Method Article
High-Throughput Screening to Obtain Crystal Hits for Protein Crystallography
In This Article
Summary
This protocol details high-throughput crystallization screening, ranging from the 1,536 microassay plate preparation to the end of a 6 week experimental time window. Details are included about the sample setup, the imaging obtained, and how users can perform analyses using an artificial intelligence-enabled graphical user interface to quickly and efficiently identify macromolecular crystallization conditions.
Abstract
X-ray crystallography is the most commonly employed technique to discern macromolecular structures, but the crucial step of crystallizing a protein into an ordered lattice amenable to diffraction remains challenging. The crystallization of biomolecules is largely experimentally defined, and this process can be labor-intensive and prohibitive to researchers at resource-limited institutions. At the National High-Throughput Crystallization (HTX) Center, highly reproducible methods have been implemented to facilitate crystal growth, including an automated high-throughput 1,536-well microbatch-under-oil plate setup designed to sample a wide breadth of crystallization parameters. Plates are monitored using state-of-the-art imaging modalities over the course of 6 weeks to provide insight into crystal growth, as well as to accurately distinguish valuable crystal hits. Furthermore, the implementation of a trained artificial intelligence scoring algorithm for identifying crystal hits, coupled with an open-source, user-friendly interface for viewing experimental images, streamlines the process of analyzing crystal growth images. Here, the key procedures and instrumentation are described for the preparation of the cocktails and crystallization plates, imaging the plates, and identifying hits in a way that ensures reproducibility and increases the likelihood of successful crystallization.
Introduction
Even in an age of tremendous progress in structural biology methods, X-ray crystallography continues to be a dependable and popular method for generating high-quality structural models of macromolecules. Over 85% of all three-dimensional structural models deposited to the Protein Data Bank (PDB) are from crystal-based structural methods (as of January, 2023).1 Furthermore, X-ray crystallography remains indispensable for solving protein-ligand structures, a crucial component of the drug discovery and development process2. Despite protein crystallization having remained the dominant structural biology technique for over half a century, methods to predict crystallization likelihood based on physical properties3 or sequence4,5 are still in their infancy.
The prediction of crystallization conditions is even more obscure; limited progress has been made to predict likely crystallization conditions even for model proteins6,7. Other studies have attempted to identify crystallization conditions based on protein homology and conditions mined from the PDB8,9,10. The predictive power to be found in the PDB is limited, however, as only the final, successful crystallization conditions are deposited, which, by necessity, misses the often extensive optimization experiments required to fine-tune crystal growth. Further, many PDB entries lack metadata containing these details, including the cocktail formulas, crystallization format, temperature, and time to crystallize11,12. Therefore, for many proteins of interest, the most accessible way to determine the crystallization conditions is experimentally, using as many conditions as possible across a wide range of chemical possibilities.
Several approaches to make crystallization screening as fruitful and thorough as possible have been explored to great effect, including sparse matrices13, incomplete factorial screening14, additives15,16, seeding17, and nucleating agents18. The National HTX Center at Hauptman-Woodward Medical Research Institute (HWI) has developed an efficient pipeline for crystallization screening using the microbatch-under-oil approach19, which utilizes automated liquid handling and imaging modalities to streamline the identification of initial crystallization conditions using comparatively minimal sample and cocktail volumes (Figure 1). The set of 1,536 unique cocktails are based on conditions previously determined to be conducive to protein crystal growth and are designed to be chemically diverse in order to sample a large range of possible crystallization conditions20,21,22. The broad sampling of crystallization conditions increases the likelihood of observing one or more crystallization leads.
Few formal analyses of how many conditions are needed for screening have appeared in the literature. One study focused on the sampling layout of different screens and found that the random sampling of components (similar to an incomplete factorial) represented the most thorough and efficient sampling method23. Another study of screening noted that there have been numerous instances when the very thorough 1,536 screen has yielded only a single crystal hit24, and a very recent study highlighted that most commercial screens undersample the crystallization space known to be associated with screening hits25. Not all crystallization leads will yield a diffraction quality crystal suitable for data collection due to inherent disorder within the crystal, diffraction limitations, or crystal flaws; therefore, casting a wider net for conditions has the additional benefit of providing alternative crystal forms for optimization.
The format of protein crystallization experiments also has an impact on the success of the screen. Vapor diffusion is the most commonly used setup for high-throughput crystallization applications and is utilized at state-of-the art crystallization centers, including the EMBL Hamburg and Institut Pasteur high-throughput screening centers26,27,28. The HTX Center uses the microbatch-under-oil method; while less commonly used, it is a robust method that minimizes the consumption of sample and crystallization cocktails20,21,22. One advantage of the microbatch-under-oil method, particularly when using a high-viscosity paraffin oil, is that only slight evaporation occurs within the drop during the experiment, meaning that the equilibrium concentration is achieved upon drop mixing. If positive crystallization results are observed in the microbatch-under-oil method, the reproduction of these conditions is typically more straightforward than in vapor diffusion setups, in which crystallization occurs at some undefined point during the equilibration between the crystallization drop and the reservoir. The reproducibility of hits is desirable for high-throughput crystallization approaches, which produce prohibitively tiny protein crystals that typically need to be optimized for single-crystal X-ray experiments.
The high-throughput crystallization screen for soluble proteins is made up of cocktails that are prepared in-house, ready-made commercial screens, and in-house-modified commercial screens22. The cocktails were initially developed using the incomplete factorial strategy using previously successful crystallization cocktails20. The reagents in the screen that are commercially available include arrays of polymers, crystallization salts, PEG, and ion combinations and screens that utilize sparse matrix and incomplete factorial approaches. There are also reagents that are modified before inclusion in the screen: an additive screen, a pH and buffer screen, an ionic liquid additive screen, and a polymer screen.
The power of known crystallization conditions and strategies has been leveraged in the 1,536 crystallization cocktails, along with the benefits of the microbatch-under-oil system to generate a pipeline that employs automated liquid handling, automated brightfield imaging, and second order nonlinear imaging of chiral crystals (SONICC). The automation of both the liquid handling and imaging provides the benefits of fewer wet lab hours and higher reproducibility. The high-throughput nature of automated crystallization screening necessitates the automation of the process of monitoring for crystal growth. These advances are achieved with state-of-the-art imaging technologies to assist in the identification of positive crystal hits. Both standard brightfield imaging of plates, as well as multi-photon methods for enhanced detection, are used via a crystal imaging system with SONICC (Figure 2). SONICC combines second harmonic generation (SHG)29 microscopy and ultraviolet two-photon excited fluorescence (UV-TPEF)30 microscopy to detect very small crystals, as well as those obscured by precipitate. The SONICC imaging informs on whether the wells contain protein (via UV-TPEF) and crystals (via SHG). Beyond the positive identification of protein crystals, additional information can also be obtained using state-of-the-art imaging methods. Cocktail-only imaging prior to sample addition serves as a negative control; these images can identify the well appearance prior to sample addition, including in terms of salt crystals and debris. Additionally, SHG and UV-TPEF imaging help differentiate protein crystals from salt crystals and can be used for visualizing protein-nucleic acid complexed material31.
High-throughput crystallization experiments undergoing repeated monitoring via imaging result in a very large volume of images needing examination. Automated crystal scoring methods have been developed to reduce the burden on the user and increase the probability of identifying positive crystal hits. The HTX Center partcipated in the development of the MAchine Recognition of Crystallization Outcomes (MARCO) scoring algorithm, a trained deep convolutional neural network architecture developed by a consortium of academic, non-profit, government, and industry partners to classify brightfield well images32. The algorithm was trained on nearly half a million brightfield images from crystallization experiments from multiple institutions using different crystallization methods and different imagers. The algorithm outputs a probabilistic score indicating whether a given image falls into four possible image classes: "crystal", "clear", "precipitate", and "other". MARCO has a reported classification accuracy of 94.5%. Crystal detection is further enhanced with software that implements the algorithm and provides a graphical user interface (GUI) for accessible and simple image viewing, enabled with the AI-enabled scoring capabilities32,33. The MARCO Polo GUI is designed to work seamlessly with the setup of the imaging and data management system in the HTX Center to identify hits in the 1,536-well screen, with human engagement to examine the output of sorted lists. Additionally, as open-source software available on GitHub, the GUI is readily available for modification to reflect the specific needs of other laboratory groups.
Here, the process of setting up a high-throughput microbatch-under-oil experiment using robotic liquid handling to deliver both the cocktail and protein is described. The HTX Center has a unique array of instrumentation and resources that are not found at other institutions, with the goal of providing screening services and educational resources to interested users. Demonstrating the methods and capabilities of robotics-enabled high-throughput techniques will enable the community to have knowledge of available technologies and make decisions for their own structure determination efforts.
Protocol
1. Preparation or purchase of cocktails for sixteen 96-well deep well blocks
- Prepare in-house-generated chemical cocktails by dispensing into 96-well deep well (DW) blocks. Use a robotic liquid handler to dispense and mix stock solutions of salts, buffers, polymers, and water.
- Prepare in-house-modified chemical cocktails by using a robotic liquid handler or multichannel pipette to add additional components to 96-well DW block screens that have been commercially purchased.
- Purchase commercially available DW blocks.
- Store labeled 96-well DW blocks at −20 °C for 12-18 months.
NOTE: The cocktails prepared in step 1.1. and 1.2. fill 10/16 96-well DW blocks, and 5/16 96-well DW blocks are used as purchased. One 96-well DW block in the screen is set up at the time of the 1,536-well plate dispensing to avoid precipitation of the additive screen (see section 3).
2. Dispensing the cocktails to 384-well plates
- Thaw the 96-well DW blocks at 4 °C overnight. Bring to room temperature (20-23 °C) before beginning the preparation of the 384-well plates.
NOTE: Room temperature is suitable for preparing the cocktail plates. The main concern in preparing these plates is to avoid precipitates, which can clog liquid-handling devices and lead to unpredictable changes in the concentrations of the cocktail ingredients. - Mix the blocks thoroughly by inversion as needed to dissolve any persistent opaque precipitate. If any wells contain precipitate, warm the blocks to 30 °C to dissolve.
- Deliver 50 µL of cocktail solution from four 96-well DW blocks to one 384-well plate using a liquid handling robot equipped with a 96 syringe or pipettor head. The four 96-well DW blocks are stamped out into the 384-well plate such that quadrants are filled (e.g., A1 of 96-DW1 to A1 of 384-plate1, A1 of 96-DW2 to B1 of 384-plate1, etc.) (Figure 3).
- Deliver 15 of the 16 96-well DW blocks to 384-well plates for storage.
- Store the 384-well plates at −20 °C for up to 6 months for use in preparing the 1,536-well plates.
3. Preparing the 1,536-well plates with oil and crystallization cocktails
- Deliver 5 µL of paraffin oil to each well of a 1,536-well plate using a robotic liquid handling system with the capacity for slow aspiration and delivery. Store the oil plates at 4 °C for up to 6 months.
- Thaw the 384-well plates from section 2 at 4 °C overnight. Invert the plates to mix the solutions and dissolve the precipitate. Incubate the plates at 30 °C to dissolve persistent precipitates.
- To prepare the additive screen components, use the final 96-well DW block containing 0.1 M HEPES pH 6.8, 30% PEG3350 to mix with the commercial additive screen by using either a liquid handling robot or a multichannel pipette.
- Prepare the additive screen solutions by dispensing a 1:1 mixture of the buffered PEG3350 solution prepared in step 3.3 and the additive screen to a final volume of 50 µL in the appropriate 384-well plate.
- Use a liquid handling robot equipped with a 384 syringe or pipettor head to deliver 200 nL of cocktail solution into each well of the 1,536-well plate. Stamp out four 384-well plates into the 1,536-well plate such that quadrants are filled (e.g., A1 of 384-plate1 to A1 of the 1,536-well plate, A2 of 384-plate1 to A3 of the 1,536-well plate, etc.) (Figure 3).
- Centrifuge the plates at 150 × g for 5 min before storing at 4 °C for up to 4 weeks.
4. Sample submission
- To submit a sample, send a reservation email prior to the reservation deadline for the upcoming screening run. Include the number of screening experiments, the name, the PI, and the institution, as well as any special handling requirements for the sample. Screening runs are conducted approximately once monthly, resulting in 12 runs yearly.
- Complete a sample submission form prior to shipping the sample.
- For new users, choose a password that will be used to download the crystallization images in section 7.
- For established users, use an existing password or change the password at this step.
- Submit the sample in a 1.5 mL tube. Ensure the macromolecule is homogeneous and adequately concentrated to promote crystallization. Use a pre-crystallization test, typically composed of ammonium sulfate or PEG 4,000, to investigate the appropriate sample concentration by observing whether the tested sample concentrations result in clear drops or precipitate34.
NOTE: Suitable quality tests that may be undertaken prior to sample submission to check for purity and homogeneity include SDS-PAGE, gel filtration, and dynamic light scattering (DLS), amongst others. Crystallization can be affected by the presence of even minor impurities. A sample volume of 500 µL is currently required to set up one 1,536-well plate. Testing is underway to decrease the sample volume requirement.- Avoid using buffer concentrations greater than 50 mM, as well as phosphates, which may crystallize within the screen.
- Avoid excessive solubilizing agents, including glycerol concentrations greater than 10% w/v.
- Package the sample to safely maintain an appropriate temperature using dry ice, wet ice, or cooling packs in a sealed container.
- Ship sample priority overnight on a Monday-Wednesday during the run.
- Email the tracking number once the sample has been shipped.
5. Sample setup in the prepared 1,536-well plates
- Unpack samples and immediately incubate at the temperature the user has requested.
- Once thawed, centrifuge the sample at 10,000 × g for 2 min at room temperature. Visually observe the sample to identify precipitation, color, and condition of the sample prior to setup.
- Warm 1,536-well plate to 23 °C and centrifuge at 150 × g for 5 min. Image the cocktail-only plate using brightfield imaging as a negative control.
NOTE: All plates are imaged with brightfield imaging prior to sample setup, which enables the identification of wells that already have crystals or debris in them prior to sample addition as a negative control. Further, it enables the identification of wells in which the crystallization cocktail has not been delivered. Warming the plate to room temperature eliminates condensation on the plate surface, leading to clear images. - Dispense 200 nL of sample to each well in the 1,536-well plate using a liquid handling robot. Centrifuge plate at 150 × g and incubate plates at 4 °C, 14 °C, or 23 °C.
NOTE: Microbatch under-oil-experiments can be set up by hand by dispensing the protein and cocktail under the desired oil. However, it is recommended to use no less than 1 µL of each protein and cocktail to achieve reproducible results.
6. Monitor 1,536-well plates for crystal formation
- After the sample has been added to the 1,536-well plates, image with brightfield imaging at day 1 and week 1, week 2, week 3, week 4, and week 6.
- Perform SONICC imaging with SHG and UV-TPEF at the 4 week time point for plates being incubated at 23 °C and at the 6 week time point for plates being incubated at 14 °C or 4 °C.
NOTE: The timing for the SONICC imaging is scheduled at the 4 week and 6 week time points for the high-throughput 1,536 microassay plate because, typically, crystals will appear by those time points. For modification to a 96-well microbatch under oil or vapor diffusion experiments, it is advisable to perform the SONICC imaging earlier in the time window. - Access the experimental images that have been automatically transferred to the user account using an internal LIMS system. Notify the users via an automated htslab email daemon that imaging has occurred.
7. Image analysis
- Retrieve the screening images from the HWI ftp site for each .rar file.
NOTE: The image output from the 1,536 screen results in a number of files containing brightfield images, SHG images, and UV-TPEF images. Each imaging modality or time point is a separate .rar file. Each .rar file, when unpacked, contains an image from each well of the 1,536-well plate at a specific time point using a specific imaging modality.- Use the FileZilla Client or other options to access the ftp data.
NOTE: The FileZilla Client is the recommended way to manage the large file transfer volume to minimize computational crashes.- If FileZilla Client needs to be installed on the user computer, download the FileZilla software.
- If FileZilla Client is already installed or upon installation, click the FileZilla icon to open the software.
- Log in to the remote ftp server from FileZilla by entering the host ftp website, username, and password.
- Download the .rar files to the desired directory.
- Use the FileZilla Client or other options to access the ftp data.
- Use the AI-enabled open-source GUI to view, score, and analyze the crystallization images.
NOTE: The GUI can be used on most Windows, Mac, and Linux operating systems (OS), and OS-specific instructions for download are located on the GitHub site. MARCO Polo is an open-source GUI that incorporates metadata from the high-throughput 1,536 crystallization screen implemented at the HTX Center. It is available for anyone to download from GitHub for modification to reflect the specific needs of other laboratory groups.- Open the .rar file in the GUI after the file has been downloaded (see Supplementary Figure S1).
- Click on Import, select Images from the dropdown menu, and then select From Rar Archive/Directory.
- Click on Browse for Folder in the popup window, and then navigate to the folder containing the images.
- Select the desired file(s), and import into the GUI by clicking on Open. Wait for the file(s) to appear in the Selected Paths window. Select one or more files to download into the GUI, and click on Import Runs.
- View the image for the first well in the window of the Slideshow Viewer in the GUI by clicking on the > symbol to the left of the sample name and then selecting the appropriate read by double-clicking on it (reads are listed by the date and type of image-brightfield, UV-TPEF, or SHG).
- Enlarge the image by resizing the whole window. The Image Details box includes information about the image, including the scoring information (empty until the read has been scored). The Cocktail Details box contains metadata about the cocktail components.
- Move to the next well by clicking on the Next button in the Navigation panel or pressing the right arrow key on the keyboard. Navigate to a specific well by entering the well number in the By Well Number window.
- View all the reads (of those imported into the GUI) by checking the Show All Dates box.
- View all the spectra (of those imported into the GUI) by checking the Show All Spectra box. Click on the Swap Spectrum button to view each spectrum image individually.
- Open the .rar file in the GUI after the file has been downloaded (see Supplementary Figure S1).
- Score the crystal images using the MARCO algorithm by first highlighting a specific run from the list on the left side of the window. Next, click on the Classify Selected Run button. View the MARCO scoring information in the Image Details window once an imaging read has been scored for all 1,536 wells.
NOTE: Classification will typically take between 2-5 min, depending on the computer speed and memory available. The algorithm generates scores that classify the contents into "crystal", "clear", "precipitate", or "other" classes. The numeric values associated with each well's classification reflect the likelihood of the well containing objects of that class.- View a subset of the scored images by ticking the desired box(es) in the Image Filtering panel and clicking on the Submit Filters button. For example, view only the images classified by MARCO as crystals by ticking the Crystals and the MARCO boxes and clicking on Submit Filters.
- Manually score the crystal images to generate the "human scored" set. Assign a score to a well by clicking on the appropriate button (the "crystal", "clear", "precipitate", or "other" buttons are located in the Classification panel at the bottom of the window). Alternatively, use the number pad on the keyboard to assign the score (1 = "crystal", 2 = "clear", 3 = "precipitate", 4 = "other"). Designate a human-scored image as a "favorite" by ticking the Favorite? box.
NOTE: View only the images classified by a human as crystals by ticking the Crystals and the Human boxes and clicking on Submit Filters. Clicking on the Favorites box in the Filtering panel further narrows down the returned images, returning only the human-scored crystal images that are also favorites. - Use the Plate Viewer tab to view multiple wells at one time. On the second Plate Viewer tab in the Controls panel, select 16, 64, or 96 images from the dropdown menu in the Images Per Plate section. Use the Image Filtering Tab to gray-out images that are not of interest. Select the Apply Filter box to filter the images.
NOTE: For example, select the "human" and "crystal" boxes, and only those wells that were scored as a crystal by a human will be easily visible.- Navigate in the Plate Viewer tab, by clicking on the Next button to view the next set of 16/64/96 images. By default, images scored as crystals are red, those scored as clear are blue, those scored as precipitate are green, and those scored as other are orange. Change the colors by using the dropdown menus.
- Select the information to be displayed on the wells by ticking various boxes on the Labels tab.
- Click on Save View to save an image file of the current view.
- Click on Swap Spectrum to toggle between brightfield, SHG, and UV-TPEF images for the multiple-well image.
- Click on Export, and select the appropriate file type from the dropdown menu to export the scored files for use in other programs.
NOTE: CSV (comma-separated values) files are compatible with spreadsheet programs such as Microsoft Excel or Google Sheets. JSON (JavaScript Object Notation) files can be opened with most text editors. PPTX (PowerPoint Presentation) can be used to display images from Polo, including a comparison of brightfield, UV-TPEF, and SHG images. Files are saved in .xtal format to be reopened in the MARCO Polo GUI.- Save an .xtal format file by clicking on File at the top of the page and then selecting either Save Run or Save Run As. Provide a file name and directory location.
- Open .xtal format files by clicking on Import and selecting Images and then From Saved Run. Browse for the appropriate file location, click on the file name, and then click on Open.
Results
The outcomes of the 1,536-well crystal screening experiment consist of seven complete brightfield image sets collected at day 0 (negative control), day 1, week 1, week 2, week 3, week 4, and week 6 (Figure 4). SONICC images are collected at the 4 week time point for plates incubated at 23 °C and at the 6 week time point for plates incubated at 4 °C or 14 °C. Altogether, once a sample has been shipped, users can anticipate having their plates set up within 1 day of arrival. The images will be uploaded as they are collected. The crystallization screening experiment concludes after 6 weeks.
The 1,536-well plate setup allows all the screening experiments to be conducted within the same plate, thus limiting sample consumption and facilitating imaging and direct comparison between imaging modalities. Representative results for the time course of crystal growth for a single cocktail condition are shown in Figure 4. Automated plate imaging throughout the course of the experiment allows the identification of both rapidly and slowly growing crystals by brightfield imaging. The UV-TPEF and SHG imaging allow cross-validation of the hits observed by brightfield imaging and indicate that the crystals observed are proteinaceous and crystalline, respectively (Figure 5A,B). Furthermore, SONICC imaging enables the identification of crystals that are visually obscured by precipitate or films (Figure 5C) or microcrystals that may otherwise be mistaken for precipitate (Figure 5D). For some crystals, a lack of SHG signal is not disqualifying, as some point groups do not produce an SHG signal35,36, as exemplified by the tetragonal thaumatin crystal in Figure 5C. Conversely, a lack of UV-TPEF signal for proteins lacking tryptophan residues should be anticipated. The observation of UV-TPEF and SHG signals also facilitates the identification of non-protein salt crystals, which will appear in brightfield and exhibit a strong positive SHG signal but will lack a UV-TPEF signal (Figure 5E).
Image analysis for the plate setup is streamlined with the MARCO Polo GUI, which also bundles the ftp data transfer from the HWI servers (as an alternative to transferring files with FileZilla). The MARCO Polo GUI allows for easily navigable plate and image viewing and performs computational image scoring using the MARCO algorithm so that the image results can be rapidly downloaded, viewed, and analyzed from the HTX Center. The MARCO scoring algorithm, as implemented in the MARCO Polo GUI, is capable of scoring images from the entire 1,536-well plate in less than 5 min. Images flagged as crystalline by the MARCO algorithm can be subsequently sorted by the Polo GUI for display. Since the MARCO algorithm was optimized for crystal identification and minimizing false negatives so as not to miss any positive hits, the scoring can result in false positive flags. Nevertheless, the ability of MARCO to limit the set of images needing to be examined by focusing attention on the wells with a high probability of containing crystals results in a substantial reduction in data processing burden for users. The convenient implementation of the algorithm in the user-friendly MARCO Polo viewing platform, with its ability to sort images based on MARCO scores, greatly improves the user's ability to analyze the dataset quickly and to accurately determine crystal hits.

Figure 1: Schematic of a high-throughput 1,536-well crystallization screening experiment performed at the HTX Center. (1) In this step, 5 µL of paraffin oil and 200 nL of cocktail are added to each well (protocol step 3.1 and step 3.5). A cartoon illustration of one well containing only oil and cocktail and a representative image are shown to the right. (2) Samples arrive at the HTX Center (protocol step 5.1). 3) In this step, 200 nL of sample is added to each well (protocol step 5.4). (4) All 1,536 wells are monitored over time using brightfield imaging, 5) as well as the UV-TPEF and SHG modalities (protocol step 6). 6) The AI-enabled open-source GUI is used to view, score, and analyze the crystallization images (protocol step 7). Abbreviations: HTX = high-throughput crystallization; UV-TPEF = UV-two-photon excited fluorescence; SHG = second harmonic generation; AI = artificial intelligence; GUI = graphical user interface. Please click here to view a larger version of this figure.
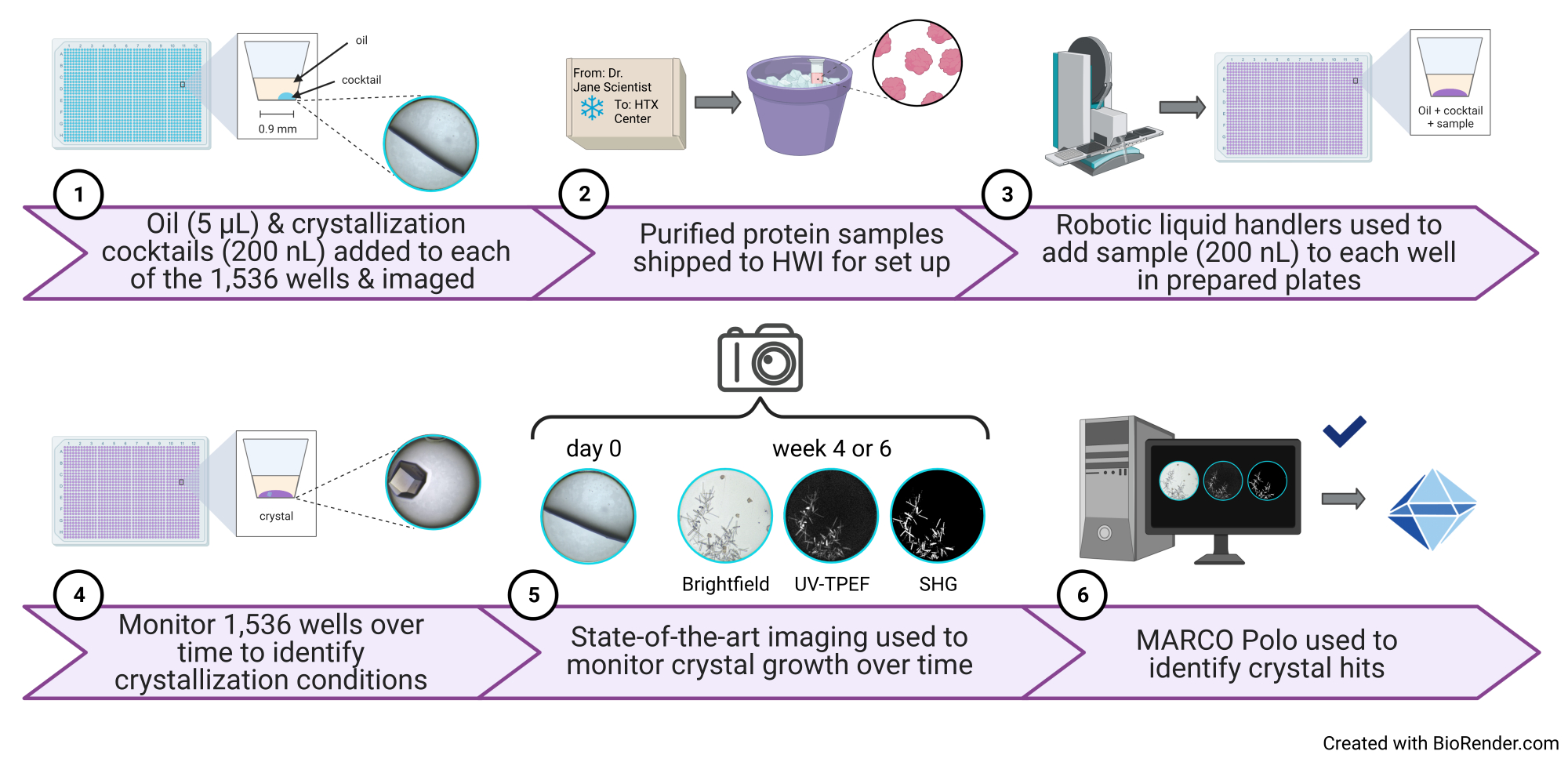{kind=link}

Figure 2: Single 1,536-well plates containing screening experiments, imaged using brightfield, UV-TPEF, and SHG imaging. The 1,536-well plates are shown with an American penny for scale (top). Each screening experiment is imaged once prior to setup and six times after sample addition with brightfield imaging (seven total brightfield image sets, left). The plates undergo UV-TPEF (center) and SHG (right) imaging at 4 weeks or 6 weeks. Abbreviations: UV-TPEF = UV-two-photon excited fluorescence; SHG = second harmonic generation. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Schematic showing how the 1,536-well plates are generated. Sixteen 96-well DW blocks are used to stamp out four 384-well plates, with each quadrant of each 384-well plate filled by dispensing crystallization cocktails. Four 96-well DW blocks fill one 384-well plate (middle). Four 384-well plates are used to stamp out the single 1,536-well plate (right). Abbreviation: DW = deep well. Please click here to view a larger version of this figure.
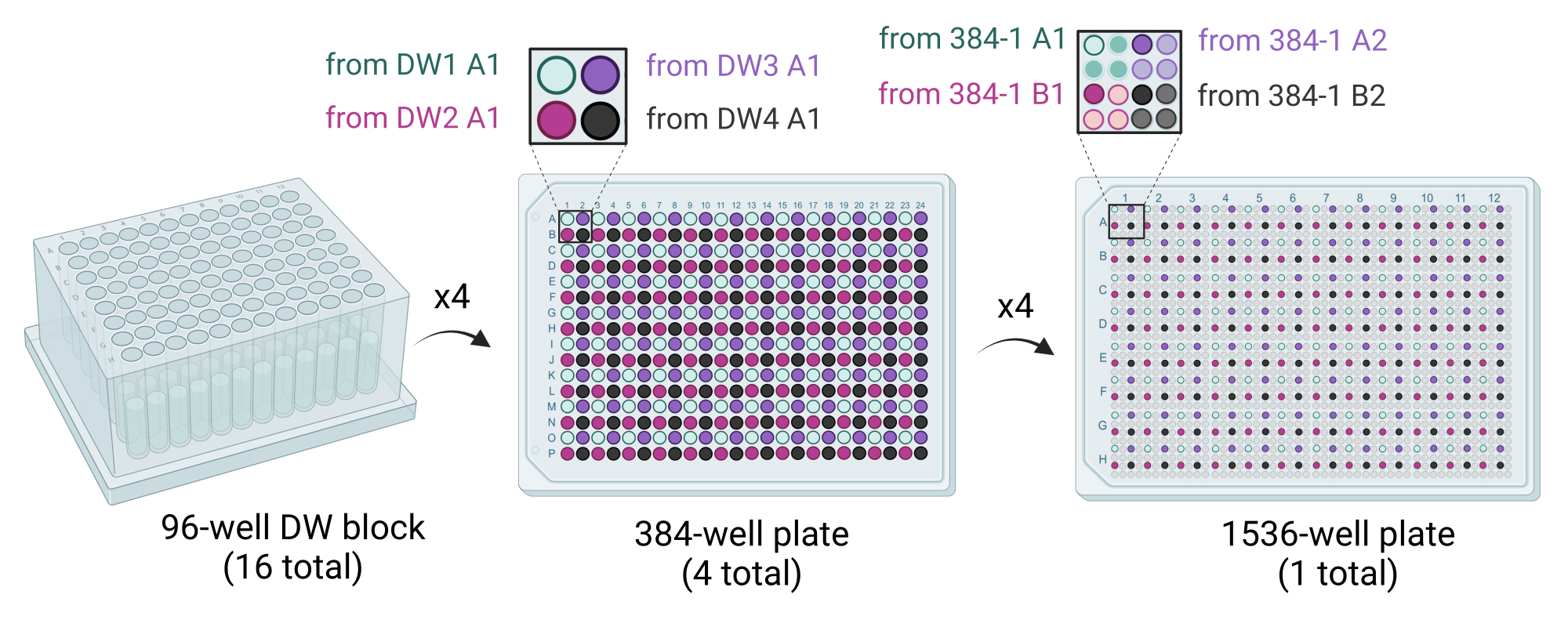{kind=link}

Figure 4: Representative time course of a single well in a 1,536-well screening experiment. Plates are imaged prior to sample setup (day 0), as well as with brightfield imaging on day 1, week 1, week 2, week 3, week 4, and week 6. The plates incubated at 23 °C are imaged with SONICC at week 4. Scale bars = 80 µm (brightfield), 200 µm (SHG, UV-TPEF). Abbreviations: SONICC = second order nonlinear imaging of chiral crystals; UV-TPEF = UV-two-photon excited fluorescence; SHG = second harmonic generation. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Representative imaging results for the HT 1,536 crystal screening experiments. Brightfield, UV-TPEF, and SHG imaging results are shown for five example wells. (A,B) Protein crystals observed by brightfield, UV-TPEF, and SHG imaging are clearly apparent in all three imaging modalities. (C) A protein crystal obscured by film in brightfield imaging is visible by UV-TPEF imaging; the crystal is not observed by SHG imaging due to point group incompatibility. (D) Example of microcrystals verified by UV-TPEF and SHG imaging that may otherwise be considered precipitate. (E) Example of salt crystals that appear crystalline by brightfield and SHG imaging but do not exhibit a UV-TPEF signal. Scale bars = 200 µm. Well diameter = 0.9 mm. Abbreviations: UV-TPEF = UV-two-photon excited fluorescence; SHG = second harmonic generation. Please click here to view a larger version of this figure.
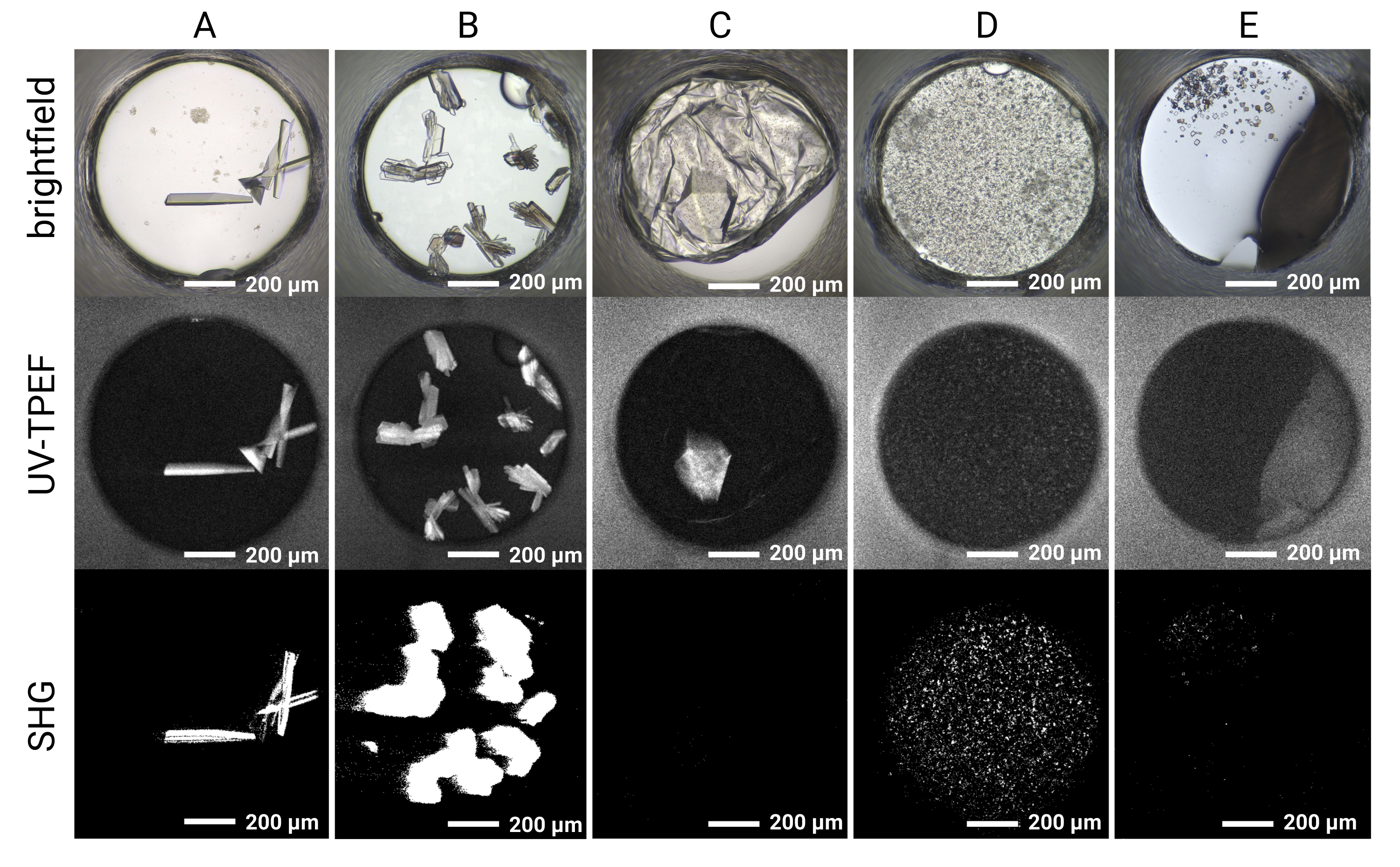{kind=link}
Supplementary Figure S1: Opening image files in MARCO Polo. Image files can be opened within the MARCO Polo GUI by navigating to the Import | Images tab at the top (a). Note that files can also be transferred via the From FTP tool directly in MARCO Polo (a) or can be transferred via FileZilla as described in protocol step 7.2. To import files that have already been downloaded, select Images | From Rar Archive/Directory. In the popup window that appears, select Browse for Folder (b), and navigate to the file directory where the plate image files are saved. Once the files are in the Selected Paths window (c), highlight a file, and click on Import Runs (d). The MARCO Polo GUI will identify the correct Cocktail File metadata to import with the images. Please click here to download this File.
{kind=link}
Discussion
The method describes a high-throughput pipeline for protein crystallization screening that requires as little as 500 µL of sample for 1,536 individual crystallization experiments in the microbatch-under-oil format. The pipeline relies on liquid-handling robotics to rapidly and reproducibly aid the experimental setup, as well as the computational image analysis resource MARCO Polo, which is customized to analyze 1,536-well plate images using the MARCO algorithm to identify and isolate crystal hits.
The small volume of individual screening drops (400 nL total with a 1:1 ratio of sample:cocktail) means that extremely small sample volumes are required to identify positive crystallization conditions. These small drop sizes necessarily produce small crystals that cannot be fished by traditional looping. Methods have been developed to harvest from the 1,536 plates37; additionally, the plates with crystals have been used directly at synchrotron sources for in situ data collection38. If a robust method for harvesting these crystals were developed, advances in synchrotron technology and micro-focused beams would further enable useful datasets to be obtained. Additionally, the crystals obtained could potentially be used as seeds for optimization efforts.
SONICC imaging is clearly advantageous in identifying both small protein crystals and protein crystals hidden beneath precipitate. Despite these advantages, not all sample types are amenable to SHG and UV-TPEF imaging. For example, proteins with few or no aromatic tryptophan residues will show an ambiguous UV-TPEF signal. Furthermore, crystals in specific space groups, including centrosymmetric groups or point group 432, will be undetected by SHG imaging. Samples with fluorophores sometimes interfere with the SHG signal, resulting in the cancellation of the signal or increased intensity, meaning careful interpretation of SHG signals is required for metal-containing proteins and proteins containing fluorescent moieties. However, in many cases, it is possible to rationalize the absence of an SHG or UV-TPEF signal, and the lack of these signals should not necessarily rule out the presence of a protein crystal.
The microbatch-under-oil format provides an alternative to the more common vapor diffusion method used for high-throughput crystallography. Importantly, the crystallization format impacts hit identification39, which provides a rationale for the use of different crystallization formats for high-throughput screening efforts. Automated imaging and SONICC-enabled modalities aid in the rapid identification of protein crystals throughout the 6 week experimental time course. Finally, the MARCO Polo GUI enables users to rapidly analyze images from 1,536 conditions to identify promising hit wells for optimization. The capabilities at the HTX Center, including the robotics-enabled high-throughput experimental setup, coupled with the state-of the-art imaging and computational tools for analyses, provide a major contribution to the structural biology community by empowering researchers to effectively address a primary bottleneck in crystal-based structural work: finding crystallization conditions.
Disclosures
The authors have no competing financial interests or other conflicts of interest.
Acknowledgements
We would like to extend our gratitude to our users for entrusting their precious samples to us for crystal screening, as well as for providing critical feedback and requests that have helped us refine and develop our resources to better serve the structural biology community. We would also like to acknowledge Ethan Holleman, Dr. Lisa J Keefe, and Dr. Erica Duguid, who drove the development of the MARCO Polo GUI. We would like to thank the HWI colleagues for their support and suggestions, especially Dr. Diana CF Monteiro. We acknowledge funding support from the National Institutes of Health, R24GM141256.
Materials
| Name | Company | Catalog Number | Comments |
| 1536 Well Imp@ct LBR LoBase | Greiner Bio-One | 790 801 | |
| Acetic acid | Hampton Research | HR2-853 | |
| AlumaSeal II Sealing Film | Hampton Research | HR8-069 | |
| Ammonium bromide | Molecular Dimensions | MD2-100-247 | |
| Ammonium chloride | Hampton Research | HR2-691 | |
| Ammonium hydroxide | Hampton Research | HR2-855 | |
| Ammonium nitrate | Hampton Research | HR2-665 | |
| Ammonium phosphate dibasic | Hampton Research | HR2-629 | |
| Ammonium phosphate monobasic | Hampton Research | HR2-555 | |
| Ammonium sulfate | Hampton Research | HR2-541 | |
| Ammonium thiocyanate | Molecular Dimensions | MD2-100-301 | |
| Bicine pH 9.0 | Hampton Research | HR2-723 | |
| Bis-tris propane pH 7.0 | Hampton Research | HR2-993-08 | |
| Calcium acetate | Hampton Research | HR2-567 | |
| Calcium chloride dihydrate | Hampton Research | HR2-557 | |
| CAPS pH 10.0 | Rigaku Reagents | none given | |
| ClearSeal Film | Hampton Research | HR4-521 | |
| Cobalt sulfate heptahydrate | Molecular Dimensions | MD2-100-42 | |
| Crystal Screen HT screen | Hampton Research | HR2-130 | |
| Formulator | Formulatrix | ||
| Glycerol | Hampton Research | HR2-623 | |
| Gryphon liquid handling robot | Art Robbins Instruments | ||
| HEPES pH 7.0 | Hampton Research | HR2-902-03 | |
| HEPES pH 7.5 | Hampton Research | HR2-902-08 | |
| HWI HTX Center sample submission form | https://hwi.buffalo.edu/high-throughput-crystallization-screening-center-sample-submission-form/ | ||
| Hydrochloric acid | Hampton Research | HR2-581 | |
| Index HT screen | Hampton Research | HR2-134 | |
| Ionic Liquid screen | Hampton Research | HR2-214 | |
| Lithium bromide | Molecular Dimensions | MD2-100-312 | |
| Lithium chloride | Hampton Research | HR2-631 | |
| Lithium sulfate monohydrate | Hampton Research | HR2-545 | |
| Magnesium acetate tetrahydrate | Hampton Research | HR2-561 | |
| Magnesium chloride hexahydrate | Hampton Research | HR2-559 | |
| Magnesium nitrate hexahydrate | Hampton Research | HR2-657 | |
| Magnesium sulfate heptahydrate | Hampton Research | HR2-821 | |
| Manganese chloride tetrahydrate | Millipore Sigma | 63535-50G | |
| Manganese sulfate monohydrate | Molecular Dimensions | MD2-100-310 | |
| MARCO Polo GUI download | https://hauptman-woodward.github.io/Marco_Polo/ | ||
| Matrix Platemate 2 x 3 liquid handling robot | Thermo Scientific | ||
| MES pH 6.0 | Hampton Research | HR2-943-09 | |
| Mosquito liquid handling robot | SPTLabtech | ||
| Paraffin Oil/White Mineral Oil Saybolt Viscosity 340-365 at 100 °F | Sigma Aldrich | PX0045-3 | |
| PEG 1000 | Hampton Research | HR2-523 | |
| PEG 2000 | Hampton Research | HR2-592 | |
| PEG 20000 | Hampton Research | HR2-609 | |
| PEG 3350 | Hampton Research | HR2-527 | |
| PEG 400 | Hampton Research | HR2-603 | |
| PEG 4000 | Hampton Research | HR2-529 | |
| PEG 6000 | Hampton Research | HR2-533 | |
| PEG 8000 | Hampton Research | HR2-535 | |
| PEG/Ion HT screen | Hampton Research | HR2-139 | |
| PEGRx HT screen | Hampton Research | HR2-086 | |
| Plate reservations | htslab@hwi.buffalo.edu | ||
| Potassium acetate | Hampton Research | HR2-671 | |
| Potassium bromide | Hampton Research | HR2-779 | |
| Potassium carbonate | Molecular Dimensions | MD2-100-311 | |
| Potassium chloride | Hampton Research | HR2-649 | |
| Potassium nitrate | Hampton Research | HR2-663 | |
| Potassium phosphate dibasic | Hampton Research | HR2-635 | |
| Potassium phosphate-monobasic | Hampton Research | HR2-553 | |
| Potassium phosphate-tribasic | Molecular Dimensions | MD2-100-309 | |
| Potassium thiocyanate | Hampton Research | HR2-695 | |
| Rock Imager 1000 with SONICC | Formulatrix | ||
| Rock Imager 54 | Formulatrix | ||
| Rubidium chloride | Millipore Sigma | R2252-10G | |
| SaltRx HT screen | Hampton Research | HR2-136 | |
| Silver Bullets screen | Hampton Research | HR2-096 | |
| Slice pH screen | Hampton Research | HR2-070 | |
| Sodium acetate pH 5.0 | Hampton Research | HR2-933-15 | |
| Sodium bromide | Hampton Research | HR2-699 | |
| Sodium chloride | Hampton Research | HR2-637 | |
| Sodium citrate pH 4.2 | Hampton Research | HR2-935-01 | |
| Sodium citrate pH 5.6 | Hampton Research | HR2-735 | |
| Sodium hydroxide | Hampton Research | HR2-583 | |
| Sodium molybdate dihydrate | Molecular Dimensions | MD2-100-207 | |
| Sodium nitrate | Hampton Research | HR2-661 | |
| Sodium phosphate monobasic | Hampton Research | HR2-551 | |
| Sodium thiosulfate pentahydrate | Molecular Dimensions | MD-100-307 | |
| StockOptions Polymer screen | Hampton Research | HR2-227 | |
| Tacsimate pH 7 | Hampton Research | HR2-755 | |
| TAPS pH 9.0 | bioWORLD | 40121071 | |
| Tris pH 8 | Hampton Research | HR2-900-11 | |
| Tris pH 8.5 | Hampton Research | HR2-725 | |
| ViaFLO 384 | Integra | ||
| ViaFLO 384 384 channel pipettor head (0.5-12.5µL) | Integra | ||
| ViaFLO 384 96 channel pipettor head (300µL) | Integra | ||
| Zinc acetate dihydrate | Hampton Research | HR2-563 |
References
- PDB data distribution by experimental method and molecular type. RCSB Protein Data Bank Available from: https://www.rcsb.org/stats/summary (2022)
- Maveyraud, L., Mourey, L. Protein X-ray crystallography and drug discovery. Molecules. 25 (5), 1030 (2020).
- Dupeux, F., Röwer, M., Seroul, G., Blot, D., Márquez, J. A. A thermal stability assay can help to estimate the crystallization likelihood of biological samples. Acta Crystallographica Section D: Biological Crystallography. 67 (11), 915-919 (2011).
- Elbasir, A., et al. DeepCrystal: A deep learning framework for sequence-based protein crystallization prediction. Bioinformatics. 35 (13), 2216-2225 (2019).
- Zucker, F. H., et al. Prediction of protein crystallization outcome using a hybrid method. Journal of Structural Biology. 171 (1), 64-73 (2010).
- George, A., Wilson, W. W. Predicting protein crystallization from a dilute solution property. Acta Crystallographica Section D: Biological Crystallography. 50 (4), 361-365 (1994).
- Jia, Y., Liu, X. -. Y. From surface self-assembly to crystallization: prediction of protein crystallization conditions. The Journal of Physical Chemistry B. 110 (13), 6949-6955 (2006).
- Slabinski, L., et al. XtalPred: A web server for prediction of protein crystallizability. Bioinformatics. 23 (24), 3403-3405 (2007).
- Abrahams, G. J., Newman, J. BLASTing away preconceptions in crystallization trials. Acta Crystallographica Section F: Structural Biology Communications. 75 (3), 184-192 (2019).
- Rosa, N., et al. Tools to ease the choice and design of protein crystallisation experiments. Crystals. 10 (2), 95 (2020).
- Newman, J., et al. On the need for an international effort to capture, share and use crystallization screening data. Acta Crystallographica Section F: Structural Biology and Crystallization Communications. 68 (3), 253-258 (2012).
- Lynch, M. L., Dudek, M. F., Bowman, S. E. A searchable database of crystallization cocktails in the PDB: analyzing the chemical condition space. Patterns. 1 (4), 100024 (2020).
- Jancarik, J., Kim, S. -. H. Sparse matrix sampling: a screening method for crystallization of proteins. Journal of Applied Crystallography. 24 (4), 409-411 (1991).
- Carter, C. W. Efficient factorial designs and the analysis of macromolecular crystal growth conditions. Methods. 1 (1), 12-24 (1990).
- McPherson, A., Cudney, B. Searching for silver bullets: An alternative strategy for crystallizing macromolecules. Journal of Structural Biology. 156 (3), 387-406 (2006).
- McPherson, A., Nguyen, C., Cudney, R., Larson, S. The role of small molecule additives and chemical modification in protein crystallization. Crystal Growth & Design. 11 (5), 1469-1474 (2011).
- Luft, J. R., DeTitta, G. T. A method to produce microseed stock for use in the crystallization of biological macromolecules. Acta Crystallographica Section D: Biological Crystallography. 55 (5), 988-993 (1999).
- Thakur, A. S., et al. Improved success of sparse matrix protein crystallization screening with heterogeneous nucleating agents. PLoS One. 2 (10), 1091 (2007).
- Chayen, N. E., Stewart, P. D. S., Blow, D. M. Microbatch crystallization under oil-a new technique allowing many small-volume crystallization trials. Journal of Crystal Growth. 122 (1-4), 176-180 (1992).
- Luft, J. R., et al. A deliberate approach to screening for initial crystallization conditions of biological macromolecules. Journal of Structural Biology. 142 (1), 170-179 (2003).
- Luft, J. R., Snell, E. H., DeTitta, G. T. Lessons from high-throughput protein crystallization screening: 10 years of practical experience. Expert Opinion on Drug Discovery. 6 (5), 465-480 (2011).
- Lynch, M. L., Snell, M. E., Potter, S. A., Snell, E. H., Bowman, S. E. 20 years of crystal hits: Progress and promise in ultrahigh-throughput crystallization screening. Acta Crystallographica Section D: Biological Crystallography. , (2023).
- Segelke, B. W. Efficiency analysis of sampling protocols used in protein crystallization screening. Journal of Crystal Growth. 232 (1-4), 553-562 (2001).
- Luft, J. R., Newman, J., Snell, E. H. Crystallization screening: the influence of history on current practice. Acta Crystallographica Section F. 70 (7), 835-853 (2014).
- Mlynek, G., Kostan, J., Leeb, S., Djinovic-Carugo, K. Tailored suits fit better: Customized protein crystallization screens. Crystal Growth & Design. 20 (2), 984-994 (2019).
- Mueller-Dieckmann, J. The open-access high-throughput crystallization facility at EMBL Hamburg. Acta Crystallographica Section D: Biological Crystallography. 62 (12), 1446-1452 (2006).
- Weber, P., et al. High-throughput crystallization pipeline at the crystallography core facility of the Institut Pasteur. Molecules. 24 (24), 4451 (2019).
- Lin, Y. What's happened over the last five years with high-throughput protein crystallization screening. Expert Opinion on Drug Discovery. 13 (8), 691-695 (2018).
- Haupert, L. M., Simpson, G. J. Screening of protein crystallization trials by second order nonlinear optical imaging of chiral crystals (SONICC). Methods. 55 (4), 379-386 (2011).
- Madden, J. T., DeWalt, E. L., Simpson, G. J. Two-photon excited UV fluorescence for protein crystal detection. Acta Crystallographica Section D: Biological Crystallography. 67 (10), 839-846 (2011).
- Fleming, A. M., et al. Second harmonic generation interrogation of the endonuclease APE1 binding interaction with G-quadruplex DNA. Analytical Chemistry. 94 (43), 15027-15032 (2022).
- Bruno, A. E., et al. Classification of crystallization outcomes using deep convolutional neural networks. PLoS One. 13 (6), 0198883 (2018).
- Holleman, E. T., Duguid, E., Keefe, L. J., Bowman, S. E. Polo: An open-source graphical user interface for crystallization screening. Journal of Applied Crystallography. 54 (2), 673-679 (2021).
- Niesen, F. H., et al. An approach to quality management in structural biology: Biophysical selection of proteins for successful crystallization. Journal of Structural Biology. 162 (3), 451-459 (2008).
- Padayatti, P., Palczewska, G., Sun, W., Palczewski, K., Salom, D. Imaging of protein crystals with two-photon microscopy. Biochemistry. 51 (8), 1625-1637 (2012).
- Haupert, L. M., DeWalt, E. L., Simpson, G. J. Modeling the SHG activities of diverse protein crystals. Acta Crystallographica Section D: Biological Crystallography. 68 (11), 1513-1521 (2012).
- Luft, J. R., Grant, T. D., Wolfley, J. R., Snell, E. H. A new view on crystal harvesting. Journal of Applied Crystallography. 47 (3), 1158-1161 (2014).
- Bruno, A. E., Soares, A. S., Owen, R. L., Snell, E. H. The use of haptic interfaces and web services in crystallography: An application for a 'screen to beam' interface. Journal of Applied Crystallography. 49 (6), 2082-2090 (2016).
- Baldock, P., Mills, V., Stewart, P. S. A comparison of microbatch and vapour diffusion for initial screening of crystallization conditions. Journal of Crystal Growth. 168 (1-4), 170-174 (1996).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved