
Method Article
Análise de uma única partícula de alta resolução a partir de imagens de crio-microscopia de elétrons usando SPHIRE
Neste Artigo
Resumo
Este artigo apresenta um protocolo para processamento de imagens cryo-EM usando o software suite SPHIRE. O presente protocolo pode ser aplicado para quase todos os projetos de EM de partículas individuais que visam a resolução quase atômica.
Resumo
SPHIRE (SPARX para Microscopia Eletrônica de Alta Resolução) é uma nova suíte de software open-source, user-friendly para o processamento semi-automático de dados de crio-microscopia eletrônica de partícula única (cryo-EM). O protocolo aqui apresentado descreve em pormenor como obter uma estrutura de resolução quase atómica a partir de filmes de micrografia crio-EM guiando os utilizadores através de todas as etapas do pipeline de determinação de estrutura de partícula única. Essas etapas são controladas a partir da nova interface gráfica SPHIRE e requerem uma intervenção mínima do usuário. Utilizando este protocolo, uma estrutura de 3,5 � de TcdA1, um complexo de toxina Tc de Photorhabdus luminescens , foi derivada de apenas 9500 part�ulas isoladas. Essa abordagem simplificada ajudará os usuários novatos sem experiência extensiva de processamento e informações estruturais a priori , para obter modelos atômicos isentos de ruído e isentos de seus complexos macromoleculares purificados em seu estado nativo.
Introdução
Após o desenvolvimento da tecnologia de detector de elétrons diretos, o notável progresso no crio-EM de partículas únicas está atualmente reformulando a biologia estrutural 1 . Comparada com a cristalografia de raios X, esta técnica requer apenas uma pequena quantidade de material proteico sem a necessidade de cristalização, ao mesmo tempo que apresenta menos restrições quanto à pureza da amostra e ainda permite a determinação de estruturas com uma resolução quase atómica. Importantemente, composições ou estados diferentes podem agora ser separados computacionalmente e a determinação da estrutura das diferentes conformações pode ser realizada num nível de detalhe sem precedentes. Recentemente, mapas de densidade de moléculas desafiadoras podem ser produzidos em resoluções que permitam a construção de modelos de novo e, portanto, uma compreensão profunda de seu modo de ação 2 , 3 , 4 , 5.
Uma grande variedade de pacotes de software de processamento de imagem estão disponíveis na comunidade 3DEM (3D Electron Microscopy) (https://en.wikibooks.org/wiki/Software_Tools_For_Molecular_Microscopy) ea maioria deles está em desenvolvimento contínuo. Foi conseguida uma resolução quase atómica para proteínas que exibem vários pesos moleculares e simetrias com vários pacotes de software diferentes, incluindo EMAN2 6 , IMAGIC 7 , FREALIGN 8 , RELION 9 , SPIDER 10 e SPARX 11 . Cada pacote requer um nível diferente de experiência do usuário e fornece um nível diferente de orientação do usuário, automação e extensibilidade. Além disso, enquanto alguns programas fornecem ambientes completos para facilitar todas as etapas de análise de imagem, outros são projetados para otimizar tarefas específicas, como o refinamento de parâmetros de alinhamento a partir de um rEstrutura de referência. Mais recentemente, várias plataformas foram desenvolvidas, incluindo APPION 12 e SCIPION 13 , que fornecem um único pipeline de processamento que integra abordagens e protocolos dos diferentes pacotes de software listados acima.
Para contribuir com o desenvolvimento atual de cryo-EM, a SPARX foi re-desenvolvida em uma nova plataforma autônoma e completa para análise de partículas únicas, chamada SPHIRE (SPARX para Microscopia Eletrônica de Alta Resolução). A fim de aumentar a acessibilidade da técnica para os novos pesquisadores no campo e lidar com a grande quantidade de dados produzidos por modernos modernos microscópios eletrônicos high-end, a linha de processamento foi redesenhado e simplificado através da introdução de um fácil de usar Interface Gráfica do Usuário (GUI) e automatizando as principais etapas do fluxo de trabalho. Além disso, novos algoritmos foram adicionados para permitir a determinação rápida, reproduzível e automatizada da estrutura de crImagens yo-EM. Além disso, a validação por reprodutibilidade foi introduzida para evitar artefatos comuns produzidos durante a análise de refinamento e heterogeneidade.
Embora o programa tenha sido amplamente modificado, suas características principais apreciadas foram mantidas: código open-source direto, o moderno design orientado a objetos e interfaces Python para todas as funções básicas. Assim, ele não foi alterado em um programa de caixa preta, permitindo aos usuários estudar e modificar facilmente o código Python, criar aplicativos adicionais ou modificar o fluxo de trabalho geral. Isso é especialmente útil para projetos não-padrão cryo-EM.
Aqui apresentamos um protocolo para a obtenção de um mapa de densidade de resolução quase atômica a partir de imagens cryo-EM usando a GUI de SPHIRE. Ele descreve detalhadamente todas as etapas necessárias para gerar um mapa de densidade a partir de filmes criados diretos cryo-EM de detector direto e não está restrito a qualquer tipo particular de macromolécula. Este protocolo pretende principalmente orientar newcOmers no campo através do fluxo de trabalho e fornecem informações importantes sobre etapas cruciais do processamento, bem como algumas das possíveis armadilhas e obstáculos. As características mais avançadas e o fundo teórico atrás de SPHIRE serão descritos em outra parte.
Protocolo
NOTA: Para seguir este protocolo, é necessário instalar SPHIRE adequadamente em um sistema com uma instalação MPI (atualmente, um cluster do Linux). Faça o download do SPHIRE e do conjunto de dados TcdA1 em http://www.sphire.mpg.de e siga as instruções de instalação: http://sphire.mpg.de/wiki/doku.php?id=howto:download. Este procedimento também instala EMAN2. O SPHIRE atualmente usa o e2boxer da EMAN2 para seleção de partículas e e2display para exibição de arquivos de imagem. Para correção de movimento com ponderação de dose dos filmes de micrografia em bruto, SPHIRE usa unblur 14 . Faça o download do programa e siga as instruções de instalação (http://grigoriefflab.janelia.org/unblur, Grigorieff lab). Para visualização interativa das estruturas resultantes, o protocolo utilizará o programa de gráficos moleculares Chimera 15 (https://www.cgl.ucsf.edu/chimera/download.html). Um bom tutorial para se familiarizar com os recursos utilizados ao longo deste protocolo pode ser fouNd aqui: https://www.cgl.ucsf.edu/chimera/data/tutorials/eman07/chimera-eman-2007.html. Instruções sobre como enviar um trabalho paralelo para um cluster a partir da GUI do SPHIRE podem ser encontradas aqui: http://sphire.mpg.de/wiki/doku.php?id=howto:submissions. A estrutura geral da GUI SPHIRE e as principais etapas do fluxo de trabalho realizado ao longo deste protocolo são ilustradas na Figura 1 .
1. PROJECT: Definir valores de parâmetro constante para este projecto
- Inicie o aplicativo SPHIRE GUI digitando " sphire &" e a tecla ENTER em uma janela de terminal.
- Ajuste os parâmetros de todo o projeto ( por exemplo, tamanho de pixel, raio de partícula e simetria) nos respectivos campos de entrada da página de configurações do projeto e depois registre esses valores para todas as etapas subseqüentes do fluxo de trabalho.
- Clique no ícone "PROJECT" no canto inferior direito do painel esquerdo para abrir a página de configurações do projeto.
- Meça o eixo mais longo de uma partícula usando a ferramenta de exibição interativa de imagem e2display.py, depois insira a metade do tamanho de partícula para "Raio de partícula de proteína". Se a medida estiver em Å, lembre-se de converter a unidade em pixels usando o tamanho do pixel ( por exemplo, se uma partícula tem 200 Å de comprimento e o tamanho do pixel é de 1,2 Å / pixel, então o eixo mais longo da partícula é 200 / 1,2 = ~ 166 pixels eo raio 166/2 = 83 pixels).
- Definir "Tamanho da caixa de partículas" para pelo menos 1,5 vezes o tamanho de partícula. Evite tamanhos janela que contenham número primo grande. Além disso, lembre-se de que o algoritmo de refinamento 3D requer atualmente um tamanho de caixa numerada par.
NOTA: A janela deve incluir uma margem para contabilizar os erros iniciais de centralização resultantes do picking (a necessidade de deslocar partículas dentro da janela) e para a região de fundo suficiente fora do limite da partícula para uma correção adequada do CTF (especialmente importante para grandes defocus values 16). - Defina "tamanho da janela CTF" para o tamanho da "caixa de partículas". Para projetos com dados de baixo contraste, use uma janela maior para obter estimativas mais suaves dos espectros de potência.
- Defina "simetria de grupo de pontos" do complexo ( por exemplo, "C5"). Se a simetria da estrutura alvo não for conhecida, deixe-a em "C1" (assimétrica). No entanto, se uma simetria de alta ordem específica for identificada mais tarde durante o processamento, altere essa configuração de simetria de acordo e repita as etapas após o alinhamento 2D com ISAC.
- Defina "massa molecular da proteína" em kDa (valor aproximado será suficiente). Pressione o botão "Registrar configurações".
2. FILME: Alinhar os quadros de cada filme Micrografia para corrigir o movimento global da amostra
- Para todas as micrografias de filme, calcule os deslocamentos x / y para todos os quadros e, em seguida, crie a sua dose-unweighted e dose-ponderada moCorrigida pela média (ver Discussão). Note-se que o primeiro é necessário apenas para a estimativa CTF porque a estimativa não funciona bem com as médias ponderadas pela dose, enquanto a última é utilizada para todas as outras etapas da determinação da estrutura.
- Clique no ícone "MOVIE" e depois no botão "Micrograph Movie Alignment". Defina "Unblur executável caminho", selecionando o arquivo executável. Defina "Input micrograph path path" selecionando uma micrografia de filme não alinhado bruto e substituindo a parte variável dos nomes de arquivo pelo curinga "*" ( por exemplo, TcdA1 _ *. Mrc). Especifique o caminho para "Diretório de saída".
- Defina "Summovie caminho executável" selecionando o arquivo executável.
- Defina "Número de fotogramas de filme" para o número de fotogramas em cada micrografia de filme. Defina a "tensão do microscópio" e "exposição por quadro" para os valores utilizados durante a coleta de dados. Por exemploSe a dose total for 60 e - / Å 2 com 20 fotogramas registados sem pré-exposição, a exposição para cada fotograma é 60/20 = 3 e - / A 2. ) Prima o botão "Run Command" para Quadros de cada filme micrografia.
NOTA: Isso criará automaticamente dois diretórios de saída contendo micrografias médias corrigidas de movimento não ponderadas e ponderadas em função da dose , respectivamente.
3. CTER: Estimar os parâmetros de defocus e astigmatismo do CTF
- Estimar os parâmetros CTF (defocus e astigmatismo, os outros são definidos pelo usuário) para cada micrografia dose-média não ponderada.
- Clique no ícone "CTER" e depois no botão "CTF Estimation". Para definir "Input micrograph path path", selecione uma micrografia corrigida por movimento não ponderada e, em seguida, substitua a parte variável dos nomes de arquivo pelo caractere curinga"*". Além disso, especifique o caminho para "Diretório de saída".
- Defina "Amplitude contrast" para o valor rotineiramente utilizado para o tipo de dados (espessura do gelo é um fator importante) e microscópio tensão no laboratório ( por exemplo, 10%). Valores típicos estão na faixa de 7 a 14% 17 .
- Definir "Abertura esférica do microscópio (Cs)" e "Voltagem do microscópio" utilizados durante a coleta de dados.
- Defina "Frequência mais baixa" e "Frequência mais elevada" da gama de pesquisa para o modelo CTF ajustando-se a 0.0285 e 0.285 Å -1 (40-4 Å), respectivamente. Pressione o botão "Executar comando" para estimar os parâmetros CTF.
NOTA: Os parâmetros CTF serão automaticamente armazenados no arquivo partres.txt no diretório de saída especificado. A estimativa CTF das 112 micrografias foi calculada em 96 núcleos e terminada após ~ 3 min no cluster Linux utilizado para obter os resultados representativos.
4. JANELA: Extrair Partículas da Média Ponderada Média Micrografias
- Escolha partículas manualmente ou automaticamente de micrografias com e2boxer 6 e criar arquivos de coordenadas, cada um contendo uma lista de partículas xy-coordenadas dentro da micrografia associada.
- Clique no ícone "WINDOW" e depois no botão "Particle Picking". Pressione o botão "Executar comando" para iniciar e2boxer 6 e escolher as partículas de cada micrografia manualmente ou automaticamente 18 (ver Discussão ). Armazene as coordenadas finais de cada micrografia no formato de arquivo EMAN1 (.box). Como alternativa, importe os arquivos de coordenadas de outros programas após convertê-los para o formato EMAN1.
- Criar pilhas de partículas extraindo imagens de partículas das micrografias ponderadas em dose (em SPHIRE, a pilha de partículas é freqüentementeSimplesmente chamada "pilha").
- Pressione o botão "Extração de Partículas". Especifique "Input micrograph path path" selecionando uma micrografia corrigida de movimento com ponderação de dose e, em seguida, substituindo a parte variável dos nomes de arquivo pelo curinga "*" ( por exemplo, TcdA1 _ *. Mrc). Da mesma forma, defina "Input path path path" selecionando um arquivo de coordenadas ( por exemplo, TcdA1 _ *. Box). Especifique o caminho para "Diretório de saída".
- Defina "fonte de parâmetros CTF" selecionando o arquivo de parâmetro CTF (partres.txt produzido na etapa 3.1). Pressione o botão "Executar comando".
- Combine as pilhas de imagens de partículas extraídas em uma única.
- Clique no botão "Particle Stack". Especifique o caminho para "Saída de pilha de imagens virtuais" usando um formato de caminho de arquivo BDB ( por exemplo, "bdb: Particles / stack", onde "Partículas" aponta para thE que contém um diretório de base de dados BDB cujo nome é sempre EMAN2DB e "stack" refere-se a uma pilha de imagens particular dentro deste banco de dados). Especifique "Input BDB stack pattern" selecionando um diretório começando com "mpi_proc" e depois substituindo a parte variável dos nomes de diretório pelo "*" ( por exemplo, Partículas / mpi_proc_000 para Partículas / mpi_proc_ *). Pressione o botão "Executar comando".
5. ISAC: Classificação de Imagens de Partículas em 2D
- Calcule médias de classe 2D alinhando partículas e agrupando-as de acordo com sua aparência 2D.
NOTA: As médias 2D resultantes têm uma relação sinal-ruído (SNR) melhorada em comparação com as imagens de partículas individuais e são assim usadas para avaliar visualmente a qualidade e heterogeneidade do conjunto de dados, bem como para separar imagens indesejáveis da pilha (Por exemplo, cristais de gelo, bordas de carbono,Agregados, fragmentos, etc. ) 19 . Além disso, serão posteriormente utilizados para determinar um modelo 3D inicial.- Clique no ícone "ISAC" e depois no botão "ISAC - 2D Clustering". Defina "Pilha de imagens de entrada" selecionando o arquivo de pilha que contém as partículas extraídas. Especifique o caminho para "Diretório de saída".
- Use 200 - 1000 para "Imagens por classe". Escolha o número apropriado considerando o número esperado de classes 2D (o número total de partículas dividido pelo número de imagens por classe). Ajuste este parâmetro dependendo da SNR e do tamanho do conjunto de dados. Aumente o número de membros por classe, caso o conjunto de dados seja excessivamente ruidoso. Diminua o número quando um número baixo de partículas está disponível.
NOTA: Devido às limitações de memória, para conjuntos de dados bastante grandes (> 100.000 partículas), divida o conjunto de dados completo em subconjuntos, execute ISAC para cada subconjunto independentemente e combineOs resultados no final. Instruções detalhadas para este cenário de processamento são fornecidas em http://www.sphire.mpg.de/wiki/doku.php. - Marque a caixa de seleção "Phase-flip". Mantenha os valores padrão para "Raio de partícula de destino" e "Tamanho de imagem de partícula de destino" para acelerar o processo encolhendo automaticamente todas as imagens de partículas com essas configurações. Pressione o botão "Executar comando" para calcular as médias de classe 2D.
NOTA: Esta etapa é computacionalmente exigente eo tempo de execução aumenta significativamente com o número de partículas e classes, bem como o raio de destino eo tamanho da imagem. Em um cluster com 96 processos, a classificação 2D de ~ 10.000 partículas terminou após cerca de 90 min.
- Exibir e inspecionar visualmente as médias resultantes do ISAC 2D para se certificar de que sua qualidade é satisfatória (ver Discussão).
- Pressione o botão "Display Data" em "UTILITIES". Definir "; Input files "selecionando o arquivo contendo as médias ISAC 2D (class_averages.hdf produzido na etapa 5.1) Pressione o botão" Executar comando "para exibir as médias de classe reprodutíveis e validadas finais fornecidas pelo ISAC.
- Crie uma nova pilha incluindo apenas os membros de partículas das médias de classes validadas.
- Pressione o botão "Criar subconjunto de pilha". Defina "Pilha de imagens de entrada" selecionando o mesmo arquivo de pilha como na etapa 5.1.1. Defina "médias ISAC" selecionando as médias ISAC 2D (class_averages.hdf produzidas na etapa 5.1). Especifique o caminho para "Diretório de saída". Pressione o botão "Executar comando".
6. VIPER: Calcule um Modelo 3D Inicial
- Selecione um pequeno conjunto de médias de classe (≥100 imagens), excluindo todas as médias de classes ruins e exibições idênticas da partícula (consulte Discussão) e use-as para calcular um representanteModelo roducible inicial usando VIPER. Lembre-se que a seleção deve conter pelo menos 60-80 médias de alta qualidade com ~ 200-500 membros cada.
- Clique no ícone "VIPER" e depois no botão "Display Data". Defina "Arquivos de entrada" selecionando as médias ISAC 2D (class_averages.hdf produzidas na etapa 5.1). Pressione o botão "Executar comando".
- Pressione o botão do meio do mouse em algum lugar na janela de gráficos do e2display, e ative o botão "DEL" na janela pop-up. Exclua todas as médias de classes ruins e exibições idênticas da partícula (ver Discussão ). Pressione o botão "Salvar" para armazenar as médias de classe 2D restantes em um novo arquivo.
- A partir das médias ISAC selecionadas, gere uma referência inicial para o refinamento 3D subseqüente.
- Clique no botão "Initial 3D Model - RVIPER". Defina "Entrada de pilha de imagens" selecionando a classe selecionadaMédias (produzidas no passo 6.1). Especifique o caminho para "Diretório de saída".
- Certifique-se de usar o mesmo valor para "Raio de partícula alvo" como ISAC etapa 5.1.3. Pressione o botão "Executar comando" para gerar um modelo 3D ab initio reprodutível.
NOTA: Esta etapa é computacionalmente exigente eo tempo de execução aumenta significativamente com o número de médias e tamanho das partículas. Em um cluster com 96 processos, este trabalho (~ 100 médias de classe) terminou após ~ 15 min.
- Verifique se o modelo 3D resultante é razoável, tendo em conta as médias de classe e, além disso, a sua integridade estrutural ( ou seja, sem peças desligadas e / ou artefactos direccionais). Para exibir o mapa, use o programa Quimera 15 . Neste ponto, efectuar uma primeira comparação com uma estrutura cristalina de uma proteína homóloga ou um domínio da proteína de interesse se existir (um exemplo é mostrado na secção RepreseResultados.
- Para o refinamento 3D subsequente, gere uma referência 3D inicial e uma máscara 3D a partir de um modelo 3D ab initio, removendo o ruído envolvente e ajustando-o para corresponder ao tamanho original do pixel.
- Clique no botão "Criar Referência 3D". Defina "Volume de entrada" selecionando o modelo 3D ab initio (average_volume.hdf produzido no passo 6.2). Especifique o caminho para "Diretório de saída".
- Defina "Fonte de proporção de resample" selecionando o arquivo de proporção de encolhimento ISAC (README_shrink_ratio.txt produzido na etapa 5.1). Pressione o botão "Executar comando".
7. MERIDIEN: Refinar o volume 3D inicial
- Refinar o volume 3D a partir do modelo 3D inicial.
- Clique no ícone "MERIDIEN", depois no botão "3D Refinement". Definir "Pilha de imagem de entrada" e "Referência 3D inicial" por seleCting a pilha de partículas eo modelo 3D ab initio (produzido no passo 5.3 e 6.4, respectivamente). Especifique o caminho para "Diretório de saída".
- Defina "Máscara 3D" selecionando o arquivo de máscara 3D (produzido na etapa 6.4). Utilize sempre uma máscara 3D, mas, especialmente numa fase inicial de análise, utilize uma máscara esférica ou uma máscara de borda suave folga ajustada à referência para evitar introduzir viés de máscara incorrecta.
- Marque a caixa de seleção "Aplicar máscara 2D rígida". Defina "Starting resolution" para um valor de frequência de corte entre 20 - 25 Å. Tenha em mente que um filtro passa-baixa com esta freqüência de corte será aplicado à estrutura 3D inicial para reduzir o viés inicial do modelo.
- Verifique as especificações do cluster usado para este processo e defina "Memória por nó" para a memória disponível em gigabytes. Pressione o botão "Executar comando" para refinar o volume 3D a partir do modelo 3D inicial de uma maneira totalmente automatizada.
NOTA: Este procedimento irá dividir o conjunto de dados em duas metades, refinar os dois modelos independentemente e produzir dois volumes brutos, cada um de apenas metade das partículas. É computacionalmente exigente e o tempo de execução aumentará significativamente com o número de partículas. Neste cluster, o refinamento meridiano terminou após ~ 2,5 h correndo em 192 processos (~ 8.000 partículas, tamanho de caixa de 352).
- Crie uma máscara 3D com bordas suaves a partir do volume refinado para a etapa de nitidez subseqüente.
- Clique no botão "Adaptive 3D Mask" botão. Defina "Volume de entrada" selecionando um dos semi-volumes não filtrados (produzido no passo 7.1). Especifique o caminho para "Máscara de saída".
- Defina um valor de "Limiar de binarização". Utilizar Quimera para se certificar de que, neste limiar particular, o ruído está claramente fora do volume de interesse na região solvente dos semi-mapas não filtrados e todas as densidades da proteína são ainda cConectados um ao outro. Pressione o botão "Executar comando" para criar a máscara 3D de borda suave.
NOTA: O corpo principal da máscara resultante (que consiste em voxels cujos valores são> 0,5) deve ajustar firmemente a estrutura da partícula mas ainda encerrar todas as densidades de interesse. A queda de borda suave deve ter pelo menos 8-10 pixels de largura.
- Mesclar os dois semi-volumes não filtrados obtidos pelo refinamento 3D. Em seguida, aguçar o volume mesclado ajustando o espectro de potência com base na função de transferência de modulação (MTF) do detector, no fator B estimado e na estimativa FSC (Correlação de Fourier Shell) da resolução.
- Selecione o botão "Sharpening". Defina "Meio volume primeiro não filtrado" e "Meio volume não filtrado segundo" selecionando os arquivos correspondentes (vol_0_unfil.hdf e vol_1_unfil.hdf produzidos no passo 7.1). Sempre use "B factor de melhoria". Normalmente, mantenha o valor padrão para eEstimar o valor do fator B do conjunto de dados de entrada usando o intervalo entre a freqüência de resolução final e 10 Å. Como alternativa, especifique um valor ad-hoc ( por exemplo, -100).
- Mantenha o valor padrão para "Frequência do filtro de passagem baixa" para aplicar um filtro baseado em FSC.
- Defina "Máscara fornecida pelo usuário" selecionando a máscara 3D (produzida na etapa 7.2). Lembre-se que a resolução relatada será determinada usando FSC com essa máscara. Pressione o botão "Executar comando" para refinar o volume 3D refinado.
- Gere o mapa de distribuição angular 3D a partir das direções de projeção de todas as partículas estimadas pela etapa de refinamento 3D acima.
- Clique no botão "Distribuição Angular". Defina "Arquivo de parâmetro de alinhamento" selecionando o arquivo (final_params.txt produzido na etapa 7.1) e pressione o botão "Executar comando".
- Inspecione visualmente o modelo 3D afiado usando ChiMera Certifique-se de que a estrutura parece razoável considerando a resolução alcançada (ver Discussão ).
- Inspecione visualmente a distribuição angular usando Quimera. Verifique se a distribuição cobre aproximadamente uniformemente todo o espaço angular 3D. Lembre-se de que, para estruturas simétricas, a distribuição é restrita dentro do triângulo assimétrico único.
8. SORT3D: Classificar a heterogeneidade em 3D focando as regiões altamente variáveis
- Calcule o mapa de variabilidade 3D da pilha de partículas usada no refinamento 3D.
- Clique no ícone "SORT3D" e depois no botão "3D Variability Estimation". Defina "Pilha de imagens de entrada" selecionando a mesma pilha de partículas filtradas fornecida para a etapa de refinamento 3D 7.1.1. Especifique o caminho para "Diretório de saída".
- Mantenha o valor padrão para "Número de projeções".
NOTA: As imagens do vizinho angularSerá usada para estimar a variância 2D em cada ângulo de projeção 3D. Quanto maior o número, menos ruidosa a estimativa, mas menor a resolução e os artefatos rotacionais são mais pronunciados. - Marque a caixa de seleção "Usar CTF". Pressione o botão "Executar comando".
- Use o mapa de variabilidade 3D para criar uma máscara de foco para a etapa de agrupamento 3D abaixo.
- Selecione o botão "Máscara binária 3D". Defina "Volume de entrada" selecionando o mapa de variabilidade 3D (produzido na etapa 8.1). Especifique o caminho do arquivo para "Máscara de saída".
- Defina "Limiar de binarização" usando a saída do campo "Nível" no "Visualizador de Volume" de Quimera. Pressione o botão "Executar comando".
- Classifique as imagens de partículas em grupos estruturais homogêneos focalizando regiões estruturalmente altamente variáveis.
- Pressione o botão "3D Clustering - RSORT3D".Defina "Diretório de refinamento 3D de entrada" selecionando o diretório de saída do refinamento 3D (produzido na etapa 7.1). Especifique o caminho para "Diretório de saída".
- Defina "Máscara 3D" selecionando a máscara 3D de borda suave (produzida no passo 7.2). Defina "Focus 3D mask" selecionando o mapa de variabilidade 3D binarizado (produzido na etapa 8.2).
- Para grandes conjuntos de dados, use pelo menos 5.000-10.000 para "Imagens por grupo". Tenha em atenção que o programa mantém sempre o número de imagens por grupo inferior a esta definição. Ajuste o valor considerando o número esperado de grupos 3D (o número total de partículas dividido pelo valor "Imagens por grupo"), o conjunto de dados, a SNR eo grau de heterogeneidade. Comece com ~ 5-10 grupos 3D iniciais, se um número suficiente de partículas estiver disponível, a menos que um número maior de estados estruturais distintos no conjunto de dados seja esperado.
- Use pelo menos 3.000-5.000 partículas para "menor tamanho do grupo".Observe que o programa irá desconsiderar os grupos que compõem um menor número de imagens do que a configuração de "menor tamanho do grupo". Pressione o botão "Executar comando" para executar o agrupamento em 3D.
NOTA: RSORT3D é subdividido em duas etapas. O primeiro passo "sort3d" classifica a heterogeneidade 3D. Em seguida, reconstrói os volumes de cada grupo estrutural homogêneo usando os parâmetros de alinhamento 3D determinados pela etapa de refinamento 3D acima. O segundo passo "rsort3d" descobre membros reprodutíveis de cada grupo realizando uma comparação bidireccional das duas execuções de classificação independentes. Em seguida, reconstrói estruturas homogêneas usando apenas as partículas reproduzidas. Em um cluster com 96 núcleos, este trabalho (~ 8.000 partículas, tamanho de caixa de 352) terminou após cerca de 3 h.
- Após o término do programa, use o Chimera para selecionar um grupo 3D homogêneo. Selecione a estrutura da resolução aparente mais alta, normalmente associada à mais poGrupo puloso. Certifique-se de que a estrutura selecionada é visualmente razoável, levando em consideração as médias de classe 2D e aspectos biológicos da proteína de interesse (ver Discussão ). Se houver outros volumes que tenham uma estrutura quase idêntica em resolução semelhante, considere-os como emergindo de um único grupo 3D homogêneo.
- Executar um refinamento local contra os membros da partícula do grupo 3D mais homogêneo (com a resolução mais alta).
- Clique no botão "Refinamento do subconjunto local". Defina "Subset text file path" selecionando o arquivo de texto contendo as IDs de partículas do grupo selecionado ( por exemplo, Cluster0.txt produzido na etapa 8.3). Defina "Diretório de refinamento 3D" selecionando o diretório de saída do refinamento 3D anterior (produzido na etapa 7.1).
- Defina "Reiniciar iteração" para aquele onde a resolução mais alta é alcançada no refinamento 3D anterior. aperte oBotão "Executar comando" para executar um refinamento local da população selecionada de partículas.
- Semelhante ao passo 7.2, crie uma máscara 3D de borda suave de um meio-volume final não filtrado reconstruído pelo refinamento do subconjunto local.
- Semelhante ao passo 7.3, mesclar dois semi-volumes finais não filtrados derivados pelo refinamento subconjunto local e aguçar o volume mesclado. No entanto, não filtre o volume mais nítido desta vez.
NOTA: Se a análise de heterogeneidade na etapa 8.4 indicar vários estados distintos em resolução comparável, pode-se querer refinar todos os diferentes estados independentemente.
9. LOCALRES: Estimativa da resolução local do volume 3D final
- Estimar a resolução local do volume 3D obtido a partir do conjunto homogêneo de partículas.
- Clique no ícone "LOCALRES" e depois no botão "Local Resolution". Definir "Primeira metade do volume" e "Segunda metade"Volume", selecionando os semi-volumes finais não filtrados do refinamento do subconjunto local (produzido na etapa 8.5). Defina "Máscara 3D" selecionando a máscara 3D de borda suave produzida na etapa 8.6 Especifique o caminho do arquivo para "Volume de saída" .
- Mantenha o valor padrão de 7 pixels para "Tamanho da janela FSC". Lembre-se de que essa configuração define o tamanho da janela onde a correlação local-espaço real é calculada; Tamanhos de janela maiores produzem mapas de resolução mais suaves à custa da resolução local.
- Mantenha o valor padrão 0.5 de "Limite de resolução" para o critério de resolução.
NOTA: Para cada voxel, o programa relatará a resolução local como a freqüência na qual o FSC local cai abaixo do limite de resolução selecionado. Um limiar inferior a 0,5 não é recomendado, porque os valores de correlação mais baixos têm alta incerteza estatística. Portanto, a resolução local correspondente variará fortemente entre voxels. - Para "OveraDefinir a resolução absoluta estimada no afiado após o refinamento do subconjunto local (passo 8.7) Pressione o botão "Executar comando" para calcular a resolução local do volume.
- Aplique o filtro local 3D ao volume afiado após o refinamento do subconjunto local usando o mapa de resolução local 3D.
- Clique no botão "Filtro Local 3D". Defina "Volume de entrada" selecionando o volume 3D afiado mas não filtrado (produzido na etapa 8.7). Da mesma forma, defina "arquivo de resolução Local" e "máscara 3D" (produzido no passo 9.1 e 8.6, respectivamente). Lembre-se que a máscara 3D define a região onde a filtragem local será aplicada. Especifique o caminho do arquivo para "Volume de saída". Pressione o botão "Executar comando" para aplicar o filtro 3D local.
- Use a quimera para inspecionar visualmente o modelo 3D final eo mapa de resolução local 3D (produzido na etapa 9,2 e 9.1, respEctivamente). Selecione a opção "Cor de superfície" para colorir o volume 3D de acordo com a resolução local. Tenha em mente que a distribuição da resolução local deve ser lisa (ver Discussão ).
Resultados
O protocolo acima descrito foi executado a partir de 112 filmes de detecção directa do componente A do complexo Tc de Photorhabdus luminescens (TcdA1) 20 , 21 , 22 . Este conjunto de dados foi registado num crio-microscópio de electrões Cs-corrigido com uma pistola de emissão de campo de alto brilho (XFEG), operado a uma tensão de aceleração de 300 kV. As imagens foram adquiridas automaticamente com uma dose total de 60 e - / Å -2 com um tamanho de pixel de 1,14 Å na escala da amostra. Após o alinhamento dos quadros de filme (Protocolo Passo 2 ), as médias corrigidas por movimento resultantes tinham anéis Thon isotrópicos estendendo-se para alta resolução ( Figura 2a ). As partículas individuais eram facilmente visíveis e bem separadas ( Figura 2b ). As partículas foram então colhidas utilizando a ferramenta swarm do e2boxerLass = "xref"> 18 ( Protocolo Passo 4.1 ). Neste caso, foi estabelecido um limiar adequado utilizando a opção mais selectiva ( Figura 2c ). As 112 micrografias digitais produziram 9.652 partículas. A maioria das imagens extraídas (Protocolo Passo 4.2 ) continha partículas bem definidas e seu tamanho de caixa era ~ 1,5 vezes maior que o tamanho de partícula, como recomendado ( Figura 2d ). Em seguida, utilizando ISAC, foi realizada uma análise de heterogeneidade 2D (Protocolo Passo 5 ). Rendia 98 médias de classe ( Figura 3a ). Utilizando estas médias de classes 2D, calculou-se um modelo ab initio utilizando VIPER (Protocolo Passo 6 ) a uma resolução intermédia ( Figura 3b ). Este modelo mostra excelente concordância com a estrutura cristalina de TcdA1 previamente resolvida a 3.9 Å de resolução 22 ( Figura 3c ). Este modelo ab initio foi usado como Para o refinamento 3D (MERIDIEN), proporcionando uma reconstrução de 3,5 Å (0,143) (Protocolo Etapa 7 ) de apenas ~ 40 000 unidades assimétricas ( Figura 4 ). Este mapa de resolução quase atômica foi obtido em 24 h, usando até 96 CPUs para as etapas do fluxo de trabalho que se beneficiam de múltiplos núcleos.
Para a análise de variabilidade 3D (passo 8 do protocolo ), apenas 2.000 imagens de partículas por grupo foram utilizadas no passo 8.3.3 ( ou seja, o processo começa com 5 grupos 3D iniciais) e 200 imagens para o menor tamanho de grupo no passo 8.3.4 devido a O pequeno número de partículas (~ 10.000). A análise revelou flexibilidade localizada principalmente na região N-terminal do complexo que contém a marca His usada para purificação ( Figura 5a ). De facto, doze res�uos N-terminais e a marca His n� foram resolvidos na estrutura cristalina previamente publicada de TcdA1"22 e esta região mais provavelmente desordenada permaneceu não resolvida na actual densidade de crio-EM, provavelmente devido à sua flexibilidade. Foi detectada uma variabilidade adicional nos domínios de ligação ao receptor e no domínio de ligação BC ( Figura 5a ). Resolução satisfatória da estrutura e o tamanho bastante pequeno do conjunto de dados, essa heterogeneidade foi decidida como tolerável e, portanto, não foi realizada uma classificação 3D focalizada 23. Finalmente, a resolução local do mapa de densidade final foi calculada ( Etapa 9.1 do Protocolo , Figura 5b ) e o mapa 3D afiado foi filtrado localmente ( Passo 9.2 do protocolo ) Um volume dessa qualidade pode ser usado para o modelo de novo usando Coot 24 ou qualquer outra ferramenta de refinamento ( Figura 6 ).

Figura 1: Processamento de Imagem usando SPHIRE. (A) A GUI do pacote de software SPHIRE. Uma etapa específica do fluxo de trabalho pode ser ativada selecionando o respectivo pictograma no lado esquerdo da GUI ("etapa do fluxo de trabalho"). Os comandos e utilitários associados a esta etapa do fluxo de trabalho aparecerão na área central da GUI. Depois de selecionar um dos comandos, os respectivos parâmetros são mostrados na área direita da GUI. Os parâmetros avançados normalmente não requerem a modificação dos valores predefinidos. ( B ) Estágios no fluxo de trabalho de processamento de imagem de uma única partícula usando a GUI SPHIRE. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Correção de Movimento e ParticExtracção. (A, b ) Micrografia digital de alta qualidade, de baixa dose, corrigida pela deriva, registrada em um defocus de 1,7 μm. Observe que os anéis de Thon isotrópicos estendem-se a uma resolução de 2,7 Å no espectro de potência (a) e as partículas bem discerníveis na imagem 2D ( b ). ( C ) Seleção de partículas usando e2boxer. Círculos verdes indicam partículas selecionadas. ( D ) Partículas brutas típicas extraídas da micrografia dose-ponderada. Barras de escala = 20 nm. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Agrupamento 2D e Geração Inicial do Modelo. (A) Galeria de médias de classe 2D, com a maioria representando vistas laterais F da partícula. Barra de escala = 20 nm. ( B ) Ab initio mapa 3D de TcdA1 obtido usando RVIPER a partir das médias de classe livre de referência. ( C ) Encaixe do corpo rígido da estrutura cristalina TcdA1 (fitas) (pdb-id 1VW1) na densidade inicial de crio-EM (cinza transparente). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: Estrutura 3D Cryo-EM de TcdA1. (A, b ) Mapa de densidade final de 3,5 Å de TcdA1 calculado usando ~ 9,500 imagens de partículas: ( a ) lado e ( b ) vista de cima. ( C ) �eas representativas da densidade cryo-EM para uma h�ice α e uma p-folha.Arge.jpg "target =" _ blank "> Clique aqui para ver uma versão maior desta figura.

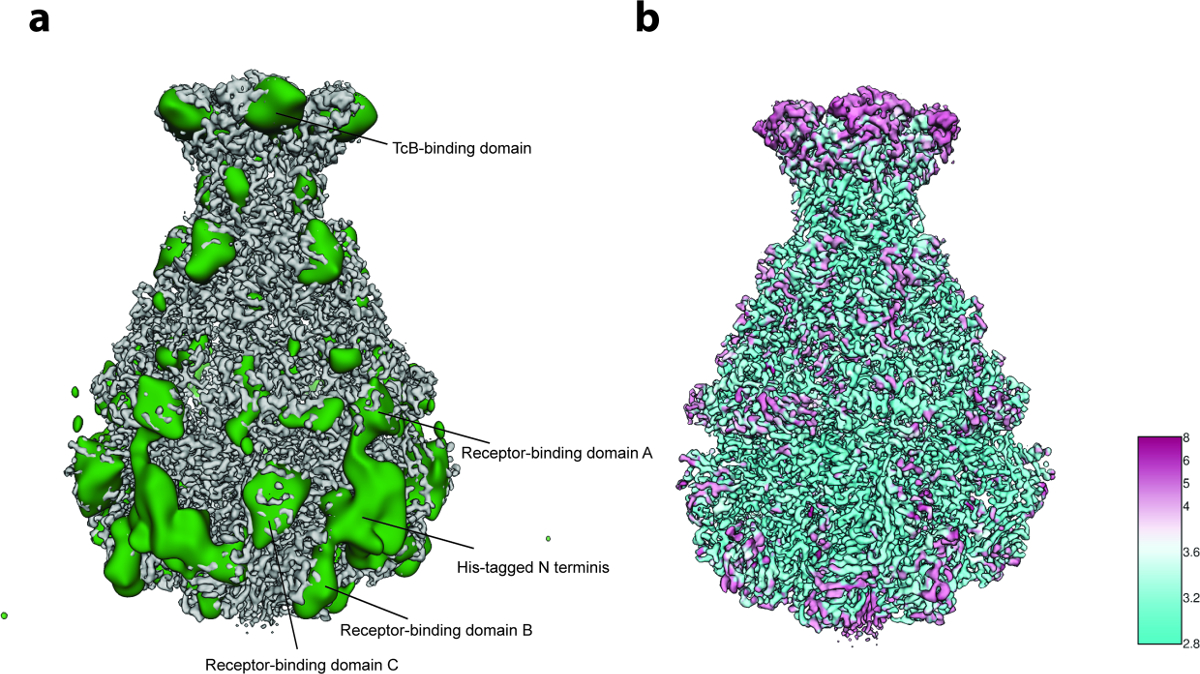
Figura 5: Análise de Variabilidade e Resolução Local. (A) Superfície do mapa cryo-EM TcdA1 afiado (cinza) eo mapa de variabilidade (verde). Para maior clareza, o mapa de variabilidade foi filtrado por passagem baixa até 30 Å. ( B ) Representação superficial do mapa cryo-EM afiado TcdA1 corado de acordo com a resolução local (Å). Observe o acordo topológico entre áreas de alta variabilidade e baixa resolução local. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 6: Modificação 3DConstrução de TcdA1 usando Coot. As regiões representativas da densidade crio-EM e o modelo atómico são mostrados para uma hélice a. O modelo atômico foi construído de novo usando Coot. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Discussão
O crio-EM de partícula única mostrou um rápido desenvolvimento nos últimos anos e forneceu numerosas estruturas de resolução atômica de complexos macromoleculares de maior significância biológica 25 . Para dar suporte ao grande número de usuários principiantes que atualmente estão entrando no campo, desenvolvemos a plataforma de análise de imagens de partículas únicas SPHIRE e apresentamos aqui um protocolo direto para todo o workflow, incluindo alinhamento de filmes, picking de partículas, estimativa CTF, modelo inicial Cálculo, análise de heterogeneidade 2D e 3D, refinamento 3D de alta resolução e estimativa e filtragem de resolução local.
O protocolo aqui descrito pretende ser um breve guia para a determinação da estrutura 3D utilizando micrografias crio-EM da proteína de interesse e com a ajuda de ferramentas computacionais fornecidas pela GUI autónoma de SPHIRE.
A principal característica do fluxo de trabalho é que a maioriaDos procedimentos precisam ser executados apenas uma vez, uma vez que se baseiam no conceito de validação por reprodutibilidade 19 e não necessitam de ajuste de parâmetros. Este mecanismo de validação automática é uma vantagem principal do SPHIRE sobre outros pacotes de software, uma vez que os resultados tendem a ser objectivos, bem como reprodutíveis e, o mais importante, obtidos a um custo computacional aceitável. O pipeline fornece além disso uma riqueza de informações de diagnóstico para usuários experientes para realizar validação e avaliação independente com métodos próprios. No entanto, um usuário iniciante que tenha, pelo menos, conhecimentos teóricos elementares em biologia estrutural e microscopia eletrônica, deve ser capaz de obter estruturas de resolução atômica usando dados próprios e os procedimentos automatizados de validação.
No entanto, a obtenção de uma estrutura de resolução quase atômica nem sempre é direta eo resultado dependerá muito da qualidade da amostra e da entradauma. Para os procedimentos aqui apresentados, presume-se que existe um número suficiente de filmes EM não alinhados de alta qualidade de alta qualidade, com as suas médias mostrando claramente partículas homogéneas e orientadas aleatoriamente. Em geral, não há restrições quanto à simetria, tamanho ou forma geral da molécula, mas um baixo peso molecular pode ser um fator limitante, especialmente quando a proteína tem uma forma globular sem traços. Normalmente, a análise de partículas maiores, bem ordenadas, com simetria de grupos pontuais elevados é menos exigente. Portanto, é altamente recomendável que usuários iniciantes executem o protocolo presente primeiro com um conjunto de dados crio-EM bem caracterizado. Os dados do tutorial SPHIRE (http: /sphire.mpg.de) ou um dos conjuntos de dados enviados pelo EMPIAR (https://www.ebi.ac.uk/pdbe/emdb/empiar/) com filmes crus são um bom ponto de partida .
Ao processar dados próprios, é muito provável que alguns conjuntos de dados ou algumas das imagens não satisfaçam certos requisitosCritérios de elegibilidade. Neste contexto, além das verificações automatizadas de estabilidade e reprodutibilidade, realizadas pelo programa para as principais etapas do fluxo de trabalho, ainda é recomendável aos usuários inspecionar visualmente os resultados em certos "pontos de verificação" do protocolo, especialmente se a reconstrução final Não é satisfatória.
A primeira inspeção visual pode ser feita no nível micrográfico após o alinhamento do filme ( etapa 2 do protocolo) e a estimativa do CTF ( etapa 3 do protocolo). As médias de movimento corrigidas resultantes devem mostrar partículas claramente discerníveis e bem separadas e seus espectros de potência devem mostrar anéis de Thon claramente visíveis e isotrópicos. A frequência espacial para a qual são visíveis define, na maioria dos casos, a maior resolução para a qual a estrutura pode, em princípio, ser determinada em última instância. Exemplos de uma média corrigida de movimento de qualidade suficiente e seu espectro de potência são mostrados na seção & #34. Resultados representativos "As imagens que podem ter um impacto negativo no resultado final podem ser removidas com a ajuda das ferramentas GUI de Drift e CTF da SPHIRE (http://sphire.mpg.de/wiki/doku.php).
No que diz respeito à triagem de partículas, o passo crucial no pipeline SPHIRE é a classificação 2D usando o ISAC ( passo 5.2 do protocolo ) . Aqui, o utilizador deve controlar que as médias de classe 2D reprodutíveis identificadas automaticamente pelo programa adoptem uma gama de orientações suficientes para cobrir quase uniformemente o espaço angular. Se a qualidade das médias de classe não for satisfatória (imagens barulhentas e / ou embaçadas) e / ou o número de médias de classes reprodutíveis for muito baixo, considere melhorar a qualidade de picking automático, otimizando a geração de imagens ou a preparação da amostra. Na maioria dos casos, não é possível calcular uma reconstrução confiável a partir de um conjunto de dados que não gere boas médias de classes 2D. Exemplos de classe 2D de alta qualidade aveSão apresentadas na secção "Resultados representativos".
Pelo menos 100 médias de classe são necessárias para obter um modelo 3D confiável inicial usando RVIPER de forma automatizada (Protocolo passo 6.1 ). Para esta etapa, o usuário deve selecionar as médias com a mais alta qualidade e incluir tantas orientações diferentes quanto possível da partícula. A qualidade do modelo inicial é fundamental para o sucesso do subsequente refinamento 3D de alta resolução.
Em outros pacotes de software, a classificação 3D às vezes é realizada para remover partículas "ruins" 8 , 9 . No entanto, em SPHIRE a maioria dessas partículas são automaticamente eliminadas já durante a classificação 2D usando ISAC. Assim, recomenda-se executar o passo computacionalmente intensivo da ordenação 3D somente se a reconstrução ea análise de variabilidade 3D indicarem heterogeneidade do conjunto de dados.
O mais importante é que o usuário deve sempre inspecionar cuidadosamente os volumes 3D resultantes cuidadosamente ( etapa 9.3 do protocolo) e confirmar que as características da respectiva densidade concordam bem com a resolução nominal. Com uma resolução <9 Å, as densidades semelhantes a vara correspondentes às hélices-a tornam-se visíveis. Com uma resolução <4,5 Å, as densidades correspondentes às cadeias nas folhas ß são normalmente bem separadas e os aminoácidos volumosos tornam-se visíveis. Um mapa de alta resolução (<3 Å) deve mostrar claramente discernível cadeias laterais, permitindo assim a construção de um modelo atômico preciso.
Os resultados obtidos até à data demonstram que, com a ajuda dos testes automatizados de reprodutibilidade do SPHIRE e de inspeções visuais mínimas, o presente protocolo é geralmente aplicável a qualquer tipo de projeto de crio-EM de partículas únicas. Resultados representativos de cada passo de processamento são mostrados para a reconstrução da toxina TcdA1 dePhotorhabdus luminescens 21 , que foi resolvido para resolução quase atômica. Mapas de densidade de qualidade semelhante podem ser usados para construir modelos atômicos confiáveis por traçado de novo backbone, bem como recíproco ou real-espaço refinamento e, assim, fornecer uma estrutura sólida estrutura para a compreensão dos mecanismos moleculares complexos.
CÓDIGOS DE ACESSO:
As coordenadas da estrutura EM e os filmes não processados foram depositados no Banco de Dados de Microscopia Electrónica e no Arquivo de Imagem Piloto de Microscopia Electrónica com os números de acesso EMD-3645 e EMPIAR-10089, respectivamente.
Divulgações
Os autores declaram que não têm interesses financeiros concorrentes.
Agradecimentos
Agradecemos a D. Roderer por nos fornecer TcdA1 micrografias. Agradecemos a Steve Ludtke pelo seu apoio contínuo à infra-estrutura EMAN2. Este trabalho foi apoiado por fundos da Sociedade Max Planck (SR) e do Conselho Europeu no âmbito do Sétimo Programa-Quadro da União Europeia (FP7 / 2007-2013) (subvenção n.º 615984) e subvenções dos Institutos Nacionais de Health R01 GM60635 para PAP).
Materiais
| Name | Company | Catalog Number | Comments |
| SPHIRE | Max Planck Institute of Molecular Physiology- Dortmund and Houston Medical School, Houston, Texas | http://sphire.mpg.de | |
| UCSF Chimera | University of California, San Francisco | http://www.cgl.ucsf.edu/chimera/ | |
| Unblur | Janelia Farm Research Campus, Ashburn | http://grigoriefflab.janelia.org/unblur | |
| Coot | MRC Laboratory of Molecular Biology, Cambridge | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ | |
| EMAN2 | Baylor College of Medicine, Houston | http://blake.bcm.edu/emanwiki/EMAN2 | |
| Computing Cluster with 1824 cores | Max Planck Institute of Molecular Physiology | Linux Cluster with 76 nodes, each with 2 Processors Xeon E5-2670v3 12C 2.30 GHz and 128 Gb RAM | |
| TITAN KRIOS electron microscope | FEI | 300 kV, Cs correction, XFEG | |
| Falcon II direct electron detector | FEI | ||
| EPU (automated data acquisition software) | FEI | https://www.fei.com/software/epu/ |
Referências
- Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nature Methods. 13 (1), 24-27 (2016).
- Liao, M., Cao, E., Julius, D., Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 504 (7478), 107-112 (2013).
- Bai, X. -. C., Yan, C., et al. An atomic structure of human γ-secretase. Nature. 525 (7568), 212-217 (2015).
- Ecken, J. V. D., Heissler, S. M., Pathan-Chhatbar, S., Manstein, D. J., Raunser, S. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature. 534 (7609), 724-728 (2016).
- von der Ecken, J., Müller, M., Lehman, W., Manstein, D. J., Penczek, P. A., Raunser, S. Structure of the F-actin-tropomyosin complex. Nature. 519 (7541), 114-117 (2015).
- Tang, G., Peng, L., et al. EMAN2: An extensible image processing suite for electron microscopy. Journal of Structural Biology. 157 (1), 38-46 (2007).
- van Heel, M., Harauz, G., Orlova, E. V., Schmidt, R., Schatz, M. A new generation of the IMAGIC image processing system. Journal of Structural Biology. 116 (1), 17-24 (1996).
- Grigorieff, N. FREALIGN: high-resolution refinement of single particle structures. Journal of Structural Biology. 157 (1), 117-125 (2007).
- Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. Journal of Structural Biology. 180 (3), 519-530 (2012).
- Shaikh, T. R., Gao, H., et al. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nature Protocols. 3 (12), 1941-1974 (2008).
- Hohn, M., Tang, G., et al. SPARX, a new environment for Cryo-EM image processing. Journal of Structural Biology. 157 (1), 47-55 (2007).
- Lander, G. C., Stagg, S. M., et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. Journal of Structural Biology. 166 (1), 95-102 (2009).
- de la Rosa-Trevìn, J. M., Quintana, A., et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. Journal of Structural Biology. 195 (1), 93-99 (2016).
- Grant, T., Grigorieff, N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife. 4, 06980 (2015).
- Pettersen, E. F., Goddard, T. D., et al. UCSF Chimera?A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25 (13), 1605-1612 (2004).
- Penczek, P. A., Fang, J., Li, X., Cheng, Y., Loerke, J., Spahn, C. M. T. CTER-rapid estimation of CTF parameters with error assessment. Ultramicroscopy. 140, 9-19 (2014).
- Frank, J. . Three-Dimensional Electron Microscopy of Macromolecular Assemblies. , (2006).
- Woolford, D., Ericksson, G., et al. SwarmPS: rapid, semi-automated single particle selection software. Journal of Structural Biology. 157 (1), 174-188 (2007).
- Yang, Z., Fang, J., Chittuluru, J., Asturias, F. J., Penczek, P. A. Iterative Stable Alignment and Clustering of 2D Transmission Electron Microscope Images. Structure/Folding and Design. 20 (2), 237-247 (2012).
- Gatsogiannis, C., Merino, F., et al. Membrane insertion of a Tc toxin in near-atomic detail. Nature Publishing Group. , (2016).
- Gatsogiannis, C., Lang, A. E., et al. A syringe-like injection mechanism in Photorhabdus luminescens toxins. Nature. 495 (7442), 520-523 (2013).
- Meusch, D., Gatsogiannis, C., et al. Mechanism of Tc toxin action revealed in molecular detail. Nature. 508 (7494), 61-65 (2014).
- Penczek, P. A., Frank, J., Spahn, C. M. T. A method of focused classification, based on the bootstrap 3D variance analysis, and its application to EF-G-dependent translocation. Journal of Structural Biology. 154 (2), 184-194 (2006).
- Emsley, P., Lohkamp, B., Scott, W. G., Cowtan, K. Features and development of Coot. Acta crystallographica. Section D, Biological crystallography. 66, 486-501 (2010).
- Callaway, E. The revolution will not be crystallized: a new method sweeps through structural biology. Nature. 525 (7568), 172-174 (2015).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados