
Method Article
SPHIRE를 사용한 전자 저온 현미경 이미지의 고해상도 단일 입자 분석
요약
이 논문은 소프트웨어 스위트 인 SPHIRE를 사용하여 저온 EM 이미지를 처리하기위한 프로토콜을 제시합니다. 현재의 프로토콜은 원 자면 해상도를 목표로하는 거의 모든 단일 파티클 EM 프로젝트에 적용될 수 있습니다.
초록
SPHIRE (고해상도 전자 현미경 용 스파크)는 단일 입자 전자 극저온 현미경 (cryo-EM) 데이터의 반자동 처리를위한 새로운 오픈 소스, 사용자 친화적 인 소프트웨어 모음입니다. 여기에 제시된 프로토콜은 단일 입자 구조 결정 파이프 라인의 모든 단계를 안내하여 Cryo-EM 현미경 사진 영화에서 시작하는 원자 가까이의 해상도 구조를 얻는 방법을 자세히 설명합니다. 이 단계는 새로운 SPHIRE 그래픽 사용자 인터페이스에서 제어되며 사용자 개입을 최소화해야합니다. 이 프로토콜을 사용하여 Photorhabdus luminescens의 Tc 독소 복합체 인 3.5Å 구조의 TcdA1이 9500 개의 단일 입자에서만 추출되었습니다. 이 간소화 된 접근 방식은 원시 상태에서 정제 된 거대 분자 복합체의 무소음 및 비 편향 원자 모델을 얻기 위해 광범위한 처리 경험과 선험적 인 구조 정보없이 초보자를 도울 것입니다.
서문
직접 전자 검출기 기술의 개발 후, 단일 입자 Cryo-EM의 현저한 진보는 현재 구조 생물학을 변형시키고 있습니다 1 . X 선 결정학에 비해이 기술은 결정질을 필요로하지 않고 단백질 물질을 소량 만 필요로하는 동시에 샘플의 순도에 대한 제한이 거의 없으며 근원 분해능에서 구조를 결정할 수 있습니다. 중요하게, 다른 조성 또는 상태를 이제 계산으로 분리 할 수 있고, 다른 형태의 구조 결정을 전례없는 수준에서 수행 할 수있다. 최근에 도전 분자의 밀도지도는 de novo 모델 구축을 가능하게하여 해상도에 따라 생성 될 수 있으므로 동작 모드 2 , 3 , 4 , 5 에 대한 깊은 이해가 가능합니다.
3DEM (3D Electron Microscopy) 커뮤니티 (https://en.wikibooks.org/wiki/Software_Tools_For_Molecular_Microscopy)에서 다양한 이미지 처리 소프트웨어 패키지를 사용할 수 있으며 대부분 개발 중입니다. EMAN2 6 , IMAGIC 7 , FREALIGN 8 , RELION 9 , SPIDER 10 및 SPARX 11을 비롯한 다양한 소프트웨어 패키지를 사용하여 다양한 분자량 및 대칭성을 나타내는 단백질에 대한 근원 분해능이 달성되었습니다. 각 패키지는 다른 수준의 사용자 전문 지식을 필요로하며 사용자 지침, 자동화 및 확장 성의 다른 수준을 제공합니다. 또한, 일부 프로그램은 이미지 분석의 모든 단계를 용이하게하기위한 완벽한 환경을 제공하지만 다른 프로그램은 알려진 작업에서 시작하는 정렬 매개 변수의 세분화와 같은 특정 작업을 최적화하도록 설계되었습니다추론 구조. 보다 최근에는 APPION 12 및 SCIPION 13 을 포함하여 위에 나열된 여러 소프트웨어 패키지의 접근 방식과 프로토콜을 통합하는 단일 처리 파이프 라인을 제공하는 여러 플랫폼이 개발되었습니다.
Cryo-EM의 현재 개발에 기여하기 위해 SPARX는 SPHIRE (고해상도 전자 현미경 검사용 SPARX)라고하는 단일 입자 분석을위한 새로운 독립형 및 완벽한 플랫폼으로 재 개발되었습니다. 현장의 새로운 연구자를위한 기술의 접근성을 높이고 현대의 완전 자동화 된 고급 전자 현미경으로 대량 생산 된 데이터에 대처하기 위해 처리 파이프 라인을 사용하기 쉽도록 다시 디자인하고 단순화했습니다 그래픽 사용자 인터페이스 (GUI) 및 워크 플로우의 주요 단계 자동화. 더욱이, 새로운 알고리즘이 추가되어 신속하고 재현성 있고 자동화 된 구조 결정을 cr요 - EM 이미지. 또한 정제 및 이질성 분석 과정에서 발생하는 공통적 인 인공물을 피하기 위해 재현성에 의한 검증이 도입되었습니다.
이 프로그램은 광범위하게 수정되었지만 직접적인 오픈 소스 코드, 현대적인 객체 지향 디자인 및 모든 기본 기능을위한 Python 인터페이스와 같은 핵심 기능이 유지되었습니다. 따라서 블랙 박스 프로그램으로 변경되지 않아 사용자가 Python 코드를 연구하고 쉽게 수정할 수 있고, 추가 응용 프로그램을 만들거나 전체 워크 플로우를 수정할 수 있습니다. 이것은 비표준 Cryo-EM 프로젝트에 특히 유용합니다.
여기서는 SPHIRE의 GUI를 사용하여 cryo-EM 이미지에서 근사 해상 밀도 맵을 얻기위한 프로토콜을 제시합니다. 그것은 원시 Cryo-EM 직접 탐지기 영화에서 밀도 맵을 생성하는 데 필요한 모든 단계를 상세히 설명하며 특정 거대 분자 유형에만 국한되지 않습니다. 이 프로토콜은 주로 newc워크 플로우를 통해 현장에서의 정보를 제공하고 처리의 중요한 단계는 물론 함정과 장애물에 대한 중요한 정보를 제공합니다. 더 많은 고급 기능과 SPHIRE의 이론적 배경은 다른 곳에서 설명 될 것이다.
프로토콜
참고 :이 프로토콜을 따르려면 SPIRE를 MPI가 설치된 시스템 (현재 Linux 클러스터)에 올바르게 설치해야합니다. http://www.sphire.mpg.de에서 SPHIRE 및 TcdA1 데이터 세트를 다운로드하고 설치 지침을 따르십시오 : http://sphire.mpg.de/wiki/doku.php?id=howto:download. 이 절차는 EMAN2도 설치합니다. SPHIRE는 현재 입자 선택에 EMAN2의 e2boxer를 사용하고 이미지 파일을 표시하기 위해 e2 디스플레이를 사용합니다. 생체 현미경 영화의 선량 가중 모션 보정의 경우, SPHIRE는 언 블러 14를 사용 합니다. 프로그램을 다운로드하고 설치 지침 (http://grigoriefflab.janelia.org/unblur, Grigorieff lab)을 따르십시오. 결과 구조의 대화 형 시각화를 위해 프로토콜은 분자 그래픽 프로그램 인 Chimera 15 (https://www.cgl.ucsf.edu/chimera/download.html)를 사용합니다. 이 프로토콜에서 사용 된 기능에 익숙해지기위한 훌륭한 튜토리얼은nd here : https://www.cgl.ucsf.edu/chimera/data/tutorials/eman07/chimera-eman-2007.html. SPHIRE GUI에서 클러스터에 병렬 작업을 제출하는 방법은 http://sphire.mpg.de/wiki/doku.php?id=howto:submissions에서 확인할 수 있습니다. SPHIRE GUI의 전반적인 구성과이 프로토콜 전반에 걸쳐 수행 된 워크 플로의 주요 단계가 그림 1 에 나와 있습니다.
1. PROJECT :이 프로젝트의 상수 매개 변수 값 설정
- 터미널 창에서 " sphire &" 를 입력하고 ENTER 키를 눌러 SPIRE GUI 응용 프로그램을 시작하십시오.
- 프로젝트 설정 페이지의 각 입력 필드에서 프로젝트 전체 매개 변수 ( 예 : 픽셀 크기, 입자 반경 및 대칭)를 조정 한 다음 워크 플로의 모든 후속 단계에 대해이 값을 등록하십시오.
- 왼쪽 패널의 오른쪽 아래에있는 "PROJECT"아이콘을 클릭하여 프로젝트 설정 페이지를 엽니 다.
- e2display.py 이미지 대화 형 디스플레이 도구를 사용하여 입자의 가장 긴 축을 측정 한 다음 "Protein particle radius"에 입자 크기의 절반을 입력하십시오. 측정 단위가 Å이면 픽셀 크기를 사용하여 단위를 픽셀로 변환해야합니다 ( 예 : 입자 크기가 200 Å이고 픽셀 크기가 1.2 Å / 픽셀 인 경우 입자의 가장 긴 축은 200 / 1.2 = ~ 166 픽셀 및 반경 166/2 = 83 픽셀).
- "입자 상자 크기"를 입자 크기의 1.5 배 이상으로 설정하십시오. 큰 소수를 포함하는 창 크기를 피하십시오. 또한 3D 미세 조정 알고리즘에는 현재 짝수 번호의 상자 크기가 필요합니다.
참고 : 창에는 피킹 (창 내부의 입자 이동 필요) 및 적절한 CTF 보정을위한 입자 경계 외부의 충분한 배경 영역 (특히 큰 초점 값에 중요)으로 인한 초기 중심 지정 오류를 설명하는 여백이 있어야합니다 16). - "CTF 창 크기"를 "입자 상자 크기"로 설정하십시오. 대비가 낮은 데이터가있는 프로젝트의 경우 큰 창을 사용하여 파워 스펙트럼을보다 부드럽게 추정하십시오.
- 복합체 ( 예 : "C5")의 "점대칭 대칭"을 설정합니다. 목표 구조의 대칭이 알려지지 않은 경우 "C1"(비대칭)으로 두십시오. 그러나 처리 중에 특정 고차 대칭이 나중에 식별되면이 대칭 설정을 적절히 변경하고 ISAC로 2D 정렬 후 단계를 반복합니다.
- "단백질 분자량"을 kDa로 설정하십시오 (대략적인 값으로 충분합니다). "Register settings"버튼을 누르십시오.
2. MOVIE : 각 Movie Micrograph의 프레임을 정렬하여 샘플의 전체 동작을 수정합니다.
- 모든 영화 현미경 사진에 대해 모든 프레임에 대해 x / y- 시프트를 계산 한 다음 도즈 - 비가 중 및 도우 - 가중치를 생성합니다.평균 보정 (Discussion 참조). CTF 추정은 선량 가중 평균에서는 잘 수행되지 않지만 후자는 구조 결정의 다른 모든 단계에서 사용되기 때문에 전자는 CTF 추정에만 필요하다는 점에 유의하십시오.
- "MOVIE"아이콘을 클릭하고 "Micrograph Movie Alignment"버튼을 클릭하십시오. 실행 파일을 선택하여 "실행 가능 경로 차단 해제"를 설정하십시오. 정렬되지 않은 원시 현미경 사진을 선택하고 파일 이름의 가변 부분을 와일드 카드 "*"( 예 : TcdA1 _ *. mrc)로 대체하여 "입력 현미경 경로 패턴"을 설정하십시오. "출력 디렉토리"에 대한 경로를 지정하십시오.
- 실행 파일을 선택하여 "Summovie executable path"를 설정하십시오.
- "영화 프레임 수"를 각 영화 현미경 사진의 프레임 수로 설정하십시오. "현미경 전압"및 "프레임 당 노출"을 데이터 수집 중에 사용되는 값으로 설정하십시오. (예 :전체 노출량이 60 e - / Å 2 이고 사전 노출없이 20 프레임을 기록한 경우 모든 프레임의 노출은 60 / 20 = 3 e - / A 2 입니다.) "명령 실행"버튼을 눌러 각 영화 현미경 사진의 프레임.
참고 : 이것은 자동으로 선량 비가 중 및 선량 가중의 모션 보정 평균 현미경 사진을 포함하는 두 개의 출력 디렉토리를 만듭니다.
3. CTER : CTF의 디 포커스 및 난시 매개 변수 추정
- 각각의 선량 - 비가 중 평균 현미경 사진에 대해 CTF 매개 변수 (초점 흐림 및 난시, 다른 것은 사용자가 설정 함)를 추정합니다.
- "CTER"아이콘을 클릭하고 "CTF Estimation"버튼을 클릭하십시오. "입력 현미경 경로 패턴"을 설정하려면 선량 - 비가 중 움직임 보정 현미경 그래프를 선택한 다음 파일 이름의 변수 부분을 와일드 카드로 대체하십시오"*". 또한 "출력 디렉터리"의 경로를 지정하십시오.
- "진폭 대비"는 실험실에서 데이터의 종류 (얼음 두께가 주요 요인 임) 및 현미경 전압 ( 예 : 10 %)에 대해 일상적으로 사용되는 값으로 설정합니다. 일반적인 값은 7 - 14 % 범위입니다.
- 데이터 수집 중에 "현미경 구면 수차 (Cs)"및 "현미경 전압"을 설정합니다.
- CTF 모델 피팅의 검색 범위의 "최저 주파수"와 "최고 주파수"를 각각 0.0285와 0.285Å -1 (40 - 4Å)로 설정하십시오. "Run command"버튼을 눌러 CTF 매개 변수를 추정하십시오.
참고 : CTF 매개 변수는 지정된 출력 디렉토리의 partres.txt 파일에 자동으로 저장됩니다. 112 개의 현미경 사진에 대한 CTF 추정은 96 개의 코어에서 계산되었고 대표 결과를 얻기 위해 사용 된 Linux 클러스터에서 ~ 3 분 후에 완료되었습니다.
4. 창 : 선량 - 가중 평균 현미경 사진에서 입자 추출
- e2boxer 6 을 사용하여 현미경 사진에서 입자를 수동 또는 자동으로 선택하고 각각 관련 현미경 사진 내의 입자 xy 좌표 목록이 포함 된 좌표 파일을 만듭니다.
- "WINDOW"아이콘을 클릭하고 "Particle Picking"버튼을 클릭하십시오. "Run command"버튼을 눌러 e2boxer 6 를 시작하고 수동 또는 자동으로 각 현미경 사진의 입자를 선택하십시오 18 ( 토론 참조). 각 현미경 사진의 최종 입자 좌표를 EMAN1 파일 형식 (.box)으로 저장하십시오. 또는 다른 프로그램에서 좌표 파일을 EMAN1 형식으로 변환 한 후 가져 오십시오.
- 선량 가중 현미경 사진에서 입자 이미지를 추출하여 입자 스택을 만듭니다 (SPHIRE에서는 입자 스택이 없습니다).단순히 "스택"이라고 부름).
- "Particle Extraction"버튼을 누르십시오. 선량 가중치가 적용된 이동 현미경 사진을 선택한 다음 파일 이름의 변수 부분을 와일드 카드 "*"( 예 : TcdA1 _ *. mrc)로 바꾸어 "입력 현미경 경로 패턴"을 지정하십시오. 마찬가지로 좌표 파일 ( 예 : TcdA1 _ *. 상자)을 선택하여 "좌표 입력 경로 패턴"을 설정하십시오. "출력 디렉토리"에 대한 경로를 지정하십시오.
- CTF 매개 변수 파일 (단계 3.1에서 생성 된 partres.txt)을 선택하여 "CTF 매개 변수 원본"을 설정하십시오. "Run command"버튼을 누르십시오.
- 추출 된 입자 이미지 스택을 하나의 이미지 스택으로 결합합니다.
- "Particle Stack"버튼을 클릭하십시오. BDB 파일 경로 형식 ( 예 : "bdb : Particles / stack", 여기서 "Particles"는 th를 가리킴)을 사용하여 "가상 이미지 스택 출력"경로를 지정하십시오e 디렉토리는 이름이 항상 EMAN2DB 인 BDB 데이터베이스 디렉토리를 포함하고 "스택"은이 데이터베이스 내의 특정 이미지 스택을 참조합니다. "mpi_proc"로 시작하는 디렉토리를 선택한 다음 디렉토리 이름의 가변 부분을 와일드 카드 "*"( 예 : Particles / mpi_proc_000 to Particles / mpi_proc_ *)로 대체하여 "Input BDB image stack pattern"을 지정하십시오. "Run command"버튼을 누르십시오.
5. ISAC : 2D에서 입자 이미지의 분류
- 입자를 정렬하고 2D 모양에 따라 클러스터링하여 2D 클래스 평균을 계산합니다.
참고 : 결과 2D 평균은 개별 입자 이미지와 비교하여 향상된 신호 대 잡음비 (SNR)를 가지므로 데이터 집합의 품질과 이질성을 시각적으로 평가하고 스택에서 바람직하지 않은 이미지를 분류하는 데 사용됩니다 ( 예 : 얼음 결정, 탄소 가장자리,응집물, 파편 등 ) 19 . 또한 초기 3D 모델을 결정하는 데 사용됩니다.- "ISAC"아이콘을 클릭하고 "ISAC - 2D 클러스터링"버튼을 클릭하십시오. 추출 된 입자가 포함 된 스택 파일을 선택하여 "입력 이미지 스택"을 설정하십시오. "출력 디렉토리"에 대한 경로를 지정하십시오.
- "클래스 당 이미지"는 200 - 1000을 사용하십시오. 예상되는 2D 클래스 수 (클래스 당 이미지 수로 나눈 총 입자 수)를 고려하여 적절한 수를 선택하십시오. SNR 및 데이터 세트의 크기에 따라이 매개 변수를 조정하십시오. 데이터 세트가 지나치게 시끄러운 경우 클래스 당 멤버 수를 늘립니다. 적은 수의 파티클을 사용할 수있는 경우 숫자를 줄입니다.
참고 : 메모리 제한 때문에 다소 큰 데이터 세트 (> 100,000 개 입자)의 경우 전체 데이터 세트를 하위 세트로 분할하고 각 하위 세트에 대해 ISAC를 개별적으로 수행 한 다음 결합결국 결과. 이 처리 시나리오에 대한 자세한 지침은 http://www.sphire.mpg.de/wiki/doku.php에 나와 있습니다. - "위상 반전"확인란을 선택하십시오. 이러한 설정으로 모든 입자 이미지를 자동으로 축소하여 프로세스 속도를 높이려면 "대상 입자 반경"및 "대상 입자 이미지 크기"의 기본값을 유지하십시오. "Run command"버튼을 눌러 2D 클래스 평균을 계산하십시오.
참고 :이 단계는 계산적으로 까다로운 작업이며 입자 및 클래스 수 및 대상 반경 및 이미지 크기에 따라 실행 시간이 크게 늘어납니다. 96 개의 프로세스가있는 클러스터에서 ~ 10,000 입자의 2D 분류는 약 90 분 후에 완료됩니다.
- 결과 ISAC 2D 평균을 표시하고 시각적으로 검사하여 품질이 만족 스러운지 확인하십시오 (토론 참조).
- "UTILITIES"아래의 "Display Data"버튼을 누릅니다. 세트 "입력 파일 "을 선택합니다 (5.1 단계에서 생성 한 class_averages.hdf)."실행 명령 "버튼을 눌러 ISAC에서 제공하는 최종 재생 가능 및 유효 클래스 평균을 표시하십시오.
- 검증 된 클래스 평균의 파티클 멤버 만 포함하는 새 스택을 만듭니다.
- "Create Stack Subset"버튼을 누릅니다. 5.1.1 절과 같은 스택 파일을 선택하여 "입력 이미지 스택"을 설정하십시오. ISAC 2D 평균 (단계 5.1에서 생성 한 class_averages.hdf)을 선택하여 "ISAC 평균"을 설정하십시오. "출력 디렉토리"에 대한 경로를 지정하십시오. "Run command"버튼을 누르십시오.
6. VIPER : 초기 3D 모델 계산
- 불량한 모든 클래스 평균과 파티클의 동일한 뷰를 삭제하고 (Discussion 참조) 클래스 평균 (100 개 이상 이미지)의 작은 세트를 선택하고 담당자를 계산하는 데 사용합니다VIPER를 이용한 초기 모델. 선택 사항에는 ~ 200-500 명의 구성원이 각각 60-80 개의 고품질 평균을 포함해야합니다.
- "VIPER"아이콘을 클릭 한 다음 "데이터 표시"버튼을 클릭하십시오. ISAC 2D 평균 (5.1 단계에서 생성 한 class_averages.hdf)을 선택하여 "입력 파일"을 설정하십시오. "Run command"버튼을 누르십시오.
- e2display의 그래픽 창에서 마우스 중간 버튼을 누르고 팝업 창에서 "DEL"버튼을 활성화하십시오. 모든 나쁜 클래스 평균과 입자의 동일한 뷰를 삭제하십시오 ( 토론 참조). "Save"버튼을 눌러 나머지 2D 클래스 평균을 새 파일에 저장하십시오.
- 선택한 ISAC 평균에서 후속 3D 상세 검색을위한 초기 참조를 생성합니다.
- "초기 3D 모델 - RVIPER"버튼을 클릭하십시오. 스크린 클래스를 선택하여 "입력 이미지 스택"을 설정하십시오.평균 (단계 6.1에서 생성). "출력 디렉토리"에 대한 경로를 지정하십시오.
- ISAC 단계 5.1.3과 같은 "대상 입자 반경"에 대해 동일한 값을 사용해야합니다. 재생 가능한 ab initio 3D 모델을 생성하려면 "Run command"버튼을 누르십시오.
참고 :이 단계는 계산적으로 까다로운 작업이며 입자의 평균 수와 크기에 따라 실행 시간이 크게 늘어납니다. 96 개의 프로세스가있는 클러스터에서이 작업 (~ 100 개의 클래스 평균)은 ~ 15 분 후에 완료되었습니다.
- 클래스 평균 및 구조적 무결성 ( 즉 , 단절된 부품 및 / 또는 방향 인공물)을 고려하여 결과 3D 모델이 합리적인지 확인하십시오. 지도를 표시하려면 Chimera 15 프로그램을 사용하십시오. 이 시점에서 상 동성 단백질의 결정 구조 또는 관심있는 단백질의 도메인과의 첫 번째 비교를 수행하십시오 (예는 Represe결과).
- 후속 3D 미세 조정을 위해 주변 노이즈를 제거하고 원본 픽셀 크기와 일치하도록 다시 스케일링하여 초기 3D 참조 및 3D 마스크를 생성합니다.
- "Create 3D Reference"버튼을 클릭하십시오. ab initio 3D 모델 (단계 6.2에서 생성 된 average_volume.hdf)을 선택하여 "입력 음량"을 설정하십시오. "출력 디렉토리"에 대한 경로를 지정하십시오.
- ISAC 축소율 파일 (5.1 절에서 생성 한 README_shrink_ratio.txt)을 선택하여 "Resample ratio source"를 설정하십시오. "Run command"버튼을 누르십시오.
7. MERIDIEN : 초기 3D 볼륨 수정
- 초기 3D 모델에서 시작하여 3D 볼륨을 미세 조정합니다.
- "MERIDIEN"아이콘을 클릭 한 다음 "3D 상세 검색"버튼을 클릭하십시오. sele로 "입력 이미지 스택"및 "초기 3D 참조"설정입자 스택과 ab initio 3D 모델 (각각 5.3 및 6.4 단계에서 생성)을 사용합니다. "출력 디렉토리"에 대한 경로를 지정하십시오.
- 6.4 단계에서 생성 한 3D 마스크 파일을 선택하여 "3D 마스크"를 설정하십시오. 항상 3D 마스크를 사용하지만, 특히 초기 분석 단계에서는 잘못된 마스킹의 편향을 피하기 위해 구형 마스크 또는 참조에 느슨하게 맞춘 부드러운 가장자리의 마스크를 사용하십시오.
- "하드 2D 마스크 적용"체크 상자를 선택하십시오. "시작 해상도"를 20 - 25Å 사이의 컷오프 주파수 값으로 설정하십시오. 이 컷오프 주파수를 가진 로우 패스 필터는 초기 모델 바이어스를 줄이기 위해 초기 3D 구조에 적용됩니다.
- 이 프로세스에 사용 된 클러스터의 사양을 확인한 다음 "노드 당 메모리"를 사용 가능한 메모리 (GB)로 설정하십시오. "실행 명령"버튼을 누르면 완전 자동으로 초기 3D 모델에서 시작하는 3D 볼륨을 다듬을 수 있습니다.
참고 :이 절차에서는 데이터 세트를 두 개의 반으로 나눠서 두 모델을 개별적으로 구체화하고 입자의 절반에서만 두 개의 원시 볼륨을 출력합니다. 계산이 까다로 우며 작업 시간이 입자 수에 따라 크게 증가합니다. 이 클러스터에서 192 프로세스 (~ 8,000 입자, 352 상자 크기)에서 ~ 2.5 시간 실행 한 후에 자오선 정제가 완료되었습니다.
- 후속하는 선명 화 단계를 위해 세련된 볼륨에서 부드러운 가장자리의 3D 마스크를 만듭니다.
- "Adaptive 3D Mask"버튼을 클릭하십시오. 필터링되지 않은 하프 볼륨 (7.1 단계에서 생성) 중 하나를 선택하여 "입력 볼륨"을 설정하십시오. "출력 마스크"에 대한 경로를 지정하십시오.
- '이진화 임계 값'값을 설정하십시오. 키메라를 사용하여이 특정 임계 값에서 노이즈가 필터링되지 않은 절반 맵의 용매 영역에서 관심있는 볼륨 외부에 분명히 존재하고 단백질의 모든 밀도가 여전히 c인지 확인하십시오서로 연결되어있다. "실행 명령"버튼을 눌러 소프트 에지 3D 마스크를 만듭니다.
참고 : 결과 마스크의 본체 (값이> 0.5 인 복셀로 구성)는 입자 구조에 꼭 맞아야하지만 관심있는 모든 밀도를 여전히 포함해야합니다. 소프트 에지 제거는 최소 8-10 픽셀 너비 여야합니다.
- 3D 정제로 얻은 두 개의 필터링되지 않은 절반 볼륨을 병합합니다. 그런 다음, 검출기의 변조 전달 함수 (MTF), 추정 된 B- 인수 및 분해능의 FSC (푸리에 셸 상관 관계) 추정에 따라 전력 스펙트럼을 조정하여 병합 된 볼륨을 선명하게하십시오.
- "선명 화"버튼을 선택하십시오. 단계 7.1에서 생성 된 해당 파일 (vol_0_unfil.hdf 및 vol_1_unfil.hdf)을 선택하여 "필터링되지 않은 첫 번째 반 볼륨"및 "두 번째 필터링되지 않은 반 볼륨"을 설정하십시오. 항상 "B 요인 강화"를 사용하십시오. 일반적으로 e를 유지하려면 기본값을 유지하십시오.최종 분해능 주파수와 10Å 사이의 범위를 사용하여 입력 데이터 세트에서 B- 인자 값을 나타냅니다. 또는 임시 값 ( 예 : -100)을 지정하십시오.
- "로우 패스 필터 주파수"의 기본값을 유지하여 FSC 기반 필터를 적용하십시오.
- 단계 7.2에서 생성 된 3D 마스크를 선택하여 "사용자 제공 마스크"를 설정하십시오. 보고 된 해결책은이 마스크로 FSC를 사용하여 결정됩니다. "실행 명령"버튼을 눌러 세련된 3D 볼륨을 선명하게하십시오.
- 위의 3D 세분화 단계에 의해 추정 된 모든 입자의 투영 방향에서 3D 각도 분포도를 생성합니다.
- "Angular Distribution"버튼을 클릭하십시오. 파일 (7.1 단계에서 생성 된 final_params.txt)을 선택하여 "Alignment parameter file"을 설정하고 "Run command"버튼을 누릅니다.
- Chi를 사용하여 예리한 3D 모델을 육안으로 검사합니다.메라. 달성 된 해결책을 고려하여 구조가 합리적으로 보이는지 확인하십시오 ( 토론 참조).
- 시각적으로 키메라를 사용하여 각도 분포를 검사합니다. 배포판이 전체 3D 각도 공간을 대략 균등하게 커버하는지 확인하십시오. 대칭 구조의 경우 고유 한 비대칭 삼각형 내에서 분포가 제한된다는 점에 유의하십시오.
8. SORT3D : 가변성이 높은 영역을 중심으로 3D 이질성 정렬
- 3D 미세 조정에 사용 된 입자 스택에서 3D 가변성 맵을 계산합니다.
- "SORT3D"아이콘을 클릭하고 "3D Variability Estimation"버튼을 클릭하십시오. 3D 정제 단계 7.1.1에 주어진 것과 동일한 스크린 파티클 스택을 선택하여 "입력 이미지 스택"을 설정하십시오. "출력 디렉토리"에 대한 경로를 지정하십시오.
- "투영 수"의 기본값을 유지하십시오.
참고 : 각 이웃의 이미지후드는 각 3D 투영 각도에서 2D 분산을 추정하는 데 사용됩니다. 숫자가 클수록 예상치는 낮지 만 해상도와 회전 인공물이 더 낮습니다. - "CTF 사용"확인란을 선택하십시오. "Run command"버튼을 누르십시오.
- 3D 변동성 맵을 사용하여 아래의 3D 클러스터링 단계에 대한 포커스 마스크를 만듭니다.
- "Binary 3D Mask"버튼을 선택하십시오. 3D 가변성 맵 (단계 8.1에서 생성)을 선택하여 "입력 음량"을 설정하십시오. "출력 마스크"의 파일 경로를 지정하십시오.
- 키메라의 "Volume Viewer"에서 "Level"필드의 출력을 사용하여 "Binarization threshold"를 설정하십시오. "Run command"버튼을 누르십시오.
- 구조적으로 매우 가변적 인 영역에 집중하여 입자 이미지를 균질 구조 그룹으로 정렬합니다.
- "3D 클러스터링 - RSORT3D"버튼을 누르십시오.3D 상세 검색 (7.1 단계에서 생성)의 출력 디렉토리를 선택하여 "3D 입체 디렉토리 입력"을 설정하십시오. "출력 디렉토리"에 대한 경로를 지정하십시오.
- 연약한 3D 마스크 (단계 7.2에서 생성)를 선택하여 "3D 마스크"를 설정하십시오. 이진화 된 3D 가변성 맵 (8.2 단계에서 생성)을 선택하여 "Focus 3D mask"를 설정하십시오.
- 대형 데이터 세트의 경우 "그룹당 이미지 수"에 5,000-10,000 이상을 사용하십시오. 이 프로그램은 그룹당 이미지 수를이 설정보다 항상 낮게 유지합니다. 예상되는 3D 그룹 수 ( "그룹당 이미지 수"값으로 나눈 입자의 총 수), 데이터 세트, SNR 및 이종 정도를 고려하여 값을 조정하십시오. 데이터 세트의 더 많은 수의 구별되는 구조적 상태가 예상되지 않는 한, 충분한 수의 파티클을 사용할 수 있다면 ~ 5-10 초기 3D 그룹부터 시작하십시오.
- "최소 그룹 크기"는 최소 3,000-5,000 개의 입자를 사용하십시오.프로그램은 "최소 그룹 크기"설정보다 적은 수의 이미지를 구성하는 그룹을 무시합니다. "실행 명령"버튼을 눌러 3D 클러스터링을 수행하십시오.
참고 : RSORT3D는 두 단계로 세분됩니다. 첫 번째 "sort3d"단계는 3D 이질성을 정렬합니다. 그런 다음 위의 3D 미세 조정 단계에서 결정된 3D 정렬 매개 변수를 사용하여 각 동질 구조 그룹의 볼륨을 재구성합니다. 두 번째 "rsort3d"단계는 두 개의 독립적 인 정렬 작업의 양방향 비교를 수행하여 각 그룹의 재현 가능한 구성원을 찾습니다. 그런 다음 재현 가능하게 할당 된 입자 만 사용하여 균질 구조를 재구성합니다. 96 코어의 클러스터에서는 약 3 시간 후에이 작업 (~ 8,000 입자, 352 상자 크기)이 완료됩니다.
- 프로그램이 끝나면 키메라를 사용하여 동종 3D 그룹을 선택하십시오. 가장 높은 겉보기 해상도의 구조를 선택하십시오. 일반적으로 가장 높은 해상도와 관련이 있습니다.pulous 그룹. 관심있는 단백질의 2D 클래스 평균 및 생물학적 측면을 고려하여 선택된 구조가 시각적으로 합리적인지 확인하십시오 ( 토론 참조). 유사한 해상도로 거의 동일한 구조를 가진 다른 볼륨이있는 경우 단일 동질 3D 그룹에서 나오는 것으로 간주하십시오.
- 가장 동 질적 인 3D 그룹의 파티클 멤버에 대해 (가장 높은 해상도로) 로컬 상세 검색을 수행합니다.
- "Local Subset Refinement"버튼을 클릭하십시오. 선택한 그룹의 파티클 ID를 포함하는 텍스트 파일을 선택하여 "서브 세트 텍스트 파일 경로"를 설정합니다 ( 예 : 8.3 단계에서 생성 한 Cluster0.txt). 이전 3D 상세 검색 (7.1 단계에서 생성)의 출력 디렉토리를 선택하여 "3D 상세 검색 디렉토리"를 설정합니다.
- "반복 반복"을 이전 3D 상세 검색에서 가장 높은 해상도를 얻은 곳으로 설정하십시오. 언론에"명령 실행"버튼을 클릭하면 선택한 입자 채우기에 대한 로컬 미세 조정을 수행 할 수 있습니다.
- 7.2 단계와 마찬가지로 로컬 서브 세트 세분화에 의해 재구성되지 않은 필터링되지 않은 최종 절반 볼륨에서 소프트 에지 3D (Soft-edged 3D) 마스크를 만듭니다.
- 7.3 단계와 마찬가지로 로컬 부분 집합 세분화에 의해 파생 된 두 개의 필터링되지 않은 마지막 절반 볼륨을 병합하고 병합 된 볼륨을 선명하게 만듭니다. 그러나 이번에는 예리한 볼륨을 필터링하지 마십시오.
주 : 단계 8.4의 이질성 분석이 비교 가능한 해상도에서 몇 가지 별개의 상태를 나타내면 모든 다른 상태를 개별적으로 다듬을 수 있습니다.
9. LOCALRES : 최종 3D 볼륨의 로컬 해상도 예측
- 균일 한 입자 집합에서 얻은 3D 볼륨의 로컬 해상도를 예측합니다.
- "LOCALRES"아이콘을 클릭하고 "Local Resolution"버튼을 클릭하십시오. "첫 번째 볼륨"및 "두 번째 볼륨(단계 8.5에서 생성 된) 로컬 서브 세트 세분화의 필터링되지 않은 최종 볼륨을 선택하여 "볼륨"을 선택합니다 8.6 단계에서 생성 된 소프트 에지 3D 마스크를 선택하여 "3D 마스크"를 설정하십시오 "출력 볼륨" .
- "FSC 창 크기"에 대한 기본값은 7 픽셀로 유지하십시오. 이 설정은 로컬 - 실제 공간 상관 관계가 계산되는 창의 크기를 정의합니다. 더 큰 창 크기는 로컬 분해능을 희생시키면서보다 매끄러운 해상도 맵을 생성합니다.
- 해상도 기준에 대해 "Resolution cut-off"의 기본값 0.5를 유지합니다.
참고 : 각 보셀에 대해 프로그램은 로컬 FSC가 선택한 해상도 임계 값 아래로 떨어지는 빈도로 로컬 해상도를보고합니다. 낮은 상관 관계 값은 높은 통계적 불확실성을 가지므로 0.5보다 낮은 임계 값은 권장하지 않습니다. 따라서 해당 로컬 해상도는 보셀간에 크게 달라집니다. - "Overall resolution "을 선택하고 로컬 부분 집합 세분화 후 선명 화에서 추정 된 절대 해상도를 설정합니다 (단계 8.7)."실행 명령 "버튼을 눌러 볼륨의 로컬 해상도를 계산합니다.
- 3D 로컬 해상도 맵을 사용하여 로컬 하위 집합 구체화 이후에 선명하게 된 볼륨에 3D 로컬 필터를 적용합니다.
- "3D 로컬 필터"버튼을 클릭하십시오. 날카롭지 만 필터링되지 않은 3D 볼륨 (단계 8.7에서 생성)을 선택하여 "입력 볼륨"을 설정하십시오. 마찬가지로 "로컬 해상도 파일"및 "3D 마스크"(각각 9.1 및 8.6 단계에서 생성)를 설정합니다. 3D 마스크는 로컬 필터링이 적용될 영역을 정의합니다. "출력 볼륨"에 대한 파일 경로를 지정하십시오. "실행 명령"버튼을 눌러 3D 로컬 필터를 적용하십시오.
- 키메라를 사용하여 최종 3D 모델과 3D 로컬 해상도 맵을 시각적으로 검사합니다 (9.2 및 9.1 단계에서 생성, resp적극적으로). "표면 색상"옵션을 선택하여 로컬 해상도에 따라 3D 볼륨을 채 웁니다. 지역 결의안 배포는 원활해야합니다 ( 토론 참조).
결과
위에서 설명한 프로토콜은 Photorhabdus luminescens Tc 복합체 (TcdA1) 20 , 21 , 22 의 A 구성 요소의 112 개의 직접 검출기 영화에서 시작하여 실행되었습니다. 이 데이터 세트는 300 kV의 가속 전압에서 작동되는 고휘도 전계 방출 총 (XFEG)이있는 Cs 보정 전자 저온 현미경으로 기록되었습니다. 이미지는 표본 규모에서 1.14 Å의 픽셀 크기에서 60 e - / Å -2 의 총 선량으로 자동 획득되었습니다. 무비 프레임 정렬 후 ( 프로토콜 단계 2 ), 결과로 얻은 모션 보정 평균은 등방성 톤 링을 고해상도까지 확장합니다 ( 그림 2a ). 개개의 입자는 쉽게 보이고 잘 분리되어있었습니다 ( 그림 2b ). 그런 다음 입자를 e2boxer의 득점 도구를 사용하여 선택했습니다.lass = "xref"> 18 ( 프로토콜 단계 4.1 ). 이 경우,보다 선별적인 옵션을 사용하여 적절한 임계 값이 설정되었습니다 ( 그림 2c ). 112 개의 디지털 현미경 사진은 9,652 개의 입자를 산출했다. 추출 된 이미지 ( 프로토콜 단계 4.2 )의 대부분은 잘 정의 된 입자를 포함하고 상자 크기는 권장 된 것처럼 입자 크기보다 1.5 배 정도 컸다 ( 그림 2d ). 다음으로 ISAC을 사용하여 2D 이질성 분석을 수행했습니다 ( 프로토콜 단계 5 ). 98 개의 클래스 평균을 산출했다 ( 그림 3a ). 이러한 2D 클래스 평균을 사용하여 중간 해상도 ( 그림 3b )에서 VIPER ( 프로토콜 단계 6 )를 사용하여 ab initio 모델을 계산했습니다. 이 모델은 3.9Å 해상도에서 이미 해결 된 TcdA1의 결정 구조와 우수한 일치를 보여준다 ( 그림 3c ). 이 ab initio 모델은 초기 온도로 사용되었다. (MERIDIEN)을 사용하여 ~ 40,000 개의 비대칭 단위만으로 3.5Å (0.143 기준) 재구성 ( 프로토콜 단계 7 )을 달성했습니다 ( 그림 4 ). 이 근 원자 분해능 맵은 24 시간 내에 획득되었으며, 다중 코어의 이점을 취하는 워크 플로우 단계에서 최대 96 개의 CPU를 사용했습니다.
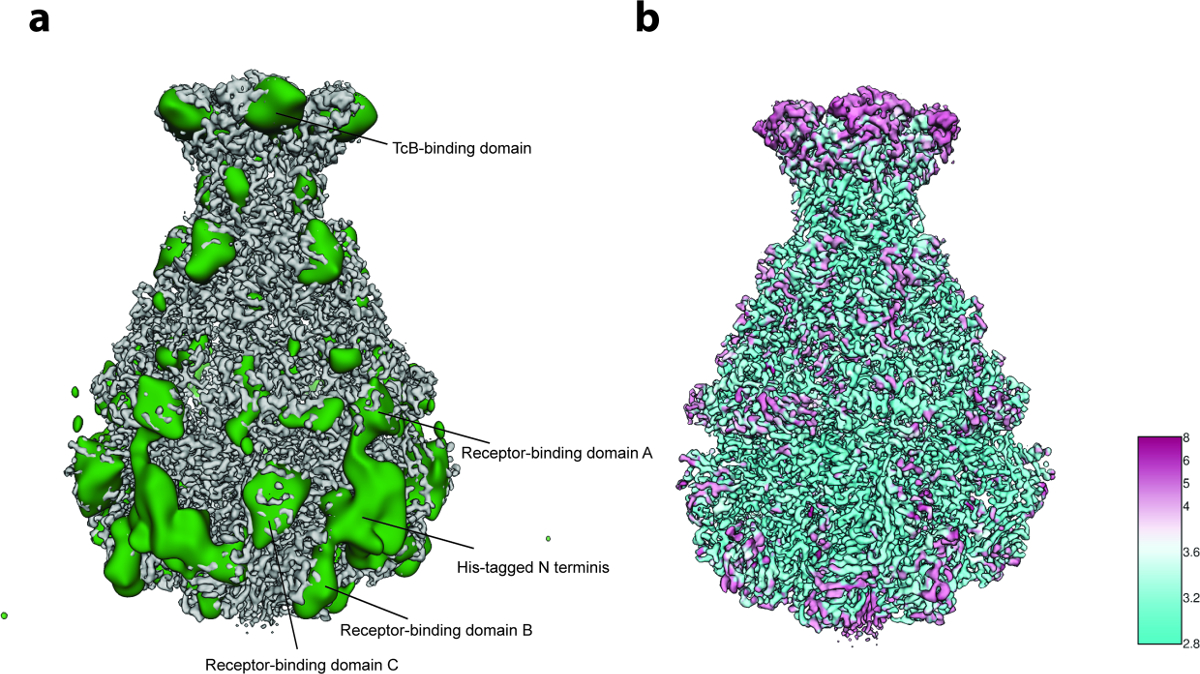
3 차원 가변성 분석 (프로토콜 단계 8)의 경우, 단계 8.3.3에서 그룹 당 2,000 개의 입자 이미지 만 사용되었으며 ( 즉 , 프로세스는 5 개의 초기 3D 그룹으로 시작 함) 8.3.4 단계에서 가장 작은 그룹 크기 인 200 개의 이미지가 적은 수의 입자 (~ 10,000). 이 분석은 주로 정제에 사용 된 His 태그를 포함하는 복합체의 N- 말단 부위에서 국부적 인 유연성을 나타냅니다 ( 그림 5a ). 실제로, 12 개의 N- 말단 잔기 및 His 태그는 이전에 공지 된 TcdA1의 결정 구조에서 분해되지 않았다이 가변 영역은 수용체 결합 영역과 BC 결합 영역에서 더 많은 가변성이 검출되었다 ( 그림 5a ). 구조의 만족스러운 해상력과 데이터 세트의 비교적 작은 크기로이 이질성은 허용 될 수있는 것으로 결정되었으므로 초점을 맞춘 3D 분류 23 은 수행되지 않았다. 마지막으로 최종 밀도지도의 로컬 해상도가 계산되었다 ( 프로토콜 단계 9.1, 그림 이 품질의 볼륨은 Coot 24 또는 다른 정제 도구 ( 그림 6 )를 사용하여 새 모델을 구축하는 데 사용할 수 있습니다.

<그림 1 : SPHIRE를 사용한 이미지 처리. ( a ) SPHIRE 소프트웨어 패키지의 GUI. 워크 플로의 특정 단계는 GUI의 왼쪽에서 각각의 그림을 선택하여 활성화 할 수 있습니다 ( "워크 플로우 단계"). 워크 플로의이 단계와 관련된 명령 및 유틸리티는 GUI의 중앙 영역에 나타납니다. 명령 중 하나를 선택하면 각 매개 변수가 GUI의 오른쪽 영역에 표시됩니다. 고급 매개 변수는 일반적으로 미리 설정된 기본값을 수정하지 않아도됩니다. ( b ) SPHIRE GUI를 이용한 단일 입자 이미지 처리의 워크 플로우 단계. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 2 : 모션 보정 및 부분르 추출. ( a , b ) 일반적인 고품질, 저용량, 드리프트 교정 된 디지털 현미경 사진은 1.7 μm의 디 포커스에서 촬영되었습니다. 파워 스펙트럼 (a)에서 2.7 Å의 해상도까지 확장되는 등방성 톤 링과 2D 이미지에서 잘 식별 가능한 입자 ( b )에 주목하십시오. ( c ) e2boxer를 이용한 입자 선택. 녹색 원은 선택된 입자를 나타냅니다. ( d ) 선량 가중 현미경 사진에서 추출한 대표적인 미립자. 스케일 바 = 20 nm. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 3 : 2D 클러스터링 및 초기 모델 생성. ( a ) 2D 등급 평균 갤러리, 대다수의 측면보기 o 입자. 스케일 바 = 20 nm. ( b ) 기준 자유 클래스 평균으로부터 RVIPER를 사용하여 얻은 TcdA1의 Ab initio 3D지도. ( c ) TcdA1 결정 구조 (리본) (pdb-id 1VW1)의 초기 Cryo-EM 밀도 (투명 회색) 로의 리지드 바디 피팅. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 4 : TcdA1의 Cryo-EM 3D 구조. ( a , b ) ~ 9,500 입자 이미지를 사용하여 계산 된 TcdA1의 최종 3.5Å 밀도지도 : ( a ) 측면 및 ( b ) 평면도. ( c ) α 헬릭스와 β 시트에 대한 cryo-EM 밀도의 대표 영역.arge.jpg "target ="_ blank ">이 그림의 확대 버전을 보려면 여기를 클릭하십시오.

그림 5 : 가변성 분석 및 로컬 해상도. ( a ) 예리한 TcdA1 저온 EM지도의 표면 (회색)과 가변성지도 (녹색). 더 명확하게하기 위해 변동성 맵은 30 Å까지 로우 패스 필터링되었습니다. ( b ) TcdA1의 표면 렌더링은 국부적 해상도 (Å)에 따라 채색 된 날카로운 Cryo-EM 맵을 나타낸다. 높은 변동성과 낮은 지역 해상도 사이의 토폴로지 협약에 유의하십시오. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 6 : 3D 모드el Coot를 이용한 TcdA1의 건물. Cryo-EM 밀도 및 원자 모델의 대표적인 영역이 α 나선 구조로 표시됩니다. 원자 모델은 Coot를 사용하여 새로 만들었습니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
토론
단일 입자 Cryo-EM은 최근 몇 년 동안 급속한 발전을 보였으며 중요한 생물학적 중요성을 지닌 고분자 복합체의 수많은 원자 분해 구조를 전달했습니다 25 . 현재 필드에 들어가고있는 많은 초보 사용자를 지원하기 위해 단일 입자 이미지 분석 플랫폼 SPHIRE 를 개발했으며 영화 정렬, 입자 선택, CTF 추정, 초기 모델을 포함한 전체 워크 플로우에 대한 워크 플로우 프로토콜을 제시합니다 계산, 2D 및 3D 이질 분석, 고해상도 3D 세분화 및 로컬 해상도 추정 및 필터링.
여기에 설명 된 프로토콜은 관심 단백질의 cryo-EM 현미경 사진과 SPHIRE의 독립 실행 형 GUI가 제공하는 전산 도구를 사용하여 3D 구조 결정에 대한 간단한 안내서로 제공됩니다.
워크 플로의 주요 특징은의 절차는 재현성에 의한 검증 개념에 의존하고 매개 변수 조정을 필요로하지 않기 때문에 단 한 번만 실행해야합니다. 이 자동 검증 메커니즘은 다른 소프트웨어 패키지보다 SPHIRE의 주된 이점입니다. 왜냐하면 결과는 객관적이며 재현성이 있으며, 가장 중요한 것은 수용 가능한 계산 비용으로 얻을 수 있기 때문입니다. 이 파이프 라인은 숙련 된 사용자가 자신의 방법으로 더욱 독립적 인 검증 및 평가를 수행 할 수 있도록 풍부한 진단 정보를 제공합니다. 그럼에도 불구하고, 구조 생물학 및 전자 현미경 검사에서 적어도 원소 이론적 배경을 가진 초보 사용자는 자체 데이터 및 자동화 된 검증 절차를 사용하여 근사 해상 구조를 얻을 수 있어야합니다.
그러나 원 자성 해상도 구조를 얻는 것이 항상 쉬운 것은 아니며 그 결과는 샘플 및 입력 데이터의 품질에 따라 크게 달라질 것입니다에이. 여기에 제시된 절차에서는 고품질의 정렬되지 않은 원시 EM 영화를 사용할 수 있으며 평균은 명확하게 식별 할 수있는 균질하고 무작위로 배향 된 단일 입자를 나타냅니다. 일반적으로 분자의 대칭성, 크기 또는 전반적인 형상에 대한 제한은 없지만, 저 분자량은 특히 단백질이 특징이없는 구형을 가졌을 때 제한 요인이 될 수 있습니다. 대개 높은 포인트 - 그룹 대칭을 갖는 더 크고 잘 정렬 된 입자의 분석은 덜 까다 롭습니다. 따라서, 초보자 사용자는 잘 특성화 된 Cryo-EM 데이터 세트를 사용하여 먼저이 프로토콜을 실행하는 것이 좋습니다. SPHIRE 자습서 데이터 (http : /sphire.mpg.de) 또는 원시 동영상이있는 EMPIAR 제출 데이터 세트 (https://www.ebi.ac.uk/pdbe/emdb/empiar/) 중 하나가 좋은 출발점입니다 .
자체 데이터를 처리 할 때 일부 데이터 세트 또는 일부 이미지가 특정 품질을 충족시키지 못할 가능성이 매우 높습니다.기준. 이러한 맥락에서 워크 플로우의 주요 단계에 대해 프로그램이 수행하는 자동화 된 안정성 및 재현성 검사 외에도 사용자가 프로토콜의 특정 "검사 점"에서 결과를 시각적으로 검사하는 것이 좋습니다. 특히 최종 재구성 만족스럽지 않다.
첫 번째 육안 검사는 영화 정렬 ( 프로토콜 단계 2 ) 및 CTF 추정 ( 프로토콜 단계 3 ) 후에 현미경 수준에서 수행 할 수 있습니다. 결과로 얻은 동작 보정 된 평균은 분명히 식별 가능하고 잘 분리 된 단일 입자를 보여야하며, 그 파워 스펙트럼은 분명하게 식별 가능하고 등방성 인 Thon 링을 보여야합니다. 가시적 인 공간 주파수는 대부분의 경우 구조가 원칙적으로 궁극적으로 결정될 수있는 가장 높은 해상도를 정의합니다. 충분한 품질과 그 파워 스펙트럼의 모션 보정 된 평균의 예는 섹션 #SPHIRE의 Drift 및 CTF 평가 GUI 도구 (http://sphire.mpg.de/wiki/doku.php)를 사용하여 최종 결과에 부정적인 영향을 줄 수있는 이상치 이미지를 제거 할 수 있습니다.
입자 선별과 관련하여 SPHIRE 파이프 라인에서 중요한 단계는 ISAC ( 프로토콜 단계 5.2) 를 사용하는 2D 분류입니다. 여기서 사용자는 프로그램에 의해 자동으로 식별 된 재현 가능한 2D 클래스 평균이 각도 공간을 준 균일하게 커버하기에 충분한 방향 범위를 채택하도록 제어해야합니다. 클래스 평균의 품질이 만족스럽지 않은 경우 (시끄러운 및 / 또는 흐릿한 이미지) 및 / 또는 재현 가능한 클래스 평균 수가 매우 낮은 경우 자동 피킹 품질을 개선하고 데이터 세트 이미징 또는 샘플 준비를 최적화하십시오. 대부분의 경우 좋은 2D 클래스 평균을 산출하지 못하는 데이터 세트로부터 신뢰할 수있는 재구성을 계산할 수 없습니다. 고품질 2D 클래스 평균의 예분노는 "대표 결과"섹션에 나와 있습니다.
자동화 된 방식으로 RVIPER를 사용하여 신뢰할 수있는 초기 3D 모델을 얻으려면 최소 100 개의 클래스 평균이 필요합니다 ( 프로토콜 단계 6.1 ). 이 단계에서 사용자는 가장 높은 품질의 평균을 선택하고 가능한 한 많은 입자의 방향을 포함시켜야합니다. 초기 모델의 품질은 이후의 고해상도 3D 개선의 성공에 중요합니다.
다른 소프트웨어 패키지에서는 "나쁜"입자를 제거하기 위해 3D 분류가 수행되기도합니다 8 , 9 . 그러나 SPHIRE에서 이러한 입자의 대부분은 ISAC을 사용하는 2D 분류 중에 이미 자동으로 제거됩니다. 따라서 재구성 및 3D 변동성 분석이 데이터 집합의 이질성을 나타내는 경우에만 3D 정렬의 계산 집약적 단계를 수행하는 것이 좋습니다.
가장 중요한 점은 사용자는 항상 조심스럽게 결과 3D 볼륨을주의 깊게 검사해야하며 ( 프로토콜 단계 9.3 ) 각 밀도의 기능이 공칭 해상도와 잘 일치하는지 확인해야합니다. <9Å의 분해능에서 α- 나선에 해당하는 막대 모양의 밀도가 보입니다. <4.5Å의 해상도에서 베타 시트의 가닥에 해당하는 밀도는 일반적으로 잘 분리되며 부피가 큰 아미노산이 보입니다. 고해상도 맵 (<3Å)은 명확한 식별이 가능한 사이드 체인을 보여야하므로 정확한 원자 모델을 구축 할 수 있습니다.
SPHIRE의 자동화 된 재현성 테스트와 최소한의 육안 검사 덕분에 현재의 프로토콜은 일반적으로 모든 유형의 단일 입자 cryo-EM 프로젝트에 적용 가능하다는 것을 입증 한 결과입니다. 각 처리 단계의 대표 결과는 다음과 같은 TcdA1 독소의 재구성을 위해 나타납니다.Photorhabdus luminescens 21 은 원자 분해능에 가까운 것으로 알려져있다. 비슷한 품질의 밀도 맵을 사용하여 신개념 또는 실제 공간의 정제뿐만 아니라 새로운 백본 추적을 통해 신뢰할 수있는 원자 모델을 구성 할 수 있으므로 복잡한 분자 메커니즘을 이해할 수있는 견고한 구조 프레임 워크를 제공합니다.
접근 코드 :
EM 구조 및 처리되지 않은 영화에 대한 좌표는 전자 현미경 데이터 뱅크 및 전자 현미경 파일럿 이미지 아카이브에 각각 EMD-3645 및 EMPIAR-10089로 기탁되었습니다.
공개
저자는 경쟁적인 금전적 이해 관계가 없다고 선언합니다.
감사의 말
우리는 TcdA1 현미경 사진을 제공 한 D. Roderer에게 감사드립니다. EMAN2 인프라를 지속적으로 지원해 준 Steve Ludtke에게 감사드립니다. 이 작업은 막스 플랑크 협회 (SR)와 유럽 연합 (EU)의 일곱 번째 프레임 워크 프로그램 (FP7 / 2007-2013) (부여 번호 615984) (SR에)에 의거 한 유럽 기금과 국립 연구소 건강 R01 GM60635에서 PAP).
자료
| Name | Company | Catalog Number | Comments |
| SPHIRE | Max Planck Institute of Molecular Physiology- Dortmund and Houston Medical School, Houston, Texas | http://sphire.mpg.de | |
| UCSF Chimera | University of California, San Francisco | http://www.cgl.ucsf.edu/chimera/ | |
| Unblur | Janelia Farm Research Campus, Ashburn | http://grigoriefflab.janelia.org/unblur | |
| Coot | MRC Laboratory of Molecular Biology, Cambridge | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ | |
| EMAN2 | Baylor College of Medicine, Houston | http://blake.bcm.edu/emanwiki/EMAN2 | |
| Computing Cluster with 1824 cores | Max Planck Institute of Molecular Physiology | Linux Cluster with 76 nodes, each with 2 Processors Xeon E5-2670v3 12C 2.30 GHz and 128 Gb RAM | |
| TITAN KRIOS electron microscope | FEI | 300 kV, Cs correction, XFEG | |
| Falcon II direct electron detector | FEI | ||
| EPU (automated data acquisition software) | FEI | https://www.fei.com/software/epu/ |
참고문헌
- Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nature Methods. 13 (1), 24-27 (2016).
- Liao, M., Cao, E., Julius, D., Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 504 (7478), 107-112 (2013).
- Bai, X. -C., Yan, C., et al. An atomic structure of human γ-secretase. Nature. 525 (7568), 212-217 (2015).
- Ecken, J. V. D., Heissler, S. M., Pathan-Chhatbar, S., Manstein, D. J., Raunser, S. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature. 534 (7609), 724-728 (2016).
- von der Ecken, J., Müller, M., Lehman, W., Manstein, D. J., Penczek, P. A., Raunser, S. Structure of the F-actin-tropomyosin complex. Nature. 519 (7541), 114-117 (2015).
- Tang, G., Peng, L., et al. EMAN2: An extensible image processing suite for electron microscopy. Journal of Structural Biology. 157 (1), 38-46 (2007).
- van Heel, M., Harauz, G., Orlova, E. V., Schmidt, R., Schatz, M. A new generation of the IMAGIC image processing system. Journal of Structural Biology. 116 (1), 17-24 (1996).
- Grigorieff, N. FREALIGN: high-resolution refinement of single particle structures. Journal of Structural Biology. 157 (1), 117-125 (2007).
- Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. Journal of Structural Biology. 180 (3), 519-530 (2012).
- Shaikh, T. R., Gao, H., et al. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nature Protocols. 3 (12), 1941-1974 (2008).
- Hohn, M., Tang, G., et al. SPARX, a new environment for Cryo-EM image processing. Journal of Structural Biology. 157 (1), 47-55 (2007).
- Lander, G. C., Stagg, S. M., et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. Journal of Structural Biology. 166 (1), 95-102 (2009).
- de la Rosa-Trevìn, J. M., Quintana, A., et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. Journal of Structural Biology. 195 (1), 93-99 (2016).
- Grant, T., Grigorieff, N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife. 4, 06980(2015).
- Pettersen, E. F., Goddard, T. D., et al. UCSF Chimera?A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25 (13), 1605-1612 (2004).
- Penczek, P. A., Fang, J., Li, X., Cheng, Y., Loerke, J., Spahn, C. M. T. CTER-rapid estimation of CTF parameters with error assessment. Ultramicroscopy. 140, 9-19 (2014).
- Frank, J. Three-Dimensional Electron Microscopy of Macromolecular Assemblies. , Oxford University Press. (2006).
- Woolford, D., Ericksson, G., et al. SwarmPS: rapid, semi-automated single particle selection software. Journal of Structural Biology. 157 (1), 174-188 (2007).
- Yang, Z., Fang, J., Chittuluru, J., Asturias, F. J., Penczek, P. A. Iterative Stable Alignment and Clustering of 2D Transmission Electron Microscope Images. Structure/Folding and Design. 20 (2), 237-247 (2012).
- Gatsogiannis, C., Merino, F., et al. Membrane insertion of a Tc toxin in near-atomic detail. Nature Publishing Group. , (2016).
- Gatsogiannis, C., Lang, A. E., et al. A syringe-like injection mechanism in Photorhabdus luminescens toxins. Nature. 495 (7442), 520-523 (2013).
- Meusch, D., Gatsogiannis, C., et al. Mechanism of Tc toxin action revealed in molecular detail. Nature. 508 (7494), 61-65 (2014).
- Penczek, P. A., Frank, J., Spahn, C. M. T. A method of focused classification, based on the bootstrap 3D variance analysis, and its application to EF-G-dependent translocation. Journal of Structural Biology. 154 (2), 184-194 (2006).
- Emsley, P., Lohkamp, B., Scott, W. G., Cowtan, K. Features and development of Coot. Acta crystallographica. Section D, Biological crystallography. 66, Pt 4 486-501 (2010).
- Callaway, E. The revolution will not be crystallized: a new method sweeps through structural biology. Nature. 525 (7568), 172-174 (2015).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유