
Method Article
Hochauflösende Einzelteilchenanalyse aus Elektronen-Kryomikroskopie-Bildern mit SPHIRE
In diesem Artikel
Zusammenfassung
Dieses Papier stellt ein Protokoll für die Verarbeitung von Kryo-EM-Bildern mit der Software-Suite SPHIRE vor. Das vorliegende Protokoll kann für fast alle Einzelteilchen-EM-Projekte angewendet werden, die auf eine nahezu atomare Auflösung abzielen.
Zusammenfassung
SPHIRE (SPARX für hochauflösende Elektronenmikroskopie) ist eine neuartige, offene, bedienerfreundliche Software-Suite für die halbautomatisierte Verarbeitung von Single-Partikel-Elektronen-Kryomikroskopie (Cryo-EM) Daten. Das hier vorgestellte Protokoll beschreibt ausführlich, wie man eine nahezu atomare Auflösungsstruktur ab Kryo-EM-Mikrograph-Filmen erhält, indem er die Benutzer durch alle Schritte der Einzelteilchenstruktur-Bestimmungs-Pipeline führt. Diese Schritte werden von der neuen grafischen Benutzeroberfläche von SPHIRE gesteuert und erfordern einen minimalen Eingriff des Benutzers. Unter Verwendung dieses Protokolls wurde eine 3,5 Å-Struktur von TcdA1, ein Tc-Toxin-Komplex aus Photorhabdus-Lumineszenen , von nur 9500 Einzelteilchen abgeleitet. Dieser stromlinienförmige Ansatz hilft Anfängern ohne umfangreiche Verarbeitungserfahrung und a priori strukturelle Informationen, um geräuscharme und unvoreingenommene Atommodelle ihrer gereinigten makromolekularen Komplexe in ihrem Heimatzustand zu erhalten.
Einleitung
Nach der Entwicklung der direkten Elektronendetektortechnologie setzt der bemerkenswerte Fortschritt bei Einzelteilchen-Kryo-EM derzeit die Strukturbiologie um 1 . Im Vergleich zur Röntgenkristallographie erfordert diese Technik nur eine geringe Menge an Proteinmaterial ohne die Notwendigkeit einer Kristallisation, während sie gleichzeitig weniger Beschränkungen hinsichtlich der Reinheit der Probe aufwirft und die Bestimmung von Strukturen bei nahezu atomarer Auflösung ermöglicht. Wichtig ist, dass verschiedene Kompositionen oder Zustände nun rechnerisch getrennt werden können und die Strukturbestimmung der verschiedenen Konformationen mit beispiellosem Detaillierungsgrad durchgeführt werden kann. In jüngster Zeit konnten Dichtekarten von anspruchsvollen Molekülen bei Auflösungen erzeugt werden, die ein de novo Modellbau und damit ein tiefes Verständnis ihrer Wirkungsweise 2 , 3 , 4 , 5 ermöglichen.
Eine Vielzahl von Bildverarbeitungs-Softwarepaketen gibt es in der 3DEM (3D Electron Microscopy) Community (https://en.wikibooks.org/wiki/Software_Tools_For_Molecular_Microscopy) und die meisten von ihnen sind kontinuierlich weiterentwickelt. Für Proteine, die verschiedene Molekulargewichte und Symmetrien mit verschiedenen Softwarepaketen aufweisen, wie EMAN2 6 , IMAGIC 7 , FREALIGN 8 , RELION 9 , SPIDER 10 und SPARX 11 , wurde eine nahezu atomare Auflösung erreicht. Jedes Paket erfordert ein anderes Niveau der Benutzer-Know-how und bietet eine andere Ebene der Benutzerführung, Automatisierung und Erweiterbarkeit. Darüber hinaus, während einige Programme komplette Umgebungen zur Erleichterung aller Schritte der Bildanalyse, andere sind entworfen, um bestimmte Aufgaben zu optimieren, wie die Verfeinerung von Ausrichtungsparametern ausgehend von einem bekannten rEferenzstruktur In jüngerer Zeit wurden mehrere Plattformen entwickelt, darunter APPION 12 und SCIPION 13 , die eine einzige Verarbeitungspipeline bereitstellen, die Ansätze und Protokolle aus den oben aufgeführten Softwarepaketen integriert.
Um zur aktuellen Entwicklung von cryo-EM beizutragen, wurde SPARX zu einer neuen eigenständigen und kompletten Plattform für die Einzelpartikelanalyse, genannt SPHIRE (SPARX für hochauflösende Elektronenmikroskopie), neu entwickelt. Um die Erreichbarkeit der Technik für neue Forscher auf dem Gebiet zu erhöhen und die große Menge an Daten, die von modernen vollautomatischen High-End-Elektronenmikroskopen produziert werden, zu bewältigen, wurde die Verarbeitungspipeline neu gestaltet und durch die Einführung einer einfach zu bedienenden Lösung vereinfacht Grafische Benutzeroberfläche (GUI) und Automatisierung der wichtigsten Schritte des Workflows. Darüber hinaus wurden neue Algorithmen hinzugefügt, um eine schnelle, reproduzierbare und automatisierte Strukturbestimmung von cr zu ermöglichenYo-EM Bilder. Darüber hinaus wurde eine Validierung durch Reproduzierbarkeit eingeführt, um gemeinsame Artefakte zu vermeiden, die während der Verfeinerung und Heterogenitätsanalyse erzeugt wurden.
Obwohl das Programm weitgehend modifiziert wurde, wurden seine geschätzten Kernmerkmale beibehalten: ein einfacher Open-Source-Code, das moderne objektorientierte Design und die Python-Schnittstellen für alle Basisfunktionen. So wurde es nicht in ein Black-Box-Programm geändert, so dass Benutzer den Python-Code studieren und einfach modifizieren, zusätzliche Anwendungen erstellen oder den gesamten Workflow ändern können. Dies ist besonders nützlich für Nicht-Standard-Kryo-EM-Projekte.
Hier stellen wir ein Protokoll zur Erlangung einer nahezu atomaren Auflösungsdichtekarte aus Kryo-EM-Bildern unter Verwendung der GUI von SPHIRE vor. Es beschreibt im Detail alle Schritte, die erforderlich sind, um eine Dichtekarte aus rohen Kryo-EM-Direktdetektorfilmen zu erzeugen, und ist nicht auf einen bestimmten Makromolekültyp beschränkt. Dieses Protokoll beabsichtigt vor allem, newc zu führenOmers im Feld durch den Workflow und liefern wichtige Informationen über entscheidende Schritte der Verarbeitung sowie einige der möglichen Fallstricke und Hindernisse. Weitergehende Funktionen und der theoretische Hintergrund hinter SPHIRE werden an anderer Stelle beschrieben.
Protokoll
HINWEIS: Um diesem Protokoll zu folgen, ist es notwendig, SPHIRE auf einem System mit einer MPI-Installation (derzeit ein Linux-Cluster) richtig zu installieren. Laden Sie SPHIRE und den TcdA1-Dataset von http://www.sphire.mpg.de herunter und folgen Sie den Installationsanweisungen: http://sphire.mpg.de/wiki/doku.php?id=howto:download. Diese Prozedur installiert auch EMAN2. SPHIRE verwendet derzeit EMAN2s e2boxer für die Partikelauswahl und e2display für die Anzeige von Bilddateien. Für die dosisgewichtete Bewegungskorrektur der Rohmikroskop-Filme verwendet SPHIRE unblur 14 . Laden Sie das Programm herunter und folgen Sie den Installationsanweisungen (http://grigoriefflab.janelia.org/unblur, Grigorieff lab). Für die interaktive Visualisierung der resultierenden Strukturen wird das Protokoll das molekulare Grafikprogramm Chimera 15 (https://www.cgl.ucsf.edu/chimera/download.html) verwenden. Ein schönes Tutorial, um sich mit den Funktionen vertraut zu machen, die während dieses Protokolls verwendet werden, können Sie seinHier: https://www.cgl.ucsf.edu/chimera/data/tutorials/eman07/chimera-eman-2007.html. Anleitung zum Einreichen eines Paralleljobs an einen Cluster aus der SPHIRE GUI finden Sie hier: http://sphire.mpg.de/wiki/doku.php?id=howto:submissions. Die Gesamtorganisation der SPHIRE GUI und die wichtigsten Schritte des Workflows, die während dieses Protokolls durchgeführt wurden, sind in Abbildung 1 dargestellt.
1. PROJEKT: Setzen Sie Konstante Parameterwerte für dieses Projekt
- Starten Sie die SPHIRE GUI-Anwendung, indem Sie " sphire &" und die ENTER-Taste an einem Terminal-Fenster eingeben.
- Passen Sie die projektweiten Parameter ( zB Pixelgröße, Partikelradius und Symmetrie) in die jeweiligen Eingabefelder der Projekteinstellungsseite an und registrieren Sie diese Werte für alle nachfolgenden Schritte des Workflows.
- Klicken Sie auf das Symbol "PROJECT" unten rechts im linken Bereich, um die Seite für die Projekteinstellungen zu öffnen.
- Messen Sie die längste Achse eines Teilchens mit dem interaktiven Anzeigeinstrument e2display.py, und geben Sie dann die Hälfte der Teilchengröße auf "Proteinpartikelradius" ein. Wenn die Messung in Å ist, denken Sie daran, die Einheit in Pixel unter Verwendung der Pixelgröße umzuwandeln ( zB wenn ein Teilchen 200 Å lang ist und die Pixelgröße 1,2 Å / Pixel beträgt, dann ist die längste Achse des Teilchens 200 / 1,2 = ~ 166 Pixel und der Radius 166/2 = 83 Pixel).
- Setzen Sie die "Partikelboxgröße" auf mindestens das 1,5fache der Partikelgröße. Vermeiden Sie Fenstergrößen mit großer Primzahl. Denken Sie auch daran, dass der 3D-Verfeinerungsalgorithmus derzeit eine geradzahlige Boxgröße benötigt.
HINWEIS: Das Fenster sollte einen Rand enthalten, um erste Zentrierfehler zu berücksichtigen, die sich aus der Kommissionierung ergeben (die Notwendigkeit, Partikel innerhalb des Fensters zu verschieben) und für den ausreichenden Hintergrundbereich außerhalb der Partikelgrenze für eine korrekte CTF-Korrektur (besonders wichtig für große Defokussierungswerte) 16). - Setzen Sie die "CTF-Fenstergröße" auf die "Partikelboxgröße". Für Projekte mit niedrigen Kontrastdaten verwenden Sie ein größeres Fenster, um glattere Schätzungen von Leistungsspektren zu erhalten.
- Set "Punktgruppen-Symmetrie" des Komplexes ( zB "C5"). Wenn die Symmetrie der Zielstruktur nicht bekannt ist, lassen Sie sie bei "C1" (asymmetrisch). Wenn jedoch während der Verarbeitung eine spezifische Symmetrie höherer Ordnung identifiziert wird, ändern Sie diese Symmetrie-Einstellung entsprechend und wiederholen Sie die Schritte nach 2D-Ausrichtung mit ISAC.
- Set "Protein-Molekülmasse" in kDa (ungefährer Wert wird genügen). Drücken Sie die Taste "Registereinstellungen".
2. MOVIE: Richten Sie die Frames jeder Movie-Mikrographur aus, um die Gesamtbewegung der Probe zu korrigieren
- Für alle Filmmikroskopische Aufnahmen berechnen Sie die x / y-Verschiebungen für alle Frames und erstellen dann ihre Dosis-ungewichteten und dosisgewichteten MoKorrigierten Durchschnitt (siehe Diskussion). Beachten Sie, dass erstere nur für die CTF-Schätzung notwendig ist, da die Schätzung mit den dosisgewichteten Durchschnitten nicht gut abläuft, während diese für alle anderen Schritte der Strukturbestimmung verwendet wird.
- Klicken Sie auf das "MOVIE" -Symbol und dann auf die Schaltfläche "Micrograph Movie Alignment". Setzen Sie "Unblur ausführbaren Pfad", indem Sie die ausführbare Datei auswählen. Setzen Sie "Input-Mikrograph-Pfad-Muster", indem Sie eine rohe, nicht ausgerichtete Filmmikroskopie auswählen und den Variablenteil der Dateinamen durch den Platzhalter "*" ( zB TcdA1 _ * .crc) ersetzen. Geben Sie den Pfad für "Ausgabeverzeichnis" an.
- Stellen Sie "Summovie executable path" ein, indem Sie die ausführbare Datei auswählen.
- Set "Anzahl der Filmrahmen" auf die Anzahl der Frames in jedem Film Mikroskop. Setzen Sie die "Mikroskopspannung" und "Per Rahmenbelichtung" auf die Werte, die bei der Datenerfassung verwendet werden. (Für exampWenn die Gesamtdosis 60 e - / Å 2 beträgt, mit 20 Bildern, die ohne Vorbelichtung aufgezeichnet wurden, beträgt die Belichtung für jeden Rahmen 60/20 = 3 e - / A 2. ) Drücken Sie die Taste "Run Command", um die Frames jeder Filmmikroskopaufnahme.
HINWEIS: Hiermit werden automatisch zwei Ausgabeverzeichnisse erstellt , die dosisungewichtete und dosisgewichtete, bewegungskorrigierte mittlere Mikrophotographien enthalten.
3. CTER: Schätzung der Defocus- und Astigmatismus-Parameter des CTF
- Schätzen Sie die CTF-Parameter (Defokussierung und Astigmatismus, die anderen werden vom Benutzer gesetzt) für jede dosisungewichtete durchschnittliche mikroskopische Aufnahme.
- Klicken Sie auf das "CTER" -Symbol und dann die Schaltfläche "CTF Schätzung". Um "Input micrograph path pattern" einzustellen, wählen Sie eine dosis-ungewichtete motion-korrigierte mikroskopische Aufnahme aus, und ersetzen Sie dann den variablen Teil der Dateinamen mit dem Platzhalter"*". Geben Sie auch den Pfad für "Ausgabeverzeichnis" an.
- Setzen Sie den "Amplitudenkontrast" auf den Wert, der routinemäßig für die Art der Daten verwendet wird (Eisdicke ist ein wichtiger Faktor) und die Mikroskopspannung im Labor ( zB 10%). Typische Werte liegen im Bereich von 7 - 14% 17 .
- Set "Mikroskop sphärische Aberration (Cs)" und "Mikroskopspannung" bei der Datenerfassung verwendet.
- Setzen Sie "Niedrigste Frequenz" und "Höchste Frequenz" des Suchbereichs für das CTF-Modell, das auf 0,0285 und 0,285 Å -1 (40 - 4 Å) passt. Drücken Sie die Taste "Run command", um die CTF-Parameter abzuschätzen.
HINWEIS: Die CTF-Parameter werden automatisch in der Datei partres.txt im angegebenen Ausgabeverzeichnis gespeichert. Die CTF-Schätzung der 112 Mikrophotographien wurde auf 96 Kerne berechnet und nach ~ 3 min auf dem Linux-Cluster beendet, um die repräsentativen Ergebnisse zu erhalten.
4. FENSTER: Partikel aus den Dosis-gewichteten mittleren Mikrophotographien extrahieren
- Teilchen manuell oder automatisch aus mikroskopischen Aufnahmen mit e2boxer 6 auswählen und Koordinatendateien erstellen, die jeweils eine Liste von Partikel-Xy-Koordinaten innerhalb der zugehörigen Mikroskopie enthalten.
- Klicken Sie auf das Symbol "WINDOW" und dann auf die Schaltfläche "Particle Picking". Drücken Sie die Taste "Run command", um e2boxer 6 zu starten und wählen Sie die Partikel jeder Mikrophotographie manuell oder automatisch 18 (siehe Diskussion ). Speichern Sie die endgültigen Partikelkoordinaten für jede mikroskopische Aufnahme im EMAN1-Dateiformat (.box). Alternativ können Sie die Koordinatendateien aus anderen Programmen importieren, nachdem Sie sie in das EMAN1-Format konvertiert haben.
- Erstellen von Partikelstapeln durch Extraktion von Partikelbildern aus den dosisgewichteten Mikrophotographien (in SPHIRE, Partikelstapel ist oftEn einfach "Stapel" genannt).
- Drücken Sie die Taste "Partikelextraktion". Geben Sie "Input micrograph path pattern" an, indem Sie eine dosisgewichtete bewegungskorrigierte mikroskopische Aufnahme auswählen und dann den variablen Teil der Dateinamen mit dem Platzhalter "*" ( zB TcdA1 _ * mrc) ersetzen. Ebenso setzen Sie "Eingabekoordinatenpfadmuster" durch Auswahl einer Koordinatendatei ( zB TcdA1 _ *.). Geben Sie den Pfad für "Ausgabeverzeichnis" an.
- Setzen Sie die "CTF-Parameterquelle", indem Sie die CTF-Parameterdatei (partres.txt in Schritt 3.1) auswählen. Drücken Sie die Taste "Run command".
- Kombinieren Sie die extrahierten Partikelbildstapel zu einem einzigen.
- Klicken Sie auf die Schaltfläche "Partikelstapel". Geben Sie den Pfad zum "virtuellen Bildstapel ausgeben" mit einem BDB-Dateipfad-Format an ( zB "bdb: Partikel / Stack", wobei "Partikel" auf thE-Verzeichnis, das ein BDB-Datenbankverzeichnis enthält, dessen Name immer EMAN2DB ist und "Stack" bezieht sich auf einen bestimmten Bildstapel in dieser Datenbank). Geben Sie "Input BDB-Bildstapelmuster" an, indem Sie ein Verzeichnis mit "mpi_proc" auswählen und dann den Variablenteil der Verzeichnisnamen mit dem Platzhalter "*" ( zB Partikel / mpi_proc_000 zu Partikel / mpi_proc_ *) ersetzen. Drücken Sie die Taste "Run command".
5. ISAC: Klassifizierung von Partikelbildern in 2D
- Berechnen Sie 2D-Klasse Mittelwerte durch Ausrichten von Partikeln und Clustering sie nach ihrem 2D-Aussehen.
HINWEIS: Die daraus resultierenden 2D-Mittelwerte haben im Vergleich zu den einzelnen Partikelbildern ein verbessertes Signal-Rausch-Verhältnis (SNR) und dienen so zur visuellen Beurteilung der Qualität und Heterogenität des Datensatzes sowie zur Beseitigung von unerwünschten Bildern aus dem Stack ( ZB Eiskristalle, Kohlenstoffkanten,Aggregate, Fragmente usw. ) 19 . Darüber hinaus werden sie später verwendet, um ein erstes 3D-Modell zu bestimmen.- Klicken Sie auf das "ISAC" -Symbol und dann die "ISAC - 2D Clustering" button.Set "Input Bild Stack" durch Auswahl der Stack Datei mit den extrahierten Partikel. Geben Sie den Pfad für "Ausgabeverzeichnis" an.
- Benutze 200 - 1000 für "Bilder pro Klasse". Wählen Sie die entsprechende Zahl unter Berücksichtigung der erwarteten Anzahl von 2D-Klassen (die Gesamtzahl der Partikel dividiert durch die Anzahl der Bilder pro Klasse). Passen Sie diesen Parameter in Abhängigkeit vom SNR und der Größe des Datensatzes an. Erhöhe die Anzahl der Mitglieder pro Klasse, falls der Datensatz übermäßig laut ist. Verringern Sie die Anzahl, wenn eine geringe Anzahl von Partikeln verfügbar ist.
HINWEIS: Aufgrund von Speicherbeschränkungen für ziemlich große Datensätze (> 100.000 Partikel), teilen Sie den vollständigen Datensatz in Teilmengen, führen Sie ISAC für jede Teilmenge unabhängig aus und kombinieren SieDie Ergebnisse am Ende. Detaillierte Anleitungen für dieses Verarbeitungsszenario finden Sie unter http://www.sphire.mpg.de/wiki/doku.php. - Aktiviere das Kontrollkästchen "Phase-Flip". Halten Sie die Standardwerte für "Zielpartikelradius" und "Zielpartikelbildgröße", um den Prozess zu beschleunigen, indem Sie alle Partikelbilder mit diesen Einstellungen automatisch schrumpfen. Drücken Sie die Taste "Run command", um die 2D-Klassenmittelwerte zu berechnen.
HINWEIS: Dieser Schritt ist rechnerisch anspruchsvoll und die Laufzeit nimmt mit der Anzahl der Partikel und Klassen sowie dem Zielradius und der Bildgröße deutlich zu. Auf einem Cluster mit 96 Prozessen beendet die 2D-Klassifikation von ~ 10.000 Partikeln nach ca. 90 min.
- Zeigen Sie die daraus resultierenden ISAC 2D-Mittelwerte visuell an, um sicherzustellen, dass ihre Qualität zufrieden stellend ist (siehe Diskussion).
- Drücken Sie die Taste "Display Data" unter "UTILITIES". Set ""Input-Dateien" durch Auswahl der Datei mit den ISAC 2D-Mittelwerten (class_averages.hdf, die in Schritt 5.1 erstellt wurden). Drücken Sie die Taste "Run command", um die endgültigen, reproduzierbaren und validierten Klassenmittelwerte von ISAC anzuzeigen.
- Erstellen Sie einen neuen Stack, der nur die Partikelmitglieder der validierten Klassenmittelwerte enthält.
- Drücken Sie die Taste "Stapel-Teilsatz erstellen". Setzen Sie "Input image stack", indem Sie dieselbe Stapeldatei wie im Schritt 5.1.1 auswählen. Setzen Sie "ISAC-Mittelwerte", indem Sie die ISAC 2D-Mittelwerte (class_averages.hdf, die in Schritt 5.1 erstellt wurden) auswählen. Geben Sie den Pfad für "Ausgabeverzeichnis" an. Drücken Sie die Taste "Run command".
6. VIPER: Berechnen Sie ein anfängliches 3D-Modell
- Wählen Sie einen kleinen Satz der Klassenmittelwerte (≥100 Bilder), indem Sie alle schlechten Klassenmittelwerte und identische Ansichten des Teilchens löschen (siehe Diskussion) und verwenden Sie diese, um einen Repräsentanten zu berechnenReduzierbares Ausgangsmodell mit VIPER. Denken Sie daran, dass die Auswahl mindestens 60-80 qualitativ hochwertige Mittelwerte mit ~ 200-500 Mitgliedern enthalten sollte.
- Klicken Sie auf das Symbol "VIPER" und dann auf die Schaltfläche "Daten anzeigen". Setzen Sie "Input-Dateien", indem Sie die ISAC 2D-Mittelwerte (class_averages.hdf, die in Schritt 5.1 erstellt wurden). Drücken Sie die Taste "Run command".
- Drücken Sie die Maustaste mittig auf das Grafikfenster des e2displays und aktivieren Sie im Popup-Fenster die Schaltfläche "DEL". Löschen Sie alle schlechten Klassenmittelwerte und identische Ansichten des Teilchens (siehe Diskussion ). Drücken Sie die Schaltfläche "Speichern", um die verbleibenden 2D-Klassenmittelwerte in eine neue Datei zu speichern.
- Aus den ausgewählten ISAC-Mitteln generieren wir eine erste Referenz für die anschließende 3D-Verfeinerung.
- Klicken Sie auf die Schaltfläche "Initial 3D Model - RVIPER". Setzen Sie "Input images stack", indem Sie die abgeschirmte Klasse auswählenMittelwerte (hergestellt in Schritt 6.1). Geben Sie den Pfad für "Ausgabeverzeichnis" an.
- Achten Sie darauf, den gleichen Wert für "Zielpartikelradius" wie ISAC Schritt 5.1.3 zu verwenden. Drücken Sie die Taste "Run command", um ein reproduzierbares ab initio 3D Modell zu erzeugen.
HINWEIS: Dieser Schritt ist rechnerisch anspruchsvoll und die Laufzeit steigt mit der Anzahl der Mittelwerte und der Größe der Partikel deutlich an. Auf einem Cluster mit 96 Prozessen beendete dieser Job (~ 100 Klasse Mittelwerte) nach ~ 15 min.
- Prüfen Sie, ob das resultierende 3D-Modell unter Berücksichtigung der Klassenmittelwerte und darüber hinaus seine strukturelle Integrität ( dh keine abgetrennten Teile und / oder Richtungsartefakte) angemessen ist. Um die Karte anzuzeigen, benutzen Sie das Programm Chimera 15 . An diesem Punkt führen Sie einen ersten Vergleich mit einer Kristallstruktur eines homologen Proteins oder einer Domäne des Proteins von Interesse, wenn es existiert (ein Beispiel ist in der Sektion Represe gezeigtNtative Ergebnisse).
- Für die anschließende 3D-Verfeinerung, generiere eine erste 3D-Referenz und eine 3D-Maske aus einem ab initio 3D-Modell, indem sie ihr Umgebungsgeräusch entfernt und es neu kalibriert, um die ursprüngliche Pixelgröße anzupassen.
- Klicken Sie auf die Schaltfläche "3D-Referenz erstellen". Stellen Sie "Input volume" ein, indem Sie das ab initio 3D Modell (average_volume.hdf in Schritt 6.2) auswählen. Geben Sie den Pfad für "Ausgabeverzeichnis" an.
- Setzen Sie die "Resample Ratio-Quelle", indem Sie die ISAC-Schrumpfungsverhältnisdatei (README_shrink_ratio.txt, die in Schritt 5.1 erzeugt wurde) auswählen. Drücken Sie die Taste "Run command".
7. MERIDIEN: Verfeinern Sie das Initial 3D Volume
- Verfeinern Sie die 3D-Lautstärke ab dem ursprünglichen 3D-Modell.
- Klicken Sie auf das "MERIDIEN" -Symbol, dann die Schaltfläche "3D Verfeinerung". Setzen Sie "Input image stack" und "Initial 3D reference" von seleCting der Partikelstapel und das ab initio 3D Modell (hergestellt in Schritt 5.3 & 6.4). Geben Sie den Pfad für "Ausgabeverzeichnis" an.
- Stellen Sie "3D-Maske" ein, indem Sie die 3D-Masken-Datei auswählen (in Schritt 6.4 erstellt). Verwenden Sie immer eine 3D-Maske, aber vor allem in einem frühen Stadium der Analyse, verwenden Sie eine sphärische Maske oder eine weichkantige Maske, die lose an der Referenz angebracht ist, um eine Vorspannung einer falschen Maskierung zu vermeiden.
- Überprüfen Sie das Kontrollkästchen "Harte 2D-Maske anwenden". Setzen Sie "Startauflösung" auf einen Cutoff-Frequenzwert zwischen 20 - 25 Å. Denken Sie daran, dass ein Tiefpassfilter mit dieser Cutoff-Frequenz auf die ursprüngliche 3D-Struktur angewendet wird, um die ursprüngliche Vorspannung zu reduzieren.
- Überprüfen Sie die Spezifikationen des Clusters, der für diesen Prozess verwendet wird, und legen Sie dann "Speicher pro Knoten" auf den verfügbaren Speicher in Gigabyte fest. Drücken Sie die Taste "Run command", um das 3D-Volume aus dem ursprünglichen 3D-Modell vollautomatisch zu verfeinern.
HINWEIS: Diese Prozedur teilt den Datensatz in zwei Hälften auf, verfeinert die beiden Modelle unabhängig und gibt zwei Rohvolumina aus, jeweils aus nur der Hälfte der Partikel. Es ist rechnerisch anspruchsvoll und die Laufzeit wird mit der Anzahl der Teilchen deutlich zunehmen. Auf diesem Cluster beendet die Meridianverfeinerung nach ~ 2,5 h auf 192 Prozessen (~ 8.000 Partikel, 352 Boxgröße).
- Erstellen Sie eine weichkantige 3D-Maske aus dem verfeinerten Volumen für den nachfolgenden Schärfschritt.
- Klicken Sie auf die Schaltfläche "Adaptive 3D Maske". Stellen Sie "Eingabevolumen" ein, indem Sie eines der ungefilterten Halbvolumina auswählen (in Schritt 7.1 erstellt). Geben Sie den Pfad für "Ausgabemaske" an.
- Setzen Sie einen Wert von "Binarisierungsschwelle". Verwenden Sie Chimäre, um sicherzustellen, dass bei dieser bestimmten Schwelle das Rauschen deutlich außerhalb des interessierenden Volumens im Lösungsmittelbereich der ungefilterten Halbkarten liegt und alle Dichten des Proteins noch c sindAufeinander angewiesen. Drücken Sie die Taste "Run command", um die 3D-Maske zu erstellen.
HINWEIS: Der Hauptkörper der resultierenden Maske (bestehend aus Voxeln, deren Werte> 0,5) sind, sollte eng an die Partikelstruktur angepasst werden, aber dennoch alle interessierenden Dichten einschließen. Der Soft-edge-Abfall sollte mindestens 8-10 Pixel breit sein.
- Füge die beiden ungefilterten Halbvolumina zusammen, die durch die 3D-Verfeinerung erhalten wurden. Dann schärfen Sie das verschmolzene Volumen durch Einstellen des Leistungsspektrums auf der Grundlage der Modulationsübertragungsfunktion (MTF) des Detektors, des geschätzten B-Faktors und der FSC (Fourier Shell Correlation) Schätzung der Auflösung.
- Wählen Sie die Schaltfläche "Schärfen". Setzen Sie "Erstes ungefiltertes Halbvolumen" und "Zweites ungefiltertes Halbvolumen" durch Auswahl der entsprechenden Dateien (vol_0_unfil.hdf und vol_1_unfil.hdf in Schritt 7.1). Verwenden Sie immer "B-Faktor-Erweiterung". Normalerweise halten Sie den Standardwert, um e zu haltenDen B-Faktor-Wert aus dem Eingabedatensatz mit dem Bereich zwischen der endgültigen Auflösung und 10 Å ausschärfen. Alternativ geben Sie einen Ad-hoc- Wert an ( zB -100).
- Halten Sie den Standardwert für "Tiefpassfilterfrequenz", um einen FSC-basierten Filter anzuwenden.
- Stellen Sie "User-provided mask" ein, indem Sie die 3D-Maske auswählen (in Schritt 7.2 erstellt). Denken Sie daran, dass die gemeldete Auflösung mit FSC mit dieser Maske bestimmt wird. Drücken Sie die Taste "Run command", um das verfeinerte 3D-Volume zu schärfen.
- Erzeugen Sie die 3D-Winkelverteilungskarte aus den Projektionsrichtungen aller Teilchen, die durch den oben beschriebenen 3D-Verfeinerungsschritt geschätzt wurden.
- Klicken Sie auf die Schaltfläche "Winkelverteilung". Setzen Sie die "Ausrichtungsparameterdatei", indem Sie die Datei (final_params.txt in Schritt 7.1) auswählen und die Schaltfläche "Befehl ausführen" drücken.
- Visuell das geschärfte 3D-Modell mit Chi untersuchenMera Vergewissern Sie sich, dass die Struktur unter Berücksichtigung der erreichten Auflösung sinnvoll erscheint (siehe Diskussion ).
- Überprüfen Sie die Winkelverteilung mit Chimäre. Vergewissern Sie sich, dass die Verteilung ungefähr gleichmäßig den gesamten 3D-Winkelraum abdeckt. Denken Sie daran, dass für symmetrische Strukturen die Verteilung innerhalb des einzigartigen asymmetrischen Dreiecks eingeschränkt ist.
8. SORT3D: 3D-Heterogenität durch Fokussierung auf die sehr variablen Regionen abgeben
- Berechnen Sie die 3D-Variabilitätskarte aus dem Partikelstapel, der in der 3D-Verfeinerung verwendet wird.
- Klicken Sie auf das Symbol "SORT3D" und dann auf die Schaltfläche "3D Variabilitätsschätzung". Setzen Sie "Input image stack", indem Sie den gleichen abgeschirmten Partikelstapel auswählen, der dem 3D-Verfeinerungsschritt 7.1.1 gegeben wurde. Geben Sie den Pfad für "Ausgabeverzeichnis" an.
- Halten Sie den Standardwert für "Anzahl der Projektionen".
ANMERKUNG: Die Bilder vom eckigen NachbarnHaube wird verwendet, um die 2D-Varianz bei jedem 3D-Projektionswinkel abzuschätzen. Je größer die Zahl, desto weniger laut ist die Schätzung, aber je niedriger die Auflösung und Rotationsartefakte sind ausgeprägter. - Kontrollieren Sie das Kontrollkästchen "CTF verwenden". Drücken Sie die Taste "Run command".
- Verwenden Sie die 3D-Variabilitätskarte, um eine Fokusmaske für den nachfolgenden 3D-Clustering-Schritt zu erstellen.
- Wählen Sie die Schaltfläche "Binäre 3D Maske". Stellen Sie "Eingabevolumen" ein, indem Sie die 3D-Variabilitätskarte auswählen (erstellt in Schritt 8.1). Geben Sie den Dateipfad für "Ausgabemaske" an.
- Setzen Sie "Binarisierungsschwelle", indem Sie die Ausgabe des Feldes "Level" im "Volume Viewer" von Chimera verwenden. Drücken Sie die Taste "Run command".
- Sortieren von Partikelbildern in homogene Strukturgruppen durch Fokussierung auf strukturell sehr variable Regionen.
- Drücken Sie die Taste "3D Clustering - RSORT3D".Stellen Sie "Input 3D-Verfeinerungsverzeichnis" ein, indem Sie das Ausgabeverzeichnis der 3D-Verfeinerung auswählen (erstellt in Schritt 7.1). Geben Sie den Pfad für "Ausgabeverzeichnis" an.
- Set "3D-Maske" durch Auswahl der weichkantigen 3D-Maske (hergestellt in Schritt 7.2). Setzen Sie "Focus 3D Maske", indem Sie die binarisierte 3D Variabilitätskarte auswählen (erstellt in Schritt 8.2).
- Für große Datensätze verwenden Sie mindestens 5.000-10.000 für "Bilder pro Gruppe". Denken Sie daran, dass das Programm immer die Anzahl der Bilder pro Gruppe niedriger als diese Einstellung hält. Passen Sie den Wert an, indem Sie die erwartete Anzahl von 3D-Gruppen berücksichtigen (die Gesamtzahl der Partikel dividiert durch den Wert "Bilder pro Gruppe"), den Datensatz, das SNR und den Grad der Heterogenität. Beginnen Sie mit ~ 5-10 anfänglichen 3D-Gruppen, wenn eine ausreichende Anzahl von Partikeln verfügbar ist, es sei denn, eine höhere Anzahl von verschiedenen strukturellen Zuständen im Datensatz wird erwartet.
- Verwenden Sie mindestens 3.000-5.000 Teilchen für "kleinste Gruppengröße".Beachten Sie, dass das Programm Gruppen mit einer geringeren Anzahl von Bildern als die Einstellung der "kleinsten Gruppengröße" ignoriert. Drücken Sie die Taste "Run command", um das 3D-Clustering durchzuführen.
HINWEIS: RSORT3D ist in zwei Stufen unterteilt. Der erste "sort3d" -Schritt zeigt 3D-Heterogenität aus. Dann rekonstruiert es die Volumina jeder homogenen Strukturgruppe unter Verwendung der 3D-Ausrichtungsparameter, die durch den oben beschriebenen 3D-Verfeinerungsschritt bestimmt wurden. Der zweite "rsort3d" -Schritt findet aus reproduzierbaren Mitgliedern jeder Gruppe heraus, indem sie einen Zwei-Wege-Vergleich der beiden unabhängigen Sortierläufe durchführt. Dann rekonstruiert er homogene Strukturen mit nur den reproduzierbar zugewiesenen Partikeln. Auf einem Cluster mit 96 Kerne beendete dieser Job (~ 8.000 Partikel, 352 Box Größe) nach ca. 3 Stunden.
- Nachdem das Programm beendet ist, verwenden Sie Chimera, um eine homogene 3D-Gruppe auszuwählen. Wählen Sie die Struktur der höchsten scheinbaren Auflösung, die typischerweise mit den meisten po verbunden istSchwere gruppe Stellen Sie sicher, dass die ausgewählte Struktur visuell vernünftig ist, indem Sie die 2D-Klassenmittelwerte und biologischen Aspekte des Proteins von Interesse berücksichtigen (siehe Diskussion ). Wenn es andere Bände gibt, die eine nahezu identische Struktur mit ähnlicher Auflösung haben, betrachten sie als aus einer einzigen homogenen 3D-Gruppe.
- Führen Sie eine lokale Verfeinerung gegen die Teilchenmitglieder der homogensten 3D-Gruppe (mit der höchsten Auflösung) durch.
- Klicken Sie auf die Schaltfläche "Local Subset Refinement". Setzen Sie "Subset text file path", indem Sie die Textdatei auswählen, die die Partikel-IDs der ausgewählten Gruppe enthält ( zB Cluster0.txt, die in Schritt 8.3 erstellt wurde). Stellen Sie "3D-Verfeinerungsverzeichnis" ein, indem Sie das Ausgabeverzeichnis der vorherigen 3D-Verfeinerung auswählen (erstellt in Schritt 7.1).
- Setzen Sie "Neustart iteration" auf diejenige, wo die höchste Auflösung in der vorherigen 3D-Verfeinerung erreicht wird. Drücken Sie die Taste"Befehl ausführen", um eine lokale Verfeinerung der ausgewählten Population von Partikeln durchzuführen.
- Ähnlich wie bei Schritt 7.2, erstellen Sie eine weichkantige 3D-Maske aus einem ungefilterten endgültigen Halbvolumen, das durch die lokale Teilmengenverfeinerung rekonstruiert wurde.
- Ähnlich wie bei Schritt 7.3 werden zwei ungefilterte endgültige Halbvolumina zusammengeführt, die durch die lokale Teilmengenverfeinerung abgeleitet werden und das zusammengeführte Volumen schärfen. Allerdings nicht filtern die geschärfte Volumen dieses Mal.
HINWEIS: Sollte die Heterogenitätsanalyse in Schritt 8.4 bei verschiedenen vergleichbaren Auflösungen mehrere verschiedene Zustände anzeigen, so möchtest du alle verschiedenen Zustände unabhängig verfassen.
9. LOCALRES: Schätzen Sie die lokale Auflösung des endgültigen 3D-Volumens
- Schätzen Sie die lokale Auflösung des 3D-Volumens aus dem homogenen Satz von Partikeln.
- Klicken Sie auf das Symbol "LOCALRES" und dann auf die Schaltfläche "Lokale Auflösung". Set "Erstes Halbvolumen" und "Zweite Hälfte"-Verstärkung "durch Auswahl der ungefilterten endgültigen Halbvolumina der lokalen Teilmengenverfeinerung (in Schritt 8.5 erstellt). Legen Sie die" 3D-Maske "fest, indem Sie die in Schritt 8.6 erzeugte Soft-Edged-3D-Maske auswählen. Geben Sie den Dateipfad für" Ausgabevolumen " .
- Halten Sie den Standardwert von 7 Pixeln für "FSC-Fenstergröße". Denken Sie daran, dass diese Einstellung die Größe des Fensters definiert, in der die lokal-reale Raum-Korrelation berechnet wird; Größere Fenstergrößen produzieren glattere Auflösungskarten auf Kosten der lokalen Auflösbarkeit.
- Halten Sie den Standardwert 0,5 von "Auflösung cut-off" für das Auflösungskriterium.
HINWEIS: Für jedes Voxel wird das Programm die lokale Auflösung als die Häufigkeit berichten, bei der der lokale FSC unter den gewählten Auflösungsschwellenwert fällt. Eine Schwelle unter 0,5 wird nicht empfohlen, da niedrigere Korrelationswerte eine hohe statistische Unsicherheit aufweisen. Daher wird die entsprechende lokale Auflösung stark variieren zwischen Voxeln. - Für "OveraLl Auflösung ", setzen Sie die absolute Auflösung, die bei der Schärfung nach der lokalen Teilmengenverfeinerung geschätzt wird (Schritt 8.7). Drücken Sie die Taste" Ausführen ", um die lokale Auflösung des Volumens zu berechnen.
- Wenden Sie den 3D-lokalen Filter auf das Volumen an, das nach der lokalen Teilmengenverfeinerung geschliffen wurde, indem Sie die 3D-Ortsauflösungskarte verwenden.
- Klicken Sie auf die Schaltfläche "3D Local Filter". Stellen Sie "Eingabevolumen" ein, indem Sie das geschärfte, aber ungefilterte 3D-Volumen auswählen (in Schritt 8.7 erstellt). Ebenso setzen Sie "Local resolution file" und "3D mask" (erstellt in Schritt 9.1 bzw. 8.6). Denken Sie daran, dass die 3D-Maske die Region definiert, in der die lokale Filterung angewendet wird. Geben Sie den Dateipfad für "Ausgabevolumen" an. Drücken Sie die Taste "Befehl ausführen", um den 3D-lokalen Filter anzuwenden.
- Verwenden Sie Chimäre, um das endgültige 3D-Modell und die 3D-Lokal-Auflösungskarte visuell zu überprüfen (erstellt in Schritt 9.2 und 9.1,Ectively). Wählen Sie die Option "Surface Color", um das 3D-Volume entsprechend der lokalen Auflösung zu färben. Denken Sie daran, dass die Verteilung der lokalen Auflösung glatt sein sollte (siehe Diskussion ).
Ergebnisse
Das oben beschriebene Protokoll wurde ausgehend von 112 direkten Detektorfilmen der A-Komponente des Photorhabdus-Lumineszenz- Tc-Komplexes (TcdA1) 20 , 21 , 22 durchgeführt. Dieser Datensatz wurde auf einem Cs-korrigierten Elektronen-Kryomikroskop mit einer High-Brightness-Feldemissions-Pistole (XFEG) aufgezeichnet, die mit einer Beschleunigungsspannung von 300 kV betrieben wurde. Die Bilder wurden automatisch mit einer Gesamtdosis von 60 e - / Å -2 bei einer Pixelgröße von 1,14 Å auf der Probenskala erfasst. Nach der Ausrichtung der Filmrahmen (Protokoll Schritt 2 ) hatten die resultierenden Bewegungskorrigierten Mittelwerte isotrope Thonringe, die sich auf eine hochauflösende Auflösung erstreckten (Abbildung 2a ). Die einzelnen Teilchen waren leicht sichtbar und gut getrennt (Abbildung 2b ). Partikel wurden dann mit dem Schwarmwerkzeug von e2boxer gepicktLass = "xref"> 18 ( Protokoll Schritt 4.1 ). In diesem Fall wurde ein geeigneter Schwellenwert unter Verwendung der selektiveren Option eingestellt (Abbildung 2c ). Die 112 digitalen Mikrophotographien lieferten 9.652 Teilchen. Die Mehrheit der extrahierten Bilder (Protokoll Schritt 4.2 ) enthielt gut definierte Partikel und ihre Kastengröße war ~ 1,5 mal größer als die Partikelgröße, wie empfohlen (Abbildung 2d ). Als nächstes wurde unter Verwendung von ISAC eine 2D-Heterogenitätsanalyse durchgeführt (Protokoll Schritt 5 ). Es lieferte 98 Klassenmittelwerte ( Abbildung 3a ). Mit diesen 2D-Klassen-Mittelwerten wurde ein ab initio- Modell mit VIPER (Protokoll Schritt 6 ) bei Zwischenauflösung berechnet (Abbildung 3b ). Dieses Modell zeigt eine ausgezeichnete Übereinstimmung mit der Kristallstruktur von TcdA1, die zuvor bei einer Auflösung von 3,9 Å gelöst wurde ( Fig. 3c ). Dieses ab initio Modell wurde als anfängliches tem verwendet Platte für die 3D-Verfeinerung (MERIDIEN), was eine Rekonstruktion von 3.5 Å (0.143 Kriterium) ( Protocol Step 7 ) von nur 40.000 asymmetrischen Einheiten ergibt (Abbildung 4 ). Diese nahezu atomare Auflösungskarte wurde innerhalb von 24 Stunden mit bis zu 96 CPUs für die Schritte des Workflows erhalten, die von mehreren Kerne profitieren.
Für die 3D-Variabilitätsanalyse ( Protokollschritt 8) wurden in Schritt 8.3.3 nur 2.000 Partikelbilder pro Gruppe verwendet ( dh der Prozess beginnt mit 5 anfänglichen 3D-Gruppen) und 200 Bilder für die kleinste Gruppengröße in Schritt 8.3.4 Die geringe Anzahl von Partikeln (~ 10.000). Die Analyse zeigte lokalisierte Flexibilität vor allem an der N-terminalen Region des Komplexes, der das für die Reinigung verwendete His-Tag enthält (Abbildung 5a ). Tatsächlich wurden zwölf N-terminale Reste und die His-Markierung nicht in der zuvor veröffentlichten Kristallstruktur von TcdA1 aufgelöst"> 22 und diese höchstwahrscheinlich ungeordnete Region blieb in der vorliegenden Kryo-EM-Dichte ungelöst, wahrscheinlich aufgrund ihrer Flexibilität. Zusätzliche Variabilität wurde an den Rezeptorbindungsdomänen und der BC-Bindungsdomäne nachgewiesen (Abbildung 5a ) Befriedigende Auflösung der Struktur und der eher geringen Größe des Datensatzes wurde diese Heterogenität als tolerierbar gewählt und daher wurde eine fokussierte 3D-Klassifikation 23 nicht durchgeführt. Schließlich wurde die lokale Auflösung der endgültigen Dichtekarte berechnet (Protokollschritt 9.1, Abb 5b ) und die geschärfte 3D-Karte wurde lokal gefiltert (Protokoll Schritt 9.2) . Ein Volumen dieser Qualität kann für das de novo Modellbau mit Coot 24 oder einem anderen Verfeinerungswerkzeug verwendet werden (Abbildung 6 ).

Abbildung 1: Bildverarbeitung mit SPHIRE. ( A ) Die GUI des SPHIRE Softwarepakets. Ein bestimmter Schritt des Workflows kann durch Auswahl des jeweiligen Piktogramms auf der linken Seite des GUI ("Workflowschritt") aktiviert werden. Die Befehle und Dienstprogramme, die mit diesem Schritt des Workflows verknüpft sind, erscheinen im zentralen Bereich der GUI. Nach Auswahl eines der Befehle werden die entsprechenden Parameter im rechten Bereich der GUI angezeigt. Fortgeschrittene Parameter erfordern in der Regel keine Änderung der voreingestellten Standardwerte. ( B ) Stufen im Workflow der Einzelteilchenbildverarbeitung mit dem SPHIRE GUI. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 2: Motion Correction und ParticLe Extraktion. ( A , b ) Typische, qualitativ hochwertige, niedrig dosierte, driftkorrigierte digitale mikroskopische Aufnahme mit einer Defokussierung von 1,7 μm. Beachten Sie die isotropen Thonringe, die sich bis zu einer Auflösung von 2,7 Å im Leistungsspektrum (a) und den gut erkennbaren Partikeln im 2D-Bild ( b ) erstrecken. ( C ) Partikelauswahl mit e2boxer. Grüne Kreise zeigen ausgewählte Partikel an. ( D ) Typische Rohteilchen, die aus der dosisgewichteten mikroskopischen Aufnahme extrahiert wurden. Maßstäbe = 20 nm. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 3: 2D-Clustering und anfängliche Modellgenerierung. ( A ) Galerie der 2D-Klasse im Durchschnitt, wobei die Mehrheit die Seitenansichten darstellt o F das Teilchen Maßstab = 20 nm. ( B ) Ab initio 3D-Karte von TcdA1, die mit RVIPER aus dem Referenz-freien Klassenmittel berechnet wurde. ( C ) starre Körperbefestigung der TcdA1-Kristallstruktur (Bänder) (pdb-id 1VW1) in die anfängliche Kryo-EM-Dichte (transparent grau). Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 4: Cryo-EM 3D Struktur von TcdA1. ( A , b ) Final 3.5 Å Dichte Karte von TcdA1 berechnet mit ~ 9.500 Partikelbildern: ( a ) Seite und ( b ) Draufsicht. ( C ) Repräsentative Bereiche der Kryo-EM-Dichte für eine α-Helix und ein β-Blatt.Arge.jpg "target =" _ blank "> Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.

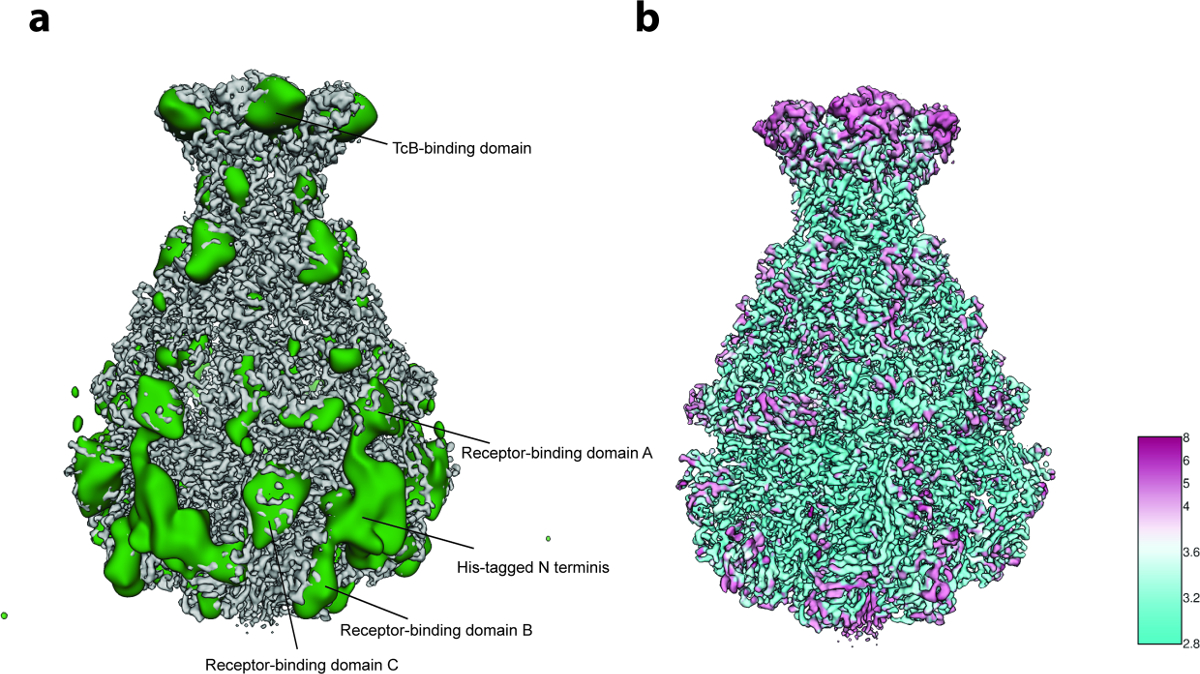
Abbildung 5: Variabilitätsanalyse und lokale Auflösung. ( A ) Oberfläche der geschärften TcdA1 Kryo-EM Karte (grau) und der Variabilitätskarte (grün). Zur besseren Übersicht wurde die Variabilitätskarte auf 30 Å tiefpaßgefiltert. ( B ) Oberflächenwiedergabe der TcdA1-geschärften Kryo-EM-Karte nach lokaler Auflösung (Å) gefärbt. Beachten Sie die topologische Vereinbarung zwischen Bereichen hoher Variabilität und niedriger lokaler Auflösung. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 6: 3D ModEl Bau von TcdA1 mit Coot. Repräsentative Bereiche der Kryo-EM-Dichte und des Atommodells sind für eine α-Helix dargestellt. Das Atommodell wurde de novo mit Coot gebaut. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}
Diskussion
Single-Partikel-Kryo-EM hat in den letzten Jahren eine rasche Entwicklung gezeigt und zahlreiche atomare Auflösungsstrukturen von makromolekularen Komplexen mit großer biologischer Bedeutung 25 geliefert. Um die große Anzahl von Anfängern, die gerade in das Feld einsteigen, zu unterstützen, haben wir die Single-Partikel- Bildanalyse-Plattform SPHIRE entwickelt und präsentieren hier ein Walk-Through-Protokoll für den gesamten Workflow inklusive Filmausrichtung, Partikelkommissionierung, CTF-Schätzung, Erstmodell Berechnung, 2D- und 3D-Heterogenitätsanalyse, hochauflösende 3D-Verfeinerung und lokale Schätzung und Filterung.
Das hier beschriebene Protokoll dient als Kurzanleitung zur 3D-Strukturbestimmung unter Verwendung von Kryo-EM-Mikrophotographien des interessierenden Proteins und mit Hilfe von Rechenwerkzeugen, die von der eigenständigen GUI von SPHIRE bereitgestellt werden.
Das Hauptmerkmal des Workflows ist, dass die meistenDer Prozeduren müssen nur einmal ausgeführt werden, da sie sich auf das Konzept der Validierung durch Reproduzierbarkeit 19 verlassen und keine Parameter-Optimierung erfordern. Dieser automatische Validierungsmechanismus ist ein Hauptvorteil von SPHIRE gegenüber anderen Softwarepaketen, da die Ergebnisse sowohl objektiv als auch reproduzierbar sind und vor allem zu einem akzeptablen Rechenaufwand erhältlich sind. Die Pipeline bietet darüber hinaus eine Fülle von Diagnoseinformationen für erfahrene Anwender, um eine unabhängige Validierung und Bewertung mit eigenen Methoden durchzuführen. Dennoch sollte ein Anfänger, der zumindest einen elementaren theoretischen Hintergrund in der Strukturbiologie und der Elektronenmikroskopie hat, nahezu atomare Auflösungsstrukturen mit eigenen Daten und den automatisierten Validierungsverfahren erhalten können.
Jedoch ist das Erhalten einer nahezu atomaren Auflösungsstruktur nicht immer einfach und das Ergebnis hängt stark von der Qualität der Probe und der Eingabe abein. Für die hier vorgestellten Verfahren wird davon ausgegangen, dass eine ausreichende Anzahl von hochwertigen, nicht ausgerichteten Roh-EM-Filmen zur Verfügung steht, deren Mittelwerte deutlich erkennbare homogene und zufällig orientierte Einzelpartikel aufweisen. Im Allgemeinen gibt es keine Beschränkungen hinsichtlich Symmetrie, Größe oder Gesamtform des Moleküls, aber ein niedriges Molekulargewicht kann ein limitierender Faktor sein, insbesondere wenn das Protein eine charakteristische globuläre Form aufweist. In der Regel ist die Analyse größerer, gut geordneter Partikel mit hoher Punktgruppen-Symmetrie weniger anspruchsvoll. Daher empfiehlt es sich für Anfänger, das aktuelle Protokoll zunächst mit einem gut charakterisierten Kryo-EM-Datensatz zu betreiben. Entweder sind die SPHIRE-Tutorialdaten (http: /sphire.mpg.de) oder einer der EMPIAR eingereichten Datensätze (https://www.ebi.ac.uk/pdbe/emdb/empiar/) mit rohen Filmen ein guter Ausgangspunkt .
Bei der Verarbeitung eigener Daten ist es sehr wahrscheinlich, dass einige Datensätze oder einige der Bilder bestimmte Quali nicht erfüllenKriterien. In diesem Zusammenhang empfiehlt es sich neben den automatisierten Stabilitäts- und Reproduzierbarkeitsprüfungen, die das Programm für wichtige Schritte des Workflows durchführt, den Nutzern weiterhin, die Ergebnisse bei bestimmten "Checkpoints" des Protokolls visuell zu untersuchen, insbesondere wenn die endgültige Rekonstruktion erfolgt Ist nicht zufriedenstellend.
Die erste visuelle Inspektion kann auf der mikroskopischen Ebene nach der Filmausrichtung (Protokollschritt 2 ) und der CTF-Schätzung durchgeführt werden (Protokollschritt 3 ). Die daraus resultierenden bewegungskorrigierten Mittelwerte sollten deutlich erkennbare und gut getrennte Einzelteilchen zeigen und ihre Leistungsspektren sollen deutlich erkennbare, isotrope Thonringe zeigen. Die räumliche Häufigkeit, zu der sie sichtbar sind, definiert in den meisten Fällen die höchste Auflösung, auf die die Struktur grundsätzlich letztlich bestimmt werden kann. Beispiele für einen bewegungskorrigierten Durchschnitt von ausreichender Qualität und sein Leistungsspektrum sind im Abschnitt & #34. Repräsentative Ergebnisse "Ausreichende Bilder, die sich negativ auf das Endergebnis auswirken könnten, können mit Hilfe von SPHIRE Drift- und CTF-Assessment-GUI-Tools (http://sphire.mpg.de/wiki/doku.php) entfernt werden.
Im Hinblick auf die Partikel-Screening ist der entscheidende Schritt in der SPHIRE-Pipeline die 2D-Klassifikation mit ISAC (Protokoll Schritt 5.2) . Hier sollte der Benutzer kontrollieren, dass die reproduzierbaren 2D-Klasse-Mittelwerte, die automatisch vom Programm identifiziert werden, eine Reihe von Orientierungen annehmen, die ausreichen, um den Winkelraum quasi gleichmäßig zu decken. Wenn die Qualität der Klassenmittelwerte nicht zufriedenstellend ist (geräuschvolle und / oder verschwommene Bilder) und / oder die Anzahl der reproduzierbaren Klassenmittelwerte sehr gering ist, sollten Sie die automatische Kommissionierqualität verbessern, die Dataset-Bildgebung oder die Probenvorbereitung optimieren. In den meisten Fällen ist es nicht möglich, eine zuverlässige Rekonstruktion aus einem Datensatz zu berechnen, der keine guten 2D-Klassenmittelwerte ergibt. Beispiele für hochwertige 2D Klasse aveWut sind im Abschnitt "Repräsentative Ergebnisse" dargestellt.
Mindestens 100 Klassenmittelwerte sind erforderlich, um ein zuverlässiges Anfangs-3D-Modell mit Hilfe von RVIPER automatisiert zu erhalten (Protokoll Schritt 6.1 ). Für diesen Schritt sollte der Benutzer die Mittelwerte mit der höchsten Qualität auswählen und möglichst viele unterschiedliche Orientierungen des Teilchens enthalten. Die Qualität des Erstmodells ist entscheidend für den Erfolg der nachfolgenden hochauflösenden 3D-Verfeinerung.
In anderen Softwarepaketen wird manchmal die 3D-Klassifikation durchgeführt, um "schlechte" Partikel 8 , 9 zu entfernen. Allerdings werden bei SPHIRE die meisten dieser Partikel bereits bei der 2D-Klassifikation mit ISAC automatisch eliminiert. So empfiehlt es sich, den rechenintensiven Schritt der 3D-Sortierung nur dann durchzuführen, wenn die Rekonstruktion und die 3D-Variabilitätsanalyse die Heterogenität des Datensatzes anzeigen.
Am wichtigsten ist, dass der Benutzer die daraus resultierenden 3D-Bände sorgfältig sorgfältig inspizieren muss (Protokoll Schritt 9.3 ) und bestätigen, dass sich die Merkmale der jeweiligen Dichte mit der nominalen Auflösung gut übereinstimmen. Bei einer Auflösung von <9 Å werden stabförmige Dichten, die α-Helices entsprechen, sichtbar. Bei einer Auflösung <4,5 Å sind Dichten, die Strängen in β-Blättern entsprechen, normalerweise gut getrennt und sperrige Aminosäuren werden sichtbar. Eine hochauflösende Karte (<3 Å) sollte deutlich erkennbare Seitenketten zeigen, so dass ein genaues Atommodell aufgebaut werden kann.
Die bisherigen Ergebnisse zeigen, dass mit Hilfe der automatischen Reproduzierbarkeitstests von SPHIRE und der minimalen visuellen Inspektionen das vorliegende Protokoll allgemein für jede Art von Einzelteilchen-Kryo-EM-Projekt anwendbar ist. Repräsentative Ergebnisse jedes Verarbeitungsschritts sind für die Rekonstruktion des TcdA1-Toxins von dargestelltPhotorhabdus luminescens 21 , die in nahezu atomare Auflösung gelöst wurde. Dichtekarten gleicher Qualität können verwendet werden, um zuverlässige Atommodelle durch de novo Backbone-Tracing sowie reziproke oder Real-Space-Verfeinerung zu konstruieren und so einen soliden strukturellen Rahmen für das Verständnis komplexer molekularer Mechanismen zu schaffen.
ACCESION CODES:
Die Koordinaten für die EM-Struktur und die unverarbeiteten Filme wurden in der Elektronenmikroskopie-Datenbank und dem Elektronenmikroskopie-Pilot-Bildarchiv unter den Zugangsnummern EMD-3645 bzw. EMPIAR-10089 hinterlegt.
Offenlegungen
Die Autoren erklären, dass sie keine konkurrierenden finanziellen Interessen haben.
Danksagungen
Wir danken D. Roderer für die Bereitstellung von TcdA1 Mikrophotographien. Wir danken Steve Ludtke für seine laufende Unterstützung der EMAN2 Infrastruktur. Diese Arbeit wurde durch Mittel der Max-Planck-Gesellschaft (nach SR) und des Europäischen Rates im Rahmen des Siebten Rahmenprogramms der Europäischen Union (RP7 / 2007-2013) (Zuschuss Nr. 615984) (nach SR) und von den nationalen Instituten von Gesundheit R01 GM60635 zu PAP).
Materialien
| Name | Company | Catalog Number | Comments |
| SPHIRE | Max Planck Institute of Molecular Physiology- Dortmund and Houston Medical School, Houston, Texas | http://sphire.mpg.de | |
| UCSF Chimera | University of California, San Francisco | http://www.cgl.ucsf.edu/chimera/ | |
| Unblur | Janelia Farm Research Campus, Ashburn | http://grigoriefflab.janelia.org/unblur | |
| Coot | MRC Laboratory of Molecular Biology, Cambridge | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ | |
| EMAN2 | Baylor College of Medicine, Houston | http://blake.bcm.edu/emanwiki/EMAN2 | |
| Computing Cluster with 1824 cores | Max Planck Institute of Molecular Physiology | Linux Cluster with 76 nodes, each with 2 Processors Xeon E5-2670v3 12C 2.30 GHz and 128 Gb RAM | |
| TITAN KRIOS electron microscope | FEI | 300 kV, Cs correction, XFEG | |
| Falcon II direct electron detector | FEI | ||
| EPU (automated data acquisition software) | FEI | https://www.fei.com/software/epu/ |
Referenzen
- Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nature Methods. 13 (1), 24-27 (2016).
- Liao, M., Cao, E., Julius, D., Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 504 (7478), 107-112 (2013).
- Bai, X. -C., Yan, C., et al. An atomic structure of human γ-secretase. Nature. 525 (7568), 212-217 (2015).
- Ecken, J. V. D., Heissler, S. M., Pathan-Chhatbar, S., Manstein, D. J., Raunser, S. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature. 534 (7609), 724-728 (2016).
- von der Ecken, J., Müller, M., Lehman, W., Manstein, D. J., Penczek, P. A., Raunser, S. Structure of the F-actin-tropomyosin complex. Nature. 519 (7541), 114-117 (2015).
- Tang, G., Peng, L., et al. EMAN2: An extensible image processing suite for electron microscopy. Journal of Structural Biology. 157 (1), 38-46 (2007).
- van Heel, M., Harauz, G., Orlova, E. V., Schmidt, R., Schatz, M. A new generation of the IMAGIC image processing system. Journal of Structural Biology. 116 (1), 17-24 (1996).
- Grigorieff, N. FREALIGN: high-resolution refinement of single particle structures. Journal of Structural Biology. 157 (1), 117-125 (2007).
- Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. Journal of Structural Biology. 180 (3), 519-530 (2012).
- Shaikh, T. R., Gao, H., et al. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nature Protocols. 3 (12), 1941-1974 (2008).
- Hohn, M., Tang, G., et al. SPARX, a new environment for Cryo-EM image processing. Journal of Structural Biology. 157 (1), 47-55 (2007).
- Lander, G. C., Stagg, S. M., et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. Journal of Structural Biology. 166 (1), 95-102 (2009).
- de la Rosa-Trevìn, J. M., Quintana, A., et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. Journal of Structural Biology. 195 (1), 93-99 (2016).
- Grant, T., Grigorieff, N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife. 4, 06980(2015).
- Pettersen, E. F., Goddard, T. D., et al. UCSF Chimera?A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25 (13), 1605-1612 (2004).
- Penczek, P. A., Fang, J., Li, X., Cheng, Y., Loerke, J., Spahn, C. M. T. CTER-rapid estimation of CTF parameters with error assessment. Ultramicroscopy. 140, 9-19 (2014).
- Frank, J. Three-Dimensional Electron Microscopy of Macromolecular Assemblies. , Oxford University Press. (2006).
- Woolford, D., Ericksson, G., et al. SwarmPS: rapid, semi-automated single particle selection software. Journal of Structural Biology. 157 (1), 174-188 (2007).
- Yang, Z., Fang, J., Chittuluru, J., Asturias, F. J., Penczek, P. A. Iterative Stable Alignment and Clustering of 2D Transmission Electron Microscope Images. Structure/Folding and Design. 20 (2), 237-247 (2012).
- Gatsogiannis, C., Merino, F., et al. Membrane insertion of a Tc toxin in near-atomic detail. Nature Publishing Group. , (2016).
- Gatsogiannis, C., Lang, A. E., et al. A syringe-like injection mechanism in Photorhabdus luminescens toxins. Nature. 495 (7442), 520-523 (2013).
- Meusch, D., Gatsogiannis, C., et al. Mechanism of Tc toxin action revealed in molecular detail. Nature. 508 (7494), 61-65 (2014).
- Penczek, P. A., Frank, J., Spahn, C. M. T. A method of focused classification, based on the bootstrap 3D variance analysis, and its application to EF-G-dependent translocation. Journal of Structural Biology. 154 (2), 184-194 (2006).
- Emsley, P., Lohkamp, B., Scott, W. G., Cowtan, K. Features and development of Coot. Acta crystallographica. Section D, Biological crystallography. 66, Pt 4 486-501 (2010).
- Callaway, E. The revolution will not be crystallized: a new method sweeps through structural biology. Nature. 525 (7568), 172-174 (2015).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten