoramidites | ChemGenes | ANP-3201, 3202, 3203, 3205 | representative, for D-RNA oligonucleotide synthesis (section 1) |
| Empty Expedite Synthesis Columns, 1µm | Glen Research | 20-0021-01 | Synthesis columns for use with Expedite DNA/RNA synthesizer (section 1) |
| EPPS, N-(2-Hydroxyethyl)piperazine-N′-(3-propanesulfonic acid), solid | MilliporeSigma | E1894 | Ribozyme reaction buffer component (section 6) |
| Ethylenediaminetetraacetic acid (EDTA), solid | MilliporeSigma | EDS | Divalent metal ion chelator for use in various buffers (sections 5–7) |
| Filters for Expedite synthesis columns | Glen Research | 20-0021-0F | Expedite-style synthesis column filters, for use with empty synthesis columns (section 1) |
| Fluorescent/phosphorescent gel scanner | Cytiva | Amersham Typhoon RGB, 29187193 | For visualizing analytical PAGE (sections 6–7) |
| Formamide, deionized | VWR Life Science | 97062 | For PAGE formamide gel loading buffer (sections 6–7) |
| Gel image quantitation software | Cytiva | ImageQuant TL | For quantifying scanned gel images (section 6) |
| Glass desiccator | MilliporeSigma | CLS3121150 | Triphosphorylation solvent storage (section 2) |
| L-RNA CPG solid supports, 1,000Å, bulk | ChemGenes | N-4691-10, N-4692-10, N-4693-10, N-4694-10 | L-RNA oligonucleotide synthesis (section 1) |
| L-RNA hammerhead template | prepared in house18 | 5′-GCGCCUCAUC AGUCGAGCC-3′ | For cross-chiral ribozyme reactions (section 7) |
| L-RNA primer | prepared in house18 | 5′-fluorescein-GGCUCGA-3′ | For cross-chiral ribozyme reactions (section 7) |
| L-RNA TOM-protected phosphoramidites | ChemGenes | OP ANP-5201, 5202, 5203, 5205 | L-RNA oligonucleotide synthesis (section 1) |
| Lyophilizer/Freeze Dryer | VirTis | Benchtop K | For concentrating oligonucleotides (section 5) |
| Magnesium Chloride Hexahydrate, solid | MilliporeSigma | M2670 | For ribozyme reactions (sections 6–7) |
| N,N-Dimethylformamide, anhydrous | MilliporeSigma | 227056 | Triphosphorylation solvent (section 2) |
| NAP-25 Desalting column (Sephadex G-25 resin) | ThermoFisher Scientific | 45000150 | Disposable gravity-flow size exclusion chromatography columns containing Sephadex G-25 resin (section 5) |
| Non-coring stainless steel needle, 20 G | ThermoFisher Scientific | 14-815-410 | Needles for piercing rubber septa (sections 2–4) |
| Oligonucleotide extinction coefficient calculator | Integrated DNA Technologies | OligoAnalyzer Tool | Nearest-Neighbor Model Short Oligonucleotide 260nm extinction coefficient calculator (section 5) |
| Oxidizer solution, 0.1 M Iodine in THF/pyridine/water | ChemGenes | RN-1456 | Triphosphorylation reagent (section 4) |
| PAGE plates | Timberrock/CBS | NGP-250-BO and NO | For PAGE (sections 5–7) |
| PAGE power supply | Bio-Rad Laboratories | PowerPac HV 1645056 | For PAGE (sections 5–7) |
| PAGE spacers and combs (analytical) | Timberrock/CBS | VGS-0725 and VGC-0714 | For PAGE (sections 6–7) |
| PAGE spacers and combs (preparative) | Timberrock/CBS | VGS-3025R and VGC-3001 | For PAGE (section 5) |
| PAGE stand | Timberrock/CBS | ASG-250 | For PAGE (sections 5–7) |
| Parafilm M | ThermoFisher Scientific | 13-374-12 (Bemis PM999) | Wax sealing film for triphosphorylation apparatus (sections 2–4) |
| PCR thermocycler | Bio-Rad Laboratories | C1000 Touch Thermalcycler | For cross-chiral ribozyme reactions (section 7) |
| PD 10 Desalting column (Sephadex G-10 resin) | MilliporeSigma | GE17-0010-01 | Disposable gravity-flow size exclusion chromatography columns containing Sephadex G-10 resin, for oligonucleotides < 15 nt (section 5) |
| Phosphor screens | Cytiva | 28956480 | For visualizing 32P-labeled RNA (section 6) |
| Phosphoramidite synthesis reagents | Glen Research | 30-3142-52, 40-4050-53, 40-4012-52, 40-4122-52, 40-4132-52, 40-4060-62 | representative, for standard RNA/DNA synthesis (section 1) |
| Polypropylene screw-cap sealable tube | MilliporeSigma | BR780752 | 1.5 mL microcentrifuge tubes with screw-cap and silicone O-ring, for safe AMA deprotection (section 5) |
| Pyridine, anhydrous | MilliporeSigma | 270970 | Triphosphorylation solvent (section 2) |
| Reverse-phase liquid chromatography/electrospray ionization mass spectrometry (RP-LC/ESI-MS) | Novatia | n/a | Commercial service for LC/MS specializing in oligonucleotides (section 5) |
| Rubber Septa (ID x OD 7.9 mm x 14 mm), white | MilliporeSigma | Z564702 | Septa for preparing triphosphorylation solvents and TBAP (section 2) |
| Self-replicator ribozyme E | prepared in house14 | 5′-GGAAGUUGUG CUCGAUUGUU ACGUAAGUAA CAGUUUGAAU GGUUGAAGUA UGAGACCGCA ACUUA-3′ | For self-replicator ribozyme reactions (section 6) |
| Self-replicator substrate A | prepared in house14 | 5′-32P-GGAAGUUGUG CUCGAUUGUU ACGUAAGUAA CAGUUUGAAU GGUUGAAGUA U-3′-OH | For self-replicator ribozyme reactions (section 6) |
| Self-replicator substrate B, transcribed | prepared in house14 | 5′-pppGAGACCGCAA CUUA-3′ | For self-replicator ribozyme reactions (section 6) |
| Small Drying Traps, 4 Å molecular sieves | ChemGenes | DMT-1975 | Drying traps for DNA/RNA synthesizer phosphoramidites and triphosphorylation reagents (sections 1–2) |
| Sodium Chloride (NaCl), solid | MilliporeSigma | S7653 | Salt for use in various buffers (sections 5–7) |
| Sodium Hydroxide (NaOH), solid | MilliporeSigma | S8045 | Salt for use in various buffers (sections 5–7) |
| Statistical data-fitting software | GraphPad | Prism | For fitting data from analytical PAGE to kinetic models (section 6) |
| Streptavidin-coated magnetic beads | ThermoFisher Scientific | 65002 | For capturing biotin-labeled RNA in cross-chiral ribozyme reactions (section 7) |
| Sucrose | MilliporeSigma | 84097 | For PAGE urea loading buffer (section 5) |
| TBE running buffer, 10x | ThermoFisher Scientific | AAJ62788K3 | For PAGE (sections 5–7) |
| Tetrabutylammonium Fluoride, 1.0 M solution in Tetrahydrofuran | Aldrich | 216143 | For removing 2′-silyl protecting groups (section 5) |
| Tetramethylethylenediamine (TEMED) | Bio-Rad Laboratories | 1610801 | For polymerizing acrylamide for PAGE (sections 5–7) |
| Tributylamine | MilliporeSigma | 90781 | Triphosphorylation reagent (section 2) |
| Tributylammonium pyrophosphate (TBAP) | MilliporeSigma | P8533 | Triphosphorylation reagent (section 2) |
| Tris base | MilliporeSigma | T6666 | Buffering agent for use in various buffers (sections 5–7) |
| TWEEN20 polysorbate detergent | MilliporeSigma | P7949 | Neutral detergent for use with magnetic beads (Section 7) |
| Urea | MilliporeSigma | U5378 | For PAGE and gel loading buffer (sections 5–7) |
| UV-Vis spectrophotometer | ThermoFisher Scientific | NanoDrop 2000, ND2000 | For measuring oligonucleotide concentrations (section 5) |
| Vacuum centrifuge | ThermoFisher Scientific | Savant Speedvac DNA130-115 Vacuum Concentrator | For removing AMA and THF (section 5) |
| Xylene cyanol | Bio-Rad Laboratories | 1610423 | For PAGE urea loading buffer (section 5) |
References
- Shuman, S. What messenger RNA capping tells us about eukaryotic evolution. Nature Reviews. Molecular Cell Biology. 3 (8), 619-625 (2002).
- Pichlmair, A., et al. RIG-I-mediated antiviral responses single-stranded RNA bearing 5'-phosphates. Science. 314 (5801), 997-1001 (2006).
- Hornung, V., et al. 5'-Triphosphate RNA is the ligand for RIG-I. Science. 314 (5801), 994-997 (2006).
- Myong, S., et al. Cytosolic viral sensor RIG-I is a 5'-triphosphate-dependent translocase on double-stranded RNA. Science. 323 (5917), 1070-1074 (2009).
- Takeuchi, O., Akira, S. Pattern recognition receptors and inflammation. Cell. 140, 805-820 (2010).
- Wang, Y., et al. Structural and functional insights into 5'-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nature Structural & Molecular Biology. 17 (7), 781-787 (2010).
- Goubau, D., et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5'-diphosphates. Nature. 514 (7522), 372-375 (2014).
- Joyce, G. F. Forty years of in vitro evolution. Angewandte Chemie. 46 (34), 6420-6436 (2007).
- Robertson, M. P., Ellington, A. D. In vitro selection of an allosteric ribozyme that transduces analytes to amplicons. Nature Biotechnology. 17 (1), 62-66 (1999).
- Robertson, M. P., Hesselberth, J. R., Ellington, A. D. Optimization and optimality of a short ribozyme ligase that joins non-Watson-Crick base pairings. RNA. 7 (4), 513-523 (2001).
- Hesselberth, J. R., Robertson, M. P., Knudsen, S. M., Ellington, A. D. Simultaneous detection of diverse analytes with an aptazyme ligase array. Analytical Biochemistry. 312 (2), 106-112 (2003).
- Lam, B. J., Joyce, G. F. Autocatalytic aptazymes enable ligand-dependent exponential amplification of RNA. Nature Biotechnology. 27 (3), 288-292 (2009).
- Lam, B. J., Joyce, G. F. An isothermal system that couples ligand-dependent catalysis to ligand-independent exponential amplification. Journal of the American Chemical Society. 133 (9), 3191-3197 (2011).
- Olea, C., Horning, D. P., Joyce, G. F. Ligand-dependent exponential amplification of a self-replicating L-RNA enzyme. Journal of the American Chemical Society. 134 (19), 8050-8053 (2012).
- Olea, C., Joyce, G. F. Real-Time Detection of a Self-Replicating RNA Enzyme. Molecules. 21 (10), (2016).
- Sczepanski, J. T., Joyce, G. F. A cross-chiral RNA polymerase ribozyme. Nature. 515 (7527), 440-442 (2014).
- Tjhung, K. F., Sczepanski, J. T., Murtfeldt, E. R., Joyce, G. F. RNA-catalyzed cross-chiral polymerization of RNA. Journal of the American Chemical Society. 142 (36), 15331-15339 (2020).
- Bare, G. A. L., Joyce, G. F. Cross-chiral, RNA-catalyzed exponential amplification of RNA. Journal of the American Chemical Society. 143 (45), 19160-19166 (2021).
- Milligan, J. F., Groebe, D. R., Witherell, G. W., Uhlenbeck, O. C. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Research. 15 (21), 8783-8798 (1987).
- Chelliserrykattil, J., Ellington, A. D. Evolution of a T7 RNA polymerase variant that transcribes 2'-O-methyl RNA. Nature Biotechnology. 22 (9), 1155-1160 (2004).
- Ibach, J., et al. Identification of a T7 RNA polymerase variant that permits the enzymatic synthesis of fully 2′-O-methyl-modified RNA. Journal of Biotechnology. 167 (3), 287-295 (2013).
- Esvelt, K. M., Carlson, J. C., Liu, D. R. A system for the continuous directed evolution of biomolecules. Nature. 472 (7344), 499-503 (2011).
- Pleiss, J. A., Derrick, M. L., Uhlenbeck, O. C. T7 RNA polymerase produces 5' end heterogeneity during in vitro transcription from certain templates. RNA. 4 (10), 1313-1317 (1998).
- Schenborn, E. T., Mierendorf, R. C. A novel transcription property of SP6 and T7 RNA polymerases: dependence on template structure. Nucleic Acids Research. 13 (17), 6223-6236 (1985).
- Martin, C. T., Muller, D. K., Coleman, J. E. Processivity in early stages of transcription by T7 RNA polymerase. Biochemistry. 27 (11), 3966-3974 (1988).
- Gholamalipour, Y., Karunanayake Mudiyanselage, A., Martin, C. T. 3' end additions by T7 RNA polymerase are RNA self-templated, distributive and diverse in character-RNA-Seq analyses. Nucleic Acids Research. 46 (18), 9253-9263 (2018).
- Vasilyev, N., Serganov, A. Preparation of short 5′-triphosphorylated oligoribonucleotides for crystallographic and biochemical studies. Nucleic Acid Crystallography: Methods and Protocols. , 11-20 (2016).
- Lebedev, A. V., Koukhareva, I. I., Beck, T., Vaghefi, M. M. Preparation of oligodeoxynucleotide 5'-triphosphates using solid support approach. Nucleosides, Nucleotides & Nucleic Acids. 20 (4-7), 1403-1409 (2001).
- Paul, N., Springsteen, G., Joyce, G. F. Conversion of a ribozyme to a deoxyribozyme through in vitro evolution. Chemistry & Biology. 13 (3), 329-338 (2006).
- Zlatev, I., et al. Efficient solid-phase chemical synthesis of 5'-triphosphates of DNA, RNA, and their analogues. Organic Letters. 12 (10), 2190-2193 (2010).
- Zlatev, I., Manoharan, M., Vasseur, J. -. J., Morvan, F. Solid-phase chemical synthesis of 5'-triphosphate DNA, RNA, and chemically modified oligonucleotides. Current Protocols in Nucleic Acid Chemistry. , (2012).
- Zlatev, I., et al. Automated parallel synthesis of 5'-triphosphate oligonucleotides and preparation of chemically modified 5'-triphosphate small interfering RNA. Bioorganic & Medicinal Chemistry. 21 (3), 722-732 (2013).
- Goldeck, M., Tuschl, T., Hartmann, G., Ludwig, J. Efficient solid-phase synthesis of pppRNA by using product-specific labeling. Angewandte Chemie. 53 (18), 4694-4698 (2014).
- Sarac, I., Meier, C. Efficient automated solid-phase synthesis of DNA and RNA 5′-triphosphates. Chemistry-A European Journal. 21 (46), 16421-16426 (2015).
- Sarac, I., Meier, C. Solid-phase synthesis of DNA and RNA 5'-O-triphosphates using cycloSal chemistry. Current Protocols in Nucleic Acid Chemistry. 64 (1), 4-67 (2016).
- Perez, J. T., et al. Influenza A virus-generated small RNAs regulate the switch from transcription to replication. Proceedings of the National Academy of Sciences of the United States of America. 107 (25), 11525-11530 (2010).
- Ludwig, J., Eckstein, F. Rapid and efficient synthesis of nucleoside 5'-0-(1-thiotriphosphates), 5'-triphosphates and 2',3'-cyclophosphorothioates using 2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one. The Journal of Organic Chemistry. 54 (3), 631-635 (1989).
- Gaur, R. K., Sproat, B. S., Krupp, G. Novel solid phase synthesis of 2'-o-methylribonucleoside 5'-triphosphates and their α-thio analogues. Tetrahedron Letters. 33 (23), 3301-3304 (1992).
- Reddy, M. P., Hanna, N. B., Farooqui, F. Fast cleavage and deprotection of oligonucleotides. Tetrahedron Letters. 35 (25), 4311-4314 (1994).
- Hogrefe, R. I., McCaffrey, A. P., Borozdina, L. U., McCampbell, E. S., Vaghefi, M. M. Effect of excess water on the desilylation of oligoribonucleotides using tetrabutylammonium fluoride. Nucleic Acids Research. 21 (20), 4739-4741 (1993).
- Pitsch, S., Weiss, P. A., Jenny, L., Stutz, A., Wu, X. Reliable chemical synthesis of oligoribonucleotides (RNA) with 2′-O-[(Triisopropylsilyl)oxy]methyl(2′-O-tom)-protected phosphoramidites. Helvetica Chimica Acta. 84 (12), 3773-3795 (2001).
- Green, M. R., Sambrook, J. Isolation of DNA fragments from polyacrylamide gels by the crush and soak method. Cold Spring Harbor Protocols. 2019 (2), (2019).
- Paul, N., Joyce, G. F. A self-replicating ligase ribozyme. Proceedings of the National Academy of Sciences of the United States of America. 99 (20), 12733-12740 (2002).
- Lincoln, T. A., Joyce, G. F. Self-sustained replication of an RNA enzyme. Science. 323 (5918), 1229-1232 (2009).
- Robertson, M. P., Joyce, G. F. Highly efficient self-replicating RNA enzymes. Chemistry & Biology. 21 (2), 238-245 (2014).
- Singh, J., et al. Synthesis of modified nucleoside oligophosphates simplified: fast, pure, and protecting group free. Journal of the American Chemical Society. 141 (38), 15013-15017 (2019).
- Bellon, L. Oligoribonucleotides with 2'-O-(tert-butyldimethylsilyl) groups. Current Protocols in Nucleic Acid Chemistry. , (2001).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved