
Method Article
High-Throughput Contractile Measurements of Hydrogel-Embedded Intact Mouse Muscle Fibers Using an Optics-Based System
* These authors contributed equally
In This Article
Summary
Skeletal muscle function can be assessed by quantifying the contractility of isolated muscle fibers, traditionally using laborious, low-throughput approaches. Here, we describe an optics-based, high-throughput method to quantify the contractility of hydrogel-embedded muscle fibers. This approach has applications for drug screening and therapeutic development.
Abstract
In vitro cell culture is a powerful tool to assess cellular processes and test therapeutic strategies. For skeletal muscle, the most common approaches involve either differentiating myogenic progenitor cells into immature myotubes or the short-term ex vivo culture of isolated individual muscle fibers. A key benefit of ex vivo culture over in vitro is the retention of the complex cellular architecture and contractile characteristics. Here, we detail an experimental protocol for the isolation of intact flexor digitorum brevis muscle fibers from mice and their subsequent ex vivo culture. In this protocol, muscle fibers are embedded in a fibrin-based and basement membrane matrix hydrogel to immobilize the fibers and maintain their contractile function. We then describe methods to assess the muscle fiber contractile function using an optics-based, high-throughput contractility system. The embedded muscle fibers are electrically stimulated to induce contractions, after which their functional properties, such as sarcomere shortening and contractile velocity, are assessed using optics-based quantification. Coupling muscle fiber culture with this system allows for high-throughput testing of the effects of pharmacological agents on contractile function and ex vivo studies of genetic muscle disorders. Finally, this protocol can also be adapted to study dynamic cellular processes in muscle fibers using live-cell microscopy.
Introduction
Advances in in vitro cell culture techniques have opened up new possibilities to study tissue regenerative capabilities, pathophysiological cellular mechanisms, and subsequent therapeutic strategies, all while using mammalian tissues under well-controlled conditions1,2,3. The use of in vitro culture systems is well established within the muscle research field4,5. In general, in vitro-differentiated immature myotubes from myogenic progenitor cells are used2,6,7,8. Although progress has been made in the differentiation protocol to generate more mature muscle fibers9, their immaturity still limits the translation of the findings to an in vivo setting1,10. One central issue in the muscle biology field is the inability of in vitro-differentiated myotubes to fully epitomize the complex intracellular structures, cell signaling processes, and extracellular interactions observed in native muscle tissue and, importantly, recapitulate the contractile forces produced by muscle fibers1,2,10,11,12. In addition, the uncoordinated contraction of myotubes during the differentiation process often results in spontaneous detachment from the culture dishes, making the contractile assessment of in vitro differentiated myotubes challenging and restricted to qualitative or semi-quantitative evaluation8,11,12,13. These limitations often necessitate regular in vivo experiments with animals, particularly if muscle contractility is a primary experimental outcome1.
An alternative to culturing in vitro-differentiated myotubes is the ex vivo culture of isolated mature muscle fibers1,14. During ex vivo culture, developmentally mature muscle tissue is excised out of the body, followed by single-cell isolation for cultivation in laboratory conditions1,14. The isolated mature muscle fibers maintain their complex cellular structures observed within the native tissue14,15, and this method opens the possibility for direct interventions, such as genetic manipulation and drug screening, in a well-defined and controllable culture environment. One of the first reports regarding skeletal muscle fiber isolation and ex vivo culture dates back to the 1930s; however, the yield of viable fibers from this protocol was low16. With the continuous optimization of the isolation procedure and culture conditions, a significant improvement in the amount of viable and functional muscle fibers is now possible14,15,17,18,19. One such improvement in the culture conditions involves coating culture dishes with extracellular matrix proteins to promote the adherence of the isolated muscle fibers on the culture dish15,18,20. Usually, laminin coating is used, since laminin is one of the most abundant elements within the extracellular matrix of muscles20,21. The optimization of the isolation procedure combined with the coating of the culture dishes have enabled the muscle research field to keep isolated viable muscle fibers with intact cellular architecture and contractile functionality in culture for short periods of time1,15,18,22.
The most conventional approach used within the muscle field to measure force and contractile capabilities is to mount individual muscle fibers between a length driver motor and a force transducer23,24. In general, the muscle fibers used for these motor-driven setups are dissected from either snap-frozen or fresh tissue, followed by permeabilization or "skinning", which allows for external calcium activation, where varying calcium concentrations are used to induce muscle fiber contraction24. While this method is the gold standard for muscle fiber contractile measurements, only a single muscle fiber can be measured at a time, making this technique a laborious and time-consuming procedure25. Furthermore, the isolation and skinning procedure of the muscle fibers disrupts the various structures involved in excitation-contraction coupling (i.e., calcium release and subsequent reuptake into the sarcoplasmic reticulum), thereby not allowing for the study of relaxation kinetics and any diseases that might affect this process26,27. An alternative to the skinned fiber preparation is using mechanical dissection to isolate intact muscle fibers, where contractile forces can be measured in response to electrical activation28; however, this approach is technically challenging and very time-consuming, resulting in low-throughput measurements. Lastly, in both skinned and intact preparations, the muscle cells are completely removed from the extracellular environment during contractile measurements24, making the investigation of the effect of the extracellular matrix composition/stiffness on muscle fiber contraction impossible24. As a result, the development of alternative methods is needed to enable muscle fiber contractility measurements of isolated intact muscle fibers in a high-throughput manner while recreating the connection between muscle fibers and the extracellular matrix.
Recently, a new optics-based approach for high-throughput muscle fiber contractility measurements was developed29. This optics-based system measures the periodicity of sarcomeres to assess the sarcomere length during contraction using high-speed imaging. Within this system, the cells stay in place in the culture dish while the optics is moved, thereby minimizing the time needed between the measurements of multiple cells29. A major advantage of using this high-throughput, optics-based approach is that it allows for developing culture conditions that are similar to those of the native tissue. An approach used to mimic native in vivo conditions is embedding cells in hydrogels30. Typically, a hydrogel is a viscoelastic material capable of maintaining its volume and shape, and hydrogels have the properties of both solid and liquid materials31. The solid part consists of polymer chains cross-linked to each other, creating a structure that looks similar to a net30,31. The material properties of hydrogels can be tuned to mimic the matrix deposition of muscles30,31. Thus, the combination of a high-throughput, optics-based system with cells embedded in hydrogels opens new possibilities to assess the effects of extracellular matrix composition and mechanical properties on muscle fiber functionality.
The overall aim of this paper is to 1) describe the methodology for the enzymatic isolation and ex vivo culture of muscle fibers in conditions mimicking the native tissue environment and 2) assess muscle fiber contractility using a high-throughput approach. We describe a detailed methodology to easily isolate a large number of single muscle fibers from the flexor digitorum brevis (FDB) muscle using enzymatic digestion. In addition, we describe a technique to embed the isolated muscle fibers in a fibrin-based hydrogel for the sake of mimicking the native environment of the muscles and improving the muscle fiber viability and contractility. We then outline a high-throughput method of measuring live muscle fiber contractions in vitro using this recently developed system. An added advantage of this embedding procedure is the immobilization of fibers during contraction, which may improve the signal-to-noise ratio of these measurements. This gel-embedding method is applicable for both single polymer and composite gel encapsulation procedures, facilitating the assessment of the effects of extracellular matrix composition on muscle fiber contractility.
Protocol
For the ex vivo contraction studies, post-mortem tissue was obtained from animals sacrificed for other approved research projects and/or breeding surplus of the VU University in accordance with the European Council Directive (2010/63/EU) by permission of the Animal Research Law of the Netherlands.
1. Material preparation
- Prepare pipettes for trituration (store in 70% ethanol in a flow cabinet until use). Cut the ends of two P1,000 tips to create different hole sizes; ensure that the hole is just big enough for the muscle to pass through without blocking the pipet. Run the cut ends through a flame so they become smooth.
- Prepare Sylgard dishes as described elsewhere32, and sterilize with 70% ethanol in advance for use in securing the paw and muscle during isolation.
NOTE: Sylgard dishes can be reused after thorough cleaning with deionized water and 70% ethanol. - Prepare the following solutions prior to the isolation procedure: dissection medium, fibrin culture medium, and muscle digestion medium (see Table 1).
NOTE: The muscle digestion medium should be filter-sterilized using a 0.22 µm filter. All the prepared solutions should be equilibrated in a standard cell culture incubator at 37 °C and 5% CO2 for at least 30 min before usage. - After the muscle digestion, prepare the following solutions to cast the hydrogel: cell mix and matrix mix (see Table 2). Combine the cell mix and matrix mix in a ratio of 1:1 for a final fibrin concentration of 2.5 mg/mL.
NOTE: Prepare the matrix on ice, and use prechilled pipette tips to prevent the premature polymerization of the basement membrane matrix. The thrombin:fibrinogen ratio of 1:10 (units per mg of fibrinogen) used here has been optimized for a polymerization time of 30 min. If the gel polymerizes within 30 min, a lower thrombin concentration should be used. See the discussion for more information.
2. FDB muscle dissection
- Euthanize the mouse by cervical dislocation. Disinfect the hind limb with 70% alcohol.
- Cut off the lower hind leg above the ankle. Cut open the skin on the dorsal side of the foot toward the toes.
NOTE: Cut off the lower hind leg at the ankle joint to prevent damage to the lower limb muscles and tibia if they are needed later. - Carefully peel the skin toward the toes. Be careful not to damage the muscle. The FDB is the most superficial muscle on the ventral side of the foot.
- Place the dissected foot in a Sylgard dish with 10 mL of prewarmed dissection medium at 37 °C. Pin the foot through the skin that is still attached to the toes, and pin the lower leg beyond the ankle.
- Carefully remove the connective tissue on top of the muscle. Cut the tendon at the heel, and lift the muscle up by its tendon.
- Cut alongside and underneath the muscle through the connective tissue. Keep cutting until the toe tendons are exposed.
- When half of the length of the three tendons are exposed, cut the tendons, and release the muscle from the foot. OPTIONAL: Trim off the fourth lateral tendon and its muscle fibers.
- Clean the connective tissue off the muscle, and transfer to a tube containing prewarmed dissection medium.
3. FDB muscle digestion
- Prepare the muscle digestion medium in accordance with Table 1.
- Transfer the FDB muscles to the muscle digestion medium using a serological pipette. Incubate in a tissue culture incubator at 37 °C and 5% CO2 for 80 min.
NOTE: This time should be optimized for each collagenase batch. The digestion is complete when the muscle starts to fray and looks enlarged. See the discussion for the optimization of the digestion time. - After the digestion, transfer the muscle to a 15 mL tube containing 3 mL of dissection medium, and incubate for 30 min before trituration.
4. FDB muscle trituration and gravity sedimentation
- Triturate the muscle using the previously prepared trituration tips (step 1.1) by pipetting the muscle, going from the largest to the smallest size. If this step takes longer than 5 min, place the muscle into the incubator for 5 min to allow it to rest.
- Triturate until the muscle fibers have mostly come off the tendon and the tendon can pass through a P200 tip. Remove the tendons.
- Add the dissociated FDB fibers to a 15 mL tube containing 10 mL of dissection medium, and allow the fibers to settle in the incubator for 20 min. Observe the pellet formed.
- Optional: Remove 10 mL of medium from the top, and repeat step 4.3. This step facilitates the removal of excess debris and associated mononucleated cells.
5. FDB fiber embedding
- Carefully remove all the medium from the top of the fiber pellet. Resuspend the cells in 875 µL of cell mix per FDB muscle (on ice).
NOTE: One FDB muscle yields enough fibers for seven wells of a 24-well plate. This density can be adjusted according to the needs of the experiment. The following gel volume (250 µL) is optimized for a 24-well format but can be scaled accordingly for other formats. - Aliquot 125 µL of the cell mix into single microcentrifuge tubes.
- One well at a time, add 125 µL of matrix mix to a cell suspension aliquot, and mix by pipetting up and down carefully, avoiding the formation of bubbles.
- Immediately transfer the final mix into a well.
NOTE: Be sure to pipet the mix into the middle of the well. Repeat step 5.3 and step 5.4 for each well. - Solidify the gels in an incubator for 30-45 min. After solidification, carefully add the culture medium to the wells.
NOTE: Fast pipetting can detach the hydrogel from the well. From this point on, the fibers can be stimulated and measured. However, in our experience, letting the fibers adjust to the culture conditions for 24 h may improve the fiber contractility. - For longer culture, replenish the culture medium by a half-change every 2 days. To do this, remove half of the culture medium, and replace it with an equal amount of fresh medium.
6. Optics-based contractile measurements
- Turn on the optics-based contractile measurement system (see the Table of Materials), fluorescence lamp, the electrical cell pacer, and the computer. Set the electrical stimulator to 1.0 Hz, 10.0 V, and a pulse duration of 5.00 ms to stimulate the isolated muscle fibers.
- Insert the plate into the measurement system. Connect the pacer to the pacing insert, and insert it into the culture plate.
- Open the program IonWizard, and open a new file by clicking on File (top left of the screen) | New.
- Check if the program is on the correct experiment, Skeletal Sarcomere, by clicking on New | Collect Experiment. To change the experiment, click on the desired experiment, and press add. For the Skeletal sarcomere experiment, apply the following settings: Sarc 20x, Average lines, single FFT, 250 Hz sampling rate, and an acquisition time of 10 s.
NOTE: The experimental settings should be prepared prior to the experiment. The settings can be adjusted to the needs of the experiment. - Adjust the temperature of the measurement system to 25 °C. Click on open cell finder under the toolbar, and wait for a new screen to pop up. On the top right of this screen, select the plate type and active wells.
NOTE: The measurements are performed at 25 °C to reduce the contraction speed of the fast-twitching FDB muscle fibers, thereby preventing undersampling of the contractile event. - Bring the fibers into focus by adjusting the focus slider bar. Alternatively, use the W key and S key for this focusing function.
- Enable the pacing to start the electrical stimulation. Observe that the fibers now start twitching.
NOTE: If no fibers are twitching, make sure all the wires are connected and the pacer is submerged completely. If after this there is still no movement, the fibers may not be responding due to overdigestion or excessive damage during trituration. - Focus the measurement area on the end of a fiber such that the sarcomeres run vertically. Make sure the sarcomeres are in focus. If the sarcomeres are in focus, a single peak is visible in the toolbar. During contraction, this peak will move to the right as the sarcomere shortens.
NOTE: The purple measurement area can be adjusted prior to starting the experiment. To ensure a proper measurement, include ~20 sarcomeres. If this peak changes shape during contraction, this may indicate that the sarcomere is obscured or out of focus during the contraction, which introduces noise. - Start the experiment by clicking on start in the toolbar. Press the Q key to start a measurement, and wait for the program to measure 10 contraction transients. If more than four transients look noise-free, accept the measurement by pressing the Z key. If the transients have too much noise, reject the measurement by pressing the X key.
- Measure between 10 fibers and 20 fibers per condition. The locations of the previously measured fibers are retained.
- Optional: Add compounds, after which fibers can be remeasured at this point.
- When the experiment is finished, press the stop button in the lower toolbar to close the cell finder window. Save the file, and start a new file.
- To analyze the data, open the program "Cytosolver desktop". Click on import, and select the file(s) to be analyzed.
- After the program has finished the analysis, look for blue, red, and gray peaks. The blue peaks are transients that are accepted by the program. The red peaks are transients that are rejected by the program, and the gray peaks are transients rejected by the user.
NOTE: The rejection criteria can be adjusted in the analysis software. In general, values are rejected if they exceed a maximum derivative limit, and transients are rejected based on curve fit R2 values of <0.95. - Click on export. Tick the following boxes: Averaged transient data and export to excel.
- When finished, turn off all the machines. Take out the culture plate, and dispose of it. Clean the pacer electrodes with deionized water and 70% ethanol.
Results
Using this protocol, single FDB muscle fibers were isolated and embedded in hydrogel. An overview of the muscle dissection procedure is shown in Figure 1. The FDB muscle is exposed with intact tendons and cut loose from the fascia. Maintaining the tendons of the muscles as fixation points minimizes potential damage to the muscle fibers during the isolation procedure. Excess connective tissue can be trimmed off to reduce debris and the growth of secondary cell types. Once the muscle has been excised and cleaned sufficiently, the muscle is enzymatically digested using collagenase, and single muscle fibers are released by trituration prior to embedding the isolated cells in hydrogels. The isolation of FDB muscle fibers provides relatively short muscle fibers, which have the advantage of being easily manipulated. Due to their size, FDB muscle fibers can be pipetted safely without tangle-induced damage and are easily embedded within a hydrogel. As single muscle fibers do not adhere to culture plates very well, embedding the fibers in hydrogel ensures that the fibers stay put during the cell culture and contractile measurements. Furthermore, the addition of a basement membrane matrix to the fibrin gel allows for interactions between the muscle fibers and the matrix, mimicking the native in vivo environment. Isolated single muscle fibers can be manipulated and maintained in culture for several days post isolation. In Figure 2, an example of isolated FDB fibers embedded in a hydrogel matrix is shown. The healthy fibers have visible sarcomeres and are stretched out straight (blue arrow), while the curved fibers are typically damaged or unviable (yellow arrow) and should be excluded from the measurements. Hypercontracted fibers appear as dark balled-up objects in the matrix (red arrow). If the isolation procedure was successful, the proportion of healthy fibers should be ~75%. A larger proportion of hypercontracted fibers usually indicates damage during the isolation procedure. The membrane of the muscle fibers can be damaged either by overdigestion of the muscle or damage to the fibers during trituration. Trituration damage mainly occurs if the muscle is underdigested and, thus, does not come apart easily.
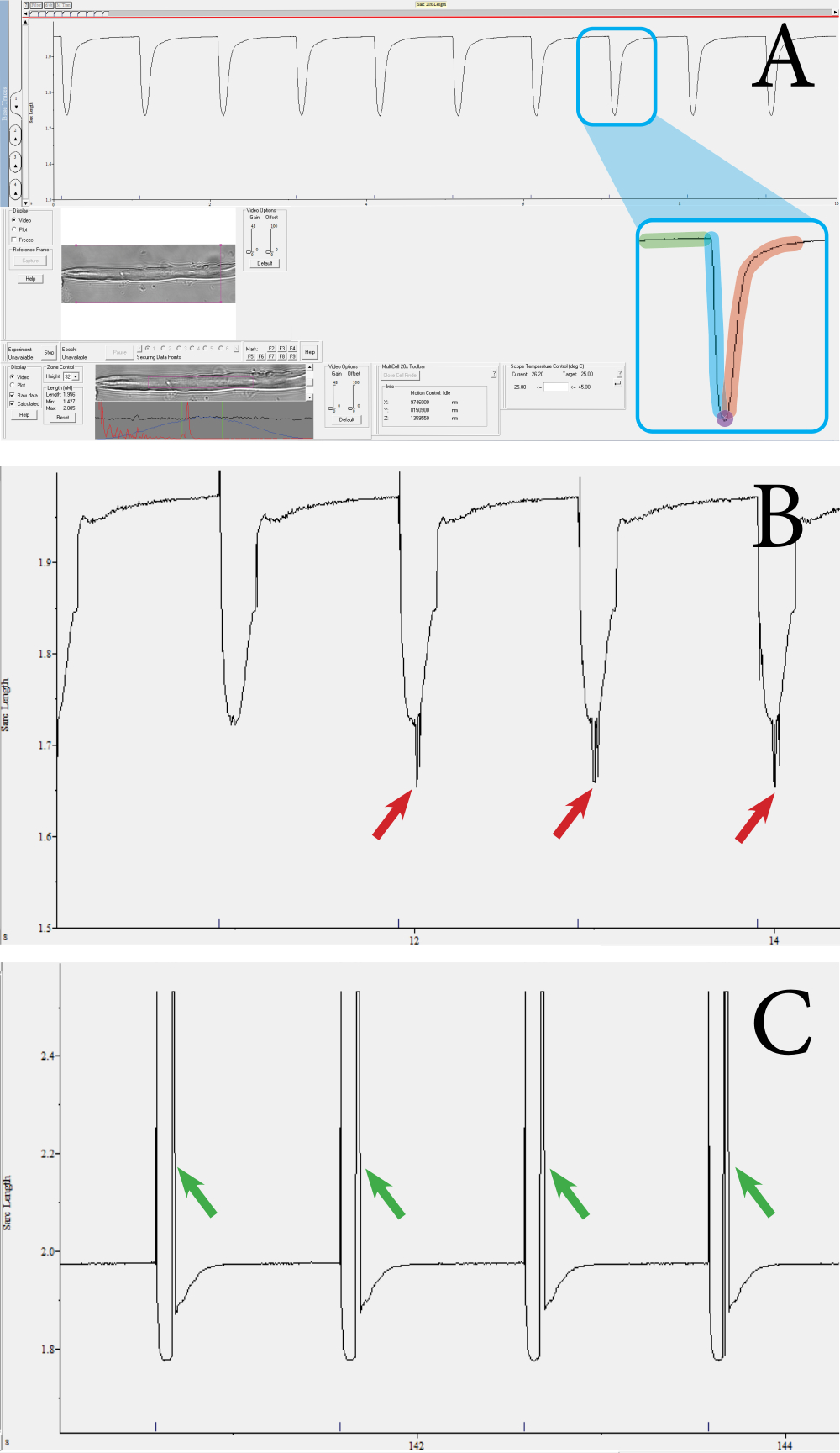
The fiber functionality and viability were assessed through contractile measurements of the muscle fibers utilizing an optics-based, high-throughput contractile measurement system. The system outputs several parameters, such as the sarcomere length, percentage of sarcomere shortening, contractile velocity, and relaxation velocity. Using the contractile measurement system, sarcomere contraction can be measured per muscle fiber. Figure 3 shows examples of muscle fiber contractions measured using the measurement system. From a single contraction transient, the following parameters are obtained: sarcomere length at baseline, contraction duration, sarcomere length at maximal contraction, and relaxation duration (Figure 3A). These parameters are used to calculate the percentage of sarcomere shortening, contraction, and relaxation velocities. Average velocity values can also be calculated from the duration and absolute shortening values, if needed. A valid contraction measurement features a straight baseline, followed by a dip to the peak, and a return to the baseline (Figure 3A). Noise, unfocused sarcomeres, or aberrant movement of the fiber can influence the measurement transient (Figure 3B,C), and these measurements can be discarded manually or will be rejected by the analysis program. In this approach, the clear visualization of the sarcomeres is important for measuring the contractions; thus, anything that reduces the visibility of the sarcomeres may introduce noise. This could occur if there is movement of the sarcomere outside of the focal plane during contraction. Measurements in which a series of contractions differ in speed or depth should also be excluded from the dataset.
The contractile data of single muscle fibers obtained with this system can be used to compare different culture conditions. The effectiveness of the system is illustrated in Figure 4. Here, we measured the contractions of FDB muscle fibers in both 2D (laminin-coated culture plates) and 3D (fibrin hydrogel) culture formats. A higher percentage of usable measurements was achieved in 3D, as embedding the fibers in the gel prevented lateral movement and other movement artifacts from influencing the measurement (Figure 4A). Embedding the fibers had no significant effect on either the sarcomere shortening or maximum contraction speed values as compared to the 2D-cultured fibers (Figure 4B). To illustrate how different matrices influence the contraction of FDB muscle fibers, we also compared this fibrin hydrogel to a pure basement membrane matrix (4 mg/mL) (Figure 4C). The reduced contractility observed in the pure basement membrane matrix was likely due to the stiffness of the gel or increased cell-matrix interactions. Fibrin concentrations of up to 7 mg/mL have also been tested, with no significant effect on contractile speed and shortening (unpublished data). The usage of this fibrin-based hydrogel ensures minimal interference with contractile parameters.

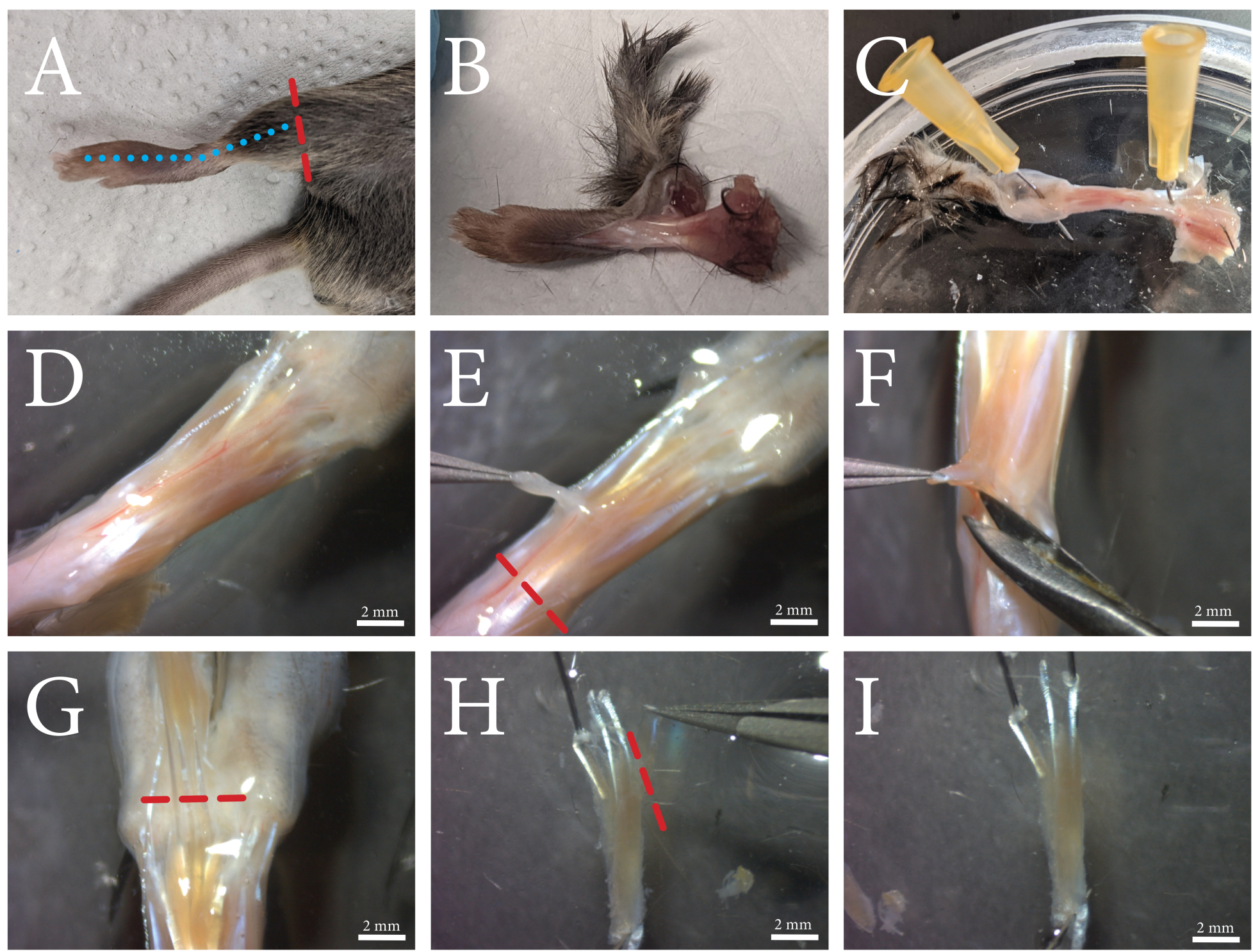
Figure 1: An overview of the FDB muscle dissection procedure. (A) The hind limb is cut above the ankle (red dashed line), and (B) the skin is removed by cutting along the top of the foot along the blue dashed line. (C) The foot is pinned through the ankle and the skin at the toes. (D) Microscopic view of the exposed muscle and its surrounding connective tissue. The fascia is visible as a white opaque layer through which the vasculature runs. (E) The fascia is removed from the muscle, and the tendon is cut along the red dashed line. (F) The FDB muscle is separated from the underlying tissue by lifting and cutting underneath and alongside the muscle. (G) When the tendons of the toes are clearly visible, the FDB is cut loose along the red dashed line. (H) The FDB is secured by the tendons, and the fourth lateral tendon and its fibers can be removed by cutting along the red dashed line. Excess fascia can now be trimmed off. (I) After cleaning, the FDB muscle is transferred to the collagenase solution. Scale bars = (D-I) 2 mm. Abbreviation: FDB = flexor digitorum brevis. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Microscopic image of FDB muscle fibers embedded in hydrogel after 24 h of culture. Blue arrow: Example of a viable FDB muscle fiber. Yellow arrow: Example of a twisted FDB muscle fiber. Twisted muscle fibers may have reduced viability and impaired contractions and, thus, should be excluded from the measurements. Red arrow: Example of a hypercontracted FDB muscle fiber. Excessive hypercontraction occurs when trituration is performed too vigorously or can be caused by collagenase overdigestion or the use of non-equilibrated culture medium. Scale bar = 100 µm. Abbreviation: FDB = flexor digitorum brevis. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Example of fiber contraction transients measured using the optics-based, high-throughput contractile system. (A) Example of a normal contraction transient. This transient is obtained from the sarcomeres enclosed by the purple square shown in the lower toolbar. The transient consists of the following components: sarcomere length at baseline (green), contraction duration (blue), sarcomere length at maximal contraction (purple), and relaxation duration (red). Parameters such as the velocity and percentage of contraction are calculated from these values. (B) Example of an inadequate contraction transient. These measurements occur when the sarcomere signal is not picked up due to noise (see red arrows). (C) Example of a contraction transient that has a movement artifact (see green arrows). Movement artifacts occur when the muscle fiber moves outside the focus during the contraction. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Representative data obtained using the optics-based high-throughput contractile system when comparing 2D versus 3D conditions and different hydrogels. (A) Percentage of accepted and rejected contraction measurements found by the data analysis program in 2D culture and in 3D culture based on 30 measurements. (B) Comparison of the maximum contraction speed in 2D-cultured and 3D-cultured muscle fibers isolated from three mice after 24 h of culture. (C) Comparison of the sarcomere shortening in 2D-cultured and 3D-cultured muscle fibers isolated from three mice after 24 h of culture. (D) Comparison of the maximum contraction speed of pure basement membrane matrix (Matrigel)-embedded and fibrin hydrogel-embedded muscle fibers isolated from three mice after 24 h of culture. (E) Comparison of the sarcomere shortening of pure basement membrane matrix (Matrigel)-embedded and fibrin hydrogel-embedded muscle fibers isolated from three mice after 24 h of culture. The data were analyzed using a Student's t-test and are shown as mean ± SD. Each data point is one muscle fiber. Please click here to view a larger version of this figure.
{kind=link}
Table 1: Composition of the dissection medium used to dissect the muscle, the fibrin culture medium used to culture the isolated muscle fibers, and the muscle digestion medium used to enzymatically digest the isolated muscles. Please click here to download this Table.
Table 2: Composition of the cell mix used to cast the hydrogels and the matrix mix used to cast the hydrogels. Please click here to download this Table.
Discussion
Here, we detail a protocol to perform the enzymatic isolation and culture of FDB muscle fibers in a 3D culture format, followed by contractile measurements using an optics-based contractile measurement system. This protocol has a number of advantages, including the 1) straight-forward and timely isolation of many intact muscle fibers from a single muscle; 2) the embedding of the muscle fibers in a tunable hydrogel matrix; 3) the performance of high-throughput contractility measurements using the optics-based system; and 4) the ability to conduct repeated measurements of the same muscle fibers following an intervention. The isolation of single live muscle fibers provides mature muscle cells that retain their contractile function. As the muscle fibers obtained from the mouse FDB are relatively small, they are easily manipulated during isolation and maintain their straight shape, allowing for downstream contractile measurements. Although the system was primarily developed to study cardiomyocyte contraction, skeletal muscle fibers contain the same contractile machinery with easily distinguishable sarcomere patterning and, thus, can also be measured using this system29. The coupling of single-cell contractile measurements with live ex vivo muscle fiber culture is a powerful tool to assess mature muscle fiber health and function in response to electrical activation.
A limitation of using dissociated muscle fibers is the lack of external forces (i.e., passive stretch) applied to the fibers, which results in lower resting sarcomere lengths compared to those found in vivo. Although a sarcomere length of 2.4-2.5 µm ensures optimal force production, the resting sarcomere length can vary greatly33. While the in vivo resting sarcomere length of the FDB has not yet been described, our own unpublished data suggest an average length of 2.2 µm. The current results show an average resting sarcomere length of ~1.95 µm in unloaded FDB fibers after 24 h in culture (Figure 3). Although this lower resting sarcomere length would result in lower force production, a length of ~1.95 µm should still produce >90% of the maximum force34. As such, these sarcomere lengths should be sufficient to determine differences in fiber function between different genetic models or following drug treatments. Additionally, the embedding of fibers in a hydrogel provides many additional points for adhesion compared to free-floating 2D cultured fibers, which would limit further sarcomere shortening over time.
One advantage of this muscle fiber isolation protocol is the usage of an easily dissected fast-twitch muscle consisting of relatively small muscle fibers when compared to other muscles, such as the extensor digitorum longus (EDL). Their smaller size makes the muscle isolation more suitable for trituration based-separation, thereby reducing the chance of pipet- or tangle-induced damage to the muscle fibers. The extracellular matrix of FDB muscles can be easily enzymatically digested with collagenase, allowing for the isolation of hundreds of muscle fibers in a short period of time. However, overdigestion can lead to damage to the muscle fibers. The overdigestion of the muscle fibers can be recognized when the muscle falls apart almost instantly when triturating the muscle or when a large proportion of the cell volume is hypercontracted during the cell seeding procedure. To prevent overdigestion of the muscle, the digestion time needs to be optimized for each collagenase batch. To test this, two FDB muscles should be digested in parallel with a staggered digestion time of 5 min between them. The digestion time with the highest yield of viable muscle fibers should be chosen. This optimization should then be performed a second time, again with 5 min of separation in digestion time. The digestion time yielding the highest viable muscle fibers should be used as the optimal digestion time for the current batch of collagenase. Another way to limit the batch-to-batch variability of collagenase would be to directly calculate the activity units per milliliter of stock solution and then reconstitute the subsequent collagenase batches to the same amount. Lastly, the digestion times may need to be optimized across different mouse strains, for example, if studying aged or diseased animals that exhibit increased extracellular matrix deposition35,36.
The possibility of pipetting viable isolated muscle fibers opens possibilities to culture FDB muscle fibers in various culture conditions. One such option is the culturing of these fibers in hydrogels to mimic the native tissue culture environment. This embedding protocol ensures that the fibers stay in place during the contractile measurements and has been optimized to allow the fibers to settle to the bottom of the plate before the gel sets. However, this protocol may need to be adjusted to accommodate differences in the thrombin and fibrinogen stocks. If the thrombin activity is too high, the gel will set prematurely, and the fibers may stay suspended at higher locations outside the focal plane of the microscope. If this happens, the thrombin:fibrinogen ratio needs to be adjusted. This can be tested by plating fibers in increasingly lower thrombin concentrations and taking note of how long the polymerization takes. Typically, this should not happen faster than 30 min. However, having a thrombin concentration that is too low may also impair the polymerization process. Another method to ensure the fibers are in the correct focal plane is to first seed them using a 2D protocol and then add a layer of hydrogel over the fibers after they have adhered to the culture plate. However, one should be aware that removing the medium from the fibers may induce hypercontraction, as they are sensitive to drying out. It is also unclear if the hydrogel will fully attach to the culture plate, and it might come loose more easily. Therefore, this embedding procedure is preferable for keeping fibers viable and in place for contraction measurements.
The usage of this protocol enables the study of the contractile dynamics of mature muscle fibers ex vivo and can be applied to both healthy mice and those carrying genetic mutations for muscular diseases. Likewise, it enables the testing of how culture conditions or the addition of compounds affect muscle fiber function. The contractile data obtained using the optics-based system give an indication of the contractile ability of live single muscle fibers, and changes in this ability can be correlated to fiber health. These data alone are, however, not sufficient to determine if these changes occur at the actin-myosin cross-bridging or calcium release stages of muscle contraction. Although we do not describe methods to measure calcium signaling in this protocol, this setup is also capable of measuring Fura-based calcium transients in contracting muscle cells29. A drawback of this system is that the FDB muscle contains only fast-twitch type IIa/IIx muscle fibers, and muscles containing slow-twitch type I fibers of this size have not yet been described37. This eliminates the ability to study fiber type-specific functioning using this method. The protocol we propose here could potentially be adapted for other muscles such as the EDL or the soleus to study fiber type differences. Due to their larger size, this protocol would need to be optimized further for these muscles. Longer fibers tend to get tangled during the gravity sedimentation step and will rupture if manipulated by pipetting, which would lead to lower yield. Due to their incompatibility with pipetting, longer fibers are, therefore, also less compatible with the gel-embedding technique. Measurements of these fibers can still be done in a 2D culture format, but the fibers may move around more during contraction due to their size, thus affecting the signal-to-noise ratio. Another limitation of this system is the inability to perform force measurements alongside the contractile measurements, such as those force measurements that can be obtained using other intact muscle fiber preparations28. However, this limitation can be circumvented by estimating the force generated by the muscle fiber. The generated force of muscle fibers can be estimated if the muscle fiber shape during contracted and relaxed states as well as the Young's modulus of the matrix are known25. Nonetheless, this optic-based system provides an easy-to-use, high-throughput approach for studying muscle contractile function and opens up a range of new possibilities for studying genetic muscle diseases and therapeutic interventions.
Disclosures
The authors have no conflicts of interest to declare.
Acknowledgements
The authors would like to thank Sylvia Bogaards, Sanna Luijcx, Valentijn Jansen, Michiel Helmes, and Emmy Manders for their technical expertise in helping develop this protocol. This work was supported by awards from the Muscular Dystrophy Association (Development Award MDA603238 to T.J.K), the Dutch Cardiovascular Alliance (Talent Grant to T.J.K), and the National Health and Medical Research Council (NHMRC, Australia; Fellowship APP1121651 to M.Y).
Materials
| Name | Company | Catalog Number | Comments |
| Aprotinin, from Bovine Lung | Thermo Scientific | AAJ63039MC | 100 mM stock solution in PBS can be stored at -20 °C. Sterilize stock solution using a 0.22 µm filter. |
| Collagenase type 2 | Worthington | 77336 | 10% (w/v) stock solution can be stored at -20 °C. Weighing collagenase should be done in a safety cabinet as inhalation is dangerous. |
| Fetal Bovine Serum | Thermo Fisher | 10500064 | |
| Fibrinogen from Bovine Plasma | Sigma Aldrich | 50-176-5054 | 20 mg/mL stock solution in PBS can be stored at -80 °C. Sterilize stock solution using a 0.22 µm filter. |
| Geltrex LDEV-Free Reduced Growth Factor Basement Membrane Matrix | Thermo Fisher | A1413201 | 4 mg/mL stock solution is prepared in MEM and stored at -20 °C. |
| Gibco MEM High glucose + pyruvate | Thermo Fisher | 11095080 | |
| Horse serum | Thermo Fisher | H1270 | |
| Matrigel GFR Membrane Matrix | Corning | CB-40230 | 4 mg/mL stock solution is prepared in MEM and stored at -20 °C. |
| Penicillin/Streptomycin | Sigma Aldrich | P4333 | |
| Serum Replacement 2 (50x) | Sigma Aldrich | S9388 | |
| Thrombin, Bovine Plasma | Thermo Scientific | AAJ63383EXP | 125 U/mL stock in PBS can be stored at -20 °C. Sterilize stock solution using a 0.22 µm filter. |
| Tranexamic Acid | Thermo Scientific | AC228042500 | 80 mM stock solution in PBS can be stored at -20 °C. Sterilize stock solution using a 0.22 µm filter. |
| Equipment | |||
| 24-well electrical stimulator | IonOptix | N/a | |
| Dumont #55 Forceps | Fine Science Tools | 11295-51 | |
| Extra Fine Bonn Scissors | Fine Science Tools | 14084-08 | |
| MultiCell Cytocypher | IonOptix | N/a | |
| MyoCam-S3 | IonOptix | N/a | |
| MyoPacer | IonOptix | N/a | |
| SYLGARD 184 silicone elastomer, Base & Curing Agent | Dow corning | N/a | |
| Vannas Spring Scissor - 25 mm Cutting Edge | Fine Science Tools | 15002-08 | |
| Software | |||
| CytoSolver | IonOptix | N/a | |
| IonWizard | IonOptix | N/a |
References
- Smith, L. R., Meyer, G. A. Skeletal muscle explants: Ex-vivo models to study cellular behavior in a complex tissue environment. Connective Tissue Research. 61 (3-4), 248-261 (2020).
- Khodabukus, A., Prabhu, N., Wang, J., Bursac, N. In vitro tissue-engineered skeletal muscle models for studying muscle physiology and disease. Advanced Healthcare Materials. 7 (15), 1701498 (2018).
- Fernandez-Costa, J. M., Fernandez-Garibay, X., Velasco-Mallorqui, F., Ramon-Azcon, J. Bioengineered in vitro skeletal muscles as new tools for muscular dystrophies preclinical studies. Journal of Tissue Engineering. 12, 2041731420981339 (2021).
- Romagnoli, C., Iantomasi, T., Brandi, M. L. Available in vitro models for human satellite cells from skeletal muscle. International Journal of Molecular Sciences. 22 (24), 13221 (2021).
- Dessauge, F., Schleder, C., Perruchot, M. -. H., Rouger, K. 3D in vitro models of skeletal muscle: Myopshere, myobundle and bioprinted muscle construct. Veterinary Research. 52 (1), 72 (2021).
- Hosoyama, T., Meyer, M. G., Krakora, D., Suzuki, M. Isolation and in vitro propagation of human skeletal muscle progenitor cells from fetal muscle. Cell Biology International. 37 (2), 191-196 (2013).
- Guo, X., et al. In vitro differentiation of functional human skeletal myotubes in a defined system. Biomaterials Science. 2 (1), 131-138 (2014).
- Denes, L. T., et al. Culturing C2C12 myotubes on micromolded gelatin hydrogels accelerates myotube maturation. Skeletal Muscle. 9 (1), 17 (2019).
- Pimentel, M. R., Falcone, S., Cadot, B., Gomes, E. R. In vitro differentiation of mature myofibers for live imaging. Journal of Visualized Experiments. (119), e55141 (2017).
- Khodabukus, A. Tissue-engineered skeletal muscle models to study muscle function, plasticity, and disease. Frontiers in Physiology. 12, 619710 (2021).
- Engler, A. J., et al. Myotubes differentiate optimally on substrates with tissue-like stiffness: Pathological implications for soft or stiff microenvironments. Journal of Cell Biology. 166 (6), 877-887 (2004).
- Huang, N. F., et al. Myotube assembly on nanofibrous and micropatterned polymers. Nano Letters. 6 (3), 537-542 (2006).
- Earle, A. J., et al. Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nature Materials. 19 (4), 464-473 (2020).
- Stange, K., Ahrens, H. E., von Maltzahn, J., Rontgen, M. Isolation and ex vivo cultivation of single myofibers from porcine muscle. In Vitro Cellular and Developmental Biology. Animal. 56 (8), 585-592 (2020).
- Ravenscroft, G., et al. Dissociated flexor digitorum brevis myofiber culture system--A more mature muscle culture system. Cell Motility and the Cytoskeleton. 64 (10), 727-738 (2007).
- Ramsey, R. W., Street, S. F. The isometric length-tension diagram of isolated skeletal muscle fibers of the frog. Journal of Cellular and Comparative Physiology. 15 (1), 11-34 (1940).
- Selvin, D., Hesse, E., Renaud, J. M. Properties of single FDB fibers following a collagenase digestion for studying contractility, fatigue, and pCa-sarcomere shortening relationship. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 308 (6), R467-R479 (2015).
- Renzini, A., et al. Culture conditions influence satellite cell activation and survival of single myofibers. European Journal of Translational Myology. 28 (2), 7567 (2018).
- Pasut, A., Jones, A. E., Rudnicki, M. A. Isolation and culture of individual myofibers and their satellite cells from adult skeletal muscle. Journal of Visualized Experiments. (73), e50074 (2013).
- Holmberg, J., Durbeej, M. Laminin-211 in skeletal muscle function. Cell Adhesion and Migration. 7 (1), 111-121 (2013).
- Stuelsatz, P., Keire, P., Yablonka-Reuveni, Z. Isolation, culture, and immunostaining of skeletal muscle myofibers from wildtype and nestin-GFP mice as a means to analyze satellite cell. Methods in Molecular Biology. 1556, 51-102 (2017).
- Alkhateeb, H., Chabowski, A., Bonen, A. Viability of the isolated soleus muscle during long-term incubation. Applied Physiology, Nutrition, and Metabolism. 31 (4), 467-476 (2006).
- Roche, S. M., Gumucio, J. P., Brooks, S. V., Mendias, C. L., Claflin, D. R. Measurement of maximum isometric force generated by permeabilized skeletal muscle fibers. Journal of Visualized Experiments. (100), e52695 (2015).
- Ottenheijm, C. A., et al. Altered myofilament function depresses force generation in patients with nebulin-based nemaline myopathy (NEM2). Journal of Structural Biology. 170 (2), 334-343 (2010).
- Rausch, M., et al. Measurement of skeletal muscle fiber contractility with high-speed traction microscopy. Biophysical Journal. 118 (3), 657-666 (2020).
- de Winter, J. M., et al. KBTBD13 is an actin-binding protein that modulates muscle kinetics. Journal of Clinical Investigation. 130 (2), 754-767 (2020).
- Wijnker, P. J. M., vander Velden, J. Mutation-specific pathology and treatment of hypertrophic cardiomyopathy in patients, mouse models and human engineered heart tissue. Biochimica et Biophysica Acta. Molecular Basis of Disease. 1866 (8), 165774 (2020).
- Cheng, A. J., Westerblad, H. Mechanical isolation, and measurement of force and myoplasmic free [Ca(2+)] in fully intact single skeletal muscle fibers. Nature Protocols. 12 (9), 1763-1776 (2017).
- Cao, L., Manders, E., Helmes, M. Automatic detection of adult cardiomyocyte for high throughput measurements of calcium and contractility. PLoS One. 16 (9), e0256713 (2021).
- Geckil, H., Xu, F., Zhang, X., Moon, S., Demirci, U. Engineering hydrogels as extracellular matrix mimics. Nanomedicine. 5 (3), 469-484 (2010).
- Lin, C. C., Anseth, K. S. PEG hydrogels for the controlled release of biomolecules in regenerative medicine. Pharmaceutical Research. 26 (3), 631-643 (2009).
- Cold Spring Harbor Protocols. Sylgard-coated coverslips and petri dishes. Cold Spring Harbor Protocols. 2022 (8), (2022).
- Moo, E. K., Fortuna, R., Sibole, S. C., Abusara, Z., Herzog, W. In vivo sarcomere lengths and sarcomere elongations are not uniform across an intact muscle. Frontiers in Physiology. 7, 187 (2016).
- Moo, E. K., Leonard, T. R., Herzog, W. The sarcomere force-length relationship in an intact muscle-tendon unit. Journal of Experimental Biology. 223, 215020 (2020).
- Schuler, S. C., et al. Extensive remodeling of the extracellular matrix during aging contributes to age-dependent impairments of muscle stem cell functionality. Cell Reports. 35 (10), 109223 (2021).
- Carberry, S., Zweyer, M., Swandulla, D., Ohlendieck, K. Proteomics reveals drastic increase of extracellular matrix proteins collagen and dermatopontin in the aged mdx diaphragm model of Duchenne muscular dystrophy. International Journal of Molecular Medicine. 30 (2), 229-234 (2012).
- Tarpey, M. D., et al. Characterization and utilization of the flexor digitorum brevis for assessing skeletal muscle function. Skeletal Muscle. 8 (1), 14 (2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved