
Method Article
Analisi di particelle singole ad alta risoluzione da immagini elettroniche a cryo-microscopia usando SPHIRE
In questo articolo
Riepilogo
Questo documento presenta un protocollo per l'elaborazione di immagini cryo-EM utilizzando la suite software SPHIRE. Il presente protocollo può essere applicato per quasi tutti i progetti di singoli particelle EM che mirano ad una risoluzione quasi atomica.
Abstract
SPHIRE (SPARX per la microscopia elettronica ad alta risoluzione) è una nuova suite software open-source per l'elaborazione semi-automatizzata di dati criomicroscopia elettronica a particelle singole (cryo-EM). Il protocollo qui descritto descrive dettagliatamente come ottenere una struttura di risoluzione quasi atomica a partire dai film di micrografia di cryo-EM guidando gli utenti attraverso tutti i passaggi della pipeline di determinazione della struttura delle particelle singole. Questi passaggi sono controllati dalla nuova interfaccia grafica SPHIRE e richiedono un intervento minimo da parte dell'utente. Utilizzando questo protocollo, una struttura di 3,5 Å di TcdA1, un complesso Tc toxin da Photorhabdus luminescens , è stato derivato da soli 9500 particelle singole. Questo approccio semplificato aiuterà gli utenti novizi senza un'estesa esperienza di elaborazione e informazioni strutturali a priori , per ottenere modelli atomici senza rumori e imparziali dei loro complessi macromolecolari purificati nel loro stato nativo.
Introduzione
Dopo lo sviluppo della tecnologia di rivelazione diretta dell'elettrone, il notevole progresso nella singola particella cryo-EM sta attualmente riformando la biologia strutturale 1 . Rispetto alla cristallografia a raggi X, questa tecnica richiede solo una piccola quantità di materiale proteico senza la necessità di cristallizzazione, mentre contemporaneamente pone minori restrizioni riguardanti la purezza del campione e consente comunque la determinazione di strutture a risoluzione quasi atomica. Importante, diverse composizioni o stati possono ora essere separati in modo computazionale e la determinazione della struttura delle diverse conformazioni può essere effettuata a livello di dettaglio senza precedenti. Recentemente, mappe di densità di molecole impegnative potrebbero essere prodotte a risoluzioni che permettono di costruire modello di de novo e quindi una profonda comprensione del loro modo di azione 2 , 3 , 4 , 5.
Una vasta gamma di pacchetti software per l'elaborazione delle immagini è disponibile nella comunità 3DEM (Microscopia Elettronica 3D) (https://en.wikibooks.org/wiki/Software_Tools_For_Molecular_Microscopy) e la maggior parte di esse è in continua evoluzione. La risoluzione quasi atomica è stata raggiunta per le proteine che presentano varie pesi molecolari e simmetrie con diversi pacchetti software diversi, tra cui EMAN2 6 , IMAGIC 7 , FREALIGN 8 , RELION 9 , SPIDER 10 e SPARX 11 . Ogni pacchetto richiede un diverso livello di competenza dell'utente e fornisce un diverso livello di guida, automazione e estensibilità dell'utente. Inoltre, mentre alcuni programmi forniscono ambienti completi per facilitare tutte le fasi dell'analisi delle immagini, altre sono progettate per ottimizzare le attività specifiche, come la perfezionamento dei parametri di allineamento a partire da un rStruttura di eference. Più di recente, sono state sviluppate diverse piattaforme, tra cui APPION 12 e SCIPION 13 , che forniscono una singola pipeline di elaborazione che integra approcci e protocolli dai vari pacchetti software elencati in precedenza.
Per contribuire allo sviluppo attuale di cryo-EM, SPARX è stato rielaborato in una nuova piattaforma autonoma e completa per l'analisi di particelle singole, denominata SPHIRE (SPARX per microscopia elettronica ad alta risoluzione). Al fine di aumentare l'accessibilità della tecnica per i nuovi ricercatori sul campo e per far fronte alla grande quantità di dati prodotti dai moderni microscopi elettronici high-end completamente automatizzati, la pipeline di trasformazione è stata ridisegnata e semplificata introducendo un facile utilizzo Interfaccia utente grafica (GUI) e automatizzazione dei passaggi principali del flusso di lavoro. Inoltre, sono stati aggiunti nuovi algoritmi per consentire la determinazione della struttura veloce, riproducibile e automatizzata dalla crImmagini yo-EM. Inoltre, è stata introdotta la convalida per riproducibilità al fine di evitare artefatti comuni prodotti durante l'analisi di raffinatezza e di eterogeneità.
Anche se il programma è stato ampiamente modificato, le sue caratteristiche fondamentali sono state mantenute: un semplice codice open-source, il moderno design orientato agli oggetti e le interfacce Python per tutte le funzioni di base. Pertanto, non è stato modificato in un programma a scatola nera, che consente agli utenti di studiare e modificare facilmente il codice Python, per creare applicazioni aggiuntive o modificare il flusso di lavoro complessivo. Ciò è particolarmente utile per progetti non standard di cryo-EM.
Qui presentiamo un protocollo per ottenere una mappa di densità vicino alla risoluzione atomica da immagini cryo-EM usando la GUI di SPHIRE. Descrive in dettaglio tutti i passaggi necessari per generare una mappa di densità dai film di rivelatore diretto crudo-EM e non è limitata a qualsiasi tipo di macromolecole particolare. Questo protocollo mira principalmente a guidare newcOmers nel campo attraverso il flusso di lavoro e fornire informazioni importanti su passaggi cruciali dell'elaborazione così come alcune delle possibili trappole e ostacoli. Le caratteristiche più avanzate e lo sfondo teorico che sta alla base di SPHIRE saranno descritti altrove.
Protocollo
NOTA: Per seguire questo protocollo, è necessario installare correttamente SPHIRE su un sistema con un'installazione MPI (attualmente un cluster Linux). Scaricare SPHIRE e il set di dati TcdA1 da http://www.sphire.mpg.de e seguire le istruzioni di installazione: http://sphire.mpg.de/wiki/doku.php?id=howto:download. Questa procedura installa anche EMAN2. SPHIRE attualmente utilizza e2boxer EMAN2 per la selezione di particelle e e2display per la visualizzazione di file di immagine. Per la correzione del movimento ponderata di dose dei filmati micrograph raw, SPHIRE utilizza unblur 14 . Scaricare il programma e seguire le istruzioni di installazione (http://grigoriefflab.janelia.org/unblur, laboratorio Grigorieff). Per la visualizzazione interattiva delle strutture risultanti, il protocollo utilizzerà il programma grafico molecolare Chimera 15 (https://www.cgl.ucsf.edu/chimera/download.html). Un tutorial piacevole per familiarizzare con le funzionalità utilizzate in tutto questo protocollo può essere fouNd qui: https://www.cgl.ucsf.edu/chimera/data/tutorials/eman07/chimera-eman-2007.html. Le istruzioni su come inviare un lavoro parallelo a un cluster dalla GUI di SPHIRE si trovano qui: http://sphire.mpg.de/wiki/doku.php?id=howto:submissions. L'organizzazione complessiva della GUI SPHIRE e le principali fasi del flusso di lavoro eseguite in questo protocollo sono illustrate nella Figura 1 .
1. PROGETTO: Imposta i valori costanti del parametro per questo progetto
- Avviare l'applicazione GUI SPHIRE digitando " sphire &" e il tasto ENTER in una finestra del terminale .
- Adattare i parametri del progetto ( ad es. Dimensioni del pixel, raggio e simmetria delle particelle) nei rispettivi campi di immissione della pagina delle impostazioni del progetto e registrare questi valori per tutti i passaggi successivi del flusso di lavoro.
- Fai clic sull'icona "PROGETTO" in basso a destra del pannello di sinistra per aprire la pagina delle impostazioni del progetto.
- Misurare l'asse più lungo di una particella utilizzando lo strumento di visualizzazione interattiva dell'immagine e2display.py, quindi inserire la metà della dimensione delle particelle in "Raggio di particelle proteine". Se la misura è in Å, tenere presente di convertire l'unità in pixel utilizzando la dimensione del pixel ( ad esempio, se una particella è lunga 200 Å e la dimensione del pixel è di 1,2 Å / pixel, allora l'asse più lungo della particella è 200 / 1.2 = ~ 166 pixel e il raggio 166/2 = 83 pixel).
- Impostare "Dimensione scatola delle particelle" almeno 1,5 volte la dimensione delle particelle. Evitare le dimensioni delle finestre contenenti grandi numeri primari. Ricorda inoltre che l'algoritmo di perfezionamento 3D attualmente richiede una dimensione casella pari a pari numero.
NOTA: La finestra dovrebbe includere un margine da tenere conto degli errori di centraggio iniziali derivanti dalla raccolta (la necessità di spostare particelle all'interno della finestra) e per la regione di fondo sufficiente al di fuori del limite delle particelle per una corretta correzione CTF (particolarmente importante per i grandi valori di defocus 16). - Impostare "la dimensione della finestra CTF" a quella di "Dimensione scatola delle particelle". Per i progetti con dati a basso contrasto, utilizzare una finestra più grande per ottenere stime più snelle degli spettri di potenza.
- Impostare la "simmetria del gruppo di punti" del complesso ( ad es. "C5"). Se la simmetria della struttura bersaglio non è nota, lasciarla a "C1" (asimmetrica). Tuttavia, se una specifica simmetria ad alto ordine viene identificata in seguito durante l'elaborazione, modificare di conseguenza questa impostazione di simmetria e ripetere i passaggi dopo l'allineamento 2D con ISAC.
- Impostare "Massa molecolare della proteina" in kDa (valore sufficiente approssimativo). Premere il pulsante "Registra impostazioni".
2. MOVIE: Allinea i fotogrammi di ogni micrografia di film per correggere il movimento complessivo del campione
- Per tutti i micrografi film, calcolare gli spostamenti x / y per tutti i fotogrammi e quindi creare il valore moMedio corretto (v. Discussione). Si noti che la prima è necessaria solo per la stima CTF perché la stima non funziona bene con le medie ponderate con la dose, mentre quest'ultimo viene utilizzato per tutte le altre fasi della determinazione della struttura.
- Fai clic sull'icona "MOVIE" e poi sul pulsante "Micrograph Movie Alignment". Impostare "Unblur percorso eseguibile" selezionando il file eseguibile. Impostare "Input micrograph path pattern" selezionando un micrograph di film non coordinato e sostituendo la parte variabile dei nomi di file con il carattere jolly "*" ( ad esempio, TcdA1 _ *. Mrc). Specificare il percorso per la "directory di output".
- Impostare "percorso eseguibile di Summovie" selezionando il file eseguibile.
- Impostare "Numero di fotogrammi cinematografici" al numero di fotogrammi in ogni micrografia di un filmato. Impostare la "tensione del microscopio" e "per esposizione al fotogramma" ai valori utilizzati durante la raccolta dei dati. (Per esempSe la dose complessiva è di 60 e - / Å 2 con 20 fotogrammi registrati senza preesposizione, l'esposizione per ogni fotogramma è 60/20 = 3 e - / A 2. Premere il pulsante "Run Command" per allineare Cornici di ogni micrografia del film.
NOTA: Questo crea automaticamente due directory di output che contengono rispettivamente micrografi mediati da movimento misurati in base alla dose e ponderati con dose .
3. CTER: stimare i parametri Defocus e Astigmatismo del CTF
- Stima i parametri CTF (defocus e astigmatismo, gli altri sono impostati dall'utente) per ogni micrografia media media non ponderata.
- Fai clic sull'icona "CTER" e poi sul pulsante "CTF Estimation". Per impostare "Input pattern pattern micrograph", selezionare un micrograph correttamente motion corrected in dose, e quindi sostituire la parte variabile dei nomi di file con il carattere jolly"*". Specificare anche il percorso per la "directory di output".
- Impostare "Contrasto di ampiezza" al valore utilizzato correntemente per il tipo di dati (spessore del ghiaccio è un fattore importante) e la tensione del microscopio in laboratorio ( ad esempio, 10%). I valori tipici sono nel range 7-14 % 17 .
- Impostare "Aberrazione sferica del microscopio (Cs)" e "Tensione microscopica" utilizzata durante la raccolta dei dati.
- Impostare rispettivamente "Frequenza più bassa" e "Frequenza più alta" dell'intervallo di ricerca per il modello CTF a 0,0285 e 0,285 Å -1 (40 - 4 Å). Premere il pulsante "Esegui comando" per stimare i parametri CTF.
NOTA: I parametri CTF verranno automaticamente memorizzati nel file partres.txt nella directory di output specificata. La stima CTF dei 112 micrografi è stata calcolata su 96 nuclei e terminata dopo ~ 3 min sul cluster Linux utilizzato per ottenere i risultati rappresentativi.
4. FINESTRA: estrarre le particelle dai micrografi mediani ponderati in dose
- Selezionare le particelle manualmente o automaticamente da micrografi con e2boxer 6 e creare file di coordinate, ognuno contenente un elenco di coordinate xy delle particelle all'interno del micrografo associato.
- Fai clic sull'icona "WINDOW" e poi sul pulsante "Particle picking". Premere il pulsante "Esegui comando" per avviare e2boxer 6 e raccogliere le particelle di ogni micrografia manualmente o automaticamente 18 (vedere Discussione ). Memorizzare le coordinate finali delle particelle per ogni micrografo nel formato di file EMAN1 (.box). In alternativa, importare i file di coordinate da altri programmi dopo la conversione nel formato EMAN1.
- Creare le pile di particelle estratte dalle immagini delle particelle dai micrografi ponderati in dose (in SPHIRE, la pila di particelle è spessoSemplicemente chiamato "stack").
- Premere il pulsante "Estrazione delle particelle". Specificare "Input micrograph path pattern" selezionando un micrograph correttamente corretto e quindi sostituendo la parte variabile dei nomi di file con il carattere jolly "*" ( es. TcdA1 _ *. Mrc). Allo stesso modo, impostare "Input Coordinate pattern path" selezionando un file di coordinate ( ad esempio, TcdA1 _ *.). Specificare il percorso per la "directory di output".
- Impostare "Source source dei parametri CTF" selezionando il file del parametro CTF (partres.txt prodotto nel passaggio 3.1). Premere il pulsante "Esegui comando".
- Unire gli stack di immagini delle particelle estratte in un singolo.
- Fai clic sul pulsante "Particle Stack". Specificare il percorso di "Output stack image virtuale" utilizzando un formato di percorso del file BDB ( ad esempio "bdb: Particles / stack", dove "Particelle"La directory contenente una directory di dati BDB la cui denominazione è sempre EMAN2DB e "stack" si riferisce ad un particolare stack di immagini all'interno di questo database. Specificare "Input BDB image stack pattern" selezionando una directory che inizia con "mpi_proc" e sostituendo la parte variabile dei nomi di directory con il carattere jolly "*" ( ad esempio, Particles / mpi_proc_000 a Particles / mpi_proc_ *). Premere il pulsante "Esegui comando".
5. ISAC: classificazione delle immagini di particelle in 2D
- Calcola le medie di classe 2D allineando le particelle e raggruppandole secondo il loro aspetto 2D.
NOTA: Le medie 2D risultanti hanno un miglior rapporto segnale / rumore (SNR) rispetto alle singole immagini di particelle e sono quindi utilizzati per valutare visivamente la qualità e l'eterogeneità del set di dati, nonché per risolvere immagini indesiderate dalla pila ( Ad esempio, cristalli di ghiaccio, bordi di carbonio,Aggregati, frammenti, ecc. ) 19 . Inoltre, verranno successivamente utilizzati per determinare un modello 3D iniziale.- Fare clic sull'icona "ISAC" e quindi sul pulsante "ISAC - 2D Clustering". Impostare "Stack immagine in ingresso" selezionando il file di stack contenente le particelle estratte. Specificare il percorso per la "directory di output".
- Utilizza 200 - 1000 per "Immagini per classe". Scegli il numero appropriato considerando il numero previsto di classi 2D (il numero totale di particelle diviso per il numero di immagini per classe). Regolare questo parametro in base alla SNR e alla dimensione del set di dati. Aumenta il numero di membri per classe nel caso in cui il set di dati sia eccessivamente rumoroso. Diminuire il numero quando è disponibile un numero limitato di particelle.
NOTA: a causa di limitazioni di memoria, per i set di dati piuttosto grandi (> 100.000 particelle), suddividere il set di dati completo in sottoinsiemi, eseguire ISAC per ogni sottoinsieme indipendentemente e combinareI risultati alla fine. Le istruzioni dettagliate per questo scenario di elaborazione sono fornite in http://www.sphire.mpg.de/wiki/doku.php. - Controllare la casella di controllo "Phase-flip". Conserva i valori predefiniti per il raggio di riferimento delle particelle e la dimensione dell'immagine delle particelle target per accelerare il processo riducendo automaticamente tutte le immagini di particelle con queste impostazioni. Premere il pulsante "Esegui comando" per calcolare le medie di classe 2D.
NOTA: Questo passaggio è computazionale e il tempo di esecuzione aumenta in modo significativo con il numero di particelle e classi, nonché il raggio di destinazione e le dimensioni dell'immagine. Su un cluster con 96 processi, la classificazione 2D di ~ 10.000 particelle finita dopo circa 90 minuti.
- Visualizzare e controllare visivamente le medie 2D ISAC risultanti per assicurarsi che la loro qualità sia soddisfacente (vedere Discussione).
- Premere il pulsante "Visualizzazione dati" sotto "UTILITIES". Impostato "; "File di input" selezionando il file contenente le medie 2D ISAC (class_averages.hdf prodotto nel passaggio 5.1). Premere il pulsante "Esegui comando" per visualizzare le medie di classe finale riproducibili e convalidate fornite da ISAC.
- Creare una nuova pila includendo solo i membri delle particelle delle medie di classe convalidate.
- Premere il pulsante "Create subset Stack". Impostare "Stack immagine in ingresso" selezionando lo stesso file di stack come nel passaggio 5.1.1. Impostare "Medie ISAC" selezionando le medie 2D ISAC (class_averages.hdf prodotte nel passaggio 5.1). Specificare il percorso per la "directory di output". Premere il pulsante "Esegui comando".
6. VIPER: Calcola un modello iniziale 3D
- Seleziona un piccolo set di medie di classe (≥ 100 immagini) eliminando tutte le medie cattive di classe e le identiche viste della particella (vedere Discussione) e li usa per calcolare un rappresentanteRoducible modello iniziale utilizzando VIPER. Ricorda che la selezione dovrebbe contenere almeno 60-80 medie di alta qualità con ~ 200-500 membri ciascuno.
- Fai clic sull'icona "VIPER" e poi sul pulsante "Visualizzazione dati". Impostare "File di input" selezionando le medie 2D ISAC (class_averages.hdf prodotti nel passaggio 5.1). Premere il pulsante "Esegui comando".
- Premere il pulsante centrale del mouse da qualche parte nella finestra grafica del e2display e attivare il pulsante "DEL" nella finestra pop-up. Eliminare tutte le medie cattive di classe e le identiche viste della particella (vedere Discussione ). Premere il pulsante "Salva" per memorizzare le restanti medie di classe 2D in un nuovo file.
- Dalle medie ISAC selezionate, generare un riferimento iniziale per la successiva raffinazione 3D.
- Fai clic sul pulsante "Modello iniziale 3D - RVIPER". Impostare "Input images stack" selezionando la classe schermataMedie (prodotte nel passaggio 6.1). Specificare il percorso per la "directory di output".
- Assicurati di utilizzare lo stesso valore per "raggio di particella target" come punto 5.1.3 di ISAC. Premere il pulsante "Esegui comando" per generare un modello ab initio riproducibile 3D.
NOTA: Questo passaggio è computazionale e il tempo di esecuzione aumenta significativamente con il numero di medie e di dimensioni delle particelle. Su un cluster con 96 processi, questo lavoro (~ 100 medie delle classi) è terminato dopo ~ 15 minuti.
- Controllare se il modello 3D risultante è ragionevole tenendo conto delle medie di classe e, in aggiunta, dell'integrità strutturale ( cioè non parti scollegate e / o artefatti direzionali). Per visualizzare la mappa, usa il programma Chimera 15 . A questo punto, eseguire un primo confronto con una struttura cristallina di una proteina omologa o un dominio della proteina di interesse se esiste (un esempio è mostrato nella sezione RipreseRisultati ntative).
- Per la successiva raffinatezza 3D, generare un riferimento 3D iniziale e una maschera 3D da un modello 3D ab initio rimuovendo il suo rumore circostante e ricondurandolo in modo che corrisponda alle dimensioni originali del pixel.
- Fai clic sul pulsante "Crea riferimento 3D". Impostare "Volume ingresso" selezionando il modello ab initio 3D (media_volume.hdf prodotto nel passaggio 6.2). Specificare il percorso per la "directory di output".
- Impostare l'origine del rapporto di resezione selezionando il file di rapporto shrink ISAC (README_shrink_ratio.txt prodotto nel passaggio 5.1). Premere il pulsante "Esegui comando".
7. MERIDIEN: Raffinare il Volume 3D iniziale
- Raffinare il volume 3D a partire dal modello 3D iniziale.
- Fai clic sull'icona "MERIDIEN", quindi sul pulsante "Raffinatura 3D". Impostare "Input image stack" e "Initial 3D reference" per seleCting la pila di particelle e il modello ab initio 3D (prodotto rispettivamente in punti 5.3 e 6.4). Specificare il percorso per la "directory di output".
- Impostare "Maschera 3D" selezionando il file maschera 3D (prodotto nel passaggio 6.4). Utilizzare sempre una maschera 3D, ma soprattutto in una fase iniziale di analisi, utilizzare una maschera sferica o una maschera con bordi morbidi appositamente montati sul riferimento per evitare di presentare una polarizzazione di mascheramento errato.
- Controllare la casella di controllo "Applica maschera 2D duro". Impostare "Risoluzione iniziale" su un valore di frequenza di intervallo tra 20 e 25 Å. Tieni presente che un filtro passa-basso con questa frequenza di taglio verrà applicato alla struttura 3D iniziale per ridurre la polarizzazione del modello iniziale.
- Controllare le specifiche del cluster utilizzato per questo processo e impostare "Memoria per nodo" nella memoria disponibile in gigabyte. Premere il pulsante "Esegui comando" per perfezionare il volume 3D a partire dal modello 3D iniziale in modo completamente automatizzato.
NOTA: Questa procedura suddividerà il set di dati in due metà, perfezionare i due modelli in modo indipendente e produrre due volumi grezzi, ciascuno da solo metà delle particelle. È computazionale e il tempo di funzionamento aumenta significativamente con il numero di particelle. Su questo cluster, la perfezionamento del meridiano finì dopo ~ 2,5 h in esecuzione su 192 processi (~ 8.000 particelle, 352 dimensioni della scatola).
- Creare una maschera 3D con bordi morbidi dal volume raffinato per la successiva fase di affilatura.
- Fai clic sul pulsante "Maschera 3D Adaptive". Impostare "Volume ingresso" selezionando uno dei metà volumi non filtrati (prodotto nel passaggio 7.1). Specificare il percorso per "Maschera di uscita".
- Impostare un valore di "soglia di binarizzazione". Usare Chimera per assicurarsi che, a questa particolare soglia, il rumore è chiaramente al di fuori del volume di interesse nella regione del solvente delle mezze mappe non filtrate e tutte le densità della proteina sono ancora cOncavano l'uno all'altro. Premere il pulsante "Esegui comando" per creare la maschera 3D a bordo soft.
NOTA: Il corpo principale della maschera risultante (costituito da voxels i cui valori sono> 0,5) dovrebbe adattarsi strettamente alla struttura delle particelle, ma ancora racchiudere tutte le densità di interesse. La caduta del soft-edge dovrebbe essere di almeno 8-10 pixel di larghezza.
- Unire i due metà volumi non filtrati ottenuti con la raffinatezza 3D. Quindi, affinare il volume fuso regolando lo spettro di potenza sulla base della funzione di trasferimento di modulazione (MTF) del rivelatore, del fattore B stimato e della stima FSC (Fourier Shell Correlation) della risoluzione.
- Selezionare il pulsante "Affilatura". Impostare "Prima metà volume non filtrata" e "Seconda metà volume non filtrata" selezionando i file corrispondenti (vol_0_unfil.hdf e vol_1_unfil.hdf prodotti nel passaggio 7.1). Utilizzare sempre "B-factor enhancement". In genere, mantenere il valore predefinito per eStimare il valore del fattore B dal dataset di input utilizzando l'intervallo tra la frequenza di risoluzione finale e 10 Å. In alternativa, specificare un valore ad-hoc ( ad esempio, -100).
- Mantenere il valore predefinito per la "frequenza del filtro passa-basso" per applicare un filtro basato su FSC.
- Impostare "Maschera fornita dall'utente" selezionando la maschera 3D (prodotto nel passaggio 7.2). Ricorda che la risoluzione riportata verrà determinata utilizzando FSC con questa maschera. Premere il pulsante "Esegui comando" per affinare il raffinato volume 3D.
- Generare la mappa di distribuzione angolare 3D dalle direzioni di proiezione di tutte le particelle stimate con il passo di raffinazione 3D di cui sopra.
- Fai clic sul pulsante "Distribuzione angolare". Impostare "File di parametri di allineamento" selezionando il file (final_params.txt prodotto nel passaggio 7.1) e premere il pulsante "Esegui comando".
- Ispezionare visivamente il modello 3D affilato usando Chimera. Assicurarsi che la struttura sia ragionevole considerando la risoluzione raggiunta (vedere Discussione ).
- Ispezionare visivamente la distribuzione angolare usando Chimera. Verificare che la distribuzione copre circa uniformemente l'intero spazio angolare 3D. Tenga presente che, per le strutture simmetriche, la distribuzione è limitata nell'ambito del triangolo asimmetrico unico.
8. SORT3D: Ordina l'eterogeneità 3D concentrandosi sulle regioni altamente variabili
- Calcola la mappa di variabilità 3D dalla pila di particelle utilizzata nel perfezionamento 3D.
- Fai clic sull'icona "SORT3D" e poi sul pulsante "Stima della variabilità 3D". Impostare "Stack immagine in ingresso" selezionando lo stesso stack di particelle schermato fornito al punto di rifinitura 3D 7.1.1. Specificare il percorso per la "directory di output".
- Conserva il valore predefinito per "Numero di proiezioni".
NOTA: le immagini dal vicino angolareLa cappa verrà utilizzata per stimare la variazione 2D ad ogni angolo di proiezione 3D. Maggiore è il numero, minore è la stima ma più bassa è la risoluzione e gli artefatti rotazionali più pronunciati. - Controllare la casella di controllo "Usa CTF". Premere il pulsante "Esegui comando".
- Utilizza la mappa di variabilità 3D per creare una maschera di messa a fuoco per la fase di clustering 3D qui sotto.
- Selezionare il pulsante "Binario 3D". Impostare "Volume ingresso" selezionando la mappa di variabilità 3D (prodotta in passaggio 8.1). Specificare il percorso del file per "Maschera di uscita".
- Impostare la "soglia di binarizzazione" utilizzando l'output del campo "Livello" nel "Visualizzatore di volume" di Chimera. Premere il pulsante "Esegui comando".
- Ordinare le particelle in gruppi strutturali omogenei concentrandosi sulle regioni strutturalmente altamente variabili.
- Premere il pulsante "Clustering 3D - RSORT3D".Impostare "Input 3D refinement directory" selezionando la directory di output del raffinamento 3D (prodotto nel passaggio 7.1). Specificare il percorso per la "directory di output".
- Impostare la "maschera 3D" selezionando la maschera 3D con bordi morbidi (prodotti nel passaggio 7.2). Impostare la maschera "Focus 3D" selezionando la mappa di variabilità 3D binarizzata (prodotta nel passaggio 8.2).
- Per i grandi set di dati, utilizzare almeno 5.000-10.000 per "Immagini per gruppo". Tenere presente che il programma mantiene sempre il numero di immagini per gruppo inferiore a questa impostazione. Regolare il valore considerando il numero previsto di gruppi 3D (il numero totale di particelle diviso per il valore "Immagini per gruppo"), il set di dati, l'SNR e il grado di eterogeneità. Iniziare con gruppi iniziali di 3-5-10 se è disponibile un numero sufficiente di particelle, a meno che non si preveda un numero elevato di stati strutturali distinti nel set di dati.
- Utilizzare almeno 3.000-5.000 particelle per "dimensione del gruppo più piccola".Si noti che il programma ignorerà i gruppi che contengono un numero inferiore di immagini rispetto all'impostazione di "dimensione del gruppo più piccolo". Premere il pulsante "Esegui comando" per eseguire il clustering 3D.
NOTA: RSORT3D è suddiviso in due fasi. Il primo passo "sort3d" elabora l'eterogeneità 3D. Quindi, ricostruisce i volumi di ciascun gruppo strutturale omogeneo usando i parametri di allineamento 3D determinati dal passo di raffinazione 3D di cui sopra. Il secondo passo "rsort3d" trova i membri riproducibili di ciascun gruppo eseguendo un confronto bidirezionale delle due sequenze di selezione indipendenti. Quindi ricostruisce strutture omogenee utilizzando solo le particelle riproducibili. Su un cluster con 96 core, questo lavoro (~ 8.000 particelle, 352 dimensioni della scatola) è terminato dopo circa 3 h.
- Al termine del programma, utilizzare Chimera per selezionare un gruppo omogeneo 3D. Seleziona la struttura della risoluzione apparente più elevata, tipicamente associata al più poGruppo pulito. Assicurarsi che la struttura selezionata sia visivamente ragionevole tenendo conto delle medie di classe 2D e degli aspetti biologici della proteina di interesse (vedere Discussione ). Se ci sono altri volumi che hanno una struttura quasi identica a risoluzione simile, li consideri come emergenti da un unico gruppo omogeneo 3D.
- Esegui una raffinatezza locale nei confronti dei membri delle particelle del gruppo 3D più omogeneo (con la massima risoluzione).
- Fai clic sul pulsante "Aggiornamento locale di sottoinsieme". Impostare il percorso del file di testo "sottoinsieme" selezionando il file di testo contenente gli ID particelle del gruppo selezionato ( ad esempio Cluster0.txt prodotto nel passaggio 8.3). Impostare la "directory di raffinazione 3D" selezionando la directory di output del precedente raffinamento 3D (prodotto nel passaggio 7.1).
- Impostare "Riavvia l'iterazione" a quella in cui è ottenuta la risoluzione più alta nella correzione 3D precedente. premi ilComando "Esegui comando" per eseguire una raffinatezza locale della popolazione selezionata di particelle.
- Simile al passaggio 7.2, creare una maschera 3D con bordi morbidi da un semestre volume finale non filtrato ricostruito dal raffinamento locale del sottotitolo.
- Simile al passaggio 7.3, unire due metà volumi finali non filtrati derivanti dal raffinamento locale del sottotitolo e affinare il volume fuso. Tuttavia, non filtrare questa volta il volume affilato.
NOTA: Se l'analisi di eterogeneità nel punto 8.4 indichi diversi stati distinti a risoluzione paragonabile, si potrebbe desiderare di affinare in modo indipendente tutti i diversi stati.
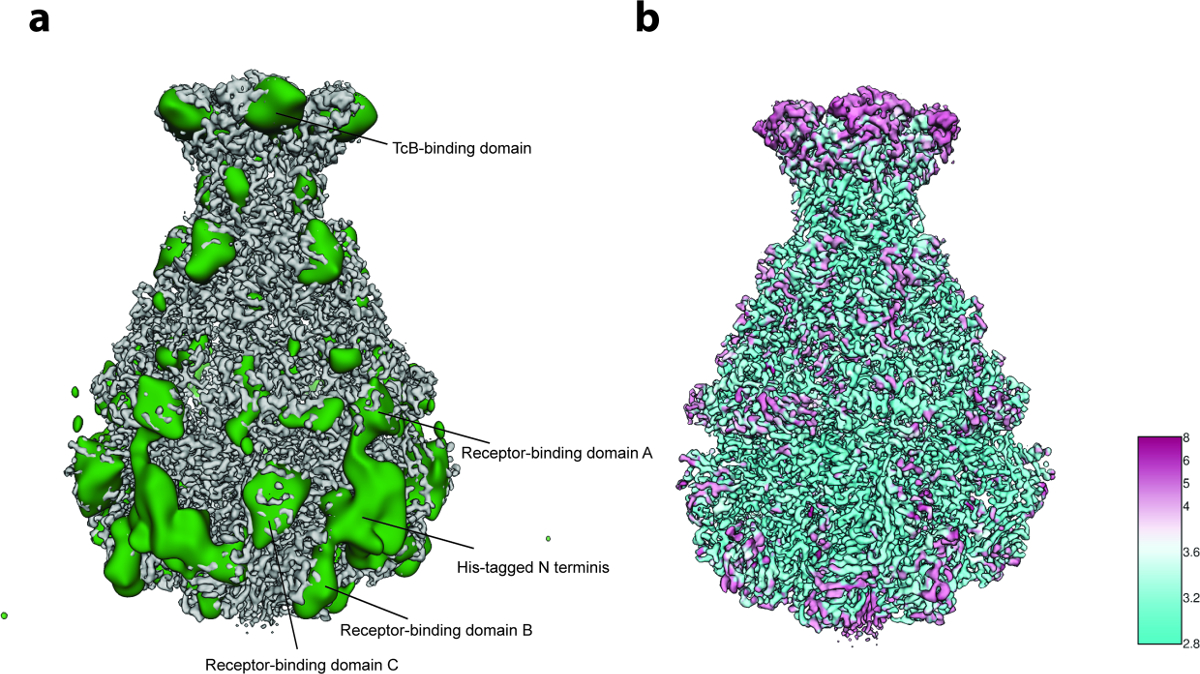
9. LOCALRES: stimare la risoluzione locale del volume finale 3D
- Stima la risoluzione locale del volume 3D ottenuto dalla serie omogenea di particelle.
- Fai clic sull'icona "LOCALRES" e poi sul pulsante "Risoluzione locale". Impostare "primo semestre" e "seconda metà"-volume "selezionando i metà volumi finali non filtrati del raffinamento del sottotitolo locale (prodotto in passaggio 8.5) Impostare la" maschera 3D "selezionando la maschera 3D a bordo morbido prodotta nel passaggio 8.6 Specificare il percorso del file" Volume di uscita " .
- Consente di mantenere il valore predefinito di 7 pixel per la "dimensione della finestra FSC". Ricorda che questa impostazione definisce la dimensione della finestra in cui viene calcolata la correlazione local-real-space; Dimensioni di grandi dimensioni producono mappe di risoluzione più snelle a scapito della risoluzione locale.
- Tenere il valore predefinito 0.5 di "Risoluzione cut-off" per il criterio di risoluzione.
NOTA: per ogni voxel, il programma comunicherà la risoluzione locale come la frequenza in cui il FSC locale scende al di sotto della soglia di risoluzione selezionata. Non è raccomandata una soglia inferiore a 0,5, in quanto i valori di correlazione inferiori hanno elevate incertezze statistiche. Pertanto, la corrispondente risoluzione locale varia fortemente tra voxels. - Per "OveraLl risoluzione ", impostare la risoluzione assoluta stimata nell'affilamento dopo il raffinamento locale del sottotitolo (passo 8.7). Premere il pulsante" Esegui comando "per calcolare la risoluzione locale del volume.
- Applicare il filtro locale 3D al volume che viene affilato dopo la raffinazione del sottotitolo locale utilizzando la mappa di risoluzione locale 3D.
- Fai clic sul pulsante "Filtro locale 3D". Impostare "Volume ingresso" selezionando il volume 3D affilato ma non filtrato (prodotto in passaggio 8.7). Allo stesso modo, impostare "File di risoluzione locale" e "Maschera 3D" (prodotti rispettivamente nei passaggi 9.1 e 8.6). Ricorda che la maschera 3D definisce la regione in cui verrà applicato il filtro locale. Specificare il percorso del file "Volume di uscita". Premere il pulsante "Esegui comando" per applicare il filtro locale 3D.
- Utilizza Chimera per ispezionare visivamente il modello 3D finale e la mappa 3D di risoluzione locale (prodotto in passaggi 9,2 e 9.1, rispcace). Selezionare l'opzione "Colore superficie" per colorare il volume 3D in base alla risoluzione locale. Tieni presente che la distribuzione della risoluzione locale dovrebbe essere liscia (vedere Discussione ).
Risultati
Il protocollo sopra descritto è stato eseguito a partire da 112 film di rivelazione diretta del componente A del complesso Tc (TcdA1) 20 , 21 , 22 di Photorhabdus luminescens . Questo set di dati è stato registrato su un criomicroscopio elettronico corretto da Cs con una pistola ad alta luminosità (XFEG), operata a una tensione di accelerazione di 300 kV. Le immagini sono state acquisite automaticamente con una dose totale di 60 e - / Å -2 ad una dimensione di pixel di 1.14 Å sulla scala del campione. Dopo l'allineamento dei frame del film ( Protocol Step 2 ), le medie corrette del movimento risultante avevano gli anelli Thon isotropici estesi ad alta risoluzione ( Figura 2a ). Le singole particelle erano facilmente visibili e ben separate ( figura 2b ). Le particelle vennero poi raccolte usando lo strumento a dondolo di e2boxerLass = "xref"> 18 ( Protocol Step 4.1 ). In questo caso è stata impostata una soglia appropriata utilizzando l'opzione più selettiva ( Figura 2c ). I 112 micrografi digitali hanno prodotto 9.652 particelle. La maggior parte delle immagini estratte ( Protocol Step 4.2 ) conteneva particelle ben definite e la loro dimensione della scatola era ~ 1,5 volte più grande della dimensione delle particelle, come raccomandato ( figura 2d ). Successivamente, utilizzando ISAC, è stata eseguita un'analisi di eterogeneità 2D (Protocollo Fase 5 ). Ha prodotto 98 medie di classe ( Figura 3a ). Utilizzando queste medie di classe 2D, è stato calcolato un modello ab initio utilizzando VIPER ( Protocol Step 6 ) alla risoluzione intermedia ( Figura 3b ). Questo modello dimostra un ottimo accordo con la struttura di cristallo di TcdA1 precedentemente risolta a 3.9 Å risoluzione 22 ( Figura 3c ). Questo modello ab initio è stato usato come un tem tem Piastra per il raffinamento 3D (MERIDIEN), con una ricostruzione di 3,5 Å (0,143 criterio) (Protocollo Step 7 ) da solo ~ 40.000 unità asimmetriche ( Figura 4 ). Questa mappa di risoluzione quasi atomica è stata ottenuta entro 24 ore, usando fino a 96 CPU per le fasi del flusso di lavoro che beneficiano di corpi multipli.
Per l'analisi della variabilità 3D (protocollo step 8), sono state usate solo 2.000 immagini di particelle per gruppo nel passaggio 8.3.3 ( cioè il processo inizia con 5 gruppi 3D iniziali) e 200 immagini per le dimensioni del gruppo più piccole nel passaggio 8.3.4 dovuto Il piccolo numero di particelle (~ 10.000). L'analisi ha rivelato flessibilità localizzata principalmente nella regione N-terminale del complesso che contiene il suo tag usato per purificazione ( Figura 5a ). Infatti, dodici residui di N-terminale e il suo tag non sono stati risolti nella struttura cristallina pubblicata precedentemente di TcdA1"> 22 e questa regione probabilmente disordinata è rimasta irrisolta nella presente densità di cri-EM, probabilmente dovuta alla sua flessibilità. Diversità aggiuntive è stata rilevata nei domini che legano il recettore e nel dominio vincolante di BC ( Figura 5a ). Una risoluzione soddisfacente della struttura e la dimensione piuttosto piccola del set di dati, questa eterogeneità è stata decisa per essere tollerabile e quindi non è stata eseguita una classificazione 3D concentrata 23. Infine, è stata calcolata la risoluzione locale della mappa di densità finale (protocollo step 9.1, figura 5b ) e la mappatura 3D affilata è stata filtrata localmente ( Protocol step 9.2) . Un volume di questa qualità può essere utilizzato per la costruzione di modelli de novo usando Coot 24 o qualsiasi altro strumento di raffinazione ( Figura 6 ).

Figura 1: elaborazione di immagini utilizzando SPHIRE. ( A ) La GUI del pacchetto software SPHIRE. Una fase specifica del flusso di lavoro può essere attivata selezionando il rispettivo pictogram sul lato sinistro della GUI ("fase di flusso di lavoro"). I comandi e le utility associati a questa fase del flusso di lavoro appariranno nell'area centrale della GUI. Dopo aver selezionato uno dei comandi, i rispettivi parametri vengono visualizzati nell'area destra della GUI. Di norma i parametri avanzati non richiedono la modifica dei valori predefiniti predefiniti. ( B ) Fasi nel flusso di lavoro dell'elaborazione delle immagini a particelle singole usando la GUI di SPHIRE. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Correzione del movimento e parteL'estrazione. ( A , b ) Micrografia digitale tipica di alta qualità, a bassa dose, corretta a drift, registrata a una defocus di 1,7 μm. Si noti che gli anelli Thon isotropici si estendono ad una risoluzione di 2,7 Å nello spettro di potenza (a) e nelle particelle ben notabili nell'immagine 2D ( b ). ( C ) Selezione delle particelle usando e2boxer. I cerchi verdi indicano particelle selezionate. ( D ) particelle grezze tipiche estratte dal micrografo ponderato con dose. Barre di scala = 20 nm. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: Clustering 2D e Generazione di modelli iniziali. ( A ) Galleria di medie di classe 2D, con la maggioranza che rappresenta le rappresentazioni laterali o La particella. Barra di scala = 20 nm. ( B ) La mappa 3D di Ab initio di TcdA1 ottenuta usando RVIPER dalle medie di classe senza riferimento. ( C ) Montaggio del corpo rigido della struttura a cristallo TcdA1 (nastri) (pdb-id 1VW1) nella densità iniziale di criomemma (grigio trasparente). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: Struttura Cryo-EM 3D di TcdA1. ( A , b ) La mappa di densità finale di 3,5 Å di TcdA1 calcolata usando ~ 9.500 immagini di particelle: ( a ) lato e ( b ) vista dall'alto. ( C ) aree rappresentative della densità di cri-EM per un α-elica e un foglio β.Arge.jpg "target =" _ blank "> Fare clic qui per visualizzare una versione più grande di questa figura.

Figura 5: Analisi della variabilità e risoluzione locale. ( A ) Superficie della mappatura cryo-EM TcdA1 affilata (grigio) e della mappa di variabilità (verde). Per una maggiore chiarezza, la mappa della variabilità è stata filtrata a 30 gradi. ( B ) Rendering superficiale della mappatura cryo-EM affilata TcdA1 colorata secondo la risoluzione locale (Å). Si noti l'accordo topologico tra aree di elevata variabilità e bassa risoluzione locale. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 6: Mod. 3DCostruzione di TcdA1 usando Coot. Le regioni rappresentative della densità di criomo-EM e del modello atomico sono mostrate per un'elica α. Il modello atomico è stato costruito de novo utilizzando Coot. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
Discussione
La singola particella cryo-EM ha mostrato un rapido sviluppo negli ultimi anni e ha fornito numerose strutture di risoluzione atomica di complessi macromolecolari di rilevante significato biologico 25 . Per supportare il gran numero di nuovi utenti che stanno attualmente entrando nel campo, abbiamo sviluppato la singola piattaforma di analisi delle immagini di particelle SPHIRE e presentato qui un protocollo di passaggio per l'intero flusso di lavoro, tra cui l'allineamento del film, la raccolta delle particelle, la stima CTF, il modello iniziale Calcolo, analisi di eterogeneità 2D e 3D, raffinatezza 3D ad alta risoluzione e stima e filtraggio di risoluzione locale.
Il protocollo qui descritto è inteso come una breve guida alla determinazione della struttura 3D utilizzando micrografi criomem-EM della proteina di interesse e con l'ausilio di strumenti di calcolo forniti dalla GUI stand-alone di SPHIRE.
La caratteristica principale del flusso di lavoro è quella piùDelle procedure devono essere eseguite solo una volta, poiché si basano sul concetto di convalida per riproducibilità 19 e non richiedono modifiche ai parametri. Questo meccanismo di convalida automatica è un vantaggio principale di SPHIRE rispetto ad altri pacchetti software poiché i risultati tendono ad essere obiettivi e riproducibili e, soprattutto, ottenibili con un costo accettabile di calcolo. La pipeline fornisce inoltre una grande quantità di informazioni diagnostiche per gli utenti esperti per condurre ulteriori convalida e valutazione indipendenti con metodi propri. Tuttavia, un utente novizio che ha almeno la priorità teorica elementare nella biologia strutturale e nella microscopia elettronica dovrebbe essere in grado di ottenere strutture di risoluzione quasi atomiche utilizzando i propri dati e le procedure di convalida automatica.
Tuttavia, ottenere una struttura di risoluzione quasi atomica non è sempre semplice e il risultato dipenderà fortemente dalla qualità del campione e dall'ingressoun. Per le procedure qui presentate, si presume che siano disponibili un numero sufficiente di filmati EM di alta qualità non coordinati, con le loro medie che mostrano particolari particelle omogenee e casuali orientate in modo chiaro. In generale, non ci sono restrizioni riguardo alla simmetria, alla dimensione o alla forma complessiva della molecola, ma un basso peso molecolare può essere un fattore limitante, specialmente quando la proteina ha una forma globulare senza caratteristiche. Di solito, l'analisi di particelle più grandi e ben ordinate con elevata simmetria di punti-gruppo è meno esigente. Pertanto, è fortemente raccomandato agli utenti novizi di eseguire il presente protocollo innanzitutto con un set di dati crio- I dati del tutorial SPHIRE (http: /sphire.mpg.de) o uno dei set di dati inviati da EMPIAR (https://www.ebi.ac.uk/pdbe/emdb/empiar/) con i filmati crudi sono un buon punto di partenza .
Quando si elaborano dati propri, è molto probabile che alcuni set di dati o alcune delle immagini non soddisfino certe qualitàI criteri. In questo contesto, oltre ai controlli automatici di stabilità e riproducibilità, eseguiti dal programma per i principali passaggi del flusso di lavoro, è ancora raccomandato agli utenti di controllare visivamente i risultati in alcuni "punti di controllo" del protocollo, soprattutto se la ricostruzione finale Non è soddisfacente.
La prima ispezione visiva può essere eseguita a livello di micrografia dopo l'allineamento del film (protocollo step 2 ) e la stima CTF (protocollo step 3 ). Le medie corrette a correzione del movimento dovrebbero mostrare particelle singole chiaramente distinguibili e ben separate e gli spettri di potenza dovrebbero mostrare chiaramente elencati isonotici Thon. La frequenza spaziale a cui sono visibili definisce nella maggior parte dei casi la più alta risoluzione a cui la struttura può in linea di principio essere determinata. Esempi di una media corretta di movimento di qualità sufficiente e dello spettro di potenza sono mostrati nella sezione & #34; Risultati rappresentativi ". Le immagini estranee che potrebbero avere un impatto negativo sul risultato finale possono essere rimosse con l'aiuto di strumenti di GUI di valutazione di Drift e CTF di SPHIRE (http://sphire.mpg.de/wiki/doku.php).
Per quanto riguarda lo screening delle particelle, il passo cruciale nella pipeline SPHIRE è la classificazione 2D che utilizza ISAC ( Protocol step 5.2) . Qui l'utente deve controllare che le medie di classe 2D riproducibili identificate automaticamente dal programma adottino una gamma di orientamenti sufficienti a coprire quasi nello spazio angolare. Se la qualità delle medie di classe non è soddisfacente (immagini rumorose e / o sfocate) e / o il numero di medie di classe riproducibili è molto basso, si consideri il miglioramento della qualità di selezione automatica, l'ottimizzazione dell'immagine dei dati o la preparazione del campione. Nella maggior parte dei casi, non è possibile calcolare una ricostruzione affidabile da un set di dati che non presenta buone medie di classe 2D. Esempi di ave di classe 2D di alta qualitàI disturbi sono riportati nella sezione "Risultati rappresentativi".
Almeno 100 medie di classe sono necessarie per ottenere un modello 3D iniziale affidabile utilizzando RVIPER in modo automatizzato (protocollo step 6.1 ). Per questo passo, l'utente deve selezionare le medie con la massima qualità e includere il maggior numero di orientamenti della particella possibile. La qualità del modello iniziale è fondamentale per il successo del successivo raffinamento 3D ad alta risoluzione.
In altri pacchetti software, talvolta viene eseguita una classificazione 3D per eliminare le particelle "cattive" 8 , 9 . Tuttavia, in SPHIRE la maggior parte di queste particelle vengono eliminate automaticamente già durante la classificazione 2D usando ISAC. Pertanto si raccomanda di eseguire il passaggio computazionale intensivo della classificazione 3D solo se la ricostruzione e l'analisi della variabilità 3D indicano l'eterogeneità del set di dati.
Ancora più importante, l'utente deve sempre accuratamente controllare con attenzione i volumi 3D risultanti (protocollo step 9.3 ) e confermare che le caratteristiche della rispettiva densità sono d'accordo con la risoluzione nominale. A una risoluzione di <9 Å, le densità ad asta corrispondenti alle α-eliche diventano visibili. A una risoluzione <4,5 Å, le densità corrispondenti ai fili in fogli β sono normalmente ben separati e gli amminoacidi ingombranti diventano visibili. Una mappa ad alta risoluzione (<3 Å) dovrebbe mostrare chiare catene laterali, permettendo così di costruire un modello atomico accurato.
I risultati ottenuti finora dimostrano che, con l'aiuto dei test di riproducibilità automatica SPHIRE e delle ispezioni visive minime, il presente protocollo è generalmente applicabile a qualsiasi tipo di progetto singolo criomem. I risultati rappresentativi di ogni fase di elaborazione sono mostrati per la ricostruzione della tossina TcdA1 diPhotorhabdus luminescens 21 , che è stato risolto a risoluzione quasi atomica. Le mappe di densità di qualità simile possono essere utilizzate per costruire modelli atomici affidabili dalla traccia di spina dorsale de novo così come la raffinatezza reciproca o reale, e quindi fornire un quadro strutturale solido per la comprensione di meccanismi molecolari complessi.
CODICI DI ACCESSIONE:
Le coordinate per la struttura EM ei film non elaborati sono stati depositati rispettivamente nella banca dati elettronica di microscopia e nell'immagine pilota di microscopia elettronica sotto i numeri di accesso EMD-3645 e EMPIAR-10089.
Divulgazioni
Gli autori dichiarano di non avere interessi finanziari concorrenti.
Riconoscimenti
Ringraziamo D. Roderer per aver fornito micrografi TcdA1. Ringraziamo Steve Ludtke per il suo costante supporto all'infrastruttura EMAN2. Questo lavoro è stato sostenuto da fondi della Società Max Planck (SR) e del Consiglio Europeo nell'ambito del Settimo programma quadro dell'Unione europea (FP7 / 2007-2013) (concedo n. 615984) (a SR) e concessione da parte degli istituti nazionali di Salute R01 GM60635 a PAP).
Materiali
| Name | Company | Catalog Number | Comments |
| SPHIRE | Max Planck Institute of Molecular Physiology- Dortmund and Houston Medical School, Houston, Texas | http://sphire.mpg.de | |
| UCSF Chimera | University of California, San Francisco | http://www.cgl.ucsf.edu/chimera/ | |
| Unblur | Janelia Farm Research Campus, Ashburn | http://grigoriefflab.janelia.org/unblur | |
| Coot | MRC Laboratory of Molecular Biology, Cambridge | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ | |
| EMAN2 | Baylor College of Medicine, Houston | http://blake.bcm.edu/emanwiki/EMAN2 | |
| Computing Cluster with 1824 cores | Max Planck Institute of Molecular Physiology | Linux Cluster with 76 nodes, each with 2 Processors Xeon E5-2670v3 12C 2.30 GHz and 128 Gb RAM | |
| TITAN KRIOS electron microscope | FEI | 300 kV, Cs correction, XFEG | |
| Falcon II direct electron detector | FEI | ||
| EPU (automated data acquisition software) | FEI | https://www.fei.com/software/epu/ |
Riferimenti
- Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nature Methods. 13 (1), 24-27 (2016).
- Liao, M., Cao, E., Julius, D., Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 504 (7478), 107-112 (2013).
- Bai, X. -C., Yan, C., et al. An atomic structure of human γ-secretase. Nature. 525 (7568), 212-217 (2015).
- Ecken, J. V. D., Heissler, S. M., Pathan-Chhatbar, S., Manstein, D. J., Raunser, S. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature. 534 (7609), 724-728 (2016).
- von der Ecken, J., Müller, M., Lehman, W., Manstein, D. J., Penczek, P. A., Raunser, S. Structure of the F-actin-tropomyosin complex. Nature. 519 (7541), 114-117 (2015).
- Tang, G., Peng, L., et al. EMAN2: An extensible image processing suite for electron microscopy. Journal of Structural Biology. 157 (1), 38-46 (2007).
- van Heel, M., Harauz, G., Orlova, E. V., Schmidt, R., Schatz, M. A new generation of the IMAGIC image processing system. Journal of Structural Biology. 116 (1), 17-24 (1996).
- Grigorieff, N. FREALIGN: high-resolution refinement of single particle structures. Journal of Structural Biology. 157 (1), 117-125 (2007).
- Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. Journal of Structural Biology. 180 (3), 519-530 (2012).
- Shaikh, T. R., Gao, H., et al. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nature Protocols. 3 (12), 1941-1974 (2008).
- Hohn, M., Tang, G., et al. SPARX, a new environment for Cryo-EM image processing. Journal of Structural Biology. 157 (1), 47-55 (2007).
- Lander, G. C., Stagg, S. M., et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. Journal of Structural Biology. 166 (1), 95-102 (2009).
- de la Rosa-Trevìn, J. M., Quintana, A., et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. Journal of Structural Biology. 195 (1), 93-99 (2016).
- Grant, T., Grigorieff, N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife. 4, 06980(2015).
- Pettersen, E. F., Goddard, T. D., et al. UCSF Chimera?A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25 (13), 1605-1612 (2004).
- Penczek, P. A., Fang, J., Li, X., Cheng, Y., Loerke, J., Spahn, C. M. T. CTER-rapid estimation of CTF parameters with error assessment. Ultramicroscopy. 140, 9-19 (2014).
- Frank, J. Three-Dimensional Electron Microscopy of Macromolecular Assemblies. , Oxford University Press. (2006).
- Woolford, D., Ericksson, G., et al. SwarmPS: rapid, semi-automated single particle selection software. Journal of Structural Biology. 157 (1), 174-188 (2007).
- Yang, Z., Fang, J., Chittuluru, J., Asturias, F. J., Penczek, P. A. Iterative Stable Alignment and Clustering of 2D Transmission Electron Microscope Images. Structure/Folding and Design. 20 (2), 237-247 (2012).
- Gatsogiannis, C., Merino, F., et al. Membrane insertion of a Tc toxin in near-atomic detail. Nature Publishing Group. , (2016).
- Gatsogiannis, C., Lang, A. E., et al. A syringe-like injection mechanism in Photorhabdus luminescens toxins. Nature. 495 (7442), 520-523 (2013).
- Meusch, D., Gatsogiannis, C., et al. Mechanism of Tc toxin action revealed in molecular detail. Nature. 508 (7494), 61-65 (2014).
- Penczek, P. A., Frank, J., Spahn, C. M. T. A method of focused classification, based on the bootstrap 3D variance analysis, and its application to EF-G-dependent translocation. Journal of Structural Biology. 154 (2), 184-194 (2006).
- Emsley, P., Lohkamp, B., Scott, W. G., Cowtan, K. Features and development of Coot. Acta crystallographica. Section D, Biological crystallography. 66, Pt 4 486-501 (2010).
- Callaway, E. The revolution will not be crystallized: a new method sweeps through structural biology. Nature. 525 (7568), 172-174 (2015).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati