
Method Article
Análisis de una sola partícula de alta resolución a partir de imágenes de crosmicroscopía electrónica usando SPHIRE
En este artículo
Resumen
Este artículo presenta un protocolo para el procesamiento de imágenes cryo-EM utilizando la suite de software SPHIRE. El presente protocolo se puede aplicar para casi todos los proyectos EM de partículas individuales que apuntan a la resolución casi atómica.
Resumen
SPHIRE (SPARX para Microscopía Electrónica de Alta Resolución) es una novedosa suite de software de código abierto y fácil de usar para el procesamiento semiautomático de datos de cryo-microscopía electrónica de partículas individuales (cryo-EM). El protocolo presentado aquí describe detalladamente cómo obtener una estructura de resolución casi atómica a partir de las películas de micrografía crio-EM guiando a los usuarios a través de todos los pasos de la tubería de determinación de estructura de una sola partícula. Estos pasos se controlan desde la nueva interfaz gráfica de usuario SPHIRE y requieren una intervención mínima del usuario. Usando este protocolo, una estructura de 3,5 Å de TcdA1, un complejo de toxina Tc de Photorhabdus luminescens , se derivó de sólo 9500 partículas individuales. Este enfoque simplificado ayudará a los usuarios principiantes sin experiencia extensa de procesamiento e información estructural a priori , para obtener modelos atómicos libres de ruido e imparcial de sus complejos macromoleculares purificados en su estado nativo.
Introducción
Después del desarrollo de la tecnología de detector de electrones directos, el notable progreso en el crio-EM de partícula única está reformando la biología estructural 1 . En comparación con la cristalografía de rayos X, esta técnica requiere sólo una pequeña cantidad de material proteico sin la necesidad de cristalización, al mismo tiempo que plantea menos restricciones con respecto a la pureza de la muestra y todavía permite la determinación de estructuras a una resolución casi atómica. Es importante destacar que diferentes composiciones o estados pueden ahora ser separados computacionalmente y la determinación de la estructura de las diferentes conformaciones puede llevarse a cabo a un nivel de detalle sin precedentes. Recientemente, los mapas de densidad de moléculas desafiantes podrían producirse en resoluciones que permitan la construcción de modelos de novo y, por tanto, una comprensión profunda de su modo de acción 2 , 3 , 4 , 5.
Una amplia variedad de paquetes de software de procesamiento de imágenes están disponibles en la comunidad 3DEM (3D Electron Microscopy) (https://en.wikibooks.org/wiki/Software_Tools_For_Molecular_Microscopy) y la mayoría de ellos están bajo desarrollo continuo. Se ha alcanzado una resolución casi atómica para proteínas que exhiben diversos pesos moleculares y simetrías con varios paquetes de software diferentes, incluyendo EMAN2 6 , IMAGIC 7 , FREALIGN 8 , RELION 9 , SPIDER 10 y SPARX 11 . Cada paquete requiere un nivel diferente de experiencia del usuario y proporciona un nivel diferente de guía del usuario, automatización y extensibilidad. Además, mientras que algunos programas proporcionan entornos completos para facilitar todas las etapas del análisis de imágenes, otros están diseñados para optimizar tareas específicas, como el refinamiento de parámetros de alineación a partir de un rEstructura de eferencia. Más recientemente, se han desarrollado varias plataformas, incluyendo APPION 12 y SCIPION 13 , que proporcionan una única tubería de procesamiento que integra enfoques y protocolos de los diferentes paquetes de software enumerados anteriormente.
Para contribuir al desarrollo actual del cryo-EM, SPARX fue re-desarrollado en una nueva plataforma autónoma y completa para análisis de partículas individuales, llamada SPHIRE (SPARX para Microscopía Electrónica de Alta Resolución). Con el fin de aumentar la accesibilidad de la técnica para los nuevos investigadores en el campo y para hacer frente a la gran cantidad de datos producidos por modernos microscopios electrónicos totalmente automatizados de gama alta, la tubería de procesamiento fue rediseñado y simplificado mediante la introducción de un fácil de usar Interfaz gráfica de usuario (GUI) y la automatización de los pasos principales del flujo de trabajo. Además, se añadieron nuevos algoritmos para permitir la determinación rápida, reproducible y automatizada de la estructura de crYo-EM imágenes. Además, se introdujo la validación por reproducibilidad para evitar artefactos comunes producidos durante el análisis de refinamiento y heterogeneidad.
A pesar de que el programa fue ampliamente modificado, se mantuvieron sus apreciadas características básicas: código abierto de código abierto, el moderno diseño orientado a objetos y interfaces Python para todas las funciones básicas. Por lo tanto, no se cambió en un programa de cuadro negro, lo que permite a los usuarios a estudiar y modificar fácilmente el código Python, para crear aplicaciones adicionales o modificar el flujo de trabajo global. Esto es especialmente útil para proyectos cryo-EM no estándar.
Aquí presentamos un protocolo para la obtención de un mapa de densidad de resolución casi atómica de imágenes cryo-EM utilizando la GUI de SPHIRE. Describe en detalle todos los pasos necesarios para generar un mapa de densidad a partir de películas de detección directa cryo-EM crudas y no está restringido a ningún tipo particular de macromolécula. Este protocolo pretende principalmente guiar a newcOmers en el campo a través del flujo de trabajo y proporcionar información importante sobre los pasos cruciales del procesamiento, así como algunos de los posibles obstáculos y obstáculos. Las características más avanzadas y los antecedentes teóricos detrás de SPHIRE serán descritos en otra parte.
Protocolo
NOTA: Para seguir este protocolo, es necesario instalar correctamente SPHIRE en un sistema con una instalación MPI (actualmente, un clúster de Linux). Descargue SPHIRE y el conjunto de datos TcdA1 de http://www.sphire.mpg.de y siga las instrucciones de instalación: http://sphire.mpg.de/wiki/doku.php?id=howto:download. Este procedimiento también instala EMAN2. SPHIRE utiliza actualmente e2boxer de EMAN2 para la selección de partículas y e2display para mostrar archivos de imagen. Para la corrección de movimiento ponderada de la dosis de las películas de micrografía sin procesar, SPHIRE utiliza unblur 14 . Descargue el programa y siga las instrucciones de instalación (http://grigoriefflab.janelia.org/unblur, Grigorieff lab). Para la visualización interactiva de las estructuras resultantes, el protocolo utilizará el programa de gráficos moleculares Chimera 15 (https://www.cgl.ucsf.edu/chimera/download.html). Un buen tutorial para familiarizarse con las características utilizadas a lo largo de este protocolo puede ser fouNd aquí: https://www.cgl.ucsf.edu/chimera/data/tutorials/eman07/chimera-eman-2007.html. Las instrucciones sobre cómo enviar un trabajo paralelo a un clúster desde la GUI de SPHIRE pueden encontrarse aquí: http://sphire.mpg.de/wiki/doku.php?id=howto:submissions. La organización general de la GUI SPHIRE y las etapas principales del flujo de trabajo realizado a lo largo de este protocolo se ilustran en la Figura 1 .
1. PROYECTO: Establecer valores de parámetros constantes para este proyecto
- Inicie la aplicación SPHIRE GUI escribiendo " sphire &" y la tecla ENTER en una ventana de terminal.
- Ajuste los parámetros de todo el proyecto ( por ejemplo, el tamaño de píxeles, el radio de partículas y la simetría) en los respectivos campos de entrada de la página de configuración del proyecto y luego registre estos valores para todos los pasos siguientes del flujo de trabajo.
- Haga clic en el icono "PROYECTO" en la parte inferior derecha del panel izquierdo para abrir la página de configuración del proyecto.
- Mida el eje más largo de una partícula usando la herramienta de visualización interactiva de imágenes e2display.py, luego ingrese la mitad del tamaño de partícula en "Radio de partículas de proteína". Si la medida es en Å, tenga en cuenta para convertir la unidad a píxeles usando el tamaño de píxel ( por ejemplo, si una partícula tiene 200 Å de longitud y el tamaño de píxel es 1,2 Å / píxel, entonces el eje más largo de la partícula es 200 / 1,2 = ~ 166 píxeles y el radio 166/2 = 83 píxeles).
- Ajuste el "tamaño de la caja de partículas" por lo menos a 1,5 veces del tamaño de partícula. Evite tamaños de ventana que contengan un número primo grande. Además, recuerde que el algoritmo de refinamiento 3D requiere actualmente un tamaño de caja numerada par.
NOTA: La ventana debe incluir un margen para dar cuenta de los errores de centrado iniciales resultantes de la selección (la necesidad de desplazar las partículas dentro de la ventana) y de la suficiente región de fondo fuera del límite de la partícula para una corrección CTF adecuada (especialmente importante para grandes defocus 16). - Ajuste el tamaño de la ventana "CTF" a "Tamaño de la caja de partículas". Para proyectos con datos de bajo contraste, utilice una ventana más grande para obtener estimaciones más suaves de los espectros de potencia.
- Establecer "simetría de grupo de puntos" del complejo ( por ejemplo, "C5"). Si no se conoce la simetría de la estructura objetivo, déjela en "C1" (asimétrica). Sin embargo, si se identifica una simetría de orden superior específica más adelante durante el procesamiento, cambie este ajuste de simetría en consecuencia y repita los pasos después de la alineación 2D con ISAC.
- Defina "Masa molecular de proteína" en kDa (el valor aproximado será suficiente). Pulse el botón "Registrar ajustes".
2. PELÍCULA: Alinee los marcos de cada micrografía para corregir el movimiento global de la muestra
- Para todas las micrografías de películas, calcule los desplazamientos x / y para todos los fotogramas y luego cree su dosis no ponderada y ponderada en la dosis mo(Ver Discusión). Obsérvese que el primero sólo es necesario para la estimación del CTF porque la estimación no funciona bien con los promedios ponderados en función de la dosis mientras que el último se utiliza para todos los otros pasos de la determinación de la estructura.
- Haga clic en el icono "MOVIE" y luego en el botón "Micrograph Movie Alignment". Establezca "Desbloquear la ruta ejecutable" seleccionando el archivo ejecutable. Establezca "Patrón de ruta de micrografía de entrada" seleccionando una micrografía de película sin alinear en bruto y reemplazando la parte variable de los nombres de archivo por el comodín "*" ( por ejemplo, TcdA1 _ *. Mrc). Especifique la ruta para "Directorio de salida".
- Establezca "ruta ejecutable de Summovie" seleccionando el archivo ejecutable.
- Establezca "Número de fotogramas de película" en el número de fotogramas de cada micrografía de película. Ajuste la "Voltaje del microscopio" y "Exposición por fotograma" a los valores utilizados durante la recolección de datos. Por ejemploSi la dosis total es de 60 e - / Å 2 con 20 fotogramas grabados sin exposición previa, la exposición para cada fotograma es 60/20 = 3 e - / A 2. ) Pulse el botón "Run Command" para alinear la Fotogramas de cada micrografía de película.
NOTA: Esto creará automáticamente dos directorios de salida que contengan micrografías de media de corrección de movimiento no ponderadas y ponderadas en función de la dosis, respectivamente.
3. CTER: Estimación de los parámetros de defocus y astigmatismo de la CTF
- Estimación de los parámetros CTF (defocus y astigmatismo, los otros son fijados por el usuario) para cada micrografía media de dosis-no ponderada.
- Haga clic en el icono "CTER" y luego en el botón "Estimación CTF". Para configurar "Patrón de trayectoria de micrografía de entrada", seleccione una micrografía corregida por movimiento no ponderada de dosis, luego reemplace la parte variable de los nombres de archivo por el comodínUnesdoc.unesco.org unesdoc.unesco.org Además, especifique la ruta de acceso para "Directorio de salida".
- Ajuste el "contraste de amplitud" al valor que se utiliza habitualmente para el tipo de datos (el grosor del hielo es un factor importante) y el voltaje del microscopio en el laboratorio ( por ejemplo, 10%). Los valores típicos están en el rango de 7 a 14% 17 .
- Ajuste "Microscopio de aberración esférica (Cs)" y "Microscopio de voltaje" utilizado durante la recopilación de datos.
- Ajuste la frecuencia más baja y la frecuencia más alta del rango de búsqueda para el modelo CTF ajustándose a 0.0285 y 0.285 Å -1 (40-4 Å), respectivamente. Pulse el botón "Ejecutar comando" para estimar los parámetros CTF.
NOTA: Los parámetros CTF se almacenarán automáticamente en el archivo partres.txt del directorio de salida especificado. La estimación CTF de las 112 micrografías se calculó en 96 núcleos y se terminó después de ~ 3 min en el grupo de Linux utilizado para obtener los resultados representativos.
4. VENTANA: extraer partículas de las micrografías medias ponderadas por dosis
- Elija partículas manualmente o automáticamente desde micrografías con e2boxer 6 y cree archivos de coordenadas, cada uno conteniendo una lista de coordenadas xy de partículas dentro de la micrografía asociada.
- Haga clic en el icono "WINDOW" y luego en el botón "Particle Picking". Pulse el botón "Ejecutar comando" para iniciar e2boxer 6 y recoger las partículas de cada micrografía manualmente o automáticamente 18 (ver Discusión ). Almacene las coordenadas finales de cada micrografía en el formato de archivo EMAN1 (.box). También puede importar los archivos de coordenadas de otros programas después de convertirlos al formato EMAN1.
- Crear pilas de partículas mediante la extracción de imágenes de partículas de la micrografía ponderada en dosis (en SPHIRE, la pila de partículas es oftSimplemente llamado "stack").
- Pulse el botón "Extracción de partículas". Especifique "Patrón de ruta de micrografía de entrada" seleccionando una micrografía corregida por movimiento ponderada en función de la dosis y luego reemplazando la parte variable de los nombres de archivo por el comodín "*" ( por ejemplo, TcdA1 _ *. Mrc). De la misma manera, establezca el "Patrón de ruta de coordenadas de entrada" seleccionando un archivo de coordenadas ( por ejemplo, TcdA1 _ *. Especifique la ruta para "Directorio de salida".
- Establezca "fuente de parámetros CTF" seleccionando el archivo de parámetros CTF (partres.txt producido en el paso 3.1). Pulse el botón "Ejecutar comando".
- Combine las pilas de imágenes de partículas extraídas en una sola.
- Haga clic en el botón "Pila de partículas". Especifique la ruta de acceso a "Salida de pila de imágenes virtuales" utilizando un formato de ruta de archivo BDB ( por ejemplo, "bdb: Particles / stack", donde "Particles" apunta a thE que contiene un directorio base de datos BDB cuyo nombre es siempre EMAN2DB y "pila" se refiere a una pila de imagen particular dentro de esta base de datos). Especifique "Input BDB stack pattern" seleccionando un directorio que comience por "mpi_proc" y luego reemplace la parte variable de los nombres de directorio con el comodín "*" ( por ejemplo, Partículas / mpi_proc_000 a Partículas / mpi_proc_ *). Pulse el botón "Ejecutar comando".
5. ISAC: Clasificación de las Imágenes de Partículas en 2D
- Calcular los promedios de la clase 2D alineando las partículas y agrupándolas según su apariencia 2D.
NOTA: Los promedios 2D resultantes tienen una relación señal / ruido (SNR) mejorada en comparación con las imágenes de partículas individuales y, por lo tanto, se utilizan para evaluar visualmente la calidad y heterogeneidad del conjunto de datos, así como para separar las imágenes indeseables de la pila ( Por ejemplo, cristales de hielo, bordes de carbono,Agregados, fragmentos, etc. ) 19 . Además, se utilizarán posteriormente para determinar un modelo 3D inicial.- Haga clic en el icono "ISAC" y luego en el botón "ISAC - 2D Clustering". Establezca "Pila de imágenes de entrada" seleccionando el archivo de pila que contiene las partículas extraídas. Especifique la ruta para "Directorio de salida".
- Utilice 200 - 1000 para "Imágenes por clase". Elija el número apropiado considerando el número esperado de clases 2D (el número total de partículas dividido por el número de imágenes por clase). Ajuste este parámetro dependiendo de la SNR y el tamaño del conjunto de datos. Aumentar el número de miembros por clase en caso de que el conjunto de datos sea excesivamente ruidoso. Disminuya el número cuando haya un número bajo de partículas disponibles.
NOTA: Debido a limitaciones de memoria, para conjuntos de datos más grandes (> 100.000 partículas), divida el conjunto de datos completo en subconjuntos, realice ISAC para cada subconjunto independientemente y combineLos resultados al final. Las instrucciones detalladas para este escenario de procesamiento se proporcionan en http://www.sphire.mpg.de/wiki/doku.php. - Marque la casilla "Phase-flip". Mantenga los valores predeterminados para "Radio de partícula objetivo" y "Tamaño de imagen de partícula objetivo" para acelerar el proceso encogiendo automáticamente todas las imágenes de partículas con estas configuraciones. Pulse el botón "Ejecutar comando" para calcular los promedios de la clase 2D.
NOTA: Este paso es exigente desde el punto de vista computacional y el tiempo de ejecución aumenta significativamente con el número de partículas y clases, así como el radio objetivo y el tamaño de la imagen. En un grupo con 96 procesos, la clasificación 2D de ~ 10.000 partículas terminó después de aproximadamente 90 minutos.
- Exhiba e inspeccione visualmente los promedios ISAC 2D resultantes para asegurarse de que su calidad es satisfactoria (ver Discusión).
- Presione el botón "Display Data" bajo "UTILITIES". Set "; Input files "(archivos de entrada) seleccionando el archivo que contiene los promedios ISAC 2D (class_averages.hdf producido en el paso 5.1) Presione el botón" Run command "para mostrar los promedios de clase reproducibles y validados finales entregados por ISAC.
- Cree una pila nueva que incluya sólo los miembros de partículas de los promedios de clase validados.
- Pulse el botón "Crear subconjunto de pila". Establezca "Pila de imágenes de entrada" seleccionando el mismo archivo de pila que en el paso 5.1.1. Establezca "promedios ISAC" seleccionando los promedios ISAC 2D (class_averages.hdf producidos en el paso 5.1). Especifique la ruta para "Directorio de salida". Pulse el botón "Ejecutar comando".
6. VIPER: Calcule un modelo 3D inicial
- Seleccione un pequeño conjunto de promedios de la clase (≥100 imágenes) eliminando todos los promedios de la clase mala y las vistas idénticas de la partícula (ver Discusión) y utilícelos para calcular un representanteRoduccible modelo inicial utilizando VIPER. Recuerde que la selección debe contener al menos 60-80 promedios de alta calidad con ~ 200-500 miembros cada uno.
- Haga clic en el icono "VIPER" y luego en el botón "Display Data". Establezca "Archivos de entrada" seleccionando los promedios ISAC 2D (class_averages.hdf producidos en el paso 5.1). Pulse el botón "Ejecutar comando".
- Presione el botón central del ratón en algún lugar de la ventana gráfica de la pantalla e2 y active el botón "SUPR" en la ventana emergente. Elimine todos los promedios de la clase mala y las vistas idénticas de la partícula (ver Discusión ). Pulse el botón "Guardar" para almacenar los promedios de clase 2D restantes en un nuevo archivo.
- A partir de los promedios ISAC seleccionados, genere una referencia inicial para el posterior refinamiento 3D.
- Haga clic en el botón "Initial 3D Model - RVIPER". Ajuste "Entrada de pila de imágenes" seleccionando la clase seleccionadaPromedios (producidos en el paso 6.1). Especifique la ruta para "Directorio de salida".
- Asegúrese de utilizar el mismo valor para "Radio de partículas objetivo" como ISAC paso 5.1.3. Pulse el botón "Ejecutar comando" para generar un modelo 3D ab initio reproducible.
NOTA: Este paso es exigente desde el punto de vista computacional y el tiempo de ejecución aumenta significativamente con el número de medias y el tamaño de las partículas. En un clúster con 96 procesos, este trabajo (~ 100 promedios de clase) terminó después de ~ 15 min.
- Compruebe si el modelo 3D resultante es razonable teniendo en cuenta los promedios de clase y además su integridad estructural ( es decir, sin piezas desconectadas y / o artefactos direccionales). Para visualizar el mapa, utilice el programa Quimera 15 . En este punto, realizar una primera comparación con una estructura cristalina de una proteína homóloga o un dominio de la proteína de interés si existe (un ejemplo se muestra en la sección RepreseResultados.
- Para el posterior refinamiento 3D, genere una referencia 3D inicial y una máscara 3D desde un modelo 3D ab initio eliminando su ruido circundante y cambiándolo de escala para que coincida con el tamaño original del píxel.
- Haga clic en el botón "Crear referencia 3D". Ajuste "Volumen de entrada" seleccionando el modelo 3D ab initio (average_volume.hdf producido en el paso 6.2). Especifique la ruta para "Directorio de salida".
- Establezca "Fuente de la proporción de remezcla" seleccionando el archivo de relación de encogimiento ISAC (README_shrink_ratio.txt producido en el paso 5.1). Pulse el botón "Ejecutar comando".
7. MERIDIEN: Refinar el Volumen Inicial 3D
- Refinar el volumen 3D a partir del modelo 3D inicial.
- Haga clic en el icono "MERIDIEN", luego en el botón "3D Refinement". Fije "pila de imagen de entrada" y "referencia 3D inicial" por seleCting la pila de partículas y el modelo ab initio 3D (producido en el paso 5.3 y 6.4, respectivamente). Especifique la ruta para "Directorio de salida".
- Ajuste la "máscara 3D" seleccionando el archivo de máscara 3D (producido en el paso 6.4). Utilice siempre una máscara 3D pero, especialmente en una etapa temprana de análisis, utilice una máscara esférica o una máscara de borde blando suelta ajustada a la referencia para evitar la introducción de sesgo de enmascaramiento incorrecto.
- Marque la casilla de verificación "Aplicar máscara dura 2D". Ajuste "Resolución inicial" a un valor de frecuencia de corte entre 20 - 25 Å. Tenga en cuenta que un filtro de paso bajo con esta frecuencia de corte se aplicará a la estructura 3D inicial para reducir el sesgo inicial del modelo.
- Compruebe las especificaciones del clúster utilizado para este proceso y, a continuación, establezca "Memoria por nodo" en la memoria disponible en gigabytes. Pulse el botón "Ejecutar comando" para refinar el volumen 3D a partir del modelo 3D inicial de una manera totalmente automatizada.
NOTA: Este procedimiento dividirá el conjunto de datos en dos mitades, refinará los dos modelos de forma independiente y generará dos volúmenes sin procesar, cada uno de solo la mitad de las partículas. Es computacionalmente exigente y el tiempo de funcionamiento aumentará significativamente con el número de partículas. En este grupo, el refinamiento del meridiano terminó después de ~ 2.5 h funcionando en 192 procesos (~ 8.000 partículas, tamaño de la caja de 352).
- Cree una máscara 3D con bordes suaves del volumen refinado para el siguiente paso de afilado.
- Haga clic en el botón "Adaptive 3D Mask". Ajuste "Volumen de entrada" seleccionando uno de los medios volúmenes no filtrados (producidos en el paso 7.1). Especifique la ruta de acceso para "Máscara de salida".
- Establezca un valor de "umbral de binarización". Utilice la quimera para asegurarse de que, en este umbral en particular, el ruido está claramente fuera del volumen de interés en la región del disolvente de la mitad de los mapas no filtrados y todas las densidades de la proteína son todavía cConectados entre sí. Pulse el botón "Ejecutar comando" para crear la máscara 3D de borde suave.
NOTA: El cuerpo principal de la máscara resultante (que consiste en voxels cuyos valores son> 0,5) debería ajustarse firmemente a la estructura de la partícula pero aún así encerrar todas las densidades de interés. La caída de borde suave debe ser de al menos 8-10 píxeles de ancho.
- Combinar los dos semi-volúmenes no filtrados obtenidos por el refinamiento 3D. A continuación, agude el volumen combinado ajustando el espectro de potencia basado en la función de transferencia de modulación (MTF) del detector, el factor B estimado y la estimación FSC (correlación de Fourier Shell) de la resolución.
- Seleccione el botón "Afilado". Seleccione "Primer volumen sin filtrar" y "Segundo volumen sin filtrar" seleccionando los archivos correspondientes (vol_0_unfil.hdf y vol_1_unfil.hdf producidos en el paso 7.1). Siempre use "B factor de mejora". Normalmente, mantenga el valor predeterminado para eEstimule el valor del factor B del conjunto de datos de entrada utilizando el rango entre la frecuencia de resolución final y 10 Å. Como alternativa, especifique un valor ad-hoc ( por ejemplo, -100).
- Mantenga el valor predeterminado de "Frecuencia del filtro de paso bajo" para aplicar un filtro basado en FSC.
- Ajuste "Máscara proporcionada por el usuario" seleccionando la máscara 3D (producida en el paso 7.2). Recuerde que la resolución reportada será determinada usando FSC con esta máscara. Pulse el botón "Ejecutar comando" para ajustar el refinado volumen 3D.
- Genere el mapa de distribución angular 3D de las direcciones de proyección de todas las partículas estimadas por el paso de refinamiento 3D anterior.
- Haga clic en el botón "Angular Distribution". Establezca "Archivo de parámetros de alineación" seleccionando el archivo (final_params.txt producido en el paso 7.1) y presione el botón "Ejecutar comando".
- Inspeccione visualmente el modelo 3D afilado usando ChiMera Asegúrese de que la estructura parece razonable teniendo en cuenta la resolución alcanzada (ver Discusión ).
- Inspeccione visualmente la distribución angular usando Quimera. Verifique que la distribución cubre aproximadamente uniformemente todo el espacio angular 3D. Tenga en cuenta que, para las estructuras simétricas, la distribución está restringida dentro del triángulo asimétrico único.
8. SORT3D: Clasificar la heterogeneidad 3D centrándose en las regiones altamente variables
- Calcular el mapa de variabilidad 3D de la pila de partículas utilizada en el refinamiento 3D.
- Haga clic en el icono "SORT3D" y luego en el botón "3D Variability Estimation". Establezca "Pila de imágenes de entrada" seleccionando la misma pila de partículas filtrada dada al paso de refinamiento 3D 7.1.1. Especifique la ruta para "Directorio de salida".
- Mantenga el valor predeterminado para "Número de proyecciones".
NOTA: Las imágenes del vecino angularHood se utilizará para estimar la varianza 2D en cada ángulo de proyección 3D. Cuanto mayor sea el número, menos ruidosa será la estimación, pero más baja será la resolución y los artefactos rotacionales más pronunciados. - Marque la casilla "Usar CTF". Pulse el botón "Ejecutar comando".
- Utilice el mapa de variabilidad 3D para crear una máscara de enfoque para el paso de agrupación 3D a continuación.
- Seleccione el botón "Máscara binaria 3D". Ajuste "Volumen de entrada" seleccionando el mapa de variabilidad 3D (producido en el paso 8.1). Especifique la ruta del archivo para "Máscara de salida".
- Establezca "umbral de binarización" utilizando la salida del campo "Nivel" en el "Visor de Volumen" de Chimera. Pulse el botón "Ejecutar comando".
- Clasificar las imágenes de partículas en grupos estructurales homogéneos centrándose en regiones estructuralmente altamente variables.
- Pulse el botón "3D Clustering - RSORT3D".Establezca "Directorio de refinamiento 3D de entrada" seleccionando el directorio de salida del refinamiento 3D (producido en el paso 7.1). Especifique la ruta para "Directorio de salida".
- Ajuste la "máscara 3D" seleccionando la máscara 3D de borde suave (producida en el paso 7.2). Ajuste "Focus 3D mask" seleccionando el mapa de variabilidad 3D binarizado (producido en el paso 8.2).
- Para conjuntos de datos grandes, utilice al menos 5.000-10.000 para "Imágenes por grupo". Tenga en cuenta que el programa mantiene siempre el número de imágenes por grupo inferior a este valor. Ajuste el valor considerando el número esperado de grupos 3D (el número total de partículas dividido por el valor "Imágenes por grupo"), el conjunto de datos, la SNR y el grado de heterogeneidad. Comience con ~ 5-10 grupos 3D iniciales, si un número suficiente de partículas está disponible, a menos que se espera un número mayor de estados estructurales distintos en el conjunto de datos.
- Utilice al menos 3.000-5.000 partículas para "Tamaño de grupo más pequeño".Tenga en cuenta que el programa ignorará los grupos que comprendan un número de imágenes inferior al de "Tamaño de grupo más pequeño". Pulse el botón "Ejecutar comando" para realizar la agrupación en 3D.
NOTA: RSORT3D se subdivide en dos pasos. El primer paso "sort3d" clasifica la heterogeneidad en 3D. A continuación, reconstruye los volúmenes de cada grupo estructural homogéneo utilizando los parámetros de alineación 3D determinados por el paso de refinamiento 3D anterior. El segundo paso "rsort3d" descubre miembros reproducibles de cada grupo realizando una comparación bidireccional de las dos secuencias de clasificación independientes. Luego, reconstruye estructuras homogéneas utilizando sólo las partículas asignadas de forma reproducible. En un clúster con 96 núcleos, este trabajo (~ 8.000 partículas, tamaño de caja de 352) terminó después de aproximadamente 3 horas.
- Una vez finalizado el programa, utilice Quimera para seleccionar un grupo 3D homogéneo. Seleccione la estructura de la resolución aparente más alta, típicamente asociada con la más poGrupo puloso. Asegúrese de que la estructura seleccionada es visualmente razonable teniendo en cuenta los promedios de la clase 2D y los aspectos biológicos de la proteína de interés (ver Discusión ). Si hay otros volúmenes que tienen una estructura casi idéntica con resolución similar, considérelos como emergentes de un único grupo 3D homogéneo.
- Realice un refinamiento local contra los miembros de partículas del grupo 3D más homogéneo (con la resolución más alta).
- Haga clic en el botón "Refinamiento del subconjunto local". Establezca "Subset text file path" seleccionando el archivo de texto que contiene las ID de partículas del grupo seleccionado ( por ejemplo, Cluster0.txt producido en el paso 8.3). Establezca "Directorio de refinamiento 3D" seleccionando el directorio de salida del refinamiento 3D anterior (producido en el paso 7.1).
- Establezca "Reiniciar la iteración" a la que obtenga la resolución más alta en el refinamiento 3D anterior. presione elBotón "Ejecutar comando" para realizar un refinamiento local de la población de partículas seleccionada.
- De forma similar al paso 7.2, cree una máscara tridimensional de borde suave de un medio volumen final no filtrado reconstruido por el refinamiento del subconjunto local.
- Similar al paso 7.3, se fusionan dos volúmenes finales no filtrados derivados del refinamiento del subconjunto local y se agudiza el volumen combinado. Sin embargo, no filtre el volumen agudo esta vez.
NOTA: Si el análisis de heterogeneidad en el paso 8.4 indica varios estados distintos con resolución comparable, es posible que desee refinar independientemente los diferentes estados.
9. LOCALES: Estimación de la Resolución Local del Volumen Final en 3D
- Estimación de la resolución local del volumen 3D obtenido del conjunto homogéneo de partículas.
- Haga clic en el icono "LOCALRES" y luego en el botón "Local Resolution". Establecer "Primera mitad de volumen" y "Segunda mitad-volumen "seleccionando los volúmenes finales no filtrados del refinamiento del subconjunto local (producido en el paso 8.5) Establezca" Máscara 3D "seleccionando la máscara 3D de borde suave producida en el paso 8.6 Especifique la ruta del archivo para" Volumen de salida " .
- Mantenga el valor predeterminado de 7 píxeles para "Tamaño de ventana FSC". Recuerde que este ajuste define el tamaño de la ventana donde se calcula la correlación local-espacio real; Los tamaños de ventana más grandes producen mapas de resolución más lisos a expensas de la capacidad de resolución local.
- Mantenga el valor predeterminado 0.5 de "Resolución de corte" para el criterio de resolución.
NOTA: Para cada voxel, el programa informará la resolución local como la frecuencia a la que el FSC local cae por debajo del umbral de resolución seleccionado. No se recomienda un umbral inferior a 0,5, ya que los valores de correlación más bajos tienen una alta incertidumbre estadística. Por lo tanto, la resolución local correspondiente variará fuertemente entre voxels. - Para "OveraLl resolution ", establezca la resolución absoluta estimada en el afilado después del refinamiento del subconjunto local (paso 8.7) Pulse el botón" Run command "para calcular la resolución local del volumen.
- Aplique el filtro 3D local al volumen agudizado después del refinamiento del subconjunto local usando el mapa de resolución local en 3D.
- Haga clic en el botón "Filtro local 3D". Ajuste "Volumen de entrada" seleccionando el volumen 3D agudo pero no filtrado (producido en el paso 8.7). De forma similar, establezca "Archivo de resolución local" y "Máscara 3D" (producido en los pasos 9.1 y 8.6, respectivamente). Recuerde que la máscara 3D define la región donde se aplicará el filtrado local. Especifique la ruta del archivo para "Volumen de salida". Pulse el botón "Ejecutar comando" para aplicar el filtro 3D local.
- Utilice Chimera para inspeccionar visualmente el modelo 3D final y el mapa de resolución local 3D (producido en los pasos 9.2 y 9.1, respEctivamente). Seleccione la opción "Color de superficie" para colorear el volumen 3D de acuerdo con la resolución local. Tenga en cuenta que la distribución de la resolución local debe ser suave (ver Discusión ).
Resultados
El protocolo descrito anteriormente se ejecutó a partir de 112 películas detectoras directas del componente A del complejo Tc de Photorhabdus luminescens (TcdA1) 20 , 21 , 22 . Este conjunto de datos se registró en un cryo-microscopio de electrones Cs corregido con una pistola de emisión de campo de alto brillo (XFEG), operado a una tensión de aceleración de 300 kV. Las imágenes se adquirieron automáticamente con una dosis total de 60 e - / Å -2 con un tamaño de píxel de 1,14 Å en la escala de muestra. Después de la alineación de los fotogramas de película (Protocolo Paso 2 ), los promedios corregidos por movimiento resultantes tenían anillos Thon isotrópicos que se extienden a alta resolución ( Figura 2a ). Las partículas individuales eran fácilmente visibles y bien separadas ( Figura 2b ). Las partículas se recogieron entonces utilizando la herramienta swarm de e2boxerLass = "xref"> 18 ( Protocolo Paso 4.1 ). En este caso, se estableció un umbral apropiado utilizando la opción más selectiva ( Figura 2c ). Las 112 micrografías digitales produjeron 9.652 partículas. La mayoría de las imágenes extraídas (Protocolo Paso 4.2 ) contenía partículas bien definidas y su tamaño de caja era ~ 1,5 veces mayor que el tamaño de partícula, como se recomienda ( Figura 2d ). A continuación, utilizando ISAC, se realizó un análisis de heterogeneidad 2D (Protocolo Paso 5 ). Se obtuvieron 98 promedios de clase ( Figura 3a ). Usando estos promedios de clase 2D, un modelo ab initio se calculó usando VIPER (Protocolo Paso 6 ) a resolución intermedia ( Figura 3b ]. Este modelo muestra un excelente acuerdo con la estructura cristalina de TcdA1 previamente resuelto a 3.9 Å de resolución 22 ( Figura 3c ]. Este modelo ab initio se utilizó como Para el refinamiento 3D (MERIDIEN), produciendo una reconstrucción de 3.5 Å (0.143 criterio) a partir de sólo ~ 40.000 unidades asimétricas ( Figura 4 ). Este mapa de resolución casi atómica se obtuvo en 24 h, utilizando hasta 96 CPUs para los pasos del flujo de trabajo que se benefician de múltiples núcleos.
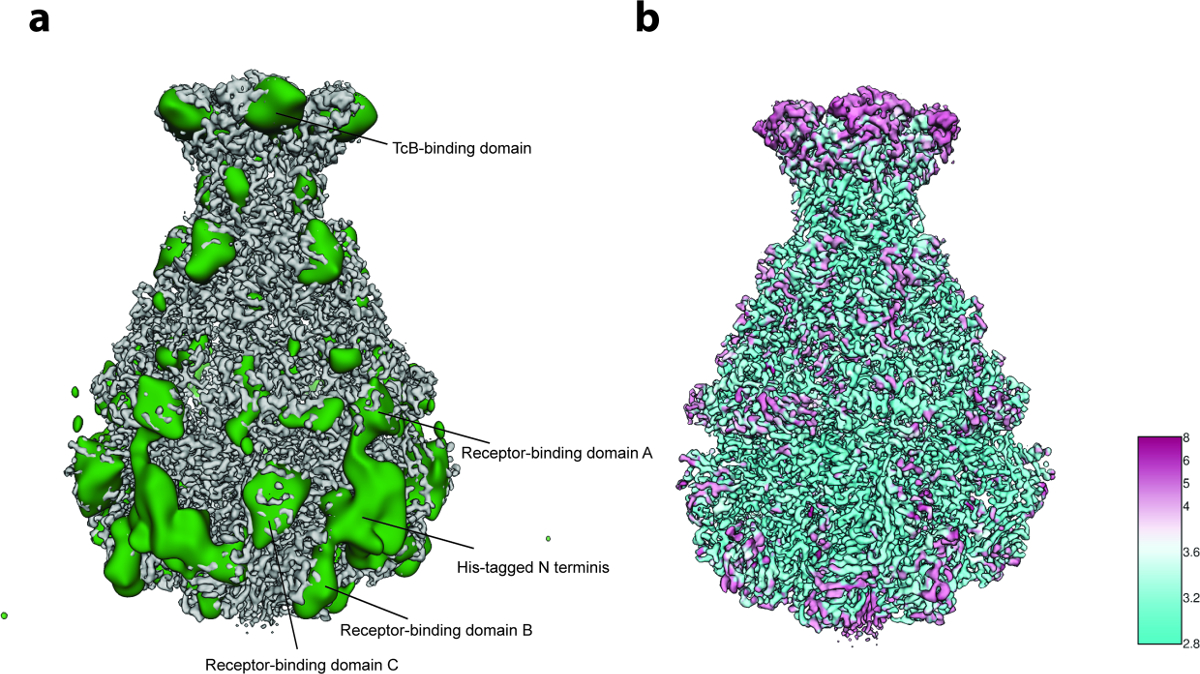
Para el análisis de variabilidad 3D (paso 8 del protocolo ), sólo se utilizaron 2.000 imágenes de partículas por grupo en el paso 8.3.3 ( es decir, el proceso comienza con 5 grupos 3D iniciales) y 200 imágenes para el tamaño de grupo más pequeño en el paso 8.3.4 debido a El pequeño número de partículas (~ 10.000). El análisis reveló flexibilidad localizada principalmente en la región N-terminal del complejo que contiene la etiqueta His utilizados para la purificación ( Figura 5a ]. De hecho, doce residuos N-terminal y la etiqueta de His no se resolvieron en la estructura cristalina publicada anteriormente de TcdA1"22 y esta región más probablemente desordenada no se resolvió en la actual densidad de crio-EM, probablemente debido a su flexibilidad.La variabilidad adicional se detectó en los dominios de unión al receptor y el dominio de unión a BC ( Figura 5a ). La resolución satisfactoria de la estructura y el tamaño bastante pequeño del conjunto de datos, se decidió que esta heterogeneidad era tolerable y por lo tanto no se realizó una clasificación 3D enfocada 23. Finalmente, se calculó la resolución local del mapa de densidad final ( Paso 9.1, 5b ) y el mapa 3D afilado fue filtrado localmente ( paso 9.2 del protocolo), y puede utilizarse un volumen de esta calidad para la construcción de modelos de novo usando Coot 24 o cualquier otra herramienta de refinamiento ( Figura 6 ).

Figura 1: Procesamiento de imágenes con SPHIRE. (A) La GUI del paquete de software SPHIRE. Se puede activar un paso específico del flujo de trabajo seleccionando el pictograma correspondiente en el lado izquierdo de la GUI ("paso de flujo de trabajo"). Los comandos y utilidades asociados con este paso del flujo de trabajo aparecerán en el área central de la GUI. Después de seleccionar uno de los comandos, los parámetros respectivos se muestran en el área derecha de la GUI. Los parámetros avanzados generalmente no requieren la modificación de los valores prefijados por defecto. ( B ) Etapas en el flujo de trabajo del procesamiento de imágenes de una sola partícula usando la GUI SPHIRE. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Corrección de movimiento y partículasExtracción. (A, b ) Micrografía digital típica de baja calidad, de baja dosis, corregida por deriva registrada a un desenfoque de 1,7 μm. Obsérvese que los anillos de Thon isotrópicos se extienden hasta una resolución de 2,7 Å en el espectro de potencia (a) y las partículas bien discernibles en la imagen 2D ( b ). ( C ) Selección de partículas utilizando e2boxer. Los círculos verdes indican las partículas seleccionadas. D ) Partículas brutas típicas extraídas de la micrografía ponderada en función de la dosis. Barras de escala = 20 nm. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Agrupamiento 2D y Generación Inicial del Modelo. (A) Galería de promedios de clase 2D, con la mayoría representando vistas laterales o F la partícula. Barra de escala = 20 nm. ( B ) Mapa Ab initio 3D de TcdA1 obtenido utilizando RVIPER a partir de los promedios de clase libres de referencia. ( C ) Ajuste del cuerpo rígido de la estructura cristalina TcdA1 (cintas) (pdb-id 1VW1) en la densidad cryo-EM inicial (gris transparente). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Estructura 3D Cryo-EM de TcdA1. (A, b ) Mapa de densidad final de 3,5 Å de TcdA1 calculado usando ~ 9,500 imágenes de partículas: ( a ) lado y ( b ) vista superior. ( C ) Áreas representativas de la densidad crio-EM para una hélice α y una hoja β.Arge.jpg "target =" _ blank "> Haga clic aquí para ver una versión más grande de esta figura.

Figura 5: Análisis de Variabilidad y Resolución Local. (A) Superficie del mapa afilado TcdA1 cryo-EM (gris) y el mapa de variabilidad (verde). Para una mayor claridad, el mapa de variabilidad se filtró a paso bajo hasta 30 Å. ( B ) Representación superficial del mapa cryo-EM afilado TcdA1 coloreado de acuerdo con la resolución local (Å). Obsérvese el acuerdo topológico entre áreas de alta variabilidad y baja resolución local. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6: Modificación 3DEl Edificio de TcdA1 utilizando Coot. Las regiones representativas de la densidad cryo-EM y el modelo atómico se muestran para una α-hélice. El modelo atómico fue construido de novo usando Coot. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
El cryo-EM de partícula única ha mostrado un rápido desarrollo en los últimos años y ha proporcionado numerosas estructuras de resolución atómica de complejos macromoleculares de mayor importancia biológica 25 . Con el fin de apoyar el gran número de usuarios principiantes que actualmente están entrando en el campo, desarrollamos la plataforma de análisis de imágenes de una sola partícula SPHIRE y presentamos aquí un protocolo para todo el flujo de trabajo incluyendo la alineación de películas, picking de partículas, CTF estimación, modelo inicial Cálculo, análisis de heterogeneidad 2D y 3D, refinamiento 3D de alta resolución y estimación y filtrado de resolución local.
El protocolo descrito aquí pretende ser una breve guía para la determinación de la estructura en 3D usando micrografías crio-EM de la proteína de interés y con la ayuda de herramientas computacionales proporcionadas por la GUI autónoma de SPHIRE.
La principal característica del flujo de trabajo es que la mayoríaDe los procedimientos sólo se deben ejecutar una vez, ya que se basan en el concepto de validación por reproducibilidad [ 19] y no requieren ajuste de parámetros. Este mecanismo de validación automática es una ventaja principal de SPHIRE sobre otros paquetes de software ya que los resultados tienden a ser objetivos, así como reproducibles y, lo más importante, obtenibles a un coste computacional aceptable. El oleoducto proporciona además una gran cantidad de información de diagnóstico para que los usuarios experimentados realicen una validación y evaluación independiente con métodos propios. Sin embargo, un usuario principiante que tenga al menos un fondo teórico elemental en biología estructural y microscopía electrónica debería ser capaz de obtener estructuras de resolución casi atómicas utilizando datos propios y los procedimientos automatizados de validación.
Sin embargo, la obtención de una estructura de resolución casi atómica no siempre es directa y el resultado dependerá en gran medida de la calidad de la muestra y de la entrada quea. Para los procedimientos presentados aquí, se supone que un número suficiente de alta calidad no alineados primas películas EM están disponibles, con sus promedios que muestran claramente discernible homogénea y aleatoriamente orientada partículas individuales. En general, no hay restricciones en cuanto a simetría, tamaño o forma general de la molécula, pero un bajo peso molecular puede ser un factor limitante, especialmente cuando la proteína tiene una forma globular sin rasgos distintivos. Por lo general, el análisis de partículas grandes y bien ordenadas con una simetría de grupos de puntos altos es menos exigente. Por lo tanto, se recomienda encarecidamente a los usuarios principiantes que ejecuten el protocolo actual primero con un conjunto de datos cryo-EM bien caracterizado. Los datos del tutorial SPHIRE (http: /sphire.mpg.de) o uno de los conjuntos de datos enviados por EMPIAR (https://www.ebi.ac.uk/pdbe/emdb/empiar/) con películas crudas son un buen punto de partida .
Al procesar datos propios, es muy probable que algunos conjuntos de datos o algunas de las imágenes no satisfagan ciertos requisitosCriterios de selección. En este contexto, además de las verificaciones automatizadas de estabilidad y reproducibilidad, realizadas por el programa para los pasos principales del flujo de trabajo, sigue siendo recomendable para los usuarios inspeccionar visualmente los resultados en ciertos "puntos de control" del protocolo, especialmente si la reconstrucción final No es satisfactoria.
La primera inspección visual puede hacerse a nivel de micrografía después de la alineación de la película ( Paso 2 del protocolo) y la estimación CTF ( paso 3 del protocolo). Los promedios corregidos por movimiento resultantes deben mostrar partículas claras y bien separadas y sus espectros de potencia deben mostrar anillos Thon claramente visibles e isotrópicos. La frecuencia espacial a la que son visibles define, en la mayoría de los casos, la resolución más alta a la que en principio se puede determinar la estructura. Ejemplos de un promedio de corrección de movimiento de calidad suficiente y su espectro de potencia se muestran en la sección & #34. Resultados representativos "Las imágenes atípicas que podrían tener un impacto negativo en el resultado final pueden eliminarse con la ayuda de las herramientas GUI de Drift y CTF de SPHIRE (http://sphire.mpg.de/wiki/doku.php).
Con respecto a la detección de partículas, la etapa crucial en la tubería SPHIRE es la clasificación 2D utilizando ISAC ( paso 5.2 del protocolo ) . En este caso, el usuario debe controlar que los promedios de clase 2D reproducibles identificados automáticamente por el programa adopten una gama de orientaciones suficientes para cubrir casi uniformemente el espacio angular. Si la calidad de los promedios de clase no es satisfactoria (imágenes ruidosas y / o borrosas) y / o el número de promedios de clase reproducibles es muy bajo, considere la posibilidad de mejorar la calidad de selección automática, optimizar la formación de imágenes o la preparación de muestras. En la mayoría de los casos, no es posible calcular una reconstrucción fiable a partir de un conjunto de datos que no genere buenos promedios de clase 2D. Ejemplos de alta calidad 2D clase aveEn la sección "Resultados representativos".
Se requieren al menos 100 promedios de clase para obtener un modelo 3D inicial confiable usando RVIPER de una manera automatizada ( Paso 6.1 del protocolo). Para este paso, el usuario debe seleccionar los promedios con la calidad más alta e incluir tantas orientaciones diferentes de la partícula como sea posible. La calidad del modelo inicial es crítica para el éxito del posterior refinamiento 3D de alta resolución.
En otros paquetes de software, la clasificación 3D se realiza a veces para eliminar las partículas "malas" 8 , 9 . Sin embargo, en SPHIRE la mayoría de estas partículas se eliminan automáticamente ya durante la clasificación 2D usando ISAC. Por lo tanto, se recomienda realizar el paso computacional intensivo de clasificación 3D sólo si la reconstrucción y el análisis de variabilidad 3D indican heterogeneidad del conjunto de datos.
Lo más importante es que el usuario debe inspeccionar cuidadosamente los volúmenes 3D resultantes cuidadosamente ( paso 9.3 del protocolo) y confirmar que las características de la densidad respectiva coinciden bien con la resolución nominal. A una resolución <9 Å, las densidades similares a las barras correspondientes a las hélices α se hacen visibles. Con una resolución <4,5 Å, las densidades correspondientes a las hebras de las láminas β suelen estar bien separadas y los voluminosos aminoácidos se vuelven visibles. Un mapa de alta resolución (<3 Å) debe mostrar claramente discernible cadenas laterales, lo que permite la construcción de un modelo atómico exacto.
Los resultados obtenidos hasta la fecha demuestran que, con la ayuda de las pruebas de reproducibilidad automatizadas de SPHIRE y las mínimas inspecciones visuales, el presente protocolo es generalmente aplicable a cualquier tipo de proyecto de crio-EM de partículas individuales. Se muestran resultados representativos de cada etapa de procesamiento para la reconstrucción de la toxina TcdA1 dePhotorhabdus luminescens 21 , que se ha resuelto hasta una resolución casi atómica. Los mapas de densidad de calidad similar se pueden utilizar para construir modelos atómicos confiables mediante el trazado de novo de la columna vertebral así como el refinamiento recíproco o real-espacio, y así proporcionar un marco estructural sólido para la comprensión de mecanismos moleculares complejos.
CÓDIGOS DE ACCESIÓN:
Las coordenadas de la estructura EM y las películas sin procesar se han depositado en el Banco de Datos de Microscopía Electrónica y en el Archivo de Imágenes Piloto de Microscopía Electrónica bajo los números de registro EMD-3645 y EMPIAR-10089, respectivamente.
Divulgaciones
Los autores declaran que no tienen intereses financieros en competencia.
Agradecimientos
Damos las gracias a D. Roderer por brindarnos las micrografías TcdA1. Damos las gracias a Steve Ludtke por su apoyo constante de la infraestructura EMAN2. Este trabajo contó con el apoyo de fondos de la Sociedad Max Planck (SR) y del Consejo Europeo en el marco del Séptimo Programa Marco de la Unión Europea (FP7 / 2007-2013) (subvención nº 615984) y de los Institutos Nacionales de Salud R01 GM60635 a PAP).
Materiales
| Name | Company | Catalog Number | Comments |
| SPHIRE | Max Planck Institute of Molecular Physiology- Dortmund and Houston Medical School, Houston, Texas | http://sphire.mpg.de | |
| UCSF Chimera | University of California, San Francisco | http://www.cgl.ucsf.edu/chimera/ | |
| Unblur | Janelia Farm Research Campus, Ashburn | http://grigoriefflab.janelia.org/unblur | |
| Coot | MRC Laboratory of Molecular Biology, Cambridge | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ | |
| EMAN2 | Baylor College of Medicine, Houston | http://blake.bcm.edu/emanwiki/EMAN2 | |
| Computing Cluster with 1824 cores | Max Planck Institute of Molecular Physiology | Linux Cluster with 76 nodes, each with 2 Processors Xeon E5-2670v3 12C 2.30 GHz and 128 Gb RAM | |
| TITAN KRIOS electron microscope | FEI | 300 kV, Cs correction, XFEG | |
| Falcon II direct electron detector | FEI | ||
| EPU (automated data acquisition software) | FEI | https://www.fei.com/software/epu/ |
Referencias
- Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nature Methods. 13 (1), 24-27 (2016).
- Liao, M., Cao, E., Julius, D., Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 504 (7478), 107-112 (2013).
- Bai, X. -C., Yan, C., et al. An atomic structure of human γ-secretase. Nature. 525 (7568), 212-217 (2015).
- Ecken, J. V. D., Heissler, S. M., Pathan-Chhatbar, S., Manstein, D. J., Raunser, S. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature. 534 (7609), 724-728 (2016).
- von der Ecken, J., Müller, M., Lehman, W., Manstein, D. J., Penczek, P. A., Raunser, S. Structure of the F-actin-tropomyosin complex. Nature. 519 (7541), 114-117 (2015).
- Tang, G., Peng, L., et al. EMAN2: An extensible image processing suite for electron microscopy. Journal of Structural Biology. 157 (1), 38-46 (2007).
- van Heel, M., Harauz, G., Orlova, E. V., Schmidt, R., Schatz, M. A new generation of the IMAGIC image processing system. Journal of Structural Biology. 116 (1), 17-24 (1996).
- Grigorieff, N. FREALIGN: high-resolution refinement of single particle structures. Journal of Structural Biology. 157 (1), 117-125 (2007).
- Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. Journal of Structural Biology. 180 (3), 519-530 (2012).
- Shaikh, T. R., Gao, H., et al. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nature Protocols. 3 (12), 1941-1974 (2008).
- Hohn, M., Tang, G., et al. SPARX, a new environment for Cryo-EM image processing. Journal of Structural Biology. 157 (1), 47-55 (2007).
- Lander, G. C., Stagg, S. M., et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. Journal of Structural Biology. 166 (1), 95-102 (2009).
- de la Rosa-Trevìn, J. M., Quintana, A., et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. Journal of Structural Biology. 195 (1), 93-99 (2016).
- Grant, T., Grigorieff, N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife. 4, 06980(2015).
- Pettersen, E. F., Goddard, T. D., et al. UCSF Chimera?A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25 (13), 1605-1612 (2004).
- Penczek, P. A., Fang, J., Li, X., Cheng, Y., Loerke, J., Spahn, C. M. T. CTER-rapid estimation of CTF parameters with error assessment. Ultramicroscopy. 140, 9-19 (2014).
- Frank, J. Three-Dimensional Electron Microscopy of Macromolecular Assemblies. , Oxford University Press. (2006).
- Woolford, D., Ericksson, G., et al. SwarmPS: rapid, semi-automated single particle selection software. Journal of Structural Biology. 157 (1), 174-188 (2007).
- Yang, Z., Fang, J., Chittuluru, J., Asturias, F. J., Penczek, P. A. Iterative Stable Alignment and Clustering of 2D Transmission Electron Microscope Images. Structure/Folding and Design. 20 (2), 237-247 (2012).
- Gatsogiannis, C., Merino, F., et al. Membrane insertion of a Tc toxin in near-atomic detail. Nature Publishing Group. , (2016).
- Gatsogiannis, C., Lang, A. E., et al. A syringe-like injection mechanism in Photorhabdus luminescens toxins. Nature. 495 (7442), 520-523 (2013).
- Meusch, D., Gatsogiannis, C., et al. Mechanism of Tc toxin action revealed in molecular detail. Nature. 508 (7494), 61-65 (2014).
- Penczek, P. A., Frank, J., Spahn, C. M. T. A method of focused classification, based on the bootstrap 3D variance analysis, and its application to EF-G-dependent translocation. Journal of Structural Biology. 154 (2), 184-194 (2006).
- Emsley, P., Lohkamp, B., Scott, W. G., Cowtan, K. Features and development of Coot. Acta crystallographica. Section D, Biological crystallography. 66, Pt 4 486-501 (2010).
- Callaway, E. The revolution will not be crystallized: a new method sweeps through structural biology. Nature. 525 (7568), 172-174 (2015).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados