Method Article
Using the Chick Embryo Brain as a Model for In Vivo and Ex Vivo Analyses of Human Glioblastoma Cell Behavior
In This Article
Summary
Chick embryos are used for studying human glioblastoma (GBM) brain tumors in ovo and in ex vivo brain slice co-cultures. GBM cell behavior can be recorded by time-lapse microscopy in ex vivo co-cultures, and both preparations can be analyzed at the experimental endpoint by detailed 3D confocal analysis.
Abstract
The chick embryo has been an ideal model system for the study of vertebrate development, particularly for experimental manipulations. Use of the chick embryo has been extended for studying the formation of human glioblastoma (GBM) brain tumors in vivo and the invasiveness of tumor cells into surrounding brain tissue. GBM tumors can be formed by injection of a suspension of fluorescently labeled cells into the E5 midbrain (optic tectum) ventricle in ovo.
Depending on the GBM cells, compact tumors randomly form in the ventricle and within the brain wall, and groups of cells invade the brain wall tissue. Thick tissue sections (350 µm) of fixed E15 tecta with tumors can be immunostained to reveal that invading cells often migrate along blood vessels when analyzed by 3D reconstruction of confocal z-stack images. Live E15 midbrain and forebrain slices (250-350 µm) can be cultured on membrane inserts, where fluorescently labeled GBM cells can be introduced into non-random locations to provide ex vivo co-cultures to analyze cell invasion, which also can occur along blood vessels, over a period of about 1 week. These ex vivo co-cultures can be monitored by widefield or confocal fluorescence time-lapse microscopy to observe live cell behavior.
Co-cultured slices then can be fixed, immunostained, and analyzed by confocal microscopy to determine whether or not the invasion occurred along blood vessels or axons. Additionally, the co-culture system can be used for investigating potential cell-cell interactions by placing aggregates of different cell types and colors in different precise locations and observing cell movements. Drug treatments can be performed on ex vivo cultures, whereas these treatments are not compatible with the in ovo system. These two complementary approaches allow for detailed and precise analyses of human GBM cell behavior and tumor formation in a highly manipulatable vertebrate brain environment.
Introduction
In vitro studies of cancer cell behaviors are often used to dissect potential mechanisms that operate during the more complex behavior that is observed during tumor formation and cell invasion in in vivo xenograft models. For example, with glioblastoma (GBM), in vitro studies have uncovered mechanisms of how L1CAM potentially operates during tumor formation and brain invasion in a novel chick embryo xenograft brain tumor model1,2,3,4,5. Although in vitro and in vivo experiments complement each other in useful ways, they leave a substantial gap in how the results can be correlated. For instance, mechanistic analyses of GBM cell motility on a dish is a highly artificial situation, and in vivo xenograft models can only reveal static time point or endpoint analyses of tumor formation and cell behavior. In vivo studies using either rodents or chick embryos do not easily lend themselves to monitoring cell behavior while the cells invade brain tissue in these xenograft models. Nevertheless, the chick embryo xenograft model has demonstrated that the adhesion protein L1CAM plays a stimulatory role in the invasive ability of human T98G GBM cells2,5.
A suitable solution to this problem can be reached by bridging both in vivo and in vitro methods using an organotypic brain slice culture model, referred to as an ex vivo model. In this ex vivo model, live brain tissue can be maintained at a thickness of several hundred microns for up to a few weeks, making it possible to implant cancer cells, observe their behavior in actual tissue over time, and then perform a more detailed marker analysis at the endpoint of the experiment.
A popular organotypic slice culture method has been to culture a several hundred micron-thick brain slice on top of a translucent or transparent porous membrane, leaving the tissue exposed to air, yet allowing nutrient media to sustain the tissue from below the membrane (refer to Stoppini et al.6). Different variations of this method have been used for different studies, including using different media or different membrane inserts. Different membrane inserts include a 30 mm diameter, porous (0.4 µm) membrane insert in a 35 mm culture dish6, and cell culture inserts (0.4 µm) for 6-well plates7. Different media include 50% MEM/HEPES + 25% heat-inactivated horse serum + 25% Hanks balanced salt solution (HBSS)8, 50% reduced serum media + 25% horse serum + 25% HBSS9, as well as others. If a translucent or transparent membrane is used along with fluorescently labeled GBM cells, then such cultures can be imaged from below using an inverted widefield or confocal fluorescence microscope10,11,12,13,14,15.
While many in vivo orthotopic brain tumor xenograft and ex vivo organotypic brain slice culture models have been established using rodents, as cited above, the chick embryo (Gallus gallus) has been underutilized for these purposes. However, the chick embryo has been demonstrated to be capable of being used as an in vivo orthotopic xenograft model for the study of both human and rat glioma invasion1,2,5. Xenografted cells into chick embryo brains have exhibited invasion patterns similar to those observed in rodent models, further supporting the use of chick embryos as an in vivo model for GBM tumor cell analysis. Chick embryos also are inexpensive, can be more easily maintained than rodents (i.e., in their egg shells in a lab incubator), and are much easier to work with, making them an attractive option for short-term in vivo GBM studies. A recent article has described the use of chick embryo brain slice cultures for the formation and growth of axons during normal brain development where the slices were viable for at least 7 days16. However, the use of such chick embryo brain slice cultures for ex vivo analysis of GBM cell behavior in a tissue environment is lacking. In this article, both the transplantation of human GBM cells and GBM stem cells (GSCs) into the early chick embryo brain in vivo, as well as the introduction of GBM cells onto live chick embryo brain slice cultures ex vivo, are described. Some representative examples of the resulting tumors and cell invasion patterns obtained from these preparations are also provided.
Protocol
No clearance or approval was needed at the University of Delaware to carry out this work.
1. GBM cell injection into chick optic tectum
- Preparation of GSCs and GBM cells for injection
- Culture GSCs in GSC media (Table 1). Culture established GBM cell lines in GBM media (Table 1).
- Rinse the cells on plates with sterile phosphate-buffered saline (PBS) and place 1 mL of trypsin solution into a 10 cm dish. Leave in a cell culture incubator for 2-3 min until the cells start to detach.
- Inactivate the trypsin by adding 10 mL of appropriate serum-containing culture media into the 10 cm dish and detach the cells by pipetting up and down. Place the cell suspension into a 15 mL conical centrifuge tube.
- Pellet the cells by centrifugation at 800 × g for 5 min at 4 °C.
- Aspirate the media from the cell pellet using a disposable fine glass pipette attached to a side arm flask and vacuum pump. Resuspend the cells in appropriate growth media to a concentration of 10,000 cells per microliter and place in a microfuge tube or small screw-top tube on ice.
- Mix the cells with a small amount of sterile 1% Fast Green FCF dye (use a ratio of 5 µL of dye/100 µL of cell suspension).
- Injection of GBM cells into E5 optic tectum in ovo. See Supplementary Figure 1.
- Incubate the fertilized chicken eggs in a humidified egg incubator, with the pointed end facing down, at 37.5 °C (the 1st day of incubation is embryonic day 0 [E0]). On the 6th day of incubation (E5), sterilize the eggshell by spraying with 70% ethanol.
- Using an egg candler, trace along the perimeter of the air space above the embryo with a pencil and cover the outlined area with transparent tape.
- Using curved scissors, gently cut around the traced area, being careful not to cut into the embryonic membranes or blood vessels, and discard the top of the eggshell.
- Place a few drops of saline or cell culture media onto the air space membrane to wet it so that it will detach easily. Using fine forceps, carefully pierce the air space membrane over the top of the embryo, remove it, and locate the chick embryo's head.
- Use fine forceps to grab the transparent amnion membrane that immediately surrounds the embryo to position the head so that the optic tectum can be injected with cells. With one hand, use the fine forceps to hold onto the amnion to keep the head in place during the injection process. Be careful not to damage the extraembryonic blood vessels in the chorioallantoic membrane on the yolk.
NOTE: The above procedure takes some practice to be able to efficiently grab the clear amnion membrane that intimately surrounds the embryo, since it is invisible until it is grabbed and pulled. Practice using fine forceps to grab the clear amnion that immediately surrounds the embryo. - Hold the head steady by gripping the amnion membrane with fine forceps. Using a glass micropipette and a pneumatic picopump, inject around 50,000 cells in 5 µL of suitable cell culture media into the optic tectum (5 µL is approximately 1/2 of a filled micropipette).
NOTE: See Cretu et al.1 for an image of an E5 embryo that has been injected with GBM cells mixed with dye. - Place a few drops of 50 mg/mL ampicillin on top of the embryo.
- Cover the hole in the top of the eggs with clear tape and leave in humidifier until E15 for dissection.
2. Dissection of brain regions from E15 embryos
NOTE: The dissection of E15 brains here for fixation is similar to that described in step 8.2 for live brain slices, but the dissection here does not have to be done under aseptic conditions.
- Cut away the tape around the top of the shell, allowing access to the embryo.
- Decapitate the embryo at the neck and place the head in a 10 cm dish with sterile calcium- and magnesium-free Tyrodes (CMF Tyrode's) solution. See Supplementary Figure 2.
- Using fine forceps, peel away the skin covering the brain to reveal the underlying dura mater. Remove the forming skull bones on the left and right sides of the brain. The forming skull bones do not yet cover the majority of the brain.
- Gently use fine pointed forceps to tear through the meninges overlying the center of the brain and peel it to each side to uncover the brain.
- Use curved forceps to scoop the brain from below and gently pull it out of its cavity.
- Dissect the brain into its parts: forebrain, tecta, cerebellum.
- Remove the overlying pia mater from the tecta using fine forceps. Note that the pia separates easily from the tecta, but not from the forebrain or cerebellum. Remove the pia from the forebrain by touching it or rolling it on a small piece of filter paper.
- Place the dissected brain regions in a small dish.
- Place the brain regions for fixation in a 24-well plate well and fix in 2% paraformaldehyde (PFA) in 0.1 M sodium cacodylate buffer. Leave for at least 24 h at 4 °C.
- After fixation, rinse the tissue in 3x PBS over at least 1 hour before embedding in agar.
3. Embedding and slicing harvested brain regions
- Prewarm a solution of 3.5% agar and 8% sucrose in PBS until molten and keep it at melting temperature.
- Using curved forceps as a scoop, gently pick up either the optic tectum or forebrain region of the chick embryo brain, and blot slightly on filter paper to remove extra liquid to ensure adhesion of the agar to the outer brain surface.
- Using a sterile transfer pipette, fill up the mold with agar solution.
NOTE: A simple mold can be made by forming aluminum foil around an appropriately sized object, such as small rectangular metal vibrating tissue slicer blocks. - Place the brain region in agar, and let the agar completely solidify.
- Remove the aluminum foil from around the embedded brain and trim excess agar around the sides using a razor blade or scalpel.
- Place a drop of cyanoacrylate glue on the stainless-steel square of the slicing tray, place the agarose block with the brain onto the glue, and let the glue bond for 1 min. Place the tray into the vibrating tissue slicer chuck, tighten, and fill the tray with enough PBS to cover the top of the agar block.
- Cut the brain slices on a vibrating tissue slicer at a 350 µm thickness using a steel razor blade.
- As brain slices are cut and float freely into the slicing tray, use a spatula to scoop up and remove the slices from the tray. Gently slide the section off the spatula and into a labeled 10 cm Petri dish with PBS so the sections float freely.
NOTE: The agar surrounding the brain slice may become detached if the brain is not sufficiently blotted with filter paper to remove excess liquid before embedding. If this occurs, the brain tissue can be gently removed from the solidified agar and re-embedded after proper blotting of excess liquid.
4. Immunostaining brain slices with tumor cells
- Using a stereo microscope equipped with epifluorescence, screen individual slices in the 10 cm dish one by one for the presence or absence of tumor cells.
- Prepare a sufficient amount of phosphate-buffered saline with 0.1% Triton X-100 and 5% normal goat serum (PBSTG) (see Table 1) for immunostaining and rinse the slices to be immunostained.
- Half-fill wells in a 24-well plate that will be used for staining brain sections with PBS.
- Trim off the corners of agar around the brain slices with the edge of a spatula or a scalpel, and gently place them into the wells containing PBS.
- Aspirate the PBS and replace with 350 µL of primary antibody staining solution in PBSTG (e.g., 2 µg/mL UJ127 in PBSTG).
- Incubate for 24 h in a cold room with gentle agitation so that the slice is seen to move freely within the well.
- After 24 h, remove the primary antibody solution and rinse 3 x 1 h with PBSTG in a cold room with agitation.
- When finished rinsing, incubate in 350 µL/well of secondary antibody staining solution in PBSTG (e.g., 1/200 dilution of biotin-GAM in PBSTG). Incubate for 20 h in a cold room with agitation.
NOTE: If foregoing the tertiary step and incubating with the fluorochrome-containing secondary antibody at this step, cover to protect from light during incubation now and then skip to step 4.11. - Remove the secondary antibody solution and rinse 3 x 1 h in PBSTG in a cold room with agitation.
- Remove the PBSTG and incubate in the tertiary solution (e.g., 1:250 dilution of Alexa Fluor 647 streptavidin in PBSTG). If desired, stain the nuclei at this step by adding 0.1 µg/mL bisbenzimide to the mixture. Incubate for 20 h in a cold room with agitation.
- Remove the tertiary solution and rinse 3 x 1 h in PBSTG in a cold room with agitation.
- Leave in PBS until ready to mount on microscope slides.
5. Mounting slices on microscope slides
- Prepare as many microscope slides as there are sections to mount.
- For each slide, place a 50 mm length strip of 10 mil (254 µm) thick vinyl electrical tape on a piece of parafilm.
- Using a 1 cm x 1 cm square hole punch, punch a hole through the center of the electrical tape and parafilm.
- Pull the tape off of the parafilm and place it on top of the microscope slide, leaving room for labeling tape on the slide.
- Using a micropipette, place one or two drops of anti-fade mounting media in the square hole in the center of the electrical tape.
- Use a curved spatula to lift the desired vibrating tissue slicer section from the PBSTG and thoroughly wick away moisture with a clean laboratory wipe or piece of filter paper.
- Touch the edge of the slice to the drop of mountant and use another spatula to gently slide the section into the mounting media.
- Cover the section with a few more drops of mountant and carefully place a 24 mm x 30 mm (#1.5 thickness) coverslip on top of the section and mountant.
- Seal the edges of the coverslip with nail polish to keep it in place.
6. Confocal microscopy of fixed brain slices
- Use widefield fluorescence to find fluorescent tumors in the mounted brain slice using the appropriate objective lens and filter set(s).
NOTE: Some tumors can be quite large and are easily visible with the 4x objective, while single cells might require the 10x objective. - Switch to confocal microscopy using an appropriate objective lens, laser(s), pinhole size, and detector settings. To follow this protocol, use a 20x objective (numerical aperture [N.A.] = 0.75) for routine imaging and a 60x oil objective (N.A. = 1.40) for high-resolution imaging.
- Set upper and lower limits of the z-axis and step size (for the specific situation according to the confocal microscope manufacturer's instructions) for acquiring optical sections. Acquire a z-stack of optical sections.
- Use the confocal microscope software to create a 3D volume render of the tumor, according to the manufacturer's instructions.
7. Spheroid preparation
- Creating poly(2-hydroxyethyl methacrylate) (poly-HEMA) plates
- Create a solution of 10 µg/mL poly-HEMA in 95% ethanol, and coat 35 mm Petri dishes (or cell culture dishes) with 1 mL of this solution.
- Let the dishes sit on a rocker uncovered overnight at room temperature to develop a coating of the dish surface.
NOTE: The solvent will evaporate and leave a translucent coating on the dish, which may appear uneven, but this will not affect the ability to make cell spheroids. - After drying, sterilize the open dishes under UV light in a biosafety cabinet for 1 h. Replace the lids after sterilization. The coated dishes are now ready for use.
- Fluorescent DiD staining for time-lapse microscopy
NOTE: This section is for staining single cells with DiD fluorescent dye to be used to make spheroids, which optimizes visualization of cell motility for live time-lapse imaging.- Suspend the cells in a serum-free culture medium.
- Add 5 µL of DiD stock/mL of cell suspension and gently mix by pipetting. Incubate for 20 min at 37 °C.
- Centrifuge the labeled cell suspension at 800 × g at 5 °C for 5 min.
- Aspirate the supernatant and resuspend the cells in warm media to rinse.
- Repeat this centrifugation and rinse step two more times.
NOTE: If desired, skip to step 7.3.6 to make spheroids immediately after this process.
- Making cell spheroids
- Warm up 0.25% trypsin/ethylenediaminetetraacetic acid (EDTA) solution to 37 °C.
- Culture GSCs in GSC media17 (Table 1) and U-118 malignant gloima (MG) cells in U-118 culture medium (see Table 1). Use a confluent 10 cm dish for the preparation of one 35 mm plate of spheroids. Add bFGF (10 ng/mL final concentration) and TGF-α (20 ng/mL final concentration) growth factors to GSCs initially and then every 3 days.
NOTE: The cells used in the current study were green GSC15-2/K72, green GSC16-4/K72, red U-118/L1LE/mCherry2x, and red U-118/1879/mCherry2x. One plate of spheroids should be sufficient for two 6-well plates of brain slices on membrane inserts. - Rinse the cells on the plates with sterile PBS and place 1 mL of trypsin solution onto a 10 cm dish. Place in the cell culture incubator for 2-3 min until the cells start to detach.
- Inactivate the trypsin by adding 10 mL of appropriate serum-containing culture media into the 10 cm dish and detach the cells by pipetting up and down. Place the cell suspension into a 15 mL conical centrifuge tube.
- Pellet the cells by centrifugation at 800 × g for 5 min at 4 °C.
- Aspirate the media from the cell pellet and resuspend the cells in 10 mL of media.
- Place 2 mL of cell suspension on each 35 mm poly-HEMA-coated dish and add 2 mL more of appropriate media to obtain 4 mL of total media per dish. If using GSCs, add growth factors.
- Incubate the cells in a cell incubator until the aggregates reach a size of 100-200 µm, which could be 1-2 days depending on the density of the cells that were plated.
8. Live chick embryo brain dissection and vibrating tissue slicer slicing
- Prep for dissection
- Prepare a 6-well polyester membrane insert plate with 1 mL of brain slice culture media (see Table 1) underneath the membrane insert.
- Sterilize the work area and tools with 70% ethanol.
- Place the 6-well insert plate on ice while the dissection is taking place.
- Prepare 100 mL of vibrating tissue slicer slicing media (Table 1) and place on ice.
- Place a vial of 4% low melt agarose in PBS in a water bath until it melts into a liquid (approximately 50 °C).
- Fill up the vibrating tissue slicer tub with ice.
- Aseptic E14/15 chick embryo brain dissection
- Using an egg candler, trace along the perimeter of the air pocket above the E14 or E15 embryo with a pencil and cover the outlined area with transparent tape.
- Using curved or fine scissors, gently cut around the traced area, being careful not to cut into the embryo membrane or blood vessels, and discard the top piece of shell.
- Using curved forceps, remove the air space membrane over the top of the embryo and locate the chick embryo's head.
- Decapitate the embryo and place the head in a 10 cm dish with cold sterile CMF solution. See Supplementary Figure 2.
- Using sterile fine forceps, peel away the skin covering the brain to reveal the underlying dura mater. Remove the forming skull bones on the left and right sides of the brain. The forming skull bones do not yet cover the majority of the brain.
- Gently use fine pointed forceps (#5 or #55) to tear through the meninges overlying the center of the brain and peel it to each side to uncover the brain.
- Using fine curved forceps, scoop the brain up from the bottom front and gently pull it out of its cavity.
- Dissect the brain into its three main parts: forebrain, midbrain (optic tecta), cerebellum. Consult an atlas of chick development, if needed.
- Remove the overlying pia mater from the tecta using fine forceps.
NOTE: The pia separates easily from the tecta, but not from the forebrain or cerebellum. The pia can be removed from the forebrain by touching it or rolling it on sterile gauze. - Place the dissected brain regions in a small sterile dish on ice.
- Embedding and slicing the brain
- Using curved forceps as a scoop, gently pick up either the optic tectum or forebrain region and blot slightly on sterile gauze to remove extra liquid to ensure adhesion of the agarose to the outer brain surface.
- Using a sterile transfer pipette, fill up the mold with low-melt agarose. Prepare a simple mold by forming aluminum foil around an appropriately sized object. The small rectangular metal vibrating tissue slicer block is routinely used as the object. See Supplementary Figure 3.
- Quickly place the brain region in agarose and let it solidify (approximately 4-5 min) on ice.
NOTE: The brain may sink to the bottom of the mold before the agarose hardens. If this occurs, wait 1 min to let the agar start to set, and then place the brain region into the agarose. Try to suspend the brain region directly in the middle of the agarose. - Remove the aluminum foil from around the embedded brain in the solidified agarose and trim excess agarose around the sides using a sterile razor blade or scalpel.
- Place a drop of cyanoacrylate glue on the stainless-steel square of the slicing dish/tray, place the agarose block with the brain, and let the glue bond for 1 min. Place the dish/tray in the vibrating tissue slicer chuck, tighten, and fill the tray with slicing media to cover the top of the agarose block.
- Cut the sections on a vibrating tissue slicer at 250-350 µm using a sapphire knife, which has been reported to result in less live tissue damage than a steel razor blade.
- As brain slices are cut and float freely into the slicing tray, use a sterile spatula to scoop up and remove the slice from the tray. Gently slide the section off of the spatula and onto a membrane insert using another sterile spatula.
NOTE: Normally, two or three brain slices can be placed onto each membrane insert, if desired. The agarose surrounding the brain slice may become detached if the brain is not sufficiently blotted with sterile gauze to remove excess liquid. If this still occurs after sufficient blotting before embedding, gently pick up the slice without the surrounding agarose and slide it onto the membrane insert. - Place the 6-well plate of brain slices on membrane inserts in the cell culture incubator at 37 °C and 5% CO2.
- The day after plating, use a sterile Pasteur pipette to aspirate the media from below the insert (there are gaps in the sides of the inserts to allow a pipette to gain access to the media below). Add 1 mL of fresh slice media to each well underneath the membrane insert. Continue to change media every other day thereafter.
- Wait a few days for the brain slices to attach firmly to the membrane inserts and appear to flatten out somewhat. This is a sign that the slices are viable and ready for the introduction of GBM cells.
9. GBM cell introduction onto brain slices
- Spheroid method
- After the cell spheroids have reached 150-200 µm in size, use a 20 µL micropipette set to 5 µL to remove one to several spheroids from their culture dish. See Supplementary Figure 3.
- Gently expel the media with spheroids onto the desired brain slice.
NOTE: The cell spheroids should be visible in the pipette tip. Using a clear pipette tip will make them easier to see in the tip. If the spheroid falls off the brain slice when the liquid is released, an ethanol-sterilized eyelash glued to a thin wood applicator stick should be used to gently nudge the spheroid back onto the brain slice. - Allow the spheroids to culture on the brain slices for 2-5 days.
NOTE: The limitation here seems to be the eventual degradation of blood vessels and brain cells in the slice. Degraded blood vessels will appear as discontinuous balls in the slice when stained for laminin.
- Biopsy punch method
- Allow the brain slices to attach by appearing to flatten out on the membrane insert (may take 2-5 days in culture).
- Thaw the cell matrix on ice.
- In the cell culture biosafety cabinet, connect a sterile 1 mm diameter biopsy punch to a vacuum aspirator tube.
- Gently touch the brain slice with the biopsy punch to create a 1 mm hole in the center of the brain slice.
NOTE: The tissue in the biopsy punch will be aspirated into the punch by the vacuum. - Prepare a cell matrix suspension by trypsinizing a 60%-70% confluent 10 cm dish of cells and resuspending in 10 mL of media; then, mix 1 mL of that suspension with 100 µL of matrix.
- Using a 20 µL micropipette, place 1 µL of the cell matrix mixture into each hole in the brain slices.
- After cell mixture placement is finished, place the dish of brain slices with embedded cells back into the incubator, and allow the matrix to solidify and the cells to potentially invade the surrounding brain slice.
10. Widefield fluorescence time-lapse microscopy
- Place removable tape around the edge of the 6-well plate to prevent evaporation of the media, leaving a small gap on one side for gas exchange.
- Place the plates in a customized culture chamber on an adjustable automated stage on an inverted epifluorescence microscope.
NOTE: The chamber was kept at atmospheric conditions of 5% CO2 and 95% air using a gas injection controller, and temperature was maintained at 37 °C with a warm air temperature controller and a temperature-controlled stage insert. See Fotos et al.18 for details of the system used here. - Using suitable microscope control software, create a time-lapse acquisition schedule that collects fluorescent images of the areas of interest once every 10 min for 20 h.
NOTE: If a green fluorescent label is used in cells (e.g., green fluorescent protein [GFP]), then use the minimum amount of blue excitation light required to visualize the cells to prevent phototoxicity. Red (e.g., mCherry) and far red (e.g., DiD) labels do not seem to have this potential problem due to longer wavelength excitation.
11. Immunostaining brain slices after time-lapse microscopy
NOTE: This immunostaining protocol is optimized for staining blood vessels with laminin and nuclei with bisbenzimide. Use appropriate antibodies for the desired molecule(s) of interest.
- Aspirate the media from below the membrane insert using a Pasteur pipette, and place 1 mL of 2% PFA in 0.1 M sodium cacodylate buffer underneath the insert and 1 mL on top of the insert to cover the brain slice. Let the slices fix overnight at 4 °C.
- Remove the fixative from below the membrane insert and any fixative remaining on the brain slice (fixative tends to leak through membrane insert into the well below).
- Remove the insert with slices from the 35 mm well and place them into a larger plastic dish.
- Prep a 24-well plate by adding 350 µL of PBS into as many wells as there are brain slices (one slice/well when immunostaining).
- Using a thin spatula, gently remove the agarose from around the brain slice without detaching the brain slice from the membrane insert. Make sure that the agarose detaches from the outer edge of the brain slice easily. If not, leave the agarose attached to the brain slice.
- Using a sharp scalpel, cut through the membrane around the brain slice until the slice with the underlying membrane is free from the rest of the insert. Pick up the membrane with the attached brain slice, using fine forceps to grab the membrane, and place it into a well of the 24-well plate in PBS.
- Rinse the slices 3x with PBS over 1 h in the cold room with constant gentle agitation or rocking, so that the slice moves within the well.
- While rinsing, prepare the primary antibody solution.
- Dilute anti-laminin to 2 µg/mL in PBSTG (see Table 1).
- Remove the PBS from the wells and incubate overnight in primary antibody solution in a cold room with gentle agitation.
- After at least 20 h of incubation, aspirate the primary antibody solution and rinse the sections 3x for 1 h in PBSTG.
- While rinsing, prepare the secondary antibody solution.
- Dilute fluorescent-GAM with the specific fluorochrome needed to a 1:200 dilution in PBSTG along with a 0.1 µg/mL concentration of bisbenzimide.
- Remove PBSTG and incubate overnight in a cold room with agitation in the fluorescent secondary antibody solution.
- Remove the secondary antibody and rinse 2x over 1 h in PBSTG and 1x over 1 h in PBS.
- Leave in PBS until ready to mount on microscope slides (section 5) and view.
Results
Presented here are multiple figures to show some representative results that were obtained from performing in vivo injections into the optic tectum (Figure 1 and Figure 2), culturing live brain slices and assessing their viability (Figure 3), creating ex vivo brain slice cultures and implanting fluorescently labeled cells using the biopsy punch method (Figure 4), generating cell spheroids by culturing cells on poly-HEMA (Figure 5), creating ex vivo brain slice co-cultures with cell spheroids and recording the invasive cell behavior using 4D confocal time-lapse microscopy (Figure 6), and analyzing invasive cell behavior from spheroids relative to blood vessels in fixed brain slice preparations (Figure 7 and Figure 8). These results are by no means exhaustive, but rather provide good examples of what can be obtained using the chick embryo brain as a xenograft model for human GBM research.
Figure 1 shows some representative results of tumors that formed in the optic tectum in vivo after the injection of GSCs expressing GFP. GSCs attach to the ventricular surface and form invasive tumors in the brain wall. GSCs clearly reside near blood vessels and appear to be migrating along them. Movies of rotating 3D volume renders of fixed and immunostained slices of in vivo GSC tumors are given in Supplementary Video S1, Supplementary Video S2, Supplementary Video S3, and Supplementary Video S4. In this experiment, four colors were used to identify five features (green GSCs, white nuclei, white blood vessels, blue integrin alpha-6, and either red Sox2 or red nestin).
Figure 2 shows some representative results of tumors that formed in the optic tectum in vivo after the injection of GSCs expressing GFP mixed with U-118/L1LE cells2 expressing mCherry due to retroviral vector transduction. These experiments revealed that as these tumors formed from a mixed-cell suspension, sorting out occurred such that GSCs resided either in the periphery or the center, while U-118 cells comprised either an inner core or an outer cortex, depending on the specific GSC line.
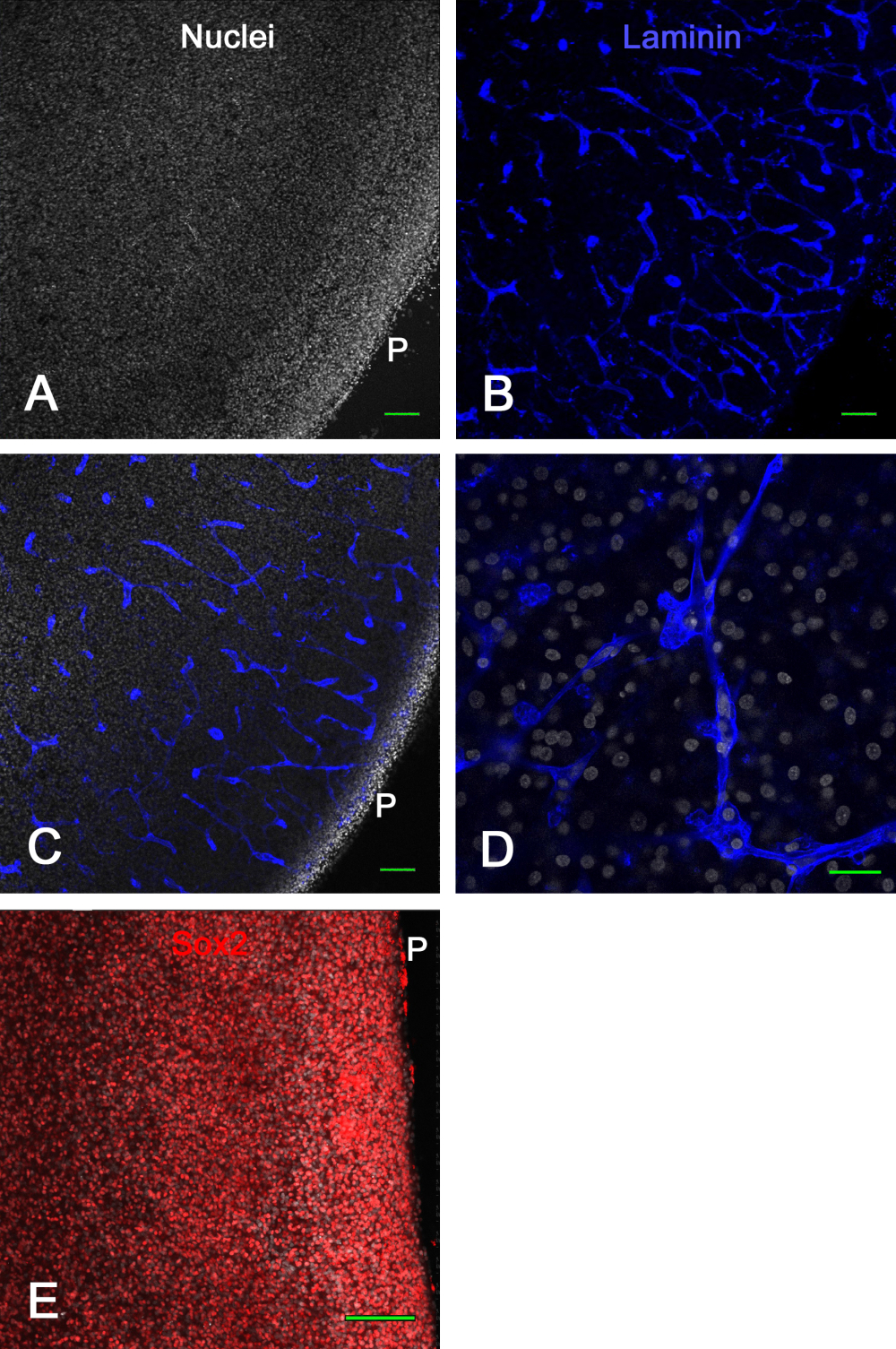
Figure 3 shows viability results of ex vivo brain slice cultures. After 1 week in culture, fixation and immunostaining for laminin revealed many intact blood vessels and the expression of Sox2, both of which were used here to demonstrate viability of the brain slice. This showed that chick embryo brain slices could be cultured on membrane inserts for approximately 2 weeks and remain viable with normal-appearing blood vessels and transcription factor expression.
Figure 4 shows the results of introducing "plugs" of red U-118/L1LE/mCherry cells (mixed with matrix) into ex vivo brain slices after creating cavities in the slices using the biopsy punch method. U-118 cells clearly invaded the brain tissue, sometimes extensively, and often along blood vessels. However, cell invasion was not uniform around the circumference of the introduced cells. Blood vessels sometimes also appeared damaged or absent in certain slices, presumably due to the added trauma of the punch method or length of time in culture. This showed that the biopsy punch/cell plug method could be used to introduce GBM cells into specific locations in a cultured ex vivo brain slice, whereafter the cells invade the brain slice.
Figure 5 shows live spheroids in culture and several examples of widefield fluorescence of live GBM cell spheroids introduced onto ex vivo brain slices for time-lapse experiments. Movies of cell invasion from the spheroids into the brain slice are given in Supplementary Video S5 and Supplementary Video S6. This showed that cell spheroids are another successful method of introducing GBM cells or GSCs onto specific locations of an ex vivo brain slice, and invasive cell behavior can be monitored by widefield fluorescence microscopy, although the resolution of individual cells can be poor.
Figure 6 shows static images of confocal time-lapse experiments of live GSC16-4/GFP and U-118/L1LE/mCherry cell invasion into brain slices. Confocal z-stack images were acquired every 10 min over a 20 h period in a multi-point time-lapse experiment. Movies of cell invasion from the spheroids into brain slices taken as confocal z-stacks over time are presented in Supplementary Video S7, Supplementary Video S8, Supplementary Video S9, Supplementary Video S10, and Supplementary Video S11. This experiment revealed that confocal time-lapse imaging was superior to widefield fluorescence for tracking individual cell invasive behavior. The U-118/L1LE cells were noticeably more invasive than the GSCs under these conditions. This is even apparent in the static images, with the GSCs being located more centrally and the U-118 cells being more dispersed.
Figure 7 shows several examples of ex vivo brain slice/spheroid preparations, where two different separately labeled spheroids (U-118/L1LE/mCherry spheroids and GSC16-4/GFP spheroids) were placed onto brain slices, grown for several days, and subsequently fixed, immunostained for laminin, and imaged by optical sectioning on a confocal microscope. This revealed that both cell types invaded the brain slice and traveled along blood vessels. When the different types of spheroids were close enough to contact each other, there seemed to be little, if any, invasion of one cell type into the spheroid of the other cell type, and the spheroids remained segregated.
Figure 8 shows several examples of ex vivo brain slice/spheroid preparations where "mixed-cell type" spheroids generated in culture using two differently labeled cell types (U-118/L1LE/mCherry mixed with GSC16-4/GFP) were placed onto brain slices, grown for several days, and subsequently fixed, immunostained for laminin, and imaged by optical sectioning on a confocal microscope. This revealed that the red U-118/L1LE/mCherry cells migrated out of the spheroids and dispersed much more evidently than the green GSC16-4/GFP cells, which tended to remain in clumps near the center of the spheroids. Additionally, U-118/L1LE/mCherry cells were also stained with DiD so that the two separate labels (mCherry and DiD) could be compared directly in the fixed ex vivo preparations. The DiD label could still be detected, even in single cells that had invaded the brain slice; however, this was as intracellular puncta.

Figure 1: Tumors at E15 resulting from injection of GSCs into E5 optic tectum in vivo. GSCs are green due to GFP expression. GSC15-2 cells are shown in panels A, C, and E, and GSC16-4 cells are shown in panels B, D, and F. (A) Low-magnification view of optic tectum with a tumor near the ventricle (V). Sox2 staining is shown in red, which stains most of the nuclei of OT cells. (B) Similar image to A but with GSC16-4 cells that also are stained for nestin in red, which can appear yellow or white in the image due to color mixing and image exposure. OT nuclei appear white due to counterstaining with bisbenzimide. (C-F) Different perspectives of volume renders generated from z-stacks using a 60x oil immersion objective. Cell nuclei appear white due to bisbenzimide staining, and some appear red in panels C and E due to immunostaining for Sox2. Red staining in panels D and F is from staining for nestin. Note that due to "Alpha Blending" for volume renders in the confocal microscope software, colors do not blend as they would using a maximum intensity projection, and the most prevalent color predominates and obscures the less intense color. Blood vessels are stained white due to immunostaining for laminin. GSC marker integrin alpha-6 staining is shown in blue, and appears punctate on GSC surfaces. Micron scales are shown along the edges of the volume renders. Videos of rotations of volume renders in panels C-F are presented in Supplementary Video S1, Supplementary Video S2, Supplementary Video S3, and Supplementary Video S4. Scale bars = 500 µm (A,B). Abbreviations: GSCs = glioblastoma stem cells; OT = optic tectum; GFP = green fluorescent protein; BV = blood vessel. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Tumors at E15 resulting from a mixture of GSCs and U-118 GBM cells injected into E5 optic tectum. GSCs are green due to GFP expression and U-118/L1LE cells are red due to mCherry expression. GSC15-2 are shown in panels A-D, and GSC16-4 are shown in panels E and F. (A) Low-magnification confocal single z-plane of a mixed cell tumor (arrow) near the ventricle. Nuclei are counterstained white with bisbenzimide. (B) Higher magnification (10x objective) of tumor shown in A with invasion of red U-118 cells into the OT near the ventricular surface. (C) A slightly different plane of optical section from that in A showing the tumor (arrow) embedded deeper into the OT wall. (D) Maximum projection (20x objective) of multiple z-planes of the tumor in C showing details of the sorted cells within the tumor. (E) Single z-plane image (20x objective) of a mixed tumor with GSC16-4 cells, showing that sorting out within the tumor occurred in an opposite pattern from GSC15-2 cells, with the green GSCs creating a thin and even cortex surrounding the red U-118 cells. The area of attachment of the tumor to the OT wall is not shown in this z-plane. Note the area of the tumor where there is a discontinuity of the GSC cortex with U-118/L1LE cells bulging through (arrow). Immunostaining for L1CAM is shown in blue. (F) Same image as in E, but showing only the green GSCs and blue L1CAM staining. Scale bars = 500 µm (A,C), 100 µm (B,D,E,F). Abbreviations: GSCs = glioblastoma stem cells; OT = optic tectum; GFP = green fluorescent protein; V = ventricle. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Viability of ex vivo optic tectum slices after 1 week in culture. E14 optic tectum slices were cultured on membrane inserts for 1 week and then fixed and immunostained. Shown in A and B are confocal images (10x objective) of a brain slice stained for nuclei with bisbenzimide (A) and immunostained for laminin (B), which clearly shows normal, intact blood vessels optically sectioned in various configurations by virtue of the laminin staining. (C) A confocal image similar to that shown in panels A and B where nuclei and laminin staining are both visible. (D) A higher-magnification (60x oil objective) confocal image showing details of nuclear and laminin staining. (E) Maximum projection image of confocal z-stack (20x objective) of brain slice stained for Sox2 transcription factor in red and total nuclei with bisbenzimide in white. Note that the majority of nuclei exhibit Sox2 staining, as shown in vivo (see Figure 1). Scale bars = 100 µm (A,B,C,E), 25 µm (D). Abbreviation: P = pial surface. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: U-118/mCherry cells placed into an ex vivo brain slice via the biopsy punch method. Cavities were created in brain slices using a 1 mm biopsy punch, and then red U-118/L1LE/mCherry cells mixed with matrix were implanted as a "plug." After several days, the brain slices were fixed, immunostained for laminin, and mounted on slides for confocal microscope analysis. Panels A and C show low-magnification, confocal, single z-plane images (4x objective) of the resulting "tumor" and surrounding cells that invaded the brain slice. (B) A volume render of a z-stack from the preparation in panel A at a higher magnification (20x objective), showing extensive invasion of U-118 cells (arrow). (D) Image shows a similar volume render of the lower part of the extensively invading cells shown in panel C. Laminin staining is shown in green, but no clear blood vessels are apparent. (E) Image shows part of a cell plug and group of cells that have invaded the brain slice, along with laminin staining for blood vessels in blue. (F) A higher magnification of the invading cells shown in panel E, and cells can clearly be seen aligned along blood vessels (arrows). All panels show white nuclear counterstaining with bisbenzimide. Scale bars = 500 µm (A,C), 100 µm (E,F). Scale for panels B and D is along the volume render axes. Abbreviation: OT = optic tectum. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Live cell spheroids in culture and widefield fluorescence images of live GBM cells in ex vivo brain slices. Shown in panels A and B are phase contrast images (using a 10x objective on an inverted microscope) of U-118/L1LE GBM cells (A) and GSCs (B) growing as spheroids (arrows). Shown in the background of panel A is the out-of-focus unevenness of the poly-HEMA coating that can occur on the cell culture dish. Shown in panels C-F are widefield fluorescence images of U-118/L1LE cell spheroids and invading cells (arrows) during a time-lapse experiment to monitor the live behavior of invasion into the ex vivo slices (using a 20x objective on a custom time-lapse microscope system18). In panels C and E, the cells are stained with the far-red fluorescent membrane dye DiD, and in panels D and F, the cells are imaged via their red mCherry expression. Scale bars = 100 µm. Videos of widefield fluorescence time-lapse experiments shown in panels C and D are located in Supplementary Video S5 and Supplementary Video S6, respectively. Abbreviations: GBM = glioblastoma; GSCs = GBM stem cells; S = spheroid. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Volume render images of confocal 4D time-lapse of live GSCs and GBM cells. Shown in all panels are the endpoint images of five different mixed cell spheroid engraftments on separate brain slices. For panels A-E, confocal z-stack images were acquired at 10 µm steps every 10 min over a 20 h period. Preparations included brain slices with implanted mixed cell spheroids of red U-118/L1LE/mCherry cells and green GSC16-4/GFP cells. Confocal images were taken while brain slices were cultured on membrane inserts in a 6-well plastic cell culture dish using an extra-long working distance (ELWD) 20x objective lens (0.45 NA), which provided the needed extra working distance. Volume renders were generated using confocal microscope software "Alpha Blending", which gives an apparent 3D effect. Time-lapse videos of these confocal volume renders over time are presented in Supplementary Video S7, Supplementary Video S8, Supplementary Video S9, Supplementary Video S10, and Supplementary Video S11. Abbreviations: GBM = glioblastoma; GSCs = GBM stem cells; GFP = green fluorescent protein; NA = numerical aperture. Please click here to view a larger version of this figure.
{kind=link}

Figure 7: Confocal images of fixed brain slices with invasive GBM cells from spheroids of different cell types. Green spheroids were composed of GSC16-4/GFP cells and red spheroids were composed of U-118/L1LE/mCherry cells. Shown in panels A-F are different views of brain slices, on which multiple red and green spheroids were cultured for several days before fixation and immunostaining for laminin (blue). Panels A-C are of the same OT slice where A was taken with a 4x objective, and panels B and C are higher magnification volume renders (20x objective) of cells that invaded the brain slice from two of the spheroids shown in panel A. Both cell types clearly invaded tissue along blood vessels. Panel D shows a volume render (20x objective) of a different brain slice where two different spheroids were located close together, and cells from both are seen migrating along the same blood vessel that is located between them (arrow). Panel E is a high-magnification (60x oil objective) volume render revealing that the green cells are migrating along the outside surface of the blood vessel, while the red cell is migrating inside the blood vessel (arrow). The inset shows a single z-plane optical section, where the red cell is clearly surrounded by blue staining of the blood vessel (arrow), and the green cell is clearly outside of the blood vessel. Scale bar in inset = 50 µm. Panel F shows a volume render (10x objective) of a forebrain slice with two closely apposed differently colored spheroids. Very little, if any, cell invasion occurred from one spheroid into the other, and a sharp boundary existed between them. Panels A, B, C, and E also show white nuclear counterstaining with bisbenzimide. Scale bar = 500 µm (A). Scales for panels B-F are along the volume render axes. Abbreviations: GBM = glioblastoma; GSCs = GBM stem cells; GFP = green fluorescent protein; OT = optic tectum. Please click here to view a larger version of this figure.
{kind=link}

Figure 8: Confocal images of fixed brain slices with invasive GBM cells from mixed cell spheroids and spheroids labeled with DiD. Panels A-D show volume renders of brain slices that contained mixed cell spheroids composed of green GSC16-4/GFP cells and red U-118/L1LE/mCherry cells. Numerous red U-118 cells dispersed from the spheroids and invaded the brain slice in all directions, whereas the green GSCs did not disperse and remained at the central locations of the spheroids. Panels E and F show an ex vivo slice preparation with red U-118/L1LE/mCherry spheroids also labeled with far-red membrane dye DiD (shown as blue). After fixation, the slice was immunostained for laminin in green. The DiD label was visible in red cells as punctate staining (arrows) and was visible even in cells that had dispersed from the spheroids along blood vessels. Nuclear counterstaining with bisbenzimide is not shown in this figure so that the other staining is more clearly visible. Scale bars = 100 µm (E,F). Abbreviations: GBM = glioblastoma; GSCs = GBM stem cells; GFP = green fluorescent protein. Please click here to view a larger version of this figure.
{kind=link}
| Medium/solution | Composition | ||
| GSC media | 1:1 mixture of DMEM/F12, 1% fetal bovine serum (FBS), 15 mM HEPES buffer, 2 mM L-glutamine, 100 µg/mL penicillin-streptomycin (pen/strep), 2% B27 supplement without vitamin A, and 2.5 µg/mL heparin. | ||
| GBM media | DMEM (high glucose), 10% FBS, pen/strep, and 2 mM L-glutamine. | ||
| Fixation buffer | 2% PFA in 0.1 M sodium cacodylate buffer | ||
| Embedding medium | 3.5% agar and 8% sucrose in PBS | ||
| PBSTG | 0.1% Triton X-100 + 5% normal goat serum (NGS) in PBS | ||
| U-118 MG cell culture medium | DMEM + 10% FBS + pen/strep + L-glutamine | ||
| brain slice culture media | 50% MEM + 25% HBSS + 25% Horse Serum + B27 + pen/strep + L-glut + 15 mM HEPES buffer | ||
| vibrating tissue slicer slicing media | Medium 199 + pen/strep + 15 mM HEPES buffer | ||
Table 1: Composition of media and buffers used in this protocol.
Supplementary Figure 1: Injection into E5 optic tectum. (A) After a hole is cut in the eggshell over the air space, and the air space membrane is wetted with saline or media, the membrane is removed with fine forceps. (B) To inject cells into the optic tectum, the amnion is pinched and held with fine forceps to position the head so that the optic tectum is accessible. Then the micropipette is inserted into the optic tectum and cells are pressure injected into it. (C) After injection of cells, a few drops of ampicillin solution are added on top of the embryo using a syringe and fine needle. Please click here to download this File.
Supplementary Figure 2: Dissection of E15 brain regions. (A) After decapitation, the E15 embryo head is placed in a dish with sterile CMF solution. (B) The skin overlying the brain is then removed using fine forceps. (C) The two skull bones are then removed from overlying the two forebrain (FB) hemispheres. (D) The connective tissue dura is then gently removed from surrounding the forebrain (FB), optic tectum, and cerebellum. (E) The entire brain is then removed from the head by gently scooping it out of the brain cavity from underneath using curved forceps. (F) Shown is the dorsal view of the entire removed brain with forebrain (FB), optic tectum (OT), and cerebellum (CB). (G) The isolated brain is then dissected into forebrain (FB), optic tectum (OT) hemispheres, and cerebellum (CB) using fine scissors. (H) The delicate connective tissue pia is then easily removed from the optic tectum (OT) hemispheres using fine forceps. Please click here to download this File.
Supplementary Figure 3: Embedding and slicing E15 optic tectum and placement of cell spheroids. (A) One optic tectum hemisphere is submerged in low melt agarose using curved forceps. (B) After the agarose hardens on ice, the block containing the optic tectum is trimmed and glued to the stainless-steel pedestal in the slicing dish/tray. (C) After the glue dries, the slicing dish/tray is placed into the chuck of the vibrating tissue slicer and filled with cold slicing media. Slices are then cut with the sapphire knife from the submerged tissue block. Cut slices will float into the dish/tray and can be removed using a spatula. (D) Cut slices are removed from the dish/tray and placed directly on membrane inserts with underlying slice culture media in a multi-well plate. (E) After cell spheroids are grown on poly-HEMA coated dishes, a spheroid is removed from the dish in a minimal amount of media using a 20 µL micropipettor. (F) The isolated spheroid is then placed directly onto the brain slice in the minimal media. (G) If the spheroid falls off of the brain slice due to flow of the media, then it can be nudged back onto the brain slice using an eyelash glued to a wooden applicator stick. Please click here to download this File.
Supplementary Video S1: Video of high-magnification volume render of a small GSC15-2 tumor at E15. GSCs are green due to GFP expression. Video corresponds to Figure 1C and shows GSC15-2 cells. The video shows rotation of a volume render generated from a z-stack using a 60x oil immersion objective. Cell nuclei appear white due to bisbenzimide staining, and some appear red due to immunostaining for Sox2. Note that due to "Alpha Blending" for volume renders in the confocal microscope software, colors do not blend as they would using a maximum intensity projection, and the most intense color predominates and obscures the less intense color. Blood vessels are stained white due to immunostaining for laminin. GSC marker integrin alpha-6 staining is shown in blue and appears punctate on green GSC surfaces. Micron scales are shown along the edges of the volume render. Please click here to download this Video.
Supplementary Video S2: Video of high-magnification volume render of small GSC16-4 tumor at E15. GSCs are green due to GFP expression. Video corresponds to Figure 1D and shows GSC16-4 cells. The video shows rotation of a volume render generated from a z-stack using a 60x oil immersion objective. Cell nuclei appear white due to bisbenzimide staining, and some GSCs appear red due to immunostaining for nestin. Note that due to "Alpha Blending" for volume renders in the confocal microscope software, colors do not blend as they would using a maximum intensity projection, and the most intense color predominates and obscures the less intense color. Blood vessels are stained white due to immunostaining for laminin. GSC marker integrin alpha-6 staining is shown in blue and appears punctate on green GSC surfaces. Micron scales are shown along the edges of the volume render. Please click here to download this Video.
Supplementary Video S3: Video of high-magnification volume renders of small GSC15-2 tumor at E15. GSCs are green due to GFP expression. Video corresponds to Figure 1E and shows GSC15-2 cells. The video shows rotation of a volume render generated from a z-stack using a 60x oil immersion objective. Cell nuclei appear white due to bisbenzimide staining, and some appear red due to immunostaining for Sox2. Note that due to "Alpha Blending" for volume renders in the confocal microscope software, colors do not blend as they would using a maximum intensity projection, and the most intense color predominates and obscures the less intense color. Blood vessels are stained white due to immunostaining for laminin. GSC marker integrin alpha-6 staining is shown in blue and appears punctate on green GSC surfaces. Micron scales are shown along the edges of the volume render. Please click here to download this Video.
Supplementary Video S4: Video of high-magnification volume renders of small GSC16-4 tumors at E15. GSCs are green due to GFP expression. Video corresponds to Figure 1F and shows GSC16-4 cells. The video shows rotation of a volume render generated from a z-stack using a 60x oil immersion objective. Cell nuclei appear white due to bisbenzimide staining, and some appear red due to immunostaining for nestin. Note that due to "Alpha Blending" for volume renders in the confocal microscope software, colors do not blend as they would using a maximum intensity projection, and the most intense color predominates and obscures the less intense color. Blood vessels are stained white due to immunostaining for laminin. GSC marker integrin alpha-6 staining is shown in blue and appears punctate on green GSC surfaces. Micron scales are shown along the edges of the volume render. Please click here to download this Video.
Supplementary Video S5: Video of live GBM cells in ex vivo brain slice. Video corresponds to Figure 5C and shows widefield fluorescence images of U-118/L1LE cell spheroids and invading cells during a time-lapse experiment to monitor the live behavior of invasion into the ex vivo slice (using a 20x objective on a custom time-lapse microscope system). The U-118/L1LE cells were stained with the far-red fluorescent membrane dye DiD. Images were acquired with a monochrome camera. Please click here to download this Video.
Supplementary Video S6: Video of live GBM cells in ex vivo brain slice. Video corresponds to Figure 5D and shows widefield fluorescence images of U-118/L1LE cell spheroids and invading cells during a time-lapse experiment to monitor the live behavior of invasion into the ex vivo slice (using a 20x objective on a custom time-lapse microscope system). The cells were imaged via their red mCherry expression. Images were acquired with a monochrome camera. Please click here to download this Video.
Supplementary Video S7: Video of volume render images of confocal 4D time-lapse of live GSCs and GBM cells. Video corresponds to Figure 6A. Confocal z-stack images were acquired at 10 µm steps every 10 min over a 20 h period. The preparation was of a brain slice with implanted mixed cell spheroids of red U-118/L1LE/mCherry cells and green GSC16-4/GFP cells. Confocal images were taken while the brain slice was cultured on a membrane insert in a 6-well plastic cell culture dish using an ELWD 20x objective lens (0.45 NA), which provided the needed extra working distance. The volume render was generated using the confocal microscope software "Alpha Blending", which gives an apparent 3D effect. Micron scales are shown along the edges of the volume render. The video is best observed by manually dragging the video progress slider in the video player back and forth to observe cell movement rather than allowing the video player to proceed at its normal slow speed. Please click here to download this Video.
Supplementary Video S8: Video of volume render images of confocal 4D time-lapse of live GSCs and GBM cells. Video corresponds to Figure 6B. Confocal z-stack images were acquired at 10 µm steps every 10 min over a 20 h period. The preparation was of a brain slice with implanted mixed cell spheroids of red U-118/L1LE/mCherry cells and green GSC16-4/GFP cells. Confocal images were taken while the brain slice was cultured on a membrane insert in a 6-well plastic cell culture dish using an ELWD 20x objective lens (0.45 NA), which provided the needed extra working distance. The volume render was generated using the confocal microscope software "Alpha Blending", which gives an apparent 3D effect. Micron scales are shown along the edges of the volume render. The video is best observed by manually dragging the video progress slider in the video player back and forth to observe cell movement rather than allowing the video player to proceed at its normal slow speed. Please click here to download this Video.
Supplementary Video S9: Video of volume render images of confocal 4D time-lapse of live GSCs and GBM cells. Video corresponds to Figure 6C. Confocal z-stack images were acquired at 10 µm steps every 10 min over a 20 h period. The preparation was of a brain slice with implanted mixed cell spheroids of red U-118/L1LE/mCherry cells and green GSC16-4/GFP cells. Confocal images were taken while the brain slice was cultured on a membrane insert in a 6-well plastic cell culture dish using an ELWD 20x objective lens (0.45 NA), which provided the needed extra working distance. The volume render was generated using the confocal microscope software "Alpha Blending", which gives an apparent 3D effect. Micron scales are shown along the edges of the volume render. The video is best observed by manually dragging the video progress slider in the video player back and forth to observe cell movement rather than allowing the video player to proceed at its normal slow speed. Please click here to download this Video.
Supplementary Video S10: Video of volume render images of confocal 4D time-lapse of live GSCs and GBM cells. Video corresponds to Figure 6D. Confocal z-stack images were acquired at 10 µm steps every 10 min over a 20 h period. The preparation was of a brain slice with implanted mixed cell spheroids of red U-118/L1LE/mCherry cells and green GSC16-4/GFP cells. Confocal images were taken while the brain slice was cultured on a membrane insert in a 6-well plastic cell culture dish using an ELWD 20x objective lens (0.45 NA), which provided the needed extra working distance. The volume render was generated using the confocal microscope software "Alpha Blending", which gives an apparent 3D effect. Micron scales are shown along the edges of the volume render. The video is best observed by manually dragging the video progress slider in the video player back and forth to observe cell movement rather than allowing the video player to proceed at its normal slow speed. Please click here to download this Video.
Supplementary Video S11: Video of volume render images of confocal 4D time-lapse of live GSCs and GBM cells. Video corresponds to Figure 6E. Confocal z-stack images were acquired at 10 µm steps every 10 min over a 20 h period. the preparation included a brain slice with implanted mixed cell spheroids of red U-118/L1LE/mCherry cells and green GSC16-4/GFP cells. Confocal images were taken while the brain slice was cultured on a membrane insert in a 6-well plastic cell culture dish using an ELWD 20x objective lens (0.45 NA), which provided the needed extra working distance. The volume render was generated using the confocal microscope software "Alpha Blending", which gives an apparent 3D effect. Micron scales are shown along the edges of the volume render. The video is best observed by manually dragging the video progress slider in the video player back and forth to observe cell movement rather than allowing the video player to proceed at its normal slow speed. Please click here to download this Video.
Discussion
Critical steps in the protocol for the injection of cells into the midbrain (optic tectum) ventricle include not damaging the blood vessels in the chorioallantoic membrane in the egg or surrounding the embryo before and during injection, although the amnion membrane immediately surrounding the embryo can be gently pulled and held to position the head when injecting the cells into the midbrain. The amnion is relatively tough and can be pulled with fine forceps to position the head and hold it steady with one hand, for the injection of cells with the other hand into the optic tectum, which is the large, round structure in the middle of the brain. Generally, the viability of injected embryos ranges from 25% to 75%, depending upon unknown factors, and practically every embryo that survives contains at least a small tumor in the optic tectum. Critical steps in generating viable brain slices include blotting the tissue of excess liquid so that the agarose adheres to the brain during slicing and to keep tissue and slices cold until placed on the membrane insert. As different cell types form spheroids differently (in speed and size), the plated cell density on poly-HEMA plates and the length of time before harvesting spheroids should be optimized for each cell type.
The work here has not been subject to a formal longitudinal study of brain slice viability. Yang et al. used chick embryo brain slice cultures similar to the ones used here and showed good viability of the slices for at least 7 days16. Previous work showed that when OT tissue was kept in suboptimal media, many pyknotic nuclei appeared in the tissue, which did not occur in the slices in the work here. Additionally, when slices degenerate in suboptimal conditions, the blood vessels fragment and appear as rows of laminin-positive spheres (not shown). Thus, although the viability here has not been checked by methods such as electrophysiology or active caspase-3 expression, none of the indicators of cell death that were seen under suboptimal culture conditions appeared here.
The OT has been focused on for in vivo brain tumor experiments because it is the most easily injected region with the largest ventricle. At E5, which is the latest day that the embryo is small enough to remain accessible on top of the yolk, injections must be made into a ventricle, as all brain regions are nothing more than a thin ventricular zone. Nevertheless, these injections result successfully in embedded tumors with cells that invade the brain parenchyma. Sometimes, resulting tumors are found in the forebrain or cerebellum, but this is not common. Ex vivo slices of E15 optic tectum have been primarily used for experiments here, so that the ex vivo co-culture results can be correlated with the in vivo injection experiments. However, forebrain slices are also suitable and have a larger surface area and a very thin ventricle compared to the optic tectum, which might make the forebrain more suitable for ex vivo co-cultures that are not being correlated with in vivo injections.
It has been demonstrated here that in vivo injections, followed by tissue fixation, vibrating tissue slicer sectioning, and immunostaining for laminin and other markers, resulted in high-resolution images of GBM cells and GSCs in brain tissue in close proximity to blood vessels. The ability to determine the interrelationships between tumor cells and blood vessels was greatly facilitated by creating 3D volume renders from z-stacks of confocal optical sections using the confocal software and manufacturer's instructions. Time-lapse imaging using widefield fluorescence microscopy of GFP, mCherry, and DiD labeled cells was possible; however, migrating cells that were in close proximity to the highly fluorescent spheroids were sometimes obscured by the "glow" from the spheroid. This undesired effect can be somewhat minimized by carefully adjusting the exposure times for collecting widefield images. Time-lapse imaging using confocal z-stacks over time (4D) eliminated the out-of-focus glow from the spheroids and resulted in sharply defined migrating cells with a dark background. This was not described in the protocol, but was carried out similarly to widefield time-lapse imaging, which was performed while brain slices were on the transparent membrane inserts in a 6-well plastic plate. Although confocal time-lapse imaging results in markedly clearer images of individual cells and their behavior, a multi-point time-lapse experiment collecting z-stacks of 10 z-planes/point, at 10 min intervals over a 20 h period, is an extensive use of the scan head galvanometers. As this could significantly decrease the lifespan of the galvanometers, this method is used judiciously.
Although the chick embryo system is very suitable for both in vivo injection and ex vivo co-culture experiments that investigate GBM cell behavior, there are several limitations to this model system. As with any xenograft system, the environment in which human cells are implanted is not the human brain, but GBM cell behavior appears to mimic that in rodent models and in human patients. After performing in vivo injection experiments on E5, tumors are normally allowed to form for 10 days, until E15. This clearly is not enough time to study all aspects of tumorigenesis and cell invasion. However, it has been demonstrated here that solid tumors form in the brain parenchyma, cells interact and rearrange themselves within the tumor, and significant brain invasion occurs both along blood vessels and diffusely within this relatively short time period. Another limitation to the in vivo chick embryo system is that it is not suitable for drug or other treatments because of the large yolk and extraembryonic circulatory system that operates during chick embryo development. Topical liquid drug treatments would result in a highly variable and unknown concentration in the brain due to diffusion away from the embryo into the much larger yolk mass. Similarly, intravenous injection of drugs into the very delicate extraembryonic circulatory system would leak or diffuse out of the blood vessels and also result in unknown concentrations in the brain. This is one of the main reasons that the ex vivo slice culture method was adopted-so that not only could cell behavior be observed and tracked via time-lapse microscopy, but also so that treatments that have been successful in altering GBM cell behavior in a dish4 could be tested in a more relevant brain tissue environment.
The development of the chick embryo orthotopic brain tumor model system is seen as a significant addition to the systems and tools available for the study of GBM tumor formation and invasive cell behavior. Fertilized chicken eggs are likely to be readily available in most areas, they are inexpensive compared to rodents, there are no animal care costs, the embryos are very resilient and resistant to infection (i.e., most work is done on a bench top), the embryos are highly manipulable and can be grown in shell-less culture19, and chick embryos are not considered vertebrate animals and so do not require IACUC approval by NIH guidelines (institutional requirements may vary). Thus, these multiple advantages make the chick embryo system very attractive if one confines their questions and experiments to those that fall within its limitations. Multiple GBM cell studies have been performed by others using the chick embryo, but these almost exclusively have utilized the chorioallantoic membrane (CAM) of the embryo20,21,22,23,24,25,26,27,28,29 and limb bud30, and not the brain. There has also been a report implanting medulloblastoma into the chick brain on E231. Undoubtedly, the use of the chick embryo as an orthotopic xenograft model system, as described here, should yield results that are much more meaningful to human GBM tumor biology than studies using the CAM.
Although these studies have only begun to fully utilize the chick embryo brain tumor model system for studies of human GBM cell and GSC behavior, it is hoped that others will extend the uses and find further potential applications. One could imagine that not only will this system uncover mechanisms that regulate GBM tumor formation and cell behavior, but will also allow preclinical testing of specific drugs and substances on specific patients' cells. For instance, if brain slice cultures were set up in advance, then tumor cells, pieces from surgical tumor resections, or patient-derived GBM organoids32 could be placed directly in ex vivo co-culture, and various treatments could be assessed in a matter of days. Similarly, dissociated patient cells could be injected directly into E5 midbrains in ovo to assess their ability to form tumors and invade brain parenchyma. Thus, it is hoped that the descriptions of the methods and representative results here will facilitate and encourage increased use of this highly underutilized system for brain cancer research.
Disclosures
None of the authors have any conflicts of interest.
Acknowledgements
This work was funded in part by a grant to D.S.G. from the National Cancer Institute (R03CA227312) and by a generous grant from the Lisa Dean Moseley Foundation. Live GBM specimens were obtained with patient consent through the Tissue Procurement Center of the Helen F. Graham Cancer Center and Research Institute. Funding to A.R. was provided by the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health (UL1TR003107). Summer undergraduate research fellowships to N.P., A.L., Z.W., and K.S. were provided by the University of Delaware Undergraduate Research Program.
Materials
| Name | Company | Catalog Number | Comments |
| 1 cm x 1 cm square hole paper punch | Birabira | N/A | |
| 1 mm biopsy punch pen | Robbins Instruments | 20335 | |
| 6 well insert plate (Corning Transwell) | Millipore Sigma | CLS3450 | |
| 9" Disposable Pasteur Pipets | Fisher Scientific | 13-678-20C | |
| 15 mL centrifuge tubes | Fisher Scientific | 05-539-12 | |
| 24 well plate | Corning Costar | 3526 | |
| 50 mL centrifuge tubes | Fisher Scientific | 05-539-9 | |
| Agar | Fisher BioReagents | BP1423-500 | for embedding fixed brains |
| Alexafluor 488-conjugated GAM IgG | Jackson Immunoresearch | 115-605-146 | |
| Alexafluor 647-conjugated GAM IgG | Jackson Immunoresearch | 115-545-146 | |
| Aluminum foil | ReynoldsWrap | N/A | |
| Ampicillin | Sigma Aldrich | A-9518 | |
| anti-integrin alpha-6 monoclonal antibody GOH3 | Santa Cruz Biotechnology | sc-19622 | |
| anti-L1CAM monoclonal antibody UJ127 | Santa Cruz Biotechnology | sc-53386 | |
| anti-laminin monoclonal antibody | Developmental Studies Hybridoma Bank | 3H11 | |
| anti-nestin monoclonal antibody 10c2 | Santa Cruz Biotechnology | sc-23927 | |
| anti-Sox2 monoclonal antibody E-4 | Santa Cruz Biotechnology | sc-365823 | |
| B27 supplement without vitamin A | GIBCO | 17504-044 | |
| bisbenzimide (Hoechst 33258) | Sigma-Aldrich | B2883 | nuclear stain |
| Cell culture incubator | Forma | standard humidified CO2 incubator | |
| Centrifuge | Beckman Coulter | ||
| Confocal microscope | Nikon Instruments | C2si+ | With custom-made cell incubator chamber |
| Confocal microscope objective lenses | Nikon Instruments | Plan Apo lenses, except S Plan Fluor ELWD 20x 0.45 NA objective lens for confocal time-lapse imaging | |
| Confocal microscope software | Nikon Instruments | NIS Elements | Version 5.2 |
| Curved foreceps | World Precision Intruments | 504478 | |
| Curved scissors | Fine Science Tools | ||
| Curved spatula | Fisher Scientific | 14-375-20 | |
| Cyanoacrylate glue | Krazy Glue | KG-585 12R | |
| D-Glucose | Millipore Sigma | G8270 | |
| DiD far red fluorescent dye | Invitrogen | V22887 | Vybrant DiD |
| DMEM | Sigma Aldrich | D5671 | |
| DMEM/F12 | Sigma Aldrich | D8437 | |
| DMSO | Sigma Aldrich | D4540 | |
| Dulbecco's Phosphate buffered saline (PBS) | Sigma Aldrich | P5493-1L | |
| egg incubator | Humidaire | ||
| electrical tape (10 mil thick/254 µm) | Scotch | N/A | |
| Ethanol 200 proof | Decon Laboratories | 2701 | |
| Fast green FCF dye | Avocado Research Chemicals | 16520 | |
| FBS | Gemini Bio-products | 900-108 | |
| filter paper | Fisher Scientific | ||
| Gauze | Dynarex | 3353 | |
| Glass Capillaries for microinjection | World Precision Instruments | TW100-4 | |
| Glycerol | Fisher BioReagents | BP228-1 | for mounting media |
| GSCs (human glioblastoma stem cells) | Not applicable | Isolated from patient GBM specimens in Galileo laboratory in GSC media and then transduced with a GFP encoding lentiviral vector. Cells used were between passage 10 and 30. | |
| Hanks Balanced Salt Solution (HBSS) | Corning | 21-020-CV | |
| Hemacytometer | Hausser scientific | ||
| Heparin | Fisher Scientific | BP2524-100 | |
| HEPES buffer | Sigma Aldrich | H0887 | |
| Horse Serum (HI) | Gibco | 26050-088 | |
| Human FGF-2 | BioVision | 4037-1000 | |
| Human TGF-α | BioVision | 4339-1000 | |
| Inverted phase contrast microscope | Nikon Instruments | TMS | for routine viewing of cultured cells |
| KCl | Fisher Scientific | BP366 | |
| KH2PO4 | Fisher Scientific | P284 | |
| Laboratory film | Parafilm | ||
| Labquake Shaker | LabIndustries | T400-110 | |
| L-Glut:Pen:Strep | Gemini Bio-products | 400-110 | |
| Low-melt agarose | Fisher Scientific | BP1360 | for embedding live brains |
| Matrix | Corning Matrigel | 354234 | |
| Medium 199 | GIBCO | 11150-059 | |
| MEM | Corning | 10-010-CV | |
| Metal vibratome block | |||
| Micropipette tips (20, 200, 1,000 µL) | Fisherbrand | ||
| Micropipettors (20, 200, 1,000 µL) | Gilson | ||
| Microscope Coverglass (no. 1.5 thickness) | Fisherbrand | 12544A | |
| NaCl | Fisher Scientific | S271 | |
| NaH2PO4 + H2O | Fisher Scientific | S369 | |
| NaHCO3 | Fisher Scientific | BP328 | |
| Normal goat serum | Millipore Sigma | 526-M | |
| N-propyl gallate | Sigma Aldrich | P3130 | for mounting media |
| Parafilm | Parafilm | ||
| Paraformaldehyde | Electron Microscopy Sciences | 15710 | |
| PBS | Sigma Aldrich | P5493-1L | |
| Pencil | |||
| Plain Microscope slides | Fisherbrand | 12-550-A3 | |
| Plastic 35 mm Petri dish | Becton Dickinson | 351008 | |
| pneumatic picopump | World Precision Intruments | PV830 | |
| Poly(2-hydroxyethyl methacrylate) (poly-HEMA) | Sigma Aldrich | P-3932 | |
| razor blade- double edge | PACE | for cutting fixed brain slices | |
| sapphire knife | Delaware Diamond Knives | for cutting live brain slices | |
| Scalpel | TruMed | 1001 | |
| Sodium cacodylate buffer 0.2 M pH 7.4 | Electron Microscopy Sciences | 11652 | |
| Specimen chamber for vibratome | custom-made | ||
| Stereo Dissecting Microscope | Nikon Instruments | SMZ1500 | Equipped with epifluorescence |
| straight foreceps | World Precision Intruments | 500233 | |
| straight scissors | Fine Science Tools | ||
| Sucrose | Mallinckrodt | 7723 | |
| Time-lapse fluorescence microscope (widefield fluorescence) | Nikon Instruments | TE2000-E | With custom-made cell incubator chamber (see Fotos et al., 2006) |
| Tissue culture dish polystyrene 100 mm | Thermo Fisher Scientific | 130182 | for cell culturing |
| Tissue culture dish polystyrene 60 mm | Becton Dickinson | 353004 | for cell culturing |
| Transfer pipette | American Central Scientific Co. | FFP011 | |
| Transparent tape | Scotch | ||
| Triton X-100 | Sigma Aldrich | T-8787 | |
| Trypsin (0.25%) + 2.21 mM EDTA | Corning | 25-053-CI | |
| U-118 MG human GBM cell line | ATCC | HTB-15 | Cells were transduced with a lentiviral vector encoding the entire ectodomain sequence of the L1CAM adhesion protein and then with lentiviral vector pUltra-hot encoding mCherry. Passage numbers are unknown. |
| Vacuum pump | Cole-Parmer | EW-07532-40 | "Air Cadet" |
| Vibrating tissue slicer | Vibratome | 3000 | for cutting live and fixed brain slices |
References
- Cretu, A., Fotos, J. S., Little, B. W., Galileo, D. S. Human and rat glioma growth, invasion, and vascularization in a novel chick embryo brain tumor model. Clinical & Experimental Metastasis. 22 (3), 225-236 (2005).
- Yang, M., et al. L1 stimulation of human glioma cell motility correlates with FAK activation. Journal of Neuro-Oncology. 105 (1), 27-44 (2011).
- Mohanan, V., Temburni, M. K., Kappes, J. C., Galileo, D. S. L1CAM stimulates glioma cell motility and proliferation through the fibroblast growth factor receptor. Clinical & Experimental Metastasis. 30 (4), 507-520 (2013).
- Anderson, H. J., Galileo, D. S. Small-molecule inhibitors of FGFR, integrins and FAK selectively decrease L1CAM-stimulated glioblastoma cell motility and proliferation. Cellular Oncology. 39 (3), 229-242 (2016).
- Pace, K. R., Dutt, R., Galileo, D. S. Exosomal L1CAM stimulates glioblastoma cell motility, proliferation, and invasiveness. International Journal of Molecular Sciences. 20 (16), 3982 (2019).
- Stoppini, L., Buchs, P. A., Muller, D. A simple method for organotypic cultures of nervous tissue. Journal of Neuroscience Methods. 37 (2), 173-182 (1991).
- Ohnishi, T., Matsumura, H., Izumoto, S., Hiraga, S., Hayakawa, T. A novel model of glioma cell invasion using organotypic brain slice culture. Cancer Research. 58 (14), 2935-2940 (1998).
- Humpel, C. Organotypic brain slice cultures: A review. Neuroscience. 305, 86-98 (2015).
- Aaberg-Jessen, C., et al. Invasion of primary glioma- and cell line-derived spheroids implanted into corticostriatal slice cultures. International Journal of Clinical and Experimental Pathology. 6 (4), 546-560 (2013).
- Matsumura, H., Ohnishi, T., Kanemura, Y., Maruno, M., Yoshimine, T. Quantitative analysis of glioma cell invasion by confocal laser scanning microscopy in a novel brain slice model. Biochemical and Biophysical Research Communications. 269 (2), 513-520 (2000).
- Ren, B., et al. Invasion and anti-invasion research of glioma cells in an improved model of organotypic brain slice culture. Tumori. 101 (4), 390-397 (2015).
- Fayzullin, A., et al. Time-lapse phenotyping of invasive glioma cells ex vivo reveals subtype-specific movement patterns guided by tumor core signaling. Experimental Cell Research. 349 (2), 199-213 (2016).
- Jensen, S. S., et al. Establishment and characterization of a tumor stem cell-based glioblastoma invasion model. PloS One. 11 (7), e0158746 (2016).
- Marques-Torrejon, M. A., Gangoso, E., Pollard, S. M. Modelling glioblastoma tumour-host cell interactions using adult brain organotypic slice co-culture. Disease Models & Mechanisms. 11 (2), 031435 (2018).
- Tamura, R., et al. Visualization of spatiotemporal dynamics of human glioma stem cell invasion. Molecular Brain. 12 (1), 45 (2019).
- Yang, C., et al. Organotypic slice culture based on in ovo electroporation for chicken embryonic central nervous system. Journal of Cellular and Molecular Medicine. 23 (3), 1813-1826 (2019).
- Murrell, W., et al. Expansion of multipotent stem cells from the adult human brain. PloS One. 8 (8), e71334 (2013).
- Fotos, J. S., et al. Automated time-lapse microscopy and high-resolution tracking of cell migration. Cytotechnology. 51 (1), 7-19 (2006).
- Tufan, A. C., Akdogan, I., Adiguzel, E. Shell-less culture of the chick embryo as a model system in the study of developmental neurobiology. Neuroanatomy. 3 (1), 8-11 (2004).
- Shoin, K., et al. Chick embryo assay as chemosensitivity test for malignant glioma. Japanese Journal of Cancer Research. 82 (10), 1165-1170 (1991).
- Hagedorn, M., et al. Accessing key steps of human tumor progression in vivo by using an avian embryo model. Proceedings of the National Academy of Sciences. 102 (5), 1643-1648 (2005).
- Balciūniene, N., et al. Histology of human glioblastoma transplanted on chicken chorioallantoic membrane. Medicina. 45 (2), 123-131 (2009).
- De Magalhães, N., et al. Applications of a new In vivo tumor spheroid based shell-less chorioallantoic membrane 3-D model in bioengineering research. Journal of Biomedical Science and Engineering. 3 (1), 20-26 (2010).
- Szmidt, M., et al. Morphology of human glioblastoma model cultured in ovo. Journal of Veterinary Research. 56 (2), 261-266 (2012).
- Jaworski, S., et al. Comparison of tumor morphology and structure from U87 and U118 glioma cells cultured on chicken embryo chorioallantoic membrane. Journal of Veterinary Research. 57 (4), 593-598 (2013).
- Yuan, Y. J., Xu, K., Wu, W., Luo, Q., Yu, J. L. Application of the chick embryo chorioallantoic membrane in neurosurgery disease. International Journal of Medical Sciences. 11 (12), 1275-1281 (2014).
- Urbańska, K., et al. The effect of silver nanoparticles (AgNPs) on proliferation and apoptosis of in ovo cultured glioblastoma multiforme (GBM) cells. Nanoscale Research Letters. 10, 98 (2015).
- DeBord, L. C., et al. The chick chorioallantoic membrane (CAM) as a versatile patient-derived xenograft (PDX) platform for precision medicine and preclinical research. American Journal of Cancer Research. 8 (8), 1642-1660 (2018).
- Han, J. M., Jung, H. J. Synergistic anticancer effect of a combination of berbamine and arcyriaflavin A against glioblastoma stem-like cells. Molecules. 27 (22), 7968 (2022).
- Ruiz-Ontañon, P., et al. Cellular plasticity confers migratory and invasive advantages to a population of glioblastoma-initiating cells that infiltrate peritumoral tissue. Stem Cells. 31 (6), 1075-1085 (2013).
- Cage, T. A., et al. Distinct patterns of human medulloblastoma dissemination in the developing chick embryo nervous system. Clinical & Experimental Metastasis. 29 (4), 371-380 (2012).
- Darrigues, E., et al. Biobanked glioblastoma patient-derived organoids as a precision medicine model to study inhibition of invasion. International Journal of Molecular Sciences. 22 (19), 10720 (2021).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved