
Method Article
Analyse de particules à haute résolution à partir de microscopie à électrons à l'aide d'images utilisant SPHIRE
Dans cet article
Résumé
Cet article présente un protocole pour le traitement des images cryo-EM à l'aide de la suite logicielle SPHIRE. Le présent protocole peut être appliqué pour presque tous les projets EM à part particulaire qui visent une résolution proche atomique.
Résumé
SPHIRE (SPARX pour la microscopie électronique à haute résolution) est une nouvelle suite logicielle open source et conviviale pour le traitement semi automatisé de données cryo-microscopie électronique à une seule particule (cryo-EM). Le protocole présenté ici décrit en détail comment obtenir une structure de résolution à proximité atomique à partir de films de micrographie cryo-EM en guidant les utilisateurs à travers toutes les étapes du pipeline de détermination de structure de particule unique. Ces étapes sont contrôlées à partir de la nouvelle interface utilisateur graphique SPHIRE et nécessitent une intervention minimale de l'utilisateur. En utilisant ce protocole, une structure de 3,5 Å de TcdA1, un complexe de toxine Tc de Photorhabdus luminescens , provient de seulement 9500 particules simples. Cette approche simplifiée aidera les utilisateurs débutants sans une expérience de traitement étendue et une information structurelle préalable, afin d'obtenir des modèles atomiques sans bruit et sans biais de leurs complexes macromoléculaires purifiés dans leur état natal.
Introduction
Après le développement de la technologie des détecteurs d'électrons directs, le progrès remarquable de la cryo-EM à une seule particule est en train de remodeler la biologie structurale 1 . Par rapport à la cristallographie aux rayons X, cette technique ne nécessite qu'une petite quantité de protéines sans avoir besoin de cristallisation, tout en posant moins de restrictions quant à la pureté de l'échantillon et permettant encore la détermination des structures à une résolution atomique proche. Il est important de noter que différentes compositions ou états peuvent maintenant être séparés par calcul et la détermination de la structure des différentes conformations peut être effectuée à un niveau de détail sans précédent. Récemment, des cartes de densité de molécules difficiles pourraient être produites à des résolutions permettant une construction de modèle de novo et une compréhension approfondie de leur mode d'action 2 , 3 , 4 , 5.
Une grande variété de progiciels de traitement d'image sont disponibles dans la communauté 3DEM (Microscopie électronique 3D) (https://en.wikibooks.org/wiki/Software_Tools_For_Molecular_Microscopy) et la plupart d'entre eux sont en développement continu. Une résolution proche atomique a été obtenue pour des protéines présentant divers poids moléculaires et symétries avec plusieurs progiciels différents, y compris EMAN2 6 , IMAGIC 7 , FREALIGN 8 , RELION 9 , SPIDER 10 et SPARX 11 . Chaque paquetage nécessite un niveau d'expertise différent et offre un niveau différent d'orientation, d'automatisation et d'extensibilité des utilisateurs. En outre, alors que certains programmes fournissent des environnements complets pour faciliter toutes les étapes de l'analyse d'image, d'autres sont conçus pour optimiser des tâches spécifiques, comme le raffinement des paramètres d'alignement à partir d'un connuStructure de la distribution. Plus récemment, plusieurs plates-formes ont été développées, dont APPION 12 et SCIPION 13 , qui fournissent un pipeline de traitement unique qui intègre des approches et des protocoles à partir des différents logiciels répertoriés ci-dessus.
Pour contribuer au développement actuel de cryo-EM, SPARX a été redéveloppé dans une nouvelle plate-forme autonome et complète pour l'analyse de particules simples, appelée SPHIRE (SPARX pour Microscopie électronique à haute résolution). Afin d'accroître l'accessibilité de la technique pour les nouveaux chercheurs sur le terrain et de faire face à la grande quantité de données produites par des microscopes électroniques modernes entièrement automatisés, le pipeline de traitement a été redessiné et simplifié en introduisant un outil facile à utiliser Interface utilisateur graphique (GUI) et automatiser les étapes principales du flux de travail. En outre, de nouveaux algorithmes ont été ajoutés pour permettre une détermination de structure rapide, reproductible et automatisée à partir de crImages yo-EM. En outre, la validation par reproductibilité a été introduite afin d'éviter les artefacts communs produits lors de l'analyse de l'affinement et de l'hétérogénéité.
Bien que le programme ait été largement modifié, ses fonctionnalités principales appréciées ont été maintenues: un code open-source simple, la conception moderne orientée objet et les interfaces Python pour toutes les fonctions de base. Ainsi, il n'a pas été transformé en un programme de boîte noire, permettant aux utilisateurs d'étudier et de modifier facilement le code Python, de créer des applications supplémentaires ou de modifier le flux de travail global. Ceci est particulièrement utile pour les projets cryo-EM non standard.
Nous présentons ici un protocole pour l'obtention d'une carte de densité de résolution atomique proche des images cryo-EM utilisant la GUI de SPHIRE. Il décrit en détail toutes les étapes nécessaires pour générer une carte de densité à partir de films crus-EM de détection directe crue et ne se limite pas à un type de macromolécule particulier. Ce protocole vise principalement à guider newcDans le domaine grâce au flux de travail et fournir des informations importantes sur les étapes cruciales du traitement ainsi que sur certains pièges et obstacles éventuels. Les fonctionnalités plus avancées et les antécédents théoriques derrière SPHIRE seront décrits ailleurs.
Protocole
REMARQUE: pour suivre ce protocole, il est nécessaire d'installer correctement SPHIRE sur un système avec une installation MPI (actuellement un cluster Linux). Téléchargez SPHIRE et l'ensemble de données TcdA1 à partir de http://www.sphire.mpg.de et suivez les instructions d'installation: http://sphire.mpg.de/wiki/doku.php?id=howto:download. Cette procédure installe également EMAN2. SPHIRE utilise actuellement e2boxer de EMAN2 pour la sélection de particules et e2display pour afficher des fichiers image. Pour la correction de mouvement dose-pondérée des films micrographiques bruts, SPHIRE utilise unblur 14 . Téléchargez le programme et suivez les instructions d'installation (http://grigoriefflab.janelia.org/unblur, laboratoire Grigorieff). Pour une visualisation interactive des structures résultantes, le protocole utilisera le programme graphique moléculaire Chimera 15 (https://www.cgl.ucsf.edu/chimera/download.html). Un joli tutoriel pour se familiariser avec les fonctionnalités utilisées dans ce protocole peut être fouEt voici: https://www.cgl.ucsf.edu/chimera/data/tutorials/eman07/chimera-eman-2007.html. Des instructions sur la façon de soumettre un travail parallèle à un cluster de la GUI de SPHIRE peuvent être trouvées ici: http://sphire.mpg.de/wiki/doku.php?id=howto:submissions. L'organisation générale de l'interface graphique de SPHIRE et les étapes principales du flux de travail effectué tout au long de ce protocole sont illustrées à la figure 1 .
1. PROJET: Définissez des valeurs de paramètres constants pour ce projet
- Démarrez l'application SPHIRE GUI en tapant " sphire &" et la touche ENTER dans une fenêtre de terminal.
- Ajustez les paramètres du projet ( par exemple, la taille des pixels, le rayon de la particule et la symétrie) dans les champs de saisie respectifs de la page des paramètres du projet, puis enregistrez ces valeurs pour toutes les étapes ultérieures du flux de travail.
- Cliquez sur l'icône "PROJET" en bas à droite du panneau de gauche pour ouvrir la page des paramètres du projet.
- Mesurez l'axe le plus long d'une particule à l'aide de l'outil d'affichage interactif d'image e2display.py, puis entrez la moitié de la taille de particule sur "Rayon de particule protéinée". Si la mesure se trouve dans Å, pensez à convertir l'unité en pixels à l'aide de la taille du pixel ( par exemple, si une particule a une longueur de 200 Å et que la taille du pixel est de 1,2 Å / pixel, l'axe le plus long de la particule est de 200 / 1,2 = ~ 166 pixels et le rayon 166/2 = 83 pixels).
- Réglez "Taille de la boîte de particules" à au moins 1,5 fois la taille de la particule. Évitez les tailles de fenêtres contenant un grand nombre premier. Aussi, rappelez-vous que l'algorithme de raffinement 3D nécessite actuellement une taille de boîte numérotée.
REMARQUE: la fenêtre doit inclure une marge pour tenir compte des erreurs de centrage initiales résultant de la sélection (la nécessité de déplacer les particules dans la fenêtre) et pour la région de fond suffisante en dehors de la limite des particules pour une correction CTF appropriée (en particulier pour les grandes valeurs de défocalisation 16). - Définissez "taille de la fenêtre CTF" à celle de "Taille de la boîte de particules". Pour les projets à faible contraste, utilisez une fenêtre plus grande pour obtenir des estimations plus fluides des spectres de puissance.
- Définissez "symétrie de groupe de points" du complexe ( par exemple, "C5"). Si la symétrie de la structure cible n'est pas connue, laissez-la à "C1" (asymétrique). Cependant, si une symétrie de haut ordre spécifique est identifiée plus tard lors du traitement, modifiez ce paramètre de symétrie en conséquence et répétez les étapes après l'alignement 2D avec ISAC.
- Réglez "Masses moléculaires protéiniques" en kDa (la valeur approximative sera suffisante). Appuyez sur le bouton "Enregistrer les paramètres".
2. FILM: Alignez les cadres de chaque microgramme de film pour corriger le mouvement global de l'échantillon
- Pour toutes les micrographies de films, calculez les décalages x / y pour tous les cadres, puis créez leur moelle dose-non pondérée et dose pondéréeMoyenne corrigée des réparations (voir Discussion). Notez que le premier n'est nécessaire que pour l'estimation du FTC, car l'estimation ne fonctionne pas bien avec les moyennes pondérées par dose, tandis que la dernière est utilisée pour toutes les autres étapes de la détermination de la structure.
- Cliquez sur l'icône "MOVIE", puis sur le bouton "Alignement du film micrographique". Définissez "Unblur exécutable path" en sélectionnant le fichier exécutable. Réglez "Schéma de chemin de micrographie d'entrée" en sélectionnant une micrographie de film non alignée brute et en remplaçant la partie variable des noms de fichiers par le caractère générique "*" ( par exemple, TcdA1 _ * .mrc). Spécifiez le chemin d'accès pour "Répertoire de sortie".
- Définissez "chemin exécutable Summovie" en sélectionnant le fichier exécutable.
- Réglez "Nombre de cadres de film" sur le nombre d'images dans chaque micrographie de film. Réglez la "tension du microscope" et "Exposition par image" aux valeurs utilisées lors de la collecte des données. (Par exempleLe, si la dose globale est de 60 e - / Å 2 avec 20 images enregistrées sans pré-exposition, l'exposition pour chaque image est 60/20 = 3 e - / A 2. ) Appuyez sur le bouton "Exécuter la commande" pour aligner Cadres de chaque micrographie de film.
REMARQUE: cela créera automatiquement deux répertoires de sortie contenant des micrographies moyennes corrigées des mouvements dose-non pondérées et dose- pondérées, respectivement.
3. CTER: estimer les paramètres de désamorçage et d'astigmatisme du FCT
- Estimez les paramètres CTF (défocalisation et astigmatisme, les autres sont définis par l'utilisateur) pour chaque micrographie moyenne dose-non pondérée.
- Cliquez sur l'icône "CTER" puis sur le bouton "Estimation CTF". Pour définir "Schéma de chemin de micrographie d'entrée", sélectionnez une micrographie correctrice de mouvement non dose-dose, puis remplacez la partie variable des noms de fichier par le caractère générique"*". De plus, spécifiez le chemin d'accès pour "Output directory".
- Réglez "contraste d'amplitude" à la valeur habituellement utilisée pour le type de données (l'épaisseur de la glace est un facteur majeur) et la tension du microscope dans le laboratoire ( par exemple, 10%). Les valeurs typiques se situent dans la plage de 7 à 14% 17 .
- Définissez "aberration sphérique du microscope (Cs)" et "tension du microscope" utilisée lors de la collecte des données.
- Réglez la fréquence «Fréquence la plus basse» et la «Fréquence la plus élevée» de la gamme de recherche pour l'ajustement du modèle CTF à 0,0285 et 0,285 Å -1 (40 - 4 Å), respectivement. Appuyez sur le bouton "Exécuter la commande" pour estimer les paramètres CTF.
REMARQUE: les paramètres CTF seront automatiquement stockés dans le fichier partres.txt dans le répertoire de sortie spécifié. L'estimation CTF des micrographies 112 a été calculée sur 96 noyaux et a été terminée après ~ 3 min sur le cluster Linux utilisé pour obtenir les résultats représentatifs.
4. FENÊTRE: extraire les particules des micrographies moyennes pondérées par dose
- Choisissez les particules manuellement ou automatiquement à partir de micrographies avec e2boxer 6 et créez des fichiers de coordonnées contenant chacun une liste de coordonnées xy de particules dans la micrographie associée.
- Cliquez sur l'icône "WINDOW", puis sur le bouton "Particle Picking". Appuyez sur le bouton "Exécuter la commande" pour démarrer e2boxer 6 et choisissez les particules de chaque micrographie manuellement ou automatiquement 18 (voir Discussion ). Conservez les coordonnées finales de chaque micrographie dans le format de fichier EMAN1 (.box). Vous pouvez également importer les fichiers de coordonnées d'autres programmes après leur conversion au format EMAN1.
- Créer des piles de particules en extrayant des images de particules des micrographies pondérées en dose (dans SPHIRE, la pile de particules est souventEn simplement appelé "pile").
- Appuyez sur le bouton "Extraction de particules". Spécifiez "Schéma de chemin de la micrographie d'entrée" en sélectionnant une micrographie corrigée du mouvement pondérée en fonction de la dose, puis en remplaçant la partie variable des noms de fichier par le caractère générique "*" ( par exemple, TcdA1 _ * .mrc). De même, définissez "Input coordonnées pattern path" en sélectionnant un fichier de coordonnées ( p. Ex., TcdA1 _ *. Box). Spécifiez le chemin d'accès pour "Répertoire de sortie".
- Définissez "source de paramètres CTF" en sélectionnant le fichier de paramètres CTF (partres.txt produit à l'étape 3.1). Appuyez sur le bouton "Exécuter la commande".
- Combinez les piles d'images de particules extraites en une seule.
- Cliquez sur le bouton "Particle Stack". Spécifiez le chemin d'accès à "Sortie de la pile d'images virtuelles" à l'aide d'un format de chemin de fichier BDB ( p. Ex., "Bdb: Particules / pile", où "Particules"Le répertoire e contenant un répertoire BDB base de données dont le nom est toujours EMAN2DB et "stack" fait référence à une pile d'images particulière dans cette base de données). Spécifiez "Input BDB image stack pattern" en sélectionnant un répertoire commençant par "mpi_proc", puis en remplaçant la partie variable des noms de répertoire par le caractère générique "*" ( par exemple, Particles / mpi_proc_000 en Particles / mpi_proc_ *). Appuyez sur le bouton "Exécuter la commande".
5. ISAC: Classification des images de particules en 2D
- Calculez les moyennes de classe 2D en alignant les particules et en les regroupant en fonction de leur apparence 2D.
REMARQUE: les moyennes en 2D résultantes ont un rapport signal sur bruit amélioré (SNR) par rapport aux images de particules individuelles et sont donc utilisées pour évaluer visuellement la qualité et l'hétérogénéité de l'ensemble de données, ainsi que pour trier les images indésirables de la pile ( P. Ex. Cristaux de glace, bords de carbone,Agrégats, fragments, etc. ) 19 . En outre, ils seront ensuite utilisés pour déterminer un modèle 3D initial.- Cliquez sur l'icône "ISAC", puis sur le bouton "ISAC - 2D Clustering". Confirmez "Input image stack" en sélectionnant le fichier pile contenant les particules extraites. Spécifiez le chemin d'accès pour "Répertoire de sortie".
- Utilisez 200 - 1000 pour "Images par classe". Choisissez le nombre approprié compte tenu du nombre attendu de classes 2D (le nombre total de particules divisé par le nombre d'images par classe). Réglez ce paramètre en fonction du SNR et de la taille de l'ensemble de données. Augmenter le nombre de membres par classe dans le cas où l'ensemble de données est excessivement bruyant. Diminuez le nombre lorsqu'un faible nombre de particules est disponible.
REMARQUE: en raison des limitations de mémoire, pour des ensembles de données assez volumineux (> 100 000 particules), divisez l'ensemble de données complet en sous-ensembles, effectuez ISAC pour chaque sous-ensemble indépendamment et combinezLes résultats à la fin. Les instructions détaillées pour ce scénario de traitement sont données dans http://www.sphire.mpg.de/wiki/doku.php. - Cochez la case "Phase-flip". Conservez les valeurs par défaut pour "Rayon de particule cible" et "Taille d'image de particule cible" afin d'accélérer le processus en réduisant automatiquement toutes les images de particules avec ces paramètres. Appuyez sur le bouton "Exécuter la commande" pour calculer les moyennes de la classe 2D.
REMARQUE: Cette étape est exigeante en termes de calcul et le temps de fonctionnement augmente de façon significative avec le nombre de particules et de classes ainsi que le rayon cible et la taille de l'image. Sur un cluster avec 96 processus, la classification 2D de ~ 10 000 particules s'est terminée après environ 90 min.
- Affichez et vérifiez visuellement les moyennes de 2 Mo de l'ISAC pour vous assurer que leur qualité est satisfaisante (voir Discussion).
- Appuyez sur le bouton "Afficher les données" sous "UTILITIES". Ensemble "; Entrer des fichiers "en sélectionnant le fichier contenant les moyennes ISAC 2D (class_averages.hdf produit à l'étape 5.1). Appuyez sur le bouton" Exécuter la commande "pour afficher les moyennes de classe reproductibles et validées finales fournies par ISAC.
- Créez une nouvelle pile incluant uniquement les membres de particules des moyennes validées.
- Appuyez sur le bouton "Créer un sous-ensemble de pile". Réglez "Input image stack" en sélectionnant le même fichier de pile que dans l'étape 5.1.1. Définissez "Moyennes ISAC" en sélectionnant les moyennes ISAC 2D (class_averages.hdf produit à l'étape 5.1). Spécifiez le chemin d'accès pour "Répertoire de sortie". Appuyez sur le bouton "Exécuter la commande".
6. VIPER: calculer un modèle 3D initial
- Sélectionnez un petit ensemble de moyennes de classe (≥100 images) en supprimant toutes les moyennes de mauvaises classes et des vues identiques de la particule (voir Discussion) et utilisez-les pour calculer un représentantModèle initial roductible utilisant VIPER. Rappelez-vous que la sélection devrait contenir au moins 60-80 moyennes de haute qualité avec ~ 200-500 membres chacun.
- Cliquez sur l'icône "VIPER", puis sur le bouton "Afficher les données". Définissez "Input files" en sélectionnant les moyennes ISAC 2D (class_averages.hdf produit à l'étape 5.1). Appuyez sur le bouton "Exécuter la commande".
- Appuyez sur le bouton du milieu de la souris quelque part sur la fenêtre graphique de l'e2display et activez le bouton "DEL" dans la fenêtre contextuelle. Supprimez toutes les mauvaises moyennes de classe et les vues identiques de la particule (voir Discussion ). Appuyez sur le bouton "Enregistrer" pour stocker les moyennes restantes de la classe 2D vers un nouveau fichier.
- À partir des moyennes ISAC sélectionnées, génère une référence initiale pour le raffinement 3D ultérieur.
- Cliquez sur le bouton "Modèle initial 3D - RVIPER". Réglez "Input images stack" en sélectionnant la classe sélectionnéeMoyennes (produites à l'étape 6.1). Spécifiez le chemin d'accès pour "Répertoire de sortie".
- Assurez-vous d'utiliser la même valeur pour "rayon de particule cible" comme ISAC étape 5.1.3. Appuyez sur le bouton "Exécuter la commande" pour générer un modèle ab initio 3D reproductible.
REMARQUE: cette étape est exigeante en termes de calcul et le temps de fonctionnement augmente de façon significative avec le nombre de moyennes et de taille des particules. Sur un cluster avec 96 processus, ce travail (~ 100 moyennes de classe) a terminé après ~ 15 min.
- Vérifiez si le modèle 3D résultant est raisonnable en tenant compte des moyennes de la classe et en plus de son intégrité structurelle ( c.-à-d. Pas de pièces déconnectées et / ou des artefacts directionnels). Pour afficher la carte, utilisez le programme Chimera 15 . À ce stade, effectuer une première comparaison avec une structure cristalline d'une protéine homologue ou un domaine de la protéine d'intérêt s'il existe (un exemple est montré dans la section ReprésérerRésultats ntatifs).
- Pour le raffinement 3D ultérieur, générez une référence 3D initiale et un masque 3D à partir d'un modèle 3D ab initio en supprimant son bruit environnant et en le redéfinissant pour correspondre à la taille de pixel d'origine.
- Cliquez sur le bouton "Créer une référence 3D". Réglez "Volume d'entrée" en sélectionnant le modèle ab initio 3D (average_volume.hdf produit à l'étape 6.2). Spécifiez le chemin d'accès pour "Répertoire de sortie".
- Réglez "Source du rapport de rééchantillonnage" en sélectionnant le fichier de rapport de réduction ISAC (README_shrink_ratio.txt produit à l'étape 5.1). Appuyez sur le bouton "Exécuter la commande".
7. MERIDIEN: Affiner le volume 3D initial
- Affinez le volume 3D à partir du modèle 3D initial.
- Cliquez sur l'icône "MERIDIEN", puis sur le bouton "Raffinage 3D". Réglez "Input image stack" et "Initial 3D reference" par seleCtion de la pile de particules et du modèle ab initio 3D (produit à l'étape 5.3 et 6.4, respectivement). Spécifiez le chemin d'accès pour "Répertoire de sortie".
- Réglez "masque 3D" en sélectionnant le fichier masque 3D (produit à l'étape 6.4). Toujours utiliser un masque 3D mais, surtout à un stade précoce de l'analyse, utiliser un masque sphérique ou un masque à bord doux qui est bien adapté à la référence pour éviter d'introduire un biais de masquage incorrect.
- Cochez la case "Appliquer le masque 2D 2". Réglez "Démarrage de la résolution" à une valeur de fréquence de coupure comprise entre 20 et 25 Â. Gardez à l'esprit qu'un filtre passe-bas avec cette fréquence de coupure sera appliqué à la structure 3D initiale pour réduire le biais initial du modèle.
- Vérifiez les spécifications du cluster utilisé pour ce processus, puis définissez "Memory per node" sur la mémoire disponible en gigaoctets. Appuyez sur le bouton "Exécuter la commande" pour affiner le volume 3D à partir du modèle 3D initial de manière entièrement automatisée.
NOTE: Cette procédure va diviser l'ensemble de données en deux moitiés, affiner les deux modèles indépendamment et produire deux volumes bruts, chacun de la moitié seulement des particules. Il est exigeant en termes de calcul et le temps de fonctionnement augmente considérablement avec le nombre de particules. Sur ce cluster, le raffinement du méridien s'est terminé après ~ 2,5 h sur 192 processus (~ 8 000 particules, 352 tailles).
- Créez un masque 3D à bord doux du volume raffiné pour l'étape d'affûtage suivante.
- Cliquez sur le bouton "Adaptive 3D Mask". Réglez "Volume d'entrée" en sélectionnant l'un des demi-volumes non filtrés (produits à l'étape 7.1). Spécifiez le chemin d'accès pour "Masque de sortie".
- Définissez une valeur de "Seuil de binarisation". Utilisez Chimera pour vous assurer que, à ce seuil particulier, le bruit est clairement en dehors du volume d'intérêt dans la région des solvants des demi-cartes non filtrées et toutes les densités de la protéine sont encore cSe sont engagés l'un l'autre. Appuyez sur le bouton "Exécuter la commande" pour créer le masque 3D de bordure souple.
REMARQUE: Le corps principal du masque résultant (composé de voxels dont les valeurs sont> 0,5) doit s'adapter étroitement à la structure des particules mais inclure toutes les densités d'intérêt. La chute du bord doux devrait avoir au moins 8-10 pixels de largeur.
- Fusionnez les deux demi-volumes non filtrés obtenus grâce au raffinement 3D. Ensuite, affûtez le volume fusionné en ajustant le spectre de puissance en fonction de la fonction de transfert de modulation (MTF) du détecteur, du facteur B estimé et de l'estimation de la résolution FSC (Fourier Shell Correlation).
- Sélectionnez le bouton "Affûtage". Réglez "Premier demi-volume non filtré" et "Deuxième demi-volume non filtré" en sélectionnant les fichiers correspondants (vol_0_unfil.hdf et vol_1_unfil.hdf produits à l'étape 7.1). Toujours utiliser "B-factor enhancement". En règle générale, conservez la valeur par défaut afin deStimule la valeur du facteur B à partir de l'ensemble de données d'entrée en utilisant la plage entre la fréquence de résolution finale et 10 Å. Sinon, spécifiez une valeur ad hoc ( p. Ex., -100).
- Gardez la valeur par défaut pour "Fréquence de filtre passe-bas" pour appliquer un filtre basé sur FSC.
- Réglez "Masque fourni par l'utilisateur" en sélectionnant le masque 3D (produit à l'étape 7.2). Rappelez-vous que la résolution signalée sera déterminée à l'aide de FSC avec ce masque. Appuyez sur le bouton "Exécuter la commande" pour affiner le volume 3D raffiné.
- Générer la carte de répartition angulaire 3D à partir des directions de projection de toutes les particules estimées par l'étape de raffinement 3D ci-dessus.
- Cliquez sur le bouton "Réduction angulaire". Définissez "Fichier de paramètres d'alignement" en sélectionnant le fichier (final_params.txt produit à l'étape 7.1), puis appuyez sur le bouton "Exécuter la commande".
- Inspecter visuellement le modèle 3D aiguisé en utilisant ChiMera. Assurez-vous que la structure semble raisonnable compte tenu de la résolution obtenue (voir Discussion ).
- Inspecter visuellement la distribution angulaire en utilisant Chimera. Vérifiez que la distribution couvre approximativement l'espace angulaire 3D entier. Gardez à l'esprit que, pour les structures symétriques, la distribution est limitée dans le triangle asymétrique unique.
8. SORT3D: trier l'hétérogénéité 3D en mettant l'accent sur les régions à forte variable
- Calculez la carte de variabilité 3D à partir de la pile de particules utilisée dans le raffinement 3D.
- Cliquez sur l'icône "SORT3D" puis sur le bouton "Estimation de la variabilité 3D". Réglez "Input image stack" en sélectionnant la même pile de particules tamisée donnée à l'étape de raffinement 3D 7.1.1. Spécifiez le chemin d'accès pour "Répertoire de sortie".
- Conservez la valeur par défaut pour "Nombre de projections".
REMARQUE: les images du voisin angulaireLe capot sera utilisé pour estimer la variance 2D à chaque angle de projection 3D. Plus le nombre est important, moins l'estimation est bruyante, mais plus la résolution et les artefacts rotatifs sont plus prononcés. - Cochez la case "Utiliser CTF". Appuyez sur le bouton "Exécuter la commande".
- Utilisez la carte de variabilité 3D pour créer un masque de mise au point pour l'étape de clustering 3D ci-dessous.
- Sélectionnez le bouton "Binary 3D Mask". Réglez "Volume d'entrée" en sélectionnant la carte de variabilité 3D (produite à l'étape 8.1). Spécifiez le chemin du fichier pour "Masque de sortie".
- Définissez "seuil de binarisation" en utilisant la sortie du champ "Niveau" dans "Volume Viewer" de Chimera. Appuyez sur le bouton "Exécuter la commande".
- Trier les particules dans des groupes structurels homogènes en se concentrant sur des régions structurellement très variables.
- Appuyez sur le bouton "3D Clustering - RSORT3D".Réglez "Input 3D refinement directory" en sélectionnant le répertoire de sortie du raffinement 3D (produit à l'étape 7.1). Spécifiez le chemin d'accès pour "Répertoire de sortie".
- Réglez "masque 3D" en sélectionnant le masque 3D à bord doux (produit à l'étape 7.2). Réglez "Focus 3D mask" en sélectionnant la carte de variabilité 3D binarisée (produite à l'étape 8.2).
- Pour les grandes séries de données, utilisez au moins 5,000-10,000 pour "Images par groupe". Gardez à l'esprit que le programme conserve toujours le nombre d'images par groupe inférieur à ce paramètre. Ajustez la valeur en considérant le nombre attendu de groupes 3D (le nombre total de particules divisé par la valeur "Images par groupe"), l'ensemble de données, le SNR et le degré d'hétérogénéité. Commencez par ~ 5-10 groupes 3D initiaux, si un nombre suffisant de particules est disponible, sauf si un nombre plus élevé d'états structurels distincts dans l'ensemble de données est attendu.
- Utilisez au moins 3 000 à 5 000 particules pour "Taille de groupe la plus petite".Notez que le programme ignorera les groupes comprenant un nombre d'images inférieur au réglage de "Taille de groupe la plus petite". Appuyez sur le bouton "Exécuter la commande" pour effectuer le clustering 3D.
REMARQUE: RSORT3D est subdivisé en deux étapes. La première étape "sort3d" détermine l'hétérogénéité 3D. Ensuite, il reconstruit les volumes de chaque groupe structurel homogène en utilisant les paramètres d'alignement 3D déterminés par l'étape de raffinement 3D ci-dessus. La deuxième étape "rsort3d" permet d'identifier les membres reproductibles de chaque groupe en effectuant une comparaison bidirectionnelle des deux courses de tri indépendantes. Ensuite, il reconstruit des structures homogènes en utilisant uniquement les particules reproduites. Sur un cluster avec 96 noyaux, ce travail (~ 8 000 particules, 352 tailles) a fini après environ 3 h.
- Une fois le programme terminé, utilisez Chimera pour sélectionner un groupe 3D homogène. Sélectionnez la structure de la résolution apparente la plus élevée, généralement associée au plusGroupe pulpeux. Assurez-vous que la structure sélectionnée est visuellement raisonnable en tenant compte des moyennes de classe 2D et des aspects biologiques de la protéine d'intérêt (voir Discussion ). Si d'autres volumes ont une structure presque identique à une résolution similaire, considérez-les comme émergeant d'un seul groupe 3D homogène.
- Effectuez un raffinement local contre les membres de particules du groupe 3D le plus homogène (avec la plus haute résolution).
- Cliquez sur le bouton "Réglage du sous-ensemble local". Définissez "Chemin du fichier texte du sous-ensemble" en sélectionnant le fichier texte contenant les ID des particules du groupe sélectionné ( par exemple, Cluster0.txt produit à l'étape 8.3). Définissez "Répertoire de raffinement 3D" en sélectionnant le répertoire de sortie du raffinement 3D précédent (produit à l'étape 7.1).
- Réglez "Itinerance de redémarrage" à celui où la plus haute résolution est obtenue dans le perfectionnement 3D précédent. appuie sur le"Exécuter la commande" pour effectuer un raffinement local de la population sélectionnée de particules.
- Similaire à l'étape 7.2, créez un masque 3D à bord doux à partir d'un demi-volume final non filtré reconstruit par le raffinement du sous-ensemble local.
- À l'instar de l'étape 7.3, fusionnez deux demi-volumes finals non filtrés dérivés du raffinement du sous-ensemble local et affinez le volume fusionné. Cependant, ne filtrez pas le volume nettoyé cette fois-ci.
NOTE: Si l'analyse d'hétérogénéité à l'étape 8.4 indique plusieurs états distincts à une résolution comparable, on peut vouloir affiner indépendamment tous les états différents.
9. LOCALRES: estimation de la résolution locale du volume 3D final
- Estimation de la résolution locale du volume 3D obtenu à partir de l'ensemble homogène de particules.
- Cliquez sur l'icône "LOCALRES", puis sur "Résolution locale". Réglez "Premier demi-volume" et "Deuxième moitié-volume "en sélectionnant les demi-volumes finaux non filtrés du raffinement du sous-ensemble local (produit à l'étape 8.5). Définissez" masque 3D "en sélectionnant le masque 3D à bord doux produit à l'étape 8.6. Spécifiez le chemin du fichier pour" Volume de sortie " .
- Gardez la valeur par défaut de 7 pixels pour "taille de fenêtre FSC". Rappelez-vous que ce paramètre définit la taille de la fenêtre où la corrélation locale-espace réel est calculée; Les grandes tailles de fenêtre produisent des cartes de résolution plus lisses au détriment de la résolution locale.
- Gardez la valeur par défaut 0.5 de "Résolution coupée" pour le critère de résolution.
REMARQUE: pour chaque voxel, le programme signalera la résolution locale comme la fréquence à laquelle le FSC local tombe en dessous du seuil de résolution sélectionné. Un seuil inférieur à 0,5 n'est pas recommandé, car les valeurs de corrélation inférieures présentent une forte incertitude statistique. Par conséquent, la résolution locale correspondra fortement entre voxels. - Pour "OveraLl resolution ", définissez la résolution absolue estimée dans l'affûtage après le raffinement du sous-ensemble local (étape 8.7). Appuyez sur le bouton" Exécuter la commande "pour calculer la résolution locale du volume.
- Appliquer le filtre local 3D au volume affiné après le raffinement du sous-ensemble local en utilisant la carte de résolution locale 3D.
- Cliquez sur le bouton "Filtre local 3D". Réglez "Volume d'entrée" en sélectionnant le volume 3D netteté mais non filtré (produit à l'étape 8.7). De même, définissez "Fichier de résolution locale" et "Masque 3D" (produit à l'étape 9.1 et 8.6, respectivement). N'oubliez pas que le masque 3D définit la région où le filtrage local sera appliqué. Spécifiez le chemin du fichier pour "Volume de sortie". Appuyez sur le bouton "Exécuter la commande" pour appliquer le filtre local 3D.
- Utilisez Chimera pour inspecter visuellement le modèle 3D final et la carte de résolution locale 3D (produite à l'étape 9,2 et 9.1, resp.Ectively). Sélectionnez l'option "Couleur de surface" pour colorer le volume 3D en fonction de la résolution locale. Gardez à l'esprit que la répartition de la résolution locale devrait être lisse (voir Discussion ).
Résultats
Le protocole décrit ci-dessus a été exécuté à partir de 112 films de détecteurs directs du composant A du complexe Photorhabdus luminescens Tc (TcdA1) 20 , 21 , 22 . Cet ensemble de données a été enregistré sur un microscope cryo à électrons corrigé en Cs avec un pistolet d'émission de champ à haute brillance (XFEG), fonctionnant à une tension d'accélération de 300 kV. Les images ont été acquises automatiquement avec une dose totale de 60 e - / Å -2 à une taille de pixel de 1,14 Å sur l'échelle de l'échantillon. Après l'alignement des cadres de film (Protocole Étape 2 ), les moyennes corrigées en fonction résultantes ont des anneaux Thon isotropes qui s'étendent en haute résolution ( Figure 2a ). Les particules individuelles étaient facilement visibles et bien séparées ( figure 2b ). Les particules ont ensuite été choisies en utilisant l'outil en essaimage de e2boxerLass = "xref"> 18 ( Protocole Étape 4.1 ). Dans ce cas, un seuil approprié a été défini en utilisant l'option plus sélective ( Figure 2c ). Les 112 micrographies numériques ont généré 9 652 particules. La majorité des images extraites (Protocole étape 4.2 ) contiennent des particules bien définies et leur taille de boîte était ~ 1,5 fois plus grande que la taille des particules, comme cela est recommandé ( figure 2d ). Ensuite, en utilisant ISAC, une analyse d'hétérogénéité 2D a été effectuée (protocole étape 5 ). Il a généré 98 moyennes de classe ( figure 3a ). En utilisant ces moyennes de classe 2D, un modèle ab initio a été calculé en utilisant VIPER (Protocole Étape 6 ) à la résolution intermédiaire ( Figure 3b ). Ce modèle montre un excellent accord avec la structure cristalline de TcdA1 précédemment résolue à 3,9 Å de résolution 22 ( Figure 3c ). Ce modèle ab initio a été utilisé comme un premier temps Plaque pour le raffinement 3D (MERIDIEN), produisant une reconstruction de 3,5 Å (0.143 critère) (protocole étape 7 ) à partir de seulement 40 000 unités asymétriques ( figure 4 ). Cette carte de résolution à proximité atomique a été obtenue dans les 24 h, en utilisant jusqu'à 96 CPU pour les étapes du flux de travail qui bénéficient de multiples noyaux.
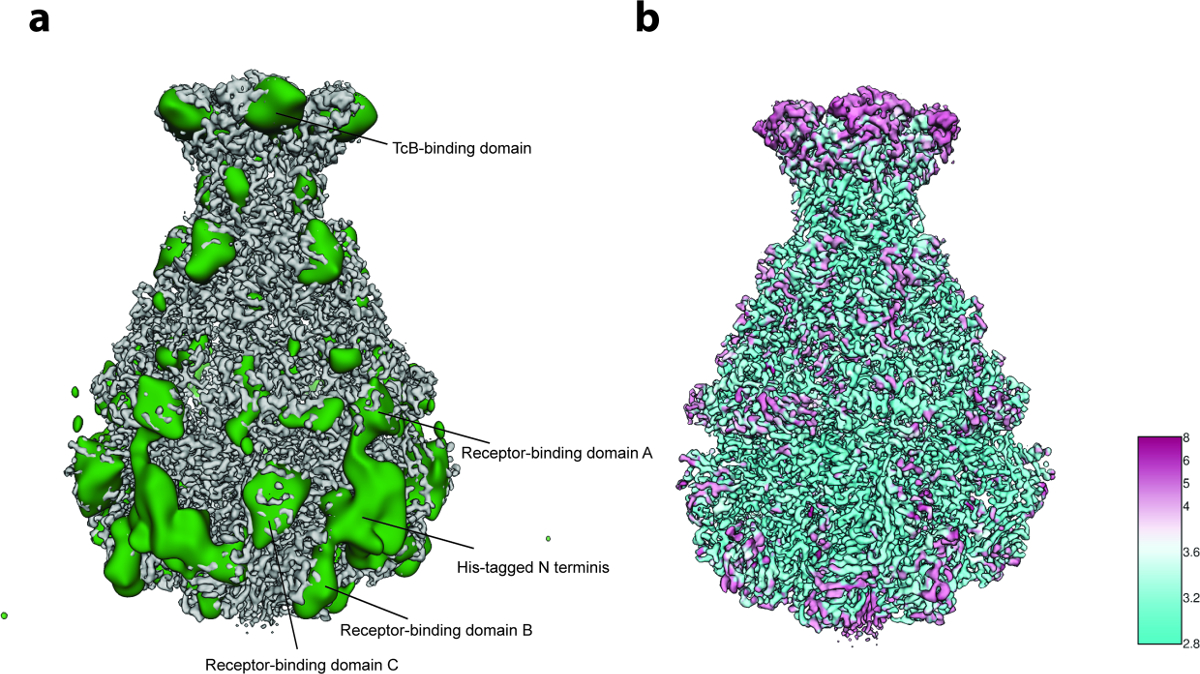
Pour l'analyse de la variabilité 3D (étape 8 du protocole ), seulement 2 000 images de particules par groupe ont été utilisées à l'étape 8.3.3 ( c'est-à-dire que le processus commence par 5 groupes 3D initiaux) et 200 images pour la plus petite taille de groupe à l'étape 8.3.4 en raison de Le petit nombre de particules (~ 10 000). L'analyse a révélé une flexibilité localisée principalement dans la région N-terminale du complexe qui contient l'étiquette His utilisé pour la purification ( Figure 5a ). En effet, douze résidus N-terminaux et l'étiquette His n'ont pas été résolus dans la structure cristalline précédemment publiée de TcdA1"> 22 et cette région très probablement désordonnée est restée non résolue dans la densité cryo-EM actuelle, probablement en raison de sa flexibilité. Une variabilité supplémentaire a été détectée aux domaines de liaison aux récepteurs et au domaine de liaison BC ( Figure 5a ). En raison de l'ensemble Une résolution satisfaisante de la structure et une taille assez petite de l'ensemble de données, cette hétérogénéité a été décidée à être tolérable et donc une classification 3D focalisée 23 n'a pas été effectuée. Enfin, la résolution locale de la carte de densité finale a été calculée (Protocole étape 9.1, Figure 5b ) et la carte 3D aiguisée a été filtrée localement (Protocole étape 9.2) . Un volume de cette qualité peut être utilisé pour le modèle de modèle de novo en utilisant Coot 24 ou tout autre outil de raffinement ( Figure 6 ).

Figure 1: Traitement d'image utilisant SPHIRE. (A) L'interface graphique du logiciel SPHIRE. Une étape spécifique du flux de travail peut être activée en sélectionnant le pictogramme respectif sur le côté gauche de l'interface graphique ("workflow step"). Les commandes et les utilitaires associés à cette étape du flux de travail apparaîtront dans la zone centrale de l'interface graphique. Après avoir sélectionné l'une des commandes, les paramètres respectifs sont affichés sur la droite de la GUI. Les paramètres avancés ne nécessitent généralement pas de modification des valeurs par défaut prédéfinies. ( B ) Etapes du flux de travail du traitement d'image à une seule particule à l'aide de la GUI de SPHIRE. Cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 2: Correction de mouvement et ParticLe Extraction. (A, b ) Micrographie numérique typique de haute qualité, à faible dose et à dérive enregistrée à un défocalisation de 1,7 μm. Notez les anneaux Thon isotropes qui s'étendent à une résolution de 2,7 Å dans le spectre de puissance (a) et les particules bien discernables dans l'image 2D ( b ). ( C ) Sélection des particules à l'aide de e2boxer. Les cercles verts indiquent des particules sélectionnées. ( D ) Particules brutes typiques extraites de la micrographie dose-pondérée. Barres d'échelle = 20 nm. Cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 3: Clustering 2D et génération initiale du modèle. (A) Galerie des moyennes de la classe 2D, la majorité représentant les vues latérales o F la particule. Barre d'échelle = 20 nm. ( B ) La carte Ab initio 3D de TcdA1 obtenue en utilisant RVIPER à partir des moyennes de classe sans référence. ( C ) Raccord rigide de la structure en cristal TcdA1 (rubans) (pdb-id 1VW1) dans la densité cryo-EM initiale (gris transparent). Cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 4: Cryo-EM Structure 3D de TcdA1. (A, b ) Carte de densité 3,5 Å finale de TcdA1 calculée en utilisant ~ 9 500 images de particules: ( a ) côté et ( b ) vue de dessus. ( C ) Les zones représentatives de la densité cryo-EM pour une α-hélice et une feuille β.Arge.jpg "target =" _ blank "> Cliquez ici pour voir une version plus grande de ce chiffre.

Figure 5: analyse de la variabilité et résolution locale. (A) Surface de la carte cryo-EM tcdA1 aiguisée (gris) et la carte de variabilité (verte). Pour une meilleure clarté, la carte de variabilité a été passée à basse altitude à 30 Å. ( B ) Rendu de surface de la carte Cryo-EM aiguisée TcdA1 colorée en fonction de la résolution locale (Å). Notez l'accord topologique entre des zones à forte variabilité et une faible résolution locale. Cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 6: Modem 3DEl Building of TcdA1 utilisant Coot. Des régions représentatives de la densité cryo-EM et du modèle atomique sont représentées pour une α-hélice. Le modèle atomique a été construit de novo à l' aide de Coot. Cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
Discussion
La cryo-EM à particules simples a connu un développement rapide au cours des dernières années et a fourni de nombreuses structures de résolution atomique de complexes macromoléculaires ayant une importance biologique majeure 25 . Afin de supporter le grand nombre d'utilisateurs débutants qui entrent actuellement sur le terrain, nous avons développé la plate-forme d'analyse d'image à une seule particule SPHIRE et présente ici un protocole de transit pour l'ensemble du flux de travail, y compris l'alignement des films, la sélection des particules, l'estimation CTF, le modèle initial Calcul, analyse de l'hétérogénéité en 2D et 3D, raffinement 3D haute résolution et estimation et filtrage de la résolution locale.
Le protocole décrit ici est conçu comme un petit guide pour la détermination de la structure 3D à l'aide de micrographies cryo-EM de la protéine d'intérêt et à l'aide d'outils informatiques fournis par la GUI autonome de SPHIRE.
La principale caractéristique du flux de travail est que la plupartDes procédures doivent être exécutées une seule fois, puisqu'elles s'appuient sur le concept de validation par reproductibilité 19 et ne nécessitent pas de modification des paramètres. Ce mécanisme de validation automatique est l'un des principaux avantages de SPHIRE sur d'autres logiciels, car les résultats ont tendance à être aussi objectifs que reproductibles et, surtout, être obtenus à un coût de calcul acceptable. Le pipeline fournit en outre une multitude d'informations de diagnostic pour les utilisateurs expérimentés afin de procéder à une validation et à une évaluation indépendantes avec leurs propres méthodes. Néanmoins, un utilisateur novice qui possède au moins des antécédents théoriques élémentaires dans la biologie structurale et la microscopie électronique devrait être en mesure d'obtenir des structures de résolution à proximité atomique en utilisant les données propres et les procédures automatisées de validation.
Cependant, l'obtention d'une structure de résolution à proximité atomique n'est pas toujours simple et le résultat dépendra fortement de la qualité de l'échantillon et de l'entrée datune. Pour les procédures présentées ici, on suppose qu'un nombre suffisant de films EM non alignés de haute qualité sont disponibles, leurs moyennes montrant des particules simples homogènes et aléatoires clairement discernables. En général, il n'y a aucune restriction concernant la symétrie, la taille ou la forme globale de la molécule, mais un faible poids moléculaire peut être un facteur limitant, en particulier lorsque la protéine a une forme globuleuse sans caractéristique. Habituellement, l'analyse de particules plus grandes et bien ordonnées avec une symétrie de groupe de points élevé est moins exigeante. Par conséquent, il est fortement recommandé aux utilisateurs débutants d'exécuter le protocole actuel d'abord avec un ensemble de données cryo-EM bien caractérisé. Les données du tutoriel SPHIRE (http: /sphire.mpg.de) ou l'un des ensembles de données envoyés par EMPIAR (https://www.ebi.ac.uk/pdbe/emdb/empiar/) avec des films bruts sont un bon point de départ .
Lors du traitement de données propres, il est très probable que certains ensembles de données ou certaines des images ne satisfassent pas certaines qualitésCritères de sélection. Dans ce contexte, en plus des contrôles automatiques de stabilité et de reproductibilité effectués par le programme pour les grandes étapes du processus, il est toujours recommandé aux utilisateurs d'inspecter visuellement les résultats à certains "points de contrôle" du protocole, surtout si la reconstruction finale N'est pas satisfaisant.
La première inspection visuelle peut être effectuée au niveau de la micrographie après l'alignement du film (Protocole étape 2 ) et l'estimation CTF (Protocole étape 3 ). Les moyennes corrigées par les mouvements résultantes devraient montrer des particules simples clairement discernables et bien séparées et leurs spectres de puissance devraient montrer des anneaux Thon isotropes clairement discernables. La fréquence spatiale à laquelle ils sont visibles définit, dans la plupart des cas, la résolution la plus élevée à laquelle la structure peut en principe être finalement déterminée. Des exemples d'une moyenne corrigée des mouvements de qualité suffisante et de son spectre de puissance sont présentés dans la section & #34; résultats représentatifs ". Les images externes qui pourraient avoir un impact négatif sur le résultat final peuvent être supprimées avec l'aide des outils de GUI d'évaluation de la dérivation et de la FCE de SPHIRE (http://sphire.mpg.de/wiki/doku.php).
En ce qui concerne le dépistage des particules, l'étape cruciale dans le pipeline SPHIRE est la classification 2D utilisant ISAC (Protocole étape 5.2) . Ici, l'utilisateur devrait contrôler que les moyennes de classe 2D reproductibles identifiées automatiquement par le programme adoptent une gamme d'orientations suffisantes pour couvrir quasi-uniformément l'espace angulaire. Si la qualité des moyennes de classe n'est pas satisfaisante (images bruyantes et / ou floues) et / ou le nombre de moyennes de classe reproductibles est très faible, envisagez d'améliorer la qualité de sélection automatique, d'optimiser l'imagerie de jeu de données ou la préparation d'échantillons. Dans la plupart des cas, il n'est pas possible de calculer une reconstruction fiable à partir d'un ensemble de données qui ne donne pas de bonnes moyennes de classe 2D. Exemples d'ave de classe 2D de haute qualitéLes rage sont présentés dans la section «Résultats représentatifs».
Au moins 100 moyennes de classe sont nécessaires pour obtenir un modèle 3D initial fiable utilisant RVIPER de manière automatisée (Protocole étape 6.1 ). Pour cette étape, l'utilisateur doit sélectionner les moyennes avec la plus haute qualité et inclure autant d'orientations différentes de la particule que possible. La qualité du modèle initial est essentielle pour la réussite du raffinement 3D ultérieur haute résolution.
Dans d'autres progiciels, la classification 3D est parfois effectuée pour éliminer les "mauvaises" particules 8 , 9 . Cependant, dans SPHIRE, la plupart de ces particules sont automatiquement éliminées déjà lors de la classification 2D en utilisant ISAC. Ainsi, il est recommandé d'effectuer l'étape de calcul du processus 3D intensif en calcul uniquement si la reconstruction et l'analyse de la variabilité 3D indiquent l'hétérogénéité de l'ensemble de données.
Plus important encore, l'utilisateur doit toujours examiner soigneusement les volumes 3D résultants (Protocole étape 9.3 ) et confirmer que les caractéristiques de la densité respective correspondent bien à la résolution nominale. À une résolution de <9 Å, des densités en forme de tige correspondant aux hélices α deviennent visibles. À une résolution <4,5 Å, les densités correspondant aux brins dans les feuilles β sont normalement bien séparées et les acides aminés volumineux deviennent visibles. Une carte haute résolution (<3 Å) devrait montrer des chaînes latérales clairement discernables, ce qui permet de construire un modèle atomique précis.
Les résultats obtenus à ce jour démontrent que, avec l'aide des tests de reproductibilité automatisés de SPHIRE et des inspections visuelles minimales, le présent protocole s'applique généralement à tout type de projet cryo-EM à part unique. Des résultats représentatifs de chaque étape de traitement sont montrés pour la reconstruction de la toxine TcdA1 dePhotorhabdus luminescens 21 , qui a été résolu à une résolution atomique proche. Des cartes de densité de qualité similaire peuvent être utilisées pour construire des modèles atomiques fiables par le tracé de squelette de novo ainsi que par un raffinement réciproque ou réel, et constituent donc un cadre structurel solide pour la compréhension de mécanismes moléculaires complexes.
CODES D'ACCESION:
Les coordonnées de la structure EM et des films non transformés ont été déposées dans la banque de données de microscopie électronique et les archives d'images de pilote de microscopie électronique sous les numéros d'accès EMD-3645 et EMPIAR-10089, respectivement.
Déclarations de divulgation
Les auteurs déclarent qu'ils n'ont pas d'intérêts financiers concurrents.
Remerciements
Nous remercions D. Roderer de nous avoir fourni des micrographies TcdA1. Nous remercions Steve Ludtke pour son soutien continu de l'infrastructure EMAN2. Ce travail a été soutenu par des fonds de la Max Planck Society (à SR) et du Conseil européen dans le cadre du septième programme-cadre de l'Union européenne (7e PC / 2007-2013) (subvention n ° 615984) (à SR) et octroi des National Institutes of Santé R01 GM60635 à PAP).
matériels
| Name | Company | Catalog Number | Comments |
| SPHIRE | Max Planck Institute of Molecular Physiology- Dortmund and Houston Medical School, Houston, Texas | http://sphire.mpg.de | |
| UCSF Chimera | University of California, San Francisco | http://www.cgl.ucsf.edu/chimera/ | |
| Unblur | Janelia Farm Research Campus, Ashburn | http://grigoriefflab.janelia.org/unblur | |
| Coot | MRC Laboratory of Molecular Biology, Cambridge | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ | |
| EMAN2 | Baylor College of Medicine, Houston | http://blake.bcm.edu/emanwiki/EMAN2 | |
| Computing Cluster with 1824 cores | Max Planck Institute of Molecular Physiology | Linux Cluster with 76 nodes, each with 2 Processors Xeon E5-2670v3 12C 2.30 GHz and 128 Gb RAM | |
| TITAN KRIOS electron microscope | FEI | 300 kV, Cs correction, XFEG | |
| Falcon II direct electron detector | FEI | ||
| EPU (automated data acquisition software) | FEI | https://www.fei.com/software/epu/ |
Références
- Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nature Methods. 13 (1), 24-27 (2016).
- Liao, M., Cao, E., Julius, D., Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 504 (7478), 107-112 (2013).
- Bai, X. -C., Yan, C., et al. An atomic structure of human γ-secretase. Nature. 525 (7568), 212-217 (2015).
- Ecken, J. V. D., Heissler, S. M., Pathan-Chhatbar, S., Manstein, D. J., Raunser, S. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature. 534 (7609), 724-728 (2016).
- von der Ecken, J., Müller, M., Lehman, W., Manstein, D. J., Penczek, P. A., Raunser, S. Structure of the F-actin-tropomyosin complex. Nature. 519 (7541), 114-117 (2015).
- Tang, G., Peng, L., et al. EMAN2: An extensible image processing suite for electron microscopy. Journal of Structural Biology. 157 (1), 38-46 (2007).
- van Heel, M., Harauz, G., Orlova, E. V., Schmidt, R., Schatz, M. A new generation of the IMAGIC image processing system. Journal of Structural Biology. 116 (1), 17-24 (1996).
- Grigorieff, N. FREALIGN: high-resolution refinement of single particle structures. Journal of Structural Biology. 157 (1), 117-125 (2007).
- Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. Journal of Structural Biology. 180 (3), 519-530 (2012).
- Shaikh, T. R., Gao, H., et al. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nature Protocols. 3 (12), 1941-1974 (2008).
- Hohn, M., Tang, G., et al. SPARX, a new environment for Cryo-EM image processing. Journal of Structural Biology. 157 (1), 47-55 (2007).
- Lander, G. C., Stagg, S. M., et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. Journal of Structural Biology. 166 (1), 95-102 (2009).
- de la Rosa-Trevìn, J. M., Quintana, A., et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. Journal of Structural Biology. 195 (1), 93-99 (2016).
- Grant, T., Grigorieff, N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife. 4, 06980(2015).
- Pettersen, E. F., Goddard, T. D., et al. UCSF Chimera?A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25 (13), 1605-1612 (2004).
- Penczek, P. A., Fang, J., Li, X., Cheng, Y., Loerke, J., Spahn, C. M. T. CTER-rapid estimation of CTF parameters with error assessment. Ultramicroscopy. 140, 9-19 (2014).
- Frank, J. Three-Dimensional Electron Microscopy of Macromolecular Assemblies. , Oxford University Press. (2006).
- Woolford, D., Ericksson, G., et al. SwarmPS: rapid, semi-automated single particle selection software. Journal of Structural Biology. 157 (1), 174-188 (2007).
- Yang, Z., Fang, J., Chittuluru, J., Asturias, F. J., Penczek, P. A. Iterative Stable Alignment and Clustering of 2D Transmission Electron Microscope Images. Structure/Folding and Design. 20 (2), 237-247 (2012).
- Gatsogiannis, C., Merino, F., et al. Membrane insertion of a Tc toxin in near-atomic detail. Nature Publishing Group. , (2016).
- Gatsogiannis, C., Lang, A. E., et al. A syringe-like injection mechanism in Photorhabdus luminescens toxins. Nature. 495 (7442), 520-523 (2013).
- Meusch, D., Gatsogiannis, C., et al. Mechanism of Tc toxin action revealed in molecular detail. Nature. 508 (7494), 61-65 (2014).
- Penczek, P. A., Frank, J., Spahn, C. M. T. A method of focused classification, based on the bootstrap 3D variance analysis, and its application to EF-G-dependent translocation. Journal of Structural Biology. 154 (2), 184-194 (2006).
- Emsley, P., Lohkamp, B., Scott, W. G., Cowtan, K. Features and development of Coot. Acta crystallographica. Section D, Biological crystallography. 66, Pt 4 486-501 (2010).
- Callaway, E. The revolution will not be crystallized: a new method sweeps through structural biology. Nature. 525 (7568), 172-174 (2015).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.