
Method Article
Evaluation and Manipulation of Neural Activity Using Two-Photon Holographic Microscopy
In This Article
Summary
We developed a two-photon holographic microscope that can visualize, assess, and manipulate neural activity using high spatiotemporal resolution, with the aim of elucidating the pathogenesis of neuropsychiatric disorders that are associated with abnormal neural activity.
Abstract
Recent advances in optical bioimaging and optogenetics have enabled the visualization and manipulation of biological phenomena, including cellular activities, in living animals. In the field of neuroscience, detailed neural activity related to brain functions, such as learning and memory, has now been revealed, and it has become feasible to artificially manipulate this activity to express brain functions. However, the conventional evaluation of neural activity by two-photon Ca2+ imaging has the problem of low temporal resolution. In addition, manipulation of neural activity by conventional optogenetics through the optic fiber can only simultaneously regulate the activity of neurons with the same genetic background, making it difficult to control the activity of individual neurons. To solve this issue, we recently developed a microscope with a high spatiotemporal resolution for biological applications by combining optogenetics with digital holographic technology that can modify femtosecond infrared laser beams. Here, we describe protocols for the visualization, evaluation, and manipulation of neural activity, including the preparation of samples and operation of a two-photon holographic microscope (Figure 1). These protocols provide accurate spatiotemporal information on neural activity, which may be useful for elucidating the pathogenesis of neuropsychiatric disorders that lead to abnormalities in neural activity.
Introduction
Two-photon Ca2+ imaging is a useful technique for the assessment of neural activity. It can be used to identify not only the neural activity required for behavior and memory in normal animals1,2 but also an abnormal neuronal activity that occurs in mouse models of neuropsychiatric disorders3,4. The technique has been used to elucidate the neural basis of brain functions. However, although it can provide high-resolution and high-quality images, its temporal resolution is lower than that of the electrophysiological method1,3.
Optogenetics has helped to innovate the way neuroscientists understand brain function5. Given the technical limitations, the majority of optogenetic research has used activation schemes with low spatial resolution, thus limiting the types of manipulations of neural activity that can be performed accordingly. However, manipulating neural activity at finer spatiotemporal scales can potentially be useful for a more complete understanding of neural computation and the pathogenesis of neuropsychiatric disorders. Spatially precise holographic technology that can shape femtosecond near infrared laser beams promises to overcome this challenge and opens up several new experimental classes that were previously impossible6,7. This technology enables neuroscientists to uncover the fundamental aspects and pathologies of sensory, cognitive, and behavioral neural codes that have been beyond reach.
Holographic projection involves the generation of desired light patterns to access individual cells and functional networks selectively. In vivo experiments require optimal light transmission to target cells in the living brain. Infrared light penetrates deeper into living tissue and can be used for nonlinear two-photon excitation (2PE)8,9,10. Thus, two-photon holographic microscopy, which combines holographic projection and 2PE, can be used to evaluate and manipulate neural activities to probe cellular and functional networks in vivo. Recent biological applications of two-photon holographic microscopy have elucidated the neural activity and circuitry required for learning in the visual cortex11,12, olfactory bulb13, and hippocampus14.
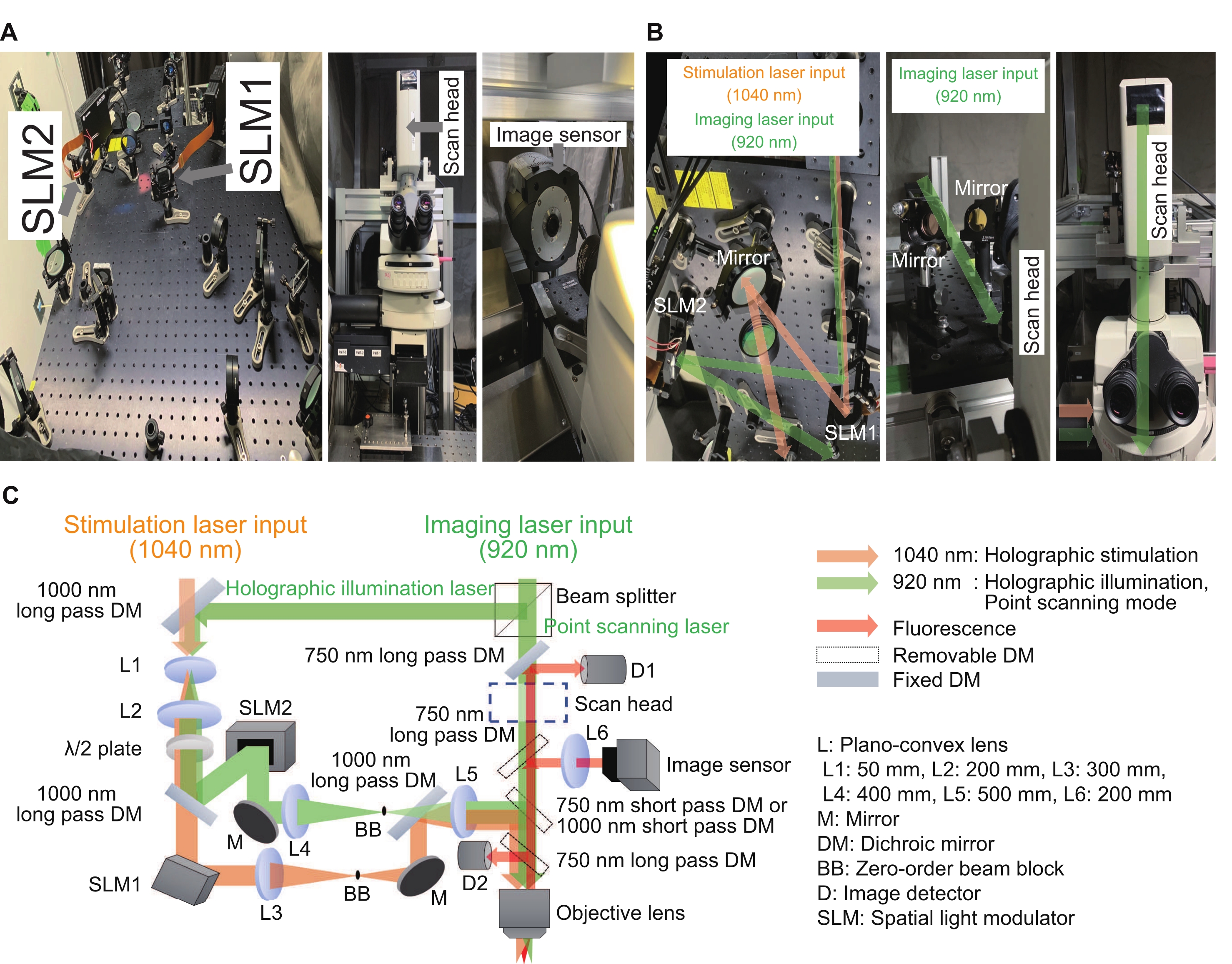
Numerous laboratories worldwide have reported exciting results and improvements using their holographic stimulation systems15,16,17,18,19,20,21,22,23. In the system described here, the holographic stimulation system can be built as an add-on device for a conventional microscope. The phase-only spatial light modulator (SLM) is the key device for modulating a plane wavefront to any shape, and the interference effect is used to control the intensity and location of the foci. Figure 2 shows holographic stimulation and imaging light paths. The first light path is for point-scanning imaging mode and consists of a scan head and image detectors. The second light path is for holographic stimulation with a 1040 nm wavelength and consists of an SLM1. The third light path is for holographic illumination with 920 nm wavelength and consists of an SLM2 and an image sensor. The holographic imaging mode can record intensities from multiple regions of interest by illuminating multiple points in the sample. In this way, the recording speed can be increased to few hundred of frames per second. To achieve point scanning imaging or holographic illumination imaging, the 920 nm laser was split into two paths by a beam splitter with a fixed ratio of 3:7. All the optical elements were aligned on an optical breadboard with dimensions of 600 mm × 600 mm. The modulated light entered through the light port on the side of the microscopic body, while the point scanning imaging light entered through the scan head at the top of the microscopic body. These lights were integrated just above the objective lens and created foci in the sample plane. In addition, the custom-made software enabled the regular workflow to be simple and consistent.
In this article, a complete protocol is presented for the use of holographic stimulation or illumination to measure neural activity and assess the functional connectivity between neurons. For demonstration purposes, we describe here a brain surgery targeting the hindlimb area of the primary somatosensory cortex (S1HL) of the mouse brain and a method to assess and manipulate neural activity using two-photon holographic microscopy. The experimental procedure is divided into four parts. First, the head plate was fixed to the skull of the mouse using dental cement. Second, a viral vector expressing jGCaMP8f or GCaMP6m-P2A-ChRmine was stereotactically injected into the S1HL. Third, the holographic stimulation or illumination system was calibrated. Fourth, after postoperative recovery and expression of these two proteins, in vivo Ca2+ imaging was performed to assess the neural activity and functional connectivity between neurons with two-photon holographic microscopy.
Protocol
All experimental protocols were approved by the Animal Care and Use Committees of Nagoya University Graduate School of Medicine (approval number: M220295-003).
1. Head plate implantation (Figure 1A)
- Administer the anesthetic (a mixture of 74 mg/kg ketamine and 10 mg/kg xylazine) intraperitoneally to anesthetize the mouse. Check the anesthetic status of the mouse frequently by assessing the pedal reflexes.
- After anesthesia, place the mouse in a stereotaxic instrument. Apply an eye ointment (see Table of Materials) to prevent the cornea from drying out when the head plate is implanted.
- Shave the surgical area and disinfect the skin with three alternating rounds of povidone-iodine or chlorhexidine scrubs followed by 70% alcohol wipes. Carefully expose the skull and clean it with cotton swabs.
NOTE: All surgical instruments should be sterilized, and all procedures should be performed accordingly. Any remaining debris (e.g., hair or dried blood) causes inflammatory reactions. Therefore, any debris should be removed under a stereoscope using cotton swabs moistened with sterile water or 70% alcohol. - Use the stereotactic coordinates-anterior and posterior = 0.5 mm, medial and lateral = 1.5 mm from bregma-to find the center of the craniotomy and label it with a marker pen.
- Place a custom-made head plate at the center of the skull. Next, apply dental cement (see Table of Materials) to firmly fix it to the skull. Apply light pressure until the head plate makes firm contact with the front and back of the skull.
NOTE: This step takes approximately 20 min to complete and is critical for reducing motion artifacts in the brain during two-photon imaging. The dimensions of the head-plate are 20 mm × 40 mm × 1 mm with a home plate shape opening with one edge 15 mm long, two adjacent sides 3 mm long, and the remaining two sides 10 mm long. - Mix together an acrylic-based dental adhesive resin cement as follows: half a spoon of powder, three drops of liquid, and one drop of catalyst (see Table of Materials). To prevent drying, apply this mixed dental adhesive resin cement to the intact skull surface of the mousewith the head plate.
- Place the mouse in a warm cage until it has recovered from anesthesia. Do not leave the mouse unattended until it has regained sufficient consciousness to maintain sternal recumbency.
2. Surgery and adeno-associated virus (AAV) injection (Figure 1B)
- Perform craniotomy or viral injection without removal of the dental adhesive resin cement from the skull 1 day after head plate implantation.
NOTE: Administer anesthesia (a mixture of 74 mg/kg ketamine and 10 mg/kg xylazine) intraperitoneally to the mouse during this procedure. - To avoid cerebral edema, administer dexamethasone sodium phosphate (1.32 mg/kg) intraperitoneally 1 h before surgery.
- Anesthetize the mouse with the head plate with 1% isoflurane anesthesia using a vaporizer (anesthesia delivery system) while maintaining body temperature with a heating pad. Apply an eye ointment to prevent corneal drying.
- Under a stereoscope, perform a circular craniotomy approximately 2 mm in diameter using a dental drill. To reduce brain damage, operate the dental drill carefully with constant slight movement and light downward pressure.
- Remove bone fragments several times using a suction system. After removing the bone fragments, use an artificial spinal fluid (ACSF) solution to remove and wash any debris remaining on the brain surface. Repeat this cleaning procedure several times to suppress inflammatory reactions.
NOTE: The ACSF solution (140 mM NaCl, 2.5 mM KCl, 5 mM HEPES, 2.0 mM CaCl2, and 1.0 mM MgCl2) was stored at 4 °C for 1 month after the reagent was dissolved and filtered (pore size = 0.22 µm). - Using a pressure injection system (see Table of Materials), set the appropriate pressure (about 10 PSI in pulses with a duration of 4 ms) to inject 500 nL of AAV solution through a glass capillary with a tip diameter of 10-20 µm (prepared with a micropipette puller) for 10 min.
- Determine whether the AAV solution is being administered to the brain by checking whether the level of the AAV solution in the glass capillary gradually decreases.
- Leave the glass capillary in place for an additional 10 min to prevent backflow. Repeat three times to administer a total of 1.5 µL of AAV solution into the brain.
- To evaluate and manipulate neural activity in layer 2/3 (L2/3) pyramidal cells, inject an AAV solution (for Ca2+ imaging: AAV2/1-Syn-jGCaMP8f-WPRE at 1.28 × 1014 vector genomes/mL, diluted 1:1 in saline; for Ca2+ imaging with optogenetics: AAV2/8-CaMKII-GCaMP6m-P2A-ChRmine-Kv2.1-WPRE at 1.73 × 1014 vector genomes/mL, diluted 1:1 in saline) into the hind paw area of the primary somatosensory cortex of wild-type mice (S1, centered at 0.5 mm posterior and 1.5 mm lateral from the bregma, 150 µm depth from the surface).
NOTE: The AAV solution should be injected at 2-3 weeks and 1-2 weeks before imaging for jGCaMP8f and GCaMP6m-P2A-ChRmine expression, respectively. - After an application of 2% (w/v) low-melting agarose to the brain surface of S1 using a micropipette, place a glass window over the craniotomy with two cover glasses. Attach the two cover glasses (small 2.0 mm diameter and large 4.5 mm diameter; see Table of Materials) with UV curable adhesive.
- Press the cover glass against the agarose while it is still liquid; this prevents the formation of air bubbles in the agarose. Seal the edges of the cranial window with dental and adhesive resin cement (Figure 1C).
- Remove the mouse from the stereotaxic instrument and return it to its cage. Place the mouse in a warm cage and do not return to the cage with other animals until it has fully recovered from anesthesia. Carefully maintain sterile conditions during survival surgery.
- For the first 72 h after surgery, check the health status of the mouse by observing general behavior. If there are any abnormalities in general behavior, subcutaneously inject anti-inflammatory and analgesic agents.
3. Preparation for the holographic stimulation or illumination system (Figure 3)
- Calibrate the holographic stimulation system by placing the surface of a red fluorescence slide (cast acrylic substrate) at the sample plane. Place the microscope in live imaging mode with a weak excitation light and run calibration_GUI.m file. Check the parameters pane and click on the Save button.
- Click the Z Scan button in the step 1 pane. It will automatically generate three random spots in all 21 axial planes, 2 µm apart from each plane.
- Move the slide bar and check the live image. Find a perfect plane where the spots appear the smallest and the brightest, and then click the Save button. This will automatically generate an offset spherical wavefront for the digital hologram.
NOTE: If you fail to find the brightest fluorescence spots, change the minimum and maximum values of the scan range and try again. - Click the Go button in the step 2 pane, and then click six spots on the left square. Check the live image. If there are six distinguishable fluorescence spots, then type their x and y axis to the edit boxes and click the Save button. This will automatically generate affine transform coefficients to coordinate calibration between the holographic stimulation and imaging system.
NOTE: The axis pair number and clicked spot number must be matched in order. If not sure, or if there are no spots in your image, please try again and generate a unique spot pattern or choose a smaller range around the center of the field of view (FOV). - Click the Scan button in the step 3 pane. It will generate 441 digital holograms to perform single spot scanning across the FOV in 21 × 21 steps.
- First, check the images while changing patterns in the list box. Then, adjust the laser power to obtain spot images within the dynamic range of the imaging device (for instance, to avoid overly saturated images).
- Subsequently, adjust the interval time in the edit box; the interval time should be more than two times the recording interval time. Finally, put the imaging device in recording mode and click the Play button. If the play completes, there will be "display OK" strings shown in the command window. Stop recording and stack up recorded sequential images using the max intensity method.
- Click Generate WM in the step 4 pane and choose a stacked image from above. Then close the calibration_GUI window. It will automatically generate a weight map to compensate for the unbalanced intensity in each spot.
NOTE: For a more detailed description, please refer to 2; the Matlab code can be downloaded from here (https://github.com/ZenKG/SLM_control).
4. Ca2+ imaging using an image sensor with holographic illumination (Figure 4)
- Place the AAV-injected mouse with a head plate under the microscope (Figure 1D).
NOTE: During this procedure, the mouse is restrained in the awake state, but is able to escape uncomfortable stimuli. - Perform two-photon imaging (point scanning mode) using a holographic microscope and a mode-locked Ti:sapphire laser tuned to 920 nm with a 25x objective (see Table of Materials).
- Turn on the commercial imaging software (see Table of Materials). In the live imaging mode, adjust the voltage of the image detector (see Table of Materials) and the power of the imaging laser to optimize the brightness of the neurons expressing jGCaMP8f. Capture images of the neurons expressing this protein.
NOTE: The intensity of the imaging laser (920 nm) is 20-30 mW. The FOV was 512 µm × 512 µm at a depth of 100-150 µm from the cortical surface. - To illuminate specific neurons expressing jGCaMP8f with holographic illumination, run the SLMcontrol.m script file. Click the Reference Image and choose the image acquired above. Then, click the Spot button to choose specific pixels on the neurons in the image by continuous mouse clicking (Figure 4A). If the selection is complete, push the Enter button on the keyboard to finalize it.
NOTE: The digital hologram is automatically calculated and displayed on the SLM. This pattern can also be revisited by clicking on a list box. The spatial resolution of a single spot generated by the SLM was approximately 1.2 µm along the transverse direction and ~8.3 µm along the optical axis. We used a high numerical aperture (1.1) objective lens to achieve more localized holographic stimulation. Table 1 summarizes previous reports and this system with respect to the spatial resolution of holographic stimulation. - To detect neural activity with high temporal resolution using an image sensor (see Table of Materials), set the exposure time, imaging area, and binning (Figure 4B) before performing image acquisition (Figure 4C).
NOTE: The intensity of the holographic illumination laser (920 nm) that continuously stimulates one neuron is 2 mW, which is sufficient to detect neural activity. For example, to achieve a frame rate of 100 Hz for imaging, the exposure time is 9 ms, the imaging area is 400 µm × 400 µm, and the binning is 4. - After the experiment, return the mouse to its home cage.
5. Two-photon imaging (point scanning mode) with optogenetics using a holographic microscope (Figure 2)
- Repeat steps 4.1 and 4.2.
- Turn on the commercial imaging software (see Table of Materials). In the live imaging mode, adjust the voltage of the image detector (see Table of Materials) and the power of the imaging laser to optimize the brightness of the neurons expressing GCaMP6m-P2A-ChRmine. Capture images of the neurons expressing these proteins (Figure 1E).
- Repeat step 4.4.
- To investigate the functional connectivity within L2/3 neurons, use an SLM to generate holographic patterns of optogenetic stimulation (ChRmine; 1,040 nm) and combine it with two-photon Ca2+ imaging (GCaMP6m; 920 nm, 512 × 512 pixels, 2 Hz or 30 Hz, 2x digital zoom, point scanning mode; Figure 4D-H).
- For this protocol, set the intensity of the imaging laser to 920 nm, at 10-20 mW, and the FOV as 256 µm × 256 µm measured at a depth of 100-150 µm from the cortical surface. Set the pixel dwell time at 1.5 µs for 2 Hz or 100 ns for 30 Hz.
- To see if a single holographic stimulus caused a calcium response in the neurons, use both 2 Hz and 30 Hz as the imaging frame rate. Set the intensity of the holographic stimulation laser (1,040 nm) that stimulates a single neuron at 10 mW, which is sufficient to induce neural activity (Figure 4D).
NOTE: The spatial resolution of a single spot generated by the SLM is approximately 1.2 µm along the transverse direction and ~8.3 µm along the optical axis. The range of accessible volume in the lateral direction is around 500 µm × 500 µm and 100 µm in the axial direction. We have further confirmed with Ca2+ imaging at 2 Hz or 30 Hz imaging frame rate that not only one neuron, but multiple neurons can be holographically stimulated simultaneously (Figure 4E).
- Perform image acquisition with the following protocol: simultaneously image the Ca2+ response at 920 nm with 10 holographic stimuli at 1,040 nm with 8 s intervals (0.125 Hz) for a duration of 50 ms after a baseline period of 10 s. After all the experiments are completed, the mice are euthanized.
NOTE: Ca2+ transients, if present, were evoked by holographic stimulation, with their peak appearing within 1 s after stimulation (Figure 4F-H).
6. Image analysis and assessment of functional connectivity (Figure 4)
- Open the raw images saved in steps 4.5 or 5.5 using ImageJ. To compensate for focal plane displacement, use the ImageJ plug-in TurboReg.
NOTE: If the correction with TurboReg is not sufficient, it is recommended to use CaImAn (http://github. com/simonsfoundation) to correct the focal plane displacement. - To assess neural activity, determine the regions of interest (ROIs) in L2/3 using an automated algorithm (CaImAn). Detect and analyze Ca2+ transients after defining baseline fluorescence intensity (F0) and the threshold value.
NOTE: F0 is the 35th percentile value of fluorescence intensity acquired during the baseline imaging period. Ca2+ transients are denoted by ΔF/F0 (ΔF = F-F0), where F is the instantaneous fluorescence signal. If the ΔF value is over 2 S.D. from F0, we evaluate a significant Ca2+ transient. - To define functional connectivity in L2/3 neurons, stimulate target neurons and measure GCaMP6m responses in stimulated and surrounding neurons, accordingly (Figure 4F-H).
Results
Representative recordings obtained using the method described here are presented. In vivo Ca2+ imaging using holographic microscopy requires 2-4 weeks to complete from head plate implantation and AAV injection to data acquisition. Therefore, to obtain stable results, it is important to reduce motion artifacts in the brain. The head plate implantation (step 1.5) and the placement of the cranial window (step 2.9) are very important steps in this process. Furthermore, it is also important to select an AAV (AAV2/8-CaMKII-GCaMP6m-P2A-ChRmine-Kv2.1) that simultaneously expresses calcium indicator and opsin in a single neuron (step 2.8).
Figure 4A shows a spot for the holographic illumination of an acquired neuron image using a custom-made MATLAB script. If holographic illumination successfully illuminated the neurons expressing jGCaMP8f, Ca2+ traces could be obtained with an image sensor (Figure 4B), as shown in Figure 4C.
Functional connectivity between neurons was evaluated using holographic stimulation (Figure 4D), as shown in Figure 4E. Because functional connectivity between neurons is one of the neural circuitry properties that is altered in the pathogenesis of a pain model mouse24, we describe a simple procedure to evaluate it. Figure 4F shows a typical image of L2/3 neurons in S1HL visualized using GCaMP6m. When one neuron (orange circle) was holographically stimulated, another neuron (red circle) was simultaneously active; thus, the number of functional connectivities between neurons was one (Figure 4G).

Figure 1: Schematic outline of the experimental procedure. (A) Fixation of the head plate to the skull. (B) Stereotactic injection of AAV into the hind paw area of the primary somatosensory cortex (S1HL). (C) Implantation of the cranial window. To assess and manipulate neural activity, in vivo Ca2+ imaging is performed in awake mice (D) with holographic stimulation (E). Flash marks indicate holographic stimulation or illumination. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: System used for the holographic microscope. (A) Images of the holographic stimulation and illumination light paths (left) near the microscope (middle) with an image sensor (right). (B) These are magnified images of holographic stimulation and illumination light paths around respective SLMs (left and right) and a point scanning light path around a scan head (middle and right). (C) A schematic of the stimulation and imaging optical paths. Phase-only SLMs are used to display digital holograms, and a beam expander (a combination of L1 and L2) and a 4f relay system (a combination of L3 and L4 for holographic stimulation and L4 and L5 for holographic illumination) are placed before and after respective SLMs to make sure each digital hologram is imaged at the exit pupil of a water immersion objective lens, with slightly under-filled image size. To suppress residual zero-order components, a beam block is placed at the intermediate plane. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Flowchart of how to calibrate the holographic stimulation or illumination system. This flowchart describes steps to calibrate a holographic stimulation or illumination system to the sample space and imaging system. Please visit step 3, preparation for the holographic stimulation or illumination system, for detailed instructions and download a sample program. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Representative results of imaging and functional connectivity using a holographic microscope. (A) To illuminate specific neurons expressing jGCaMP8f or GCaMP6m-P2A-ChRmine, the image of neurons is captured, and then a spot is formed on the neuron using a custom-made MATLAB script. (B) The setting of the image sensor (exposure time, imaging area, and binning). (C) Representative image and traces of neurons expressing jGCaMP8f in 100 Hz imaging with holographic illumination and an image sensor. (D) This graph shows the neural response to holographic stimulation (1,040 nm) at each laser power (data from GCaMP6m-P2A-ChRmine expressing neurons at 2 Hz imaging frame rate [n = 16]). Error bars indicate the standard error of the mean. (E) Representative Ca2+ traces during holographic stimulation (blue vertical lines) of 10 different neurons at 2 Hz (left) and 30 Hz (right) imaging frame rates. In 2 Hz and 30 Hz Ca2+ traces, the same color indicates the same neuron. (F) Schematic diagram evaluating functional connections between neurons. When the orange neuron is stimulated, the red neurons respond at the same time, indicating that there is functional connectivity between these neurons. (G) A typical image of S1HL neurons expressing GCaMP6m in WT. Scale bar = 10 µm. (H) Typical Ca2+ traces during holographic stimulation (blue vertical lines) at 2 Hz (upper) and 30 Hz (lower) imaging frame rates. The stimulated neuron is circled in orange, responding neurons are circled in red, and non-responding neurons are circled in grey. Neural responsiveness to holographic stimulation can be detected at both 2 Hz and 30 Hz imaging speeds. Please click here to view a larger version of this figure.
{kind=link}
| Our set-up 6 | Prakash, R. et al. 25 | Marshel, J. H. et al. 12 | Robinson, N. T. M. et al. 14 | |
| Lateral resolution | 1.2 μm | 1.27 μm | ― | 2.22 μm |
| Axial resolution | 8.3 μm | 56.86 μm | 15.5 μm | 10.26 μm |
| Objective lens/Numerical Aperture | 25x/1.1 | 20x/0.5 | 16x/0.8 | 16x/0.8 |
Table 1: Summary of previous reports and this system for the spatial resolution of holographic stimulation. The lateral resolution, axial resolution, and the objective lens used during the measurement are described.
Discussion
To understand brain function, it is necessary to accurately assess the neural circuitry underlying the brain function by extracting the dynamics of neural activity. Furthermore, it is essential to identify how this neural circuitry is altered to elucidate the pathogenesis of neuropsychiatric disorders. Indeed, it is known that neural activity is elevated in mouse models of Alzheimer's disease4 and fragile X syndrome26 and mice with impaired white matter function3. Moreover, in a mouse model of inflammatory pain, increased synchronization of neural activity and functional connectivity between neurons is associated with symptoms24. Two-photon holographic microscopy allows us to simultaneously observe the activity of individual neurons and the functional connections between neurons, which is necessary for understanding neural circuitry. We used a 25x objective lens with numerical aperture = 1.1 with wavelengths of 1,040 nm. The theoretical point spread function is a Gaussian distribution with full width at half maximum of 0.5 µm laterally and 1.7 µm axially. However, the actual numerical aperture is less than 1.1, and the measured spot size on a fluorescence bead is 1.2 µm laterally, and 8.3 µm axially. Given that the neuron diameter is approximately 15 µm and the calibration error is within 3 µm, the targeting is generally good. However, cells in the axial direction may be affected by the longer spot size6. Here, we described the viral injection, surgery, calibration of holographic stimulation or illumination systems, and imaging protocols for evaluating and manipulating neural activity in live mice using our microscopy system.
It takes 2-4 weeks to complete all experimental procedures, from head plate implantation and virus injection to data acquisition for in vivo Ca2+ imaging using holographic microscopy. The process is complex and laborious, and the ultimate success of the experiment depends on multiple factors, including the condition of the cranial window, which is affected by postoperative inflammation, the proper choice of Ca2+ indicator and opsin, and whether motion artifacts in the acquired images can be corrected. In particular, two steps are important for a successful outcome. The first concerns head plate fixation and surgery; it is crucial that the head plate be firmly fixed to the mouse head with dental cement. Furthermore, it is important to repeatedly clean bone fragments and clotted blood using cold ACSF during surgery. Since adherence to this procedure reduces inflammation, we successfully observed the dynamics of microglia, the cells responsible for the brain's immune system, without activating their processes and spines or the microstructures of neurons27,28. The second issue is the evaluation and manipulation of neural activity. We chose AAV2/8-CaMKII-GCaMP6m-P2A-ChRmine-Kv2.1 to express the Ca2+ indicator and opsin in one neuron simultaneously. This is because it is difficult to efficiently infect one neuron with different AAV types. Another reason for this choice was that ChRmine can efficiently activate neural activity at 1,040 nm using a two-photon laser12. Recently, it has been reported that ChRmine, by mutating its structure based on structural information obtained by cryo-electron microscopy, improves its function29, which is considered useful for target function analysis in the field of neuroscience. In light of these issues, it is necessary to share effective methods for evaluating and manipulating neurons when reading neural activity and writing information using holographic microscopy.
Recent advances in imaging and optogenetics have revealed the detailed neural activity involved in brain functions such as learning and memory, and it is possible to artificially manipulate this neural activity to express brain functions30. However, conventional methods for the manipulation of neural activity are highly invasive on account of the insertion of optical fibers in the brain and because a group of opsin-expressing cells is simultaneously stimulated, making it impossible to manipulate neural activity with temporal and spatial precision. Our method can manipulate neural activity by stimulating only specific neurons in the brain, thus enabling manipulation of neural activity with specific stimulation patterns and high spatiotemporal resolution. Furthermore, it is important to note that functional connectivity between neurons can only be evaluated in a small number of neurons using brain slice experiments31; however, this technique allows simultaneous evaluation of multiple neurons in living animals.
One of the major limitations of current holographic microscopes is the need to fix the mouse head, which limits the mouse's behavior. Recently, a miniaturized two-photon microscope was developed32, and with the further miniaturization of the device, in vivo Ca2+ imaging with holographic stimulation may be possible in free-moving mice. In addition, the potential of this microscope could be expanded by improving the temporal resolution of imaging and combining it with highly sensitive voltage-sensitive fluorescent proteins33.
Disclosures
The authors have no conflicts of interest to declare.
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research on Innovative Areas (19H04753, 19H05219, and 25110732 to H. W.), Grants-in-Aid for Transformative Research Areas (A) (20H05899 to H. W., 20H05886 to O. M., and 21H05587 to D. K.), Fostering Joint International Research (B) (20KK0170 to H. W.), Grant-in-Aid for Scientific Research (B) (18H02598 to H. W.), Grant-in-Aid for Scientific Research (A) (21H04663 to O. M.), Grant-in-Aid for Early-Career Scientists (20K15193 to X. Q.), JST CREST Grant Number JPMJCR1755, Japan, and JST A-STEP Grant Number JPMJTR204C.
Materials
| Name | Company | Catalog Number | Comments |
| 25x Objective | Nikon | N25X-APO-MP | Objective |
| A1MP | Nikon | A1MP | Microscope |
| AnesII | Bio machinery | AnesII | Anesthesia delivery system |
| C2 plus | Nikon | C2 plus | Microscope |
| DECADRON Phosphate Injection | Aspen | 21N024 | Avoid cerebral edema |
| Dental Drill | Jota | C1.HP.005 | Dental drill |
| Electric Microinjector | NARISHIGE | IM-31 | Pressure injection system |
| FEATHERS | FEARGER | FA-10 | Shaving |
| G-CEM ONE ADHESIVE ENHANCING PRIMER | GC | 2110271 | Resin cement primer for dental adhesion |
| G-CEM ONE neo | GC | 43093 | Resin cement for dental adhesion |
| Glass Capillary with Filament | NARISHIGE | GDC-1 | Glass capillary |
| Image Detector | Hamamatsu | H10770PA-40 | GaAsP photocathode photomultiplier tube |
| Imaging Software | Nikon | NISelements | Imaging software |
| Isoflurane Inhalation Solution | Pfizer | 229KAR | Anesthetics |
| iXon EMCCD Camera | Andor | iXon Life 888 | Image sensor |
| Ketamine | daiitisannkyou | s9-018506 | Anesthetics |
| Leica-M60 | Leica | M60 | Stereoscope |
| Linicon | Linicon | LV-125 | Vacuum pump |
| Mode-locked Ti:sapphire Chameleon Ultra II laser | Coherent | Chameleon Discovery NX | Femtosecond laser |
| Mos-Cure | U-VIX | mini 365 | Portable LED UV Light Source |
| PEN Bright | SHOFU INC. | PEN Bright | Dental light curing unit |
| Puller | SUTTER instaument | P-97 | Puller |
| Stereotaxic Instrument (for Mice) | NARISHIGE | SR-6M-H | Stereotaxic instrument |
| Stereotaxic Micromanipulator | NARISHIGE | SM-15R | Stereotaxic micromanipulator |
| Super-Bond CATALYST V | SUN MEDICAL | 8070 | Dental adhesive resin cement |
| Super-Bond Dental Adhesive Monomer | SUN MEDICAL | 8071 | Dental adhesive monomer |
| Super-Bond Teeth Color Polymer Powder | SUN MEDICAL | 145052000 | Teeth color polymer powder |
| Tarivid Ophthalmic Ointment 0.3% | Santen Pharmaceutical | TRN3952 | Eye ointment |
| UlTIMATE XL | NSK | Y141446 | Dental laboratory micromotor control unit |
| UV Curing Optical Adhesives | THORLABS | NOA61 | UV Curing Optical Adhesives |
| Xylazine | Bayer | KP0F2BK | Anesthetics |
References
- Masamizu, Y., et al. Two distinct layer-specific dynamics of cortical ensembles during learning of a motor task. Nature Neuroscience. 17 (7), 987-994 (2014).
- Peters, A. J., Chen, S. X., Komiyama, T. Emergence of reproducible spatiotemporal activity during motor learning. Nature. 510 (7504), 263-267 (2014).
- Kato, D., et al. Motor learning requires myelination to reduce asynchrony and spontaneity in neural activity. Glia. 68 (1), 193-210 (2020).
- Busche, M. A., et al. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nature Neuroscience. 22 (1), 57-64 (2019).
- Boyden, E. S., Zhang, F., Bamberg, E., Nagel, G., Deisseroth, K. Millisecond-timescale, genetically targeted optical control of neural activity. Nature Neuroscience. 8 (9), 1263-1268 (2005).
- Quan, X., Kato, D., Daria, V., Matoba, O., Wake, H. Holographic microscope and its biological application. Neuroscience Research. 179, 57-64 (2022).
- Adesnik, H., Abdeladim, L. Probing neural codes with two-photon holographic optogenetics. Nature Neuroscience. 24 (10), 1356-1366 (2021).
- Denk, W., Strickler, J. H., Webb, W. W. Two-photon laser scanning fluorescence microscopy. Science. 248 (4951), 73-76 (1990).
- Helmchen, F., Denk, W. Deep tissue two-photon microscopy. Nature Methods. 2 (12), 932-940 (2005).
- Svoboda, K., Denk, W., Kleinfeld, D., Tank, D. W. In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature. 385 (6612), 161-165 (1997).
- Carrillo-Reid, L., Han, S., Yang, W., Akrouh, A., Yuste, R. Controlling visually guided behavior by holographic recalling of cortical ensembles. Cell. 178 (2), 447-457 (2019).
- Marshel, J. H., et al. Cortical layer-specific critical dynamics triggering perception. Science. 365 (6453), (2019).
- Gill, J. V., et al. Precise holographic manipulation of olfactory circuits reveals coding features determining perceptual detection. Neuron. 108 (2), 382-393 (2020).
- Robinson, N. T. M., et al. Targeted activation of hippocampal place cells drives memory-guided spatial behavior. Cell. 183 (6), 1586-1599 (2020).
- Packer, A. M., Russell, L. E., Dalgleish, H. W., Häusser, M. Simultaneous all-optical manipulation and recording of neural circuit activity with cellular resolution in vivo. Nature Methods. 12 (2), 140-146 (2015).
- Mardinly, A. R., et al. Precise multimodal optical control of neural ensemble activity. Nature Neuroscience. 21 (6), 881-893 (2018).
- Yang, W., Carrillo-Reid, L., Bando, Y., Peterka, D. S., Yuste, R. Simultaneous two-photon imaging and two-photon optogenetics of cortical circuits in three dimensions. Elife. 7, 32671 (2018).
- Forli, A., et al. Two-photon bidirectional control and imaging of neuronal excitability with high spatial resolution in vivo. Cell Reports. 22 (11), 3087-3098 (2018).
- Pégard, N. C., et al. Three-dimensional scanless holographic optogenetics with temporal focusing (3D-SHOT). Nature Communications. 8 (1), 1228 (2017).
- Dal Maschio, M., Donovan, J. C., Helmbrecht, T. O., Baier, H. Linking neurons to network function and behavior by two-photon holographic optogenetics and volumetric imaging. Neuron. 94 (4), 774-789 (2017).
- Russell, L. E., et al. All-optical interrogation of neural circuits in behaving mice. Nature Protocols. 17 (7), 1579-1620 (2022).
- Oron, D., Papagiakoumou, E., Anselmi, F., Emiliani, V. Two-photon optogenetics. Progress in Brain Research. 196, 119-143 (2012).
- Hernandez, O., et al. Three-dimensional spatiotemporal focusing of holographic patterns. Nature Communications. 7, 11928 (2016).
- Okada, T., et al. Pain induces stable, active microcircuits in the somatosensory cortex that provide a therapeutic target. Science Advances. 7 (12), 8261 (2021).
- Prakash, R., et al. Two-photon optogenetic toolbox for fast inhibition, excitation and bistable modulation. Nature Methods. 9 (12), 1171-1179 (2012).
- Gonçalves, J. T., Anstey, J. E., Golshani, P., Portera-Cailliau, C. Circuit level defects in the developing neocortex of Fragile X mice. Nature Neuroscience. 16 (7), 903-909 (2013).
- Akiyoshi, R., et al. Microglia enhance synapse activity to promote local network synchronization. eNeuro. 5 (5), (2018).
- Haruwaka, K., et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nature Communications. 10 (1), 5816 (2019).
- Kishi, K. E., et al. Structural basis for channel conduction in the pump-like channelrhodopsin ChRmine. Cell. 185 (4), 672-689 (2022).
- Deisseroth, K. Optogenetics: 10 years of microbial opsins in neuroscience. Nature Neuroscience. 18 (9), 1213-1225 (2015).
- Ko, H., et al. The emergence of functional microcircuits in visual cortex. Nature. 496 (7443), 96-100 (2013).
- Zong, W., et al. Large-scale two-photon calcium imaging in freely moving mice. Cell. 185 (7), 1240-1256 (2022).
- Villette, V., et al. ultrafast two-photon imaging of a high-gain voltage indicator in awake behaving mice. Cell. 179 (7), 1590-1608 (2019).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved