
Method Article
使用SPHIRE的电子冷冻显微镜图像的高分辨率单粒子分析
摘要
本文介绍了使用软件套件SPHIRE处理低温EM图像的协议。目前的协议可以应用于几乎所有瞄准近原子分辨率的单粒子EM项目。
摘要
SPHIRE(用于高分辨率电子显微镜的SPARX)是用于半自动处理单粒子电子冷冻显微镜(cryo-EM)数据的新型开源,用户友好的软件套件。这里介绍的协议详细介绍了如何通过引导用户通过单个粒子结构确定管道的所有步骤从cryo-EM显微照片电影开始获得近原子分辨率结构。这些步骤由新的SPHIRE图形用户界面进行控制,并需要最少的用户干预。使用该方案,来自Photorhabdus luminescens的TcdA1( Tc毒素复合物)的结构来自仅9500个单一颗粒。这种简化的方法将帮助没有广泛的处理经验和先验结构信息的新手用户获得其纯天然状态的纯化大分子复合物的无噪声和无偏倚的原子模型。
引言
直接电子探测器技术的发展,单粒子低温电动机的显着进步正在重塑结构生物学1 。与X射线晶体学相比,该技术只需要少量的蛋白质材料,而不需要结晶,同时对样品的纯度提出更少的限制,并且仍允许测定近原子分辨率下的结构。重要的是,不同的组成或状态现在可以在计算上分离,不同构象的结构确定可以以前所未有的细节进行。最近,挑战性分子的密度图可以通过允许从头模型建立的决议产生,从而深入了解其作用模式2,3,4,5。
3DEM(3D电子显微镜)社区(https://en.wikibooks.org/wiki/Software_Tools_For_Molecular_Microscopy)中提供了各种各样的图像处理软件包,大多数都在不断的开发中。已经通过几种不同的软件包,包括EMAN2 6 ,IMAGIC 7 ,FREALIGN 8 ,RELION 9 ,SPIDER 10和SPARX 11 ,显示各种分子量和对称性的蛋白质已经实现了近原子分辨率。每个包需要不同级别的用户专业知识,并提供不同级别的用户指导,自动化和可扩展性。此外,虽然一些程序提供了完整的环境来促进图像分析的所有步骤,但是其他程序被设计为优化特定任务,例如从已知的r开始的对准参数的改进参考结构。最近,已经开发了几个平台,包括APPION 12和SCIPION 13 ,它提供了一个整合来自上面列出的不同软件包的方法和协议的单一处理流程。
为了有助于目前低温EM的发展,SPARX被重新开发成为一个新的独立和完整的单粒子分析平台,称为SPHIRE(用于高分辨率电子显微镜的SPARX)。为了增加该领域新研究者的技术的可及性,并且为了应对现代全自动高端电子显微镜产生的大量数据,通过引入易于使用的方法重新设计和简化了处理管道图形用户界面(GUI),并自动执行工作流程的主要步骤。此外,添加了新的算法以允许从cr获得快速,可重现和自动化的结构确定yo-EM图像。此外,引入了通过重现性的验证,以避免在细化和异质性分析期间产生的常见伪像。
虽然该程序被广泛修改,但它的核心功能得到了维护:简单的开源代码,现代面向对象的设计和所有基本功能的Python接口。因此,它没有改变为黑盒子程序,使用户能够学习和轻松地修改Python代码,创建其他应用程序或修改整个工作流程。这对于非标准的低温电力项目尤其有用。
这里我们提出一种使用SPHIRE的GUI从cryo-EM图像获得近原子分辨率密度图的协议。它详细描述了从原始的cryo-EM直接检测器电影生成密度图所需的所有步骤,并不限于任何特定的大分子类型。这个协议主要是指引newc通过工作流程在实地工作,并提供关于处理的关键步骤以及一些可能的陷阱和障碍的重要信息。更高级的功能和SPHIRE背后的理论背景将在其他地方描述。
研究方案
注意:要遵循此协议,必须在具有MPI安装的系统(目前为Linux群集)上正确安装SPHIRE。从http://www.sphire.mpg.de下载SPHIRE和TcdA1数据集,并按照安装说明进行操作:http://sphire.mpg.de/wiki/doku.php?id=howto:download。此过程也安装EMAN2。 SPHIRE目前使用EMAN2的e2boxer进行粒子选择和e2显示来显示图像文件。对于原始显微照片电影的剂量加权运动校正,SPHIRE使用unblur 14 。下载程序并按照安装说明进行操作(http://grigoriefflab.janelia.org/unblur,Grigorieff实验室)。为了对结果进行交互式可视化,协议将使用分子图形程序Chimera 15 (https://www.cgl.ucsf.edu/chimera/download.html)。熟悉本协议中使用的功能的一个很好的教程可以是fou在这里:https://www.cgl.ucsf.edu/chimera/data/tutorials/eman07/chimera-eman-2007.html。有关如何从SPHIRE GUI将并行作业提交到集群的说明,请参见:http://sphire.mpg.de/wiki/doku.php?id=howto:submissions。 SPHIRE GUI的整体组织以及整个协议中执行的工作流程的主要步骤如图1所示 。
1.项目:设置本项目的常量参数值
- 通过在终端窗口中键入" sphire&"和ENTER键启动SPHIRE GUI应用程序。
- 在项目设置页面的相应输入字段中调整项目范围的参数( 例如,像素大小,粒子半径和对称性),然后为工作流程的所有后续步骤注册这些值。
- 点击左侧面板右下方的"PROJECT"图标打开项目设置页面。
- 使用e2display.py图像交互显示工具测量粒子的最长轴,然后将粒子的一半输入"蛋白质粒子半径"。如果测量值为Å,请记住使用像素大小将单位转换为像素( 例如,如果粒子长度为200埃,像素尺寸为1.2埃/像素,则粒子的最长轴为200 / 1.2 =〜166像素,半径166/2 = 83像素)。
- 将"粒子大小"设置为粒径的至少1.5倍。避免包含大素数的窗口大小。另外,请记住,3D细化算法目前需要偶数的盒子大小。
注意:窗口应包含一个余量,以解决由于采摘(需要在窗口中移动颗粒)而产生的初始居中误差以及颗粒边界之外的足够的背景区域进行适当的CTF校正(对于大的离焦值尤其重要) 16)。 - 将"CTF窗口大小"设置为"粒子大小"。对于具有低对比度数据的项目,使用较大的窗口来获得更平滑的功率谱估计。
- 设定复合体的"点组对称性"( 例如 "C5")。如果目标结构的对称性未知,则将其保留为"C1"(不对称)。然而,如果在处理期间稍后识别特定的高阶对称性,则相应地改变该对称性设置,并且在与ISAC进行2D对准之后重复步骤。
- 将"蛋白质分子质量"设置为kDa(近似值就足够了)。按"注册设置"按钮。
2.电影:对齐每个电影显微镜的框架,以校正样品的整体运动
- 对于所有电影显微照片,计算所有帧的x / y移位,然后创建其剂量未加权和剂量加权修正后的平均值(见讨论)。注意,前者仅对于CTF估计是必要的,因为估计与剂量加权平均值不能很好地进行,而后者用于结构确定的所有其他步骤。
- 点击"电影"图标,然后点击"显微照片对齐"按钮。通过选择可执行文件设置"Unblur可执行路径"。通过选择原始未对齐的电影显微照片并用通配符"*"替换文件名的可变部分( 例如, TcdA1 _ *。mrc)来设置"输入显微图像路径模式"。指定"输出目录"的路径。
- 通过选择可执行文件设置"Summovie可执行路径"。
- 将"电影帧数"设置为每个电影显微照片中的帧数。将"显微镜电压"和"每帧曝光"设置为数据采集期间使用的值。 (例如:le,如果总体剂量为60 e - /Å2,20帧记录无预曝光,每帧曝光为60/20 = 3 e - / A 2 )按"运行命令"按钮对齐每个电影的照片显示。
注意:这将自动创建两个输出目录,分别包含 剂量未加权和剂量加权的运动校正平均显微照片。
3. CTER:估计CTF的散焦和散光参数
- 对于每个剂量未加权的平均显微照片,估计CTF参数(散焦和散光;其他由用户设定)。
- 单击"CTER"图标,然后单击"CTF估计"按钮。要设置"输入显微图像路径模式",请选择剂量未加权的运动校正显微照片,然后用通配符替换文件名的可变部分"*"。另外,指定"输出目录"的路径。
- 将"幅度对比度"设置为实际使用的数据类型(冰厚度是主要因素)和显微镜电压( 例如 10%)。典型值在7 - 14%范围内17 。
- 设置数据采集期间使用的"显微镜球面像差(Cs)"和"显微镜电压"。
- 将CTF模型的搜索范围的"最低频率"和"最高频率"分别设置为0.0285和0.285Å -1 (40 - 4Å)。按"运行命令"按钮来估计CTF参数。
注意:CTF参数将自动存储在指定输出目录中的partres.txt文件中。在96个核心上计算出112个显微照片的CTF估计值,并在Linux集群上〜3分钟后完成,用于获得代表性的结果。
4.窗口:从剂量加权平均显微照片提取颗粒
- 用e2boxer 6从显微照片手动或自动选择颗粒,并创建坐标文件,每个文件包含相关显微照片中的粒子xy坐标列表。
- 点击"窗口"图标,然后点击"粒子拾取"按钮。按"运行命令"按钮启动e2boxer 6 ,手动或自动选择每个显微照片的粒子18 (参见讨论 )。将每个显微照片的最终粒子坐标存储在EMAN1文件格式(.box)中。或者,在将它们转换为EMAN1格式后,从其他程序导入坐标文件。
- 通过从剂量加权显微照片中提取粒子图像来创建粒子堆栈(在SPHIRE中,粒子堆栈是简称"堆栈")。
- 按"粒子提取"按钮。通过选择剂量加权运动校正的显微照片,然后用通配符"*"替换文件名的可变部分( 例如, TcdA1 _ * .mrc)来指定"输入显微图像路径图案"。同样,通过选择坐标文件( 例如, TcdA1 _ *。box)来设置"输入坐标路径模式"。指定"输出目录"的路径。
- 通过选择CTF参数文件(步骤3.1中生成的partres.txt)设置"CTF参数源"。按"运行命令"按钮。
- 将提取的粒子图像堆叠合并成一个。
- 点击"粒子堆栈"按钮。使用BDB文件路径格式( 例如, "bdb:粒子/堆栈")指定"输出虚拟映像堆栈"的路径,其中"粒子"指向e目录中包含名称始终为EMAN2DB的BDB数据库目录,"stack"是指此数据库中的特定映像堆栈)。通过选择以"mpi_proc"开头的目录指定"输入BDB图像堆栈模式",然后用通配符"*"替换目录名称的变量部分( 例如, Particles / mpi_proc_000到Particles / mpi_proc_ *)。按"运行命令"按钮。
ISAC:2D中粒子图像的分类
- 通过对齐粒子并根据其2D外观将其聚类,计算2D类平均值。
注意:所得到的2D平均值与单个粒子图像相比具有改进的信噪比(SNR),因此用于可视化地评估数据集的质量和异质性,以及从堆栈中分类不期望的图像( 例如,冰晶,碳边,聚集,碎片等 ) 19 。此外,它们将被随后用于确定初始3D模型。- 单击"ISAC"图标,然后单击"ISAC - 2D聚类"按钮。通过选择包含提取的粒子的堆栈文件,输入"输入图像堆栈"。指定"输出目录"的路径。
- 对于"每个图像"使用200 - 1000。选择适当的数量,考虑到2D类的预期数量(总数量除以每个类别的图像数量)。根据SNR和数据集的大小调整此参数。如果数据集过于嘈杂,请增加每个成员的数量。当有少量颗粒可用时减少数量。
注意:由于内存限制,对于相当大的数据集(> 100,000个粒子),将完整的数据集拆分为子集,为每个子集独立地执行ISAC,并结合结果结束了。有关此处理方案的详细说明,请参见http://www.sphire.mpg.de/wiki/doku.php。 - 检查"相位翻转"复选框。保持"目标粒子半径"和"目标粒子图像大小"的默认值,以便通过使用这些设置自动收缩所有粒子图像来加快过程。按"运行命令"按钮计算2D类平均值。
注意:此步骤在计算上要求很高,随着粒子数量和类别以及目标半径和图像大小的增加,运行时间会显着增加。在具有96个过程的群集上,约10,000个粒子的2D分类在约90分钟后完成。
- 显示和目视检查所得到的ISAC 2D平均值,以确保其质量令人满意(参见讨论)。
- 按"实用程序"下的"显示数据"按钮。设置";通过选择包含ISAC 2D平均值(步骤5.1中生成的class_averages.hdf)的文件,输入文件"按"运行命令"按钮显示由ISAC提供的最终可再现和验证的类别平均值。
- 创建一个新的堆栈,只包含验证类平均值的粒子成员。
- 按"创建堆叠子集"按钮。通过选择与5.1.1中相同的堆栈文件来设置"输入图像堆栈"。通过选择ISAC 2D平均值(步骤5.1中生成的class_averages.hdf)设置"ISAC平均值"。指定"输出目录"的路径。按"运行命令"按钮。
VIPER:计算初始3D模型
- 通过删除粒子的所有不良类别平均值和相同视图来选择一组小的平均值(≥100图像)(参见讨论),并使用它们来计算代表使用VIPER的可变初始模型。请记住,选择应至少包含60-80个高质量平均值,每个成员约200-500个。
- 点击"VIPER"图标,再点击"显示数据"按钮。通过选择ISAC 2D平均值(步骤5.1中生成的class_averages.hdf)设置"输入文件"。按"运行命令"按钮。
- 在e2显示图形窗口的某处按鼠标中键,并在弹出的窗口中激活"DEL"按钮。删除粒子的所有坏类平均值和相同视图(参见讨论 )。按"保存"按钮将剩余的2D类平均值存储到新文件中。
- 从所选的ISAC平均值中,为后续的3D细化生成初始参考。
- 点击"初始3D模型 - RVIPER"按钮。通过选择筛选的类来设置"输入图像堆栈"平均值(在步骤6.1中生成)。指定"输出目录"的路径。
- 确保使用相同的值作为ISAC步骤5.1.3的"目标粒子半径"。按"运行命令"按钮生成可重复的从头开始的 3D模型。
注意:此步骤在计算上要求很高,运行时间随着粒子数的平均值和大小而显着增加。在具有96个进程的集群上,该作业(〜100类平均)在〜15分钟后完成。
- 通过考虑类别平均值以及其结构完整性( 即没有断开的部件和/或方向假象),检查所得到的3D模型是否合理。要显示地图,请使用程序Chimera 15 。此时,如果存在目的蛋白质的同源蛋白或结构域的晶体结构,则进行第一次比较(一个例子显示在Represe结果)。
- 对于随后的3D细化,通过去除其周围的噪声并重新缩放以便与原始像素大小相匹配,从起始3D模型生成初始3D参考和3D掩模。
- 点击"创建3D参考"按钮。通过选择ab initio 3D模型(步骤6.2中生成的average_volume.hdf)设置"输入音量"。指定"输出目录"的路径。
- 通过选择ISAC收缩率文件(步骤5.1中生成的README_shrink_ratio.txt)设置"重采样比率源"。按"运行命令"按钮。
7. MERIDIEN:优化初始3D体积
- 从初始3D模型开始,优化3D体积。
- 点击"MERIDIEN"图标,然后点击"3D细化"按钮。通过sele设置"输入图像堆栈"和"初始3D参考"调整粒子堆栈和从头的 3D模型(分别在步骤5.3和6.4中生成)。指定"输出目录"的路径。
- 通过选择3D蒙版文件(步骤6.4中生成)设置"3D蒙版"。始终使用3D蒙版,但特别是在分析的早期阶段,使用宽松配合参考的球形蒙版或软边蒙版,以避免引入不正确掩蔽的偏差。
- 选中"应用硬2D面具"复选框。将"起始分辨率"设置在20 - 25之间的截止频率值。请记住,具有该截止频率的低通滤波器将应用于初始3D结构以减少初始模型偏差。
- 检查此过程使用的集群规范,然后将"每个节点的内存"设置为可用内存(以GB为单位)。按"运行命令"按钮,以完全自动化的方式从初始3D模型开始优化3D体积。
注意:此过程将数据集分成两半,独立细化两个模型,并输出两个原始卷,每个只有一半的粒子。在计算上要求很高,运行时间会随着粒子数量的增加而显着增加。在这个群集上,经过192次处理(约8,000个粒子,352个盒子大小)运行约2.5小时之后,子午线细化完成。
- 从精炼的体积创建一个软边3D面具,用于随后的锐化步骤。
- 点击"自适应3D蒙版"按钮。通过选择一个未过滤的半容量(步骤7.1中生成)来设置"输入音量"。指定"输出掩码"的路径。
- 设置"二值化阈值"值。使用嵌合体确保在这个特定的阈值下,未经过滤的半图谱的溶剂区域中的噪声显然超出感兴趣体积,蛋白质的所有密度仍然为c彼此连接。按"运行命令"按钮创建软边3D面具。
注意:所得掩模的主体(由体素值大于0.5的体素组成)应紧密配合颗粒结构,但仍包含所有感兴趣的密度。软边缘掉落应至少为8-10像素宽。
- 合并通过3D细化获得的两个未过滤的半体积。然后,通过基于检测器的调制传递函数(MTF),估计的B因子和FSC(傅里叶壳相关)估计的分辨率来调整功率谱来锐化合并的体积。
- 选择"锐化"按钮。通过选择相应的文件(步骤7.1中生成的vol_0_unfil.hdf和vol_1_unfil.hdf),设置"第一个未过滤的半容量"和"第二个未过滤的半容量"。始终使用"B因子增强"。通常,保持默认值为e使用最终分辨率和10Å之间的范围,从输入数据集中调整B因子值。或者,指定ad-hoc值( 例如, -100)。
- 保持"低通滤波频率"的默认值,以应用基于FSC的滤波器。
- 通过选择3D面具(步骤7.2中生成)设置"用户提供的面具"。请记住,报告的分辨率将使用带有该掩码的FSC确定。按"运行命令"按钮来锐化精细的3D体积。
- 从上述3D细化步骤估计的所有粒子的投影方向生成3D角度分布图。
- 点击"角度分布"按钮。通过选择文件(步骤7.1中生成的final_params.txt)设置"对齐参数文件",然后按"运行命令"按钮。
- 使用Chi目视检查锐化的3D模型梅拉。考虑到解决方案,确保结构看起来是合理的(参见讨论 )。
- 使用嵌合体目视检查角度分布。验证分布大致均匀地覆盖整个3D角度空间。记住,对于对称结构,分布在独特的不对称三角形内受到限制。
8. SORT3D:通过关注高度可变区域对3D异质性进行排序
- 从3D细化中使用的粒子堆计算3D变异图。
- 点击"SORT3D"图标,然后点击"3D Variability Estimation"。通过选择给予3D细化步骤7.1.1的相同的筛选粒子堆,设置"输入图像堆栈"。指定"输出目录"的路径。
- 保留"投影数量"的默认值。
注意:来自角邻居的图像引擎盖将用于估计每个3D投影角度的2D方差。数字越大,估计噪声越小,分辨率和旋转伪像越低。 - 选中"使用CTF"复选框。按"运行命令"按钮。
- 使用3D变异图创建下面的3D聚类步骤的聚焦蒙版。
- 选择"二进制3D蒙版"按钮。通过选择3D可变性图(步骤8.1中生成)设置"输入音量"。指定"输出掩码"的文件路径。
- 通过使用嵌入式"Volume Viewer"中"Level"字段的输出设置"二值化阈值"。按"运行命令"按钮。
- 通过关注结构上高度可变的区域,将粒子图像分解成均匀的结构基团。
- 按"3D聚类 - RSORT3D"按钮。通过选择3D细化的输出目录(步骤7.1中生成)设置"输入3D细化目录"。指定"输出目录"的路径。
- 通过选择软边3D面具(步骤7.2中生成)设置"3D面具"。通过选择二值化的3D变异图(步骤8.2中生成)设置"聚焦3D面具"。
- 对于大型数据集,对于"每组图像"使用至少5,000-10,000。请记住,程序总是保持每组的图像数量低于此设置。通过考虑预期的3D组数(粒子总数除以"每组图像"值),数据集,SNR和异质性程度来调整该值。如果有足够数量的粒子可用,则开始〜5-10个初始3D组,除非预期数据集中具有更多数量的不同结构状态。
- 使用至少3,000-5,000粒子作为"最小组大小"。请注意,程序将忽略包含比"最小组大小"设置少的图像组。按"运行命令"按钮执行3D聚类。
注意:RSORT3D分为两个步骤。第一个"sort3d"步骤排除了3D异质性。然后,使用由上述3D细化步骤确定的3D对准参数来重构每个均匀结构组的体积。第二个"rsort3d"步骤通过执行两个独立排序运行的双向比较,发现每个组的可重复成员。然后,它仅使用可重复分配的粒子重构均匀结构。在具有96个核心的集群中,这项工作(约8,000个粒子,352个盒子大小)在约3小时后完成。
- 程序完成后,使用嵌套来选择均匀的3D组。选择最高表观分辨率的结构,通常与最多的po相关联可怜的团体通过考虑到所关注的蛋白质的2D类别平均值和生物学方面,确保选择的结构在视觉上是合理的(参见讨论 )。如果在类似分辨率下有其他体积具有几乎相同结构的结构,则认为它们从单个均匀3D组中出现。
- 对最均匀的3D组(具有最高分辨率)的粒子成员进行局部细化。
- 点击"本地子集优化"按钮。通过选择包含所选组的粒子ID的文本文件( 例如,在步骤8.3中生成的Cluster0.txt)来设置"子集文本文件路径"。通过选择先前3D细化的输出目录(步骤7.1中生成)来设置"3D细化目录"。
- 将"重新启动迭代"设置为在之前的3D细化中实现最高分辨率的迭代。按"运行命令"按钮执行所选粒子群的局部细化。
- 与步骤7.2类似,从由本地子集细化重建的未过滤的最终半容量创建软边3D面具。
- 与步骤7.3类似,合并由本地子集细化导出的两个未过滤的最终半体积,并锐化合并的体积。但是,这次不要过滤锐化的音量。
注意:如果步骤8.4中的异质性分析在可比较的分辨率下表示几种不同的状态,则可能希望独立地改进所有不同的状态。
9. LOCALRES:估算最终3D体积的本地分辨率
- 估计从均匀的粒子集获得的3D体积的局部分辨率。
- 点击"LOCALRES"图标,然后点击"本地分辨率"按钮。设置"上半部分"和"下半部分"- 通过选择本地子集细化的未过滤的最终半体积(在步骤8.5中生成),通过选择步骤8.6中产生的软边3D面具设置"3D掩模",指定"输出体积"的文件路径, 。
- 保持"FSC窗口大小"的默认值7像素。请记住,此设置定义了计算局部 - 空间相关性的窗口的大小;较大的窗口大小以牺牲本地可解析性为代价产生更平滑的分辨率图。
- 保持"分辨率截止"的默认值0.5为分辨率标准。
注意:对于每个体素,程序会将本地分辨率报告为本地FSC低于所选分辨率阈值的频率。不建议阈值低于0.5,因为较低的相关值具有较高的统计学不确定性。因此,相应的局部分辨率将在体素之间变化很大。 - 对于"Overa"ll分辨率",在本地子集细化后设置在锐化中估计的绝对分辨率(步骤8.7)。按"运行命令"按钮计算卷的本地分辨率。
- 通过使用3D局部分辨率图,将本地子集细化后的3D局部滤镜应用于锐化的体积。
- 点击"3D Local Filter"按钮。通过选择锐化但未过滤的3D体积(步骤8.7中生成)设置"输入音量"。类似地,设置"本地分辨率文件"和"3D掩码"(分别在步骤9.1和8.6中生成)。请记住,3D遮罩定义将应用本地过滤的区域。指定"输出卷"的文件路径。按"运行命令"按钮应用3D本地过滤器。
- 使用嵌合体来目视检查最终的3D模型和3D局部分辨率图(在步骤9,2和9.1中分别生成)ectively)。选择"表面颜色"选项,根据本地分辨率对3D体积进行着色。请记住,地方决议的分配应该顺利(参见讨论 )。
结果
从Photorhabdus发光体 Tc复合体(TcdA1) 20,21,22的A分量的112个直接检测器电影开始执行上述协议。将该数据集记录在具有高亮度场致发射枪(XFEG)的Cs校正电子冷冻显微镜上,其在300kV的加速电压下工作。以样品量表上的像素尺寸为1.14Å的总剂量为60 e - /Å -2自动获取图像。在电影帧对准( 协议步骤2 )之后,所得到的运动校正平均值具有扩展到高分辨率的各向同性Thon环( 图2a )。单个颗粒容易可见并且分离良好( 图2b )。然后使用e2boxer的群组工具挑选粒子lass ="xref"> 18( 协议步骤4.1 )。在这种情况下,使用更具选择性的选项设置适当的阈值( 图2c )。 112幅数字显微照片产生了9,652颗颗粒。大部分提取的图像( 方案步骤4.2 )包含明确定义的颗粒,并且它们的盒尺寸是粒径的约1.5倍,如推荐的( 图2d )。接下来,使用ISAC,进行2D异质性分析( 方案步骤5 )。它产生98个班级平均数( 图3a )。使用这些2D类平均值,使用VIPER( 方案步骤6 )以中间分辨率计算出一个从头模型( 图3b )。该模型与先前在3.9Å分辨率22 ( 图3c )中解析的TcdA1的晶体结构显示出极好的一致性。这个从头模型被用作初始模式 (MERIDIEN),仅从〜40,000个不对称单位产生3.5(λ3.3标准)重建( 方案步骤7 )( 图4 )。这个近原子分辨率图是在24小时内获得的,使用多达96个CPU用于从多个内核中受益的工作流程。
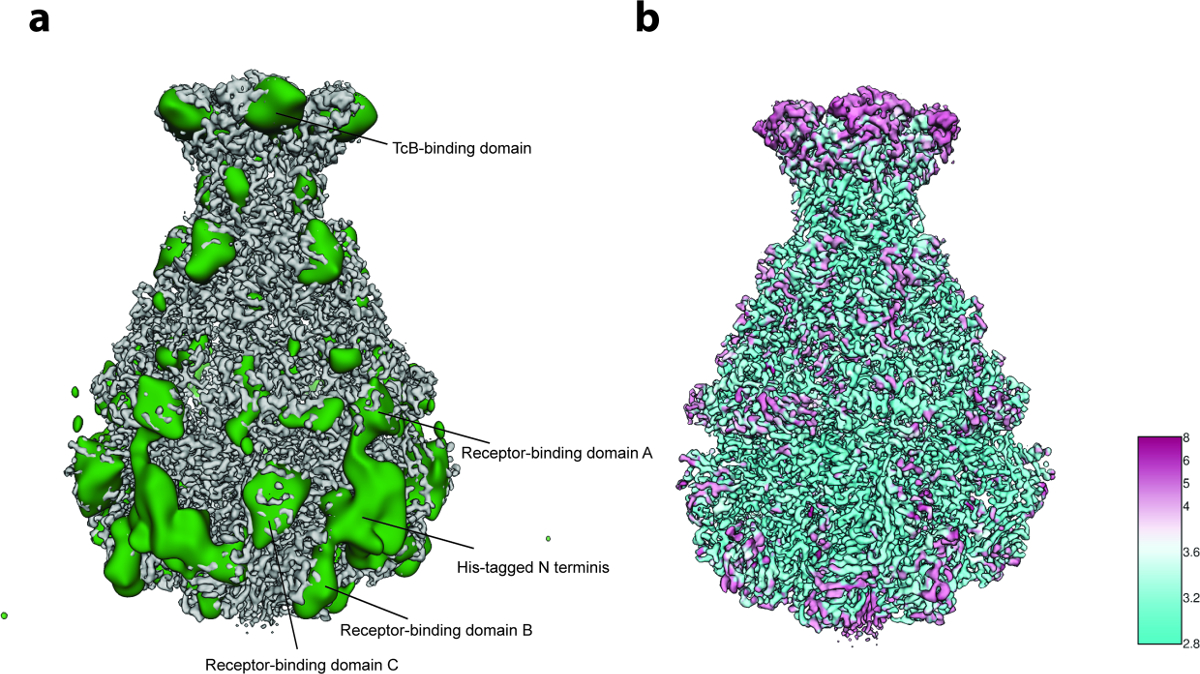
对于3D变异性分析(协议步骤8),在步骤8.3.3中使用每组只有2,000个粒子图像( 即 ,过程从5个初始3D组开始),并且在步骤8.3.4中由200个图像组成的最小组大小少量颗粒(〜10,000)。分析显示局部柔韧性主要在复合物的N末端区域,其含有用于纯化的His标签( 图5a )。事实上,在先前公布的TcdA1晶体结构中,12个N-末端残基和His标签未被解析"22,这个最可能无序的区域在目前的cryo-EM密度上仍未得到解决,这可能是由于其灵活性。在受体结合结构域和BC结合结构域处检测到另外的变异性( 图5a )结构的满意分辨率和数据集的相当小的尺寸,这种异质性被决定是可以容忍的,因此不执行聚焦的3D分类23。最后,计算最终密度图的局部分辨率( 协议步骤9.1,图5b ),并且锐化的3D地图被本地过滤( 协议步骤9.2) 。该质量的体积可以用于使用Coot 24或任何其他细化工具( 图6 )的从头模型构建。

图1:使用SPHIRE的图像处理。 ( a )SPHIRE软件包的GUI。可以通过选择GUI左侧的相应图标("工作流步骤")来激活工作流程的特定步骤。与该工作流程步骤相关联的命令和实用程序将显示在GUI的中央区域。选择其中一个命令后,相应的参数显示在GUI的右侧区域。高级参数通常不需要修改预设的默认值。 ( b )使用SPHIRE GUI进行单粒子图像处理的工作流阶段。 请点击此处查看此图的较大版本。
{kind=link}

图2:运动校正和Partic提取。 ( a , b )在散焦为1.7μm时记录的典型高品质,低剂量,漂移校正数字显微镜。注意在功率谱(a)中分辨率为2.7埃的各向同性Thon环和2D图像( b )中的可辨别的颗粒。 ( c )使用e2boxer进行粒子选择。绿色圆圈表示选定的粒子。 ( d )从剂量加权显微照片提取的典型原始颗粒。比例尺= 20nm。 请点击此处查看此图的较大版本。
{kind=link}

图3:2D聚类和初始模型生成。 ( a )2D类平均画廊,多数代表侧视图o f粒子。比例尺= 20nm。 ( b )使用RVIPER从无参考平均值中获取的TcdA1的从头开始的 3D地图。 ( c )将TcdA1晶体结构(带)(pdb-id 1VW1)刚体拟合为初始低温 - EM密度(透明灰色)。 请点击此处查看此图的较大版本。
{kind=link}

图4:TcdA1的Cryo-EM三维结构。 ( a , b )使用〜9,500粒子图像计算的TcdA1的最终3.5Å密度图:( a )侧面和( b )顶视图。 ( c )α螺旋和β片的低温 - EM密度的代表性领域。arge.jpg"target ="_ blank">请点击此处查看此图的较大版本。

图5:变异性分析和局部分辨率。 ( a )锐化的TcdA1冷冻 - EM图(灰色)和变异性图(绿色)的表面。为了更好的清晰度,将可变性图低通滤波至30埃。 ( b )根据局部分辨率(Å)对TcdA1进行锐化的低温电子地图的表面渲染。注意高变异性和低局部分辨率的区域之间的拓扑一致性。 请点击此处查看此图的较大版本。
{kind=link}

图6:3D Mod使用Coot构建TcdA1。低密度EM密度和原子模型的代表性区域显示为α-螺旋。原子模型是使用Coot 从头建造的。 请点击此处查看此图的较大版本。
{kind=link}
讨论
单粒子低温电动机近年来呈现出快速发展的趋势,并提供了许多具有重要生物学意义的大分子复合物的原子分辨率结构25 。为了支持当前正在进入该领域的大量新手用户,我们开发了单粒子图像分析平台SPHIRE ,并为此展示了包括电影对齐,粒子采集,CTF估计,初始模型在内的整个工作流程的漫游协议计算,2D和3D异质性分析,高分辨率3D细化和局部分辨率估计和滤波。
这里描述的协议旨在作为使用感兴趣的蛋白质的cryo-EM显微照片以及由SPHIRE的独立GUI提供的计算工具的帮助的3D结构确定的简短指南。
工作流的主要特点是大部分的程序只需要运行一次,因为它们依赖于通过重复性19进行验证的概念,并且不需要参数调整。这种自动验证机制是SPHIRE与其他软件包的主要优点,因为结果往往是客观的和可重复的,最重要的是可以以可接受的计算成本获得。该管道还为经验丰富的用户提供了丰富的诊断信息,以自己的方法进行进一步的独立验证和评估。然而,在结构生物学和电子显微镜中至少具有元素理论背景的新手用户应该能够使用自己的数据和自动验证程序获得近原子分辨率结构。
然而,获得近原子分辨率结构并不总是直接的,结果将高度取决于样本的质量和输入数据一个。对于这里给出的程序,假设有足够数量的高质量未对齐的原始EM电影可用,其平均值显示清晰可辨的均匀和随机取向的单个颗粒。通常,对于分子的对称性,大小或整体形状没有限制,但是低分子量可能是限制因素,特别是当蛋白质具有无特征的球形形状时。通常,具有高点组对称性的较大,有序粒子的分析要求较低。因此,强烈建议新手用户首先使用精确表征的cryo-EM数据集来运行本协议。 SPHIRE教程数据(http:/sphire.mpg.de)或EMPIAR提交的数据集之一(https://www.ebi.ac.uk/pdbe/emdb/empiar/)与原始电影是一个很好的起点。
在处理自己的数据时,很可能某些数据集或某些图像不能满足某些要求标准。在这方面,除了由程序对工作流程的主要步骤执行的自动化稳定性和再现性检查之外,仍然建议用户在协议的某些"检查点"处目视检查结果,特别是如果最终重建不满意
在电影对准( 协议步骤2 )和CTF估计( 协议步骤3 )之后,可以在显微照片级别进行第一次视觉检查。所得到的运动校正平均值应显示清晰可辨和分离好的单个颗粒,其功率谱应显示清晰可辨的各向同性Thon环。它们可见的空间频率在大多数情况下定义了最终决定结构原则上最高的分辨率。具有足够质量的运动校正平均值及其功率谱的示例显示在"代表性的结果"。可以借助SPHIRE的Drift和CTF评估GUI工具(http://sphire.mpg.de/wiki/doku.php),去除可能对最终结果产生负面影响的异常图像。
关于粒子筛选,SPHIRE流程中的关键步骤是使用ISAC的2D分类( 协议步骤5.2) 。这里,用户应该控制由程序自动识别的可再现的2D类平均采用足够准确地均匀地覆盖角度空间的取向范围。如果课程平均质量不令人满意(嘈杂和/或模糊的图像)和/或可再现的班级数量非常低,考虑提高自动采摘质量,优化数据集成像或样品制备。在大多数情况下,不可能从不产生良好2D类别平均值的数据集中计算可靠的重构。高品质2D课程的例子"代表性结果"一节中显示了愤怒。
需要至少100级平均值以自动方式获得可靠的初始3D模型( Protocol步骤6.1 )。对于此步骤,用户应选择具有最高质量的平均值,并包括尽可能多的不同粒子取向。初始模型的质量对于随后的高分辨率3D细化的成功至关重要。
在其他软件包中,有时执行3D分类以去除"坏"粒子8,9 。然而,在SPHIRE中,大多数这些粒子已经在使用ISAC的2D分类期间被自动消除。因此,如果重建和3D变异性分析指示数据集的异质性,则建议仅执行计算密集的3D排序步骤。
最重要的是,用户应该仔细仔细检查所得到的3D体积( 协议步骤9.3 ),并确认相应密度的特征与标称分辨率一致。分辨率<9Å,对应于α-螺旋的棒状密度变得可见。在分辨率<4.5Å时,对应于β-折叠中的链的密度通常很好地分离并且体积大的氨基酸变得可见。高分辨率图(<3Å)应显示清晰可辨的侧链,从而允许构建准确的原子模型。
迄今获得的结果表明,在SPHIRE的自动重现性测试和最小目视检查的帮助下,本协议通常适用于任何类型的单粒子低温 - EM项目。显示每个处理步骤的代表性结果,用于重建TcdA1毒素Photorhabdus luminescens 21 ,已被解决为近原子分辨率。类似质量的密度图可用于通过从头主干跟踪以及相互或实际空间细化构建可靠的原子模型,从而为理解复杂的分子机制提供坚实的结构框架。
访问代码:
EM结构和未处理电影的坐标已分别存放在电子显微镜数据库和电子显微镜导频图像存档中,登录号为EMD-3645和EMPIAR-10089。
披露声明
作者宣称他们没有竞争的经济利益。
致谢
我们感谢D. Roderer为我们提供了TcdA1显微照片。我们感谢Steve Ludtke持续支持EMAN2基础设施。这项工作得到了马克斯·普朗克学会(SR)和欧洲理事会根据欧盟第七框架计划(FP7 / 2007-2013)(授权号615984)(授予SR)的资助,并获得了美国国家研究机构健康R01 GM60635至PAP)。
材料
| Name | Company | Catalog Number | Comments |
| SPHIRE | Max Planck Institute of Molecular Physiology- Dortmund and Houston Medical School, Houston, Texas | http://sphire.mpg.de | |
| UCSF Chimera | University of California, San Francisco | http://www.cgl.ucsf.edu/chimera/ | |
| Unblur | Janelia Farm Research Campus, Ashburn | http://grigoriefflab.janelia.org/unblur | |
| Coot | MRC Laboratory of Molecular Biology, Cambridge | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ | |
| EMAN2 | Baylor College of Medicine, Houston | http://blake.bcm.edu/emanwiki/EMAN2 | |
| Computing Cluster with 1824 cores | Max Planck Institute of Molecular Physiology | Linux Cluster with 76 nodes, each with 2 Processors Xeon E5-2670v3 12C 2.30 GHz and 128 Gb RAM | |
| TITAN KRIOS electron microscope | FEI | 300 kV, Cs correction, XFEG | |
| Falcon II direct electron detector | FEI | ||
| EPU (automated data acquisition software) | FEI | https://www.fei.com/software/epu/ |
参考文献
- Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nature Methods. 13 (1), 24-27 (2016).
- Liao, M., Cao, E., Julius, D., Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 504 (7478), 107-112 (2013).
- Bai, X. -. C., Yan, C., et al. An atomic structure of human γ-secretase. Nature. 525 (7568), 212-217 (2015).
- Ecken, J. V. D., Heissler, S. M., Pathan-Chhatbar, S., Manstein, D. J., Raunser, S. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature. 534 (7609), 724-728 (2016).
- von der Ecken, J., Müller, M., Lehman, W., Manstein, D. J., Penczek, P. A., Raunser, S. Structure of the F-actin-tropomyosin complex. Nature. 519 (7541), 114-117 (2015).
- Tang, G., Peng, L., et al. EMAN2: An extensible image processing suite for electron microscopy. Journal of Structural Biology. 157 (1), 38-46 (2007).
- van Heel, M., Harauz, G., Orlova, E. V., Schmidt, R., Schatz, M. A new generation of the IMAGIC image processing system. Journal of Structural Biology. 116 (1), 17-24 (1996).
- Grigorieff, N. FREALIGN: high-resolution refinement of single particle structures. Journal of Structural Biology. 157 (1), 117-125 (2007).
- Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. Journal of Structural Biology. 180 (3), 519-530 (2012).
- Shaikh, T. R., Gao, H., et al. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nature Protocols. 3 (12), 1941-1974 (2008).
- Hohn, M., Tang, G., et al. SPARX, a new environment for Cryo-EM image processing. Journal of Structural Biology. 157 (1), 47-55 (2007).
- Lander, G. C., Stagg, S. M., et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. Journal of Structural Biology. 166 (1), 95-102 (2009).
- de la Rosa-Trevìn, J. M., Quintana, A., et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. Journal of Structural Biology. 195 (1), 93-99 (2016).
- Grant, T., Grigorieff, N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife. 4, 06980 (2015).
- Pettersen, E. F., Goddard, T. D., et al. UCSF Chimera?A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25 (13), 1605-1612 (2004).
- Penczek, P. A., Fang, J., Li, X., Cheng, Y., Loerke, J., Spahn, C. M. T. CTER-rapid estimation of CTF parameters with error assessment. Ultramicroscopy. 140, 9-19 (2014).
- Frank, J. . Three-Dimensional Electron Microscopy of Macromolecular Assemblies. , (2006).
- Woolford, D., Ericksson, G., et al. SwarmPS: rapid, semi-automated single particle selection software. Journal of Structural Biology. 157 (1), 174-188 (2007).
- Yang, Z., Fang, J., Chittuluru, J., Asturias, F. J., Penczek, P. A. Iterative Stable Alignment and Clustering of 2D Transmission Electron Microscope Images. Structure/Folding and Design. 20 (2), 237-247 (2012).
- Gatsogiannis, C., Merino, F., et al. Membrane insertion of a Tc toxin in near-atomic detail. Nature Publishing Group. , (2016).
- Gatsogiannis, C., Lang, A. E., et al. A syringe-like injection mechanism in Photorhabdus luminescens toxins. Nature. 495 (7442), 520-523 (2013).
- Meusch, D., Gatsogiannis, C., et al. Mechanism of Tc toxin action revealed in molecular detail. Nature. 508 (7494), 61-65 (2014).
- Penczek, P. A., Frank, J., Spahn, C. M. T. A method of focused classification, based on the bootstrap 3D variance analysis, and its application to EF-G-dependent translocation. Journal of Structural Biology. 154 (2), 184-194 (2006).
- Emsley, P., Lohkamp, B., Scott, W. G., Cowtan, K. Features and development of Coot. Acta crystallographica. Section D, Biological crystallography. 66, 486-501 (2010).
- Callaway, E. The revolution will not be crystallized: a new method sweeps through structural biology. Nature. 525 (7568), 172-174 (2015).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。