
Method Article
使用Cytofast和上游聚类方法流和细胞溅的高维细胞测量数据的可视化和量化
摘要
Cytofast是一种可视化工具,用于分析聚类输出。Cytofast可用于比较两种聚类方法:FlowSOM和Cytosplore。Cytofast可以快速生成质量细胞测量数据的定量和定性概览,并突出不同聚类算法之间的主要差异。
摘要
大规模细胞学生成的数据的复杂性使得需要新的工具来快速可视化分析结果。聚类方法,如细胞分裂或FlowSOM用于细胞簇的可视化和识别。对于下游分析,新开发的R包Cytofast可以生成聚类方法结果的快速可视化。Cytofast考虑细胞簇的表型特征,计算细胞簇丰度,然后定量比较组。该协议解释了Cytofast在肿瘤挑战后进行免疫治疗(PD-L1封锁)后,在肿瘤挑战下使用基于免疫系统在肿瘤微环境(即自然杀手[NK]细胞反应)的量大细胞测量数据的应用。演示了Cytofast与FlowSOM和Cytosplore的有用性。Cytofast可快速生成与群相关的免疫细胞簇的视觉表现,并与免疫系统组成相关。聚类分析中观察到差异,但两种聚类方法都可以看到组之间的分离。Cytofast直观地显示 PD-L1 治疗诱导的模式,包括更高丰度的激活 NK 细胞子集,表示激活标记的强度较高(即 CD54 或 CD11c)。
引言
大规模细胞测定(按飞行时间进行细胞测量,或CyTOF)允许在数百万个单细胞中检测广泛的细胞内或细胞外生物标志物。质量细胞测量数据的高维性需要某些分析工具,如SPADE 1、FlowMap2、FlowSOM 3、表象机4、VorteX 5和支架图6。此外,还开发了基于维数减少的各种技术(即主要分量分析[PCA]7、t分布随机邻域嵌入[t-SNE]8、分层随机邻域嵌入[HSNE]9、均匀的多维近似和投影[UMAP]10和扩散贴图11),以提高高维数据集的速度、解释和可视化。
对高维流量和质量细胞学数据的下游分析往往缺乏自动过程来对集群频率和与临床结果的联系进行统计测试。此前,我们开发了一种基于R的工作流程,称为Cytofast12,它允许通过Cytosplore或FlowSOM对聚类技术进行可视化和定量的下游分析。
此处描述的协议澄清了Cytofast在 R 中的使用,并说明了如何生成定量和定性热图和图形。此外,它有助于确定观察到的免疫表型和临床结果之间的联系。本报告还描述了使用两种不同的聚类过程对特定质量细胞测定数据集的分析:FlowSOM和Cytosplore。通过两种聚类方法使用Cytofast,相应地表明NK细胞的活化表型受PD-L1免疫检查点封锁的影响。
研究方案
所有动物实验均获得LUMC动物实验委员会的批准,并根据LUMC的动物实验准则按照荷兰和欧洲委员会的指导方针执行。
注:对于实验设置,C57BL/6小鼠在右侧用鼠结肠肿瘤MC38进行皮下接种,浓度为0.3 x 106细胞/200 μL的磷酸盐缓冲盐水(PBS)。10天后,当肿瘤明显时,小鼠接受PD-L1阻断抗体(克隆MIH-5,200μg/小鼠,腹内注射)或模拟治疗。肿瘤在PD-L1注射后3天后被切除,经过前体处理,并由CyTOF质量细胞测定用38个标记13进行分析。
1. 数据分析设备和软件
注:使用计算机(Windows 7 或更高版本)和处理器 I5 在 2.4 GHz 或等效,已安装内存 RAM 6 GB 和 10 GB 的可用硬盘空间。R包Cytofast使用现有的函数:主要是流核,热图,和ggplot。将在 R 中执行的命令行包含在协议中。R 指令的资源可在https://education.rstudio.com/找到。
- 要安装Cytofast软件包,请启动 R(版本"3.6"),然后输入以下代码来安装生物导体版本 3.9:
如果(!需要命名空间("生物管理器",安静 = TRUE))
安装.软件包("生物管理器")
生物管理器::安装("细胞快速") - 通过运行以下内容,确保包在所需的环境中加载:
库(细胞快)
2. 创建群集
注:为了展示两种聚类方法Cytosplore和FlowSOM与细胞快,NK细胞(CD161+)在PD-L1治疗后3天在肿瘤微环境中进行分析。
- 群集执行Cytosplore
- 下载并安装Cytosplore后,托管在 Cytosplore.org>,通过单击"文件" 上传 .fcs 文件(补充文件 1.1*1.8 [细胞分裂文件]) 在细胞中打开 FCS 文件。在提示时单击"添加唯一示例标记作为通道",并选择双曲弧形变换的系数(默认为 5),从而添加唯一的示例标记作为通道。
- 选择运行 H-SNE,运行 HSNE 级别 3,然后等待生成地图。
注:此步骤可能需要一些时间,具体取决于分析的细胞数量和选择的 HSNE 级别。 - 在第一个 HSNE 级别上,检查 CD161 阳性的细胞。选择 CD161+单元格,然后右键单击"缩放到选择"。在第二个级别上,重复此过程,以仅使用 CD161+事件达到第三级。
- 生成最后一个 tSNE 映射后,通过右键单击 tSNE 映射并选择"保存群集"来保存由 Cytosplore定义的聚类。根据Cytosplore提示选择输出文件的目录并记下此位置,因为此目录随后将用于将 .fcs 文件加载到 R 中。
注:可以通过更改西格玛值手动更改子集数。西格玛值默认设置为 30;但是,实际子集数取决于输入。在这里,Cytosplore检测到 10 个不同的子集,每个文件表示一个子集。 - 重命名输出文件时,请使用简单名称(仅限字符),这将使识别和进一步处理更加容易。通过选择"保存"保存输出文件。
注:保存后,Cytosplore正在创建一个文件夹,该文件夹包含 .fcs 文件,每个文件对应于Cytosplore中标识的群集。下一步将在Cytofast的帮助下将文件加载到R中。此处,生成的输出文件在补充文件 2.1_2.10(细胞分裂输出文件)中提供。 - 将细胞分裂生成的输出文件加载到 R 中,具有指定的功能:read CytosploreFCS。
dirFCS <- "C:\用户\用户名_桌面_研究"
cfData <- 读取 CytosploreFCS(dir = dirFCS,名称 = "描述") - 通过删除某些参数(如"时间"和"背景")来清理数据。检查列与其不必要参数相关的位置,并将其从矩阵中删除。
高名(cfData@expr)
cfData@expr [lt;- cfData@expr],-c(3,4,6,8:10,46:49,51:54)]
注:通过读取生成的矩阵的列名称,可以看到不必要的列。通过再次运行 colnames(cfData@expr),确保仅获得所需的参数。 - 重新排列标记,以便首先显示沿袭标记,然后显示功能标记。
cfData@expr <- cfData@expr],c(1,2,3,35,36,31,9,10,18,8,37,20,
29,40,5,30,33,11,34,14,19,
32,28,6,7,4,12,13,17,16,15,
21,22,24,25,26,27,38,39)]
注:步骤 2.1.8 是可选的。 - 通过上传包含临床信息的电子表格元文件(补充文件3 ),将元数据文件链接到来自Cytosplore的生成数据。
库(读本)
元 <- read_excel("C:\用户\用户名\桌面\sample_id.xlsx")
cfData@samples <- 数据.帧(元)
注:由细胞分裂者执行的聚类现已完成。Cytosplore的替代聚类选项是FlowSOM,第 2.2 节对此进行了介绍。执行两个聚类步骤之一后,继续可视化步骤(第 3 节)。
- 群集执行FlowSOM
- 通过运行以下命令在 R 中首次安装FlowSOM:
如果(!需要命名空间("生物管理器",安静 = TRUE))
安装.软件包("生物管理器")
生物经理::安装("FlowSOM")
库(流) - 使用类似方法安装 flowCore 包,并通过运行以下方法将其加载到环境中:
如果(!需要命名空间("生物管理器",安静 = TRUE))
安装.软件包("生物管理器")
生物管理器:安装("流核心")
库(流) - 使用 read.flowSet 函数在 R 中加载补充文件 4.1_4.8(FCS FlowSOM 输入)中提供的原始数据,这些数据以前在 CD161+ 事件上封闭。
fcs_raw <- 读取.flowSet(路径}C:\用户\用户名_桌面\群集",模式 = ".fcs",转换 = FALSE,truncate_max_range = FALSE,种子 =123) - 选择相关的生物标记(删除"背景"或"时间"),选择正确的列,并转换数据在弧形5的方式如下代码所示(此处,删除列1,2,4,5,6,17,21,24,25,34,35,37,38,51,不对应于任何生物标记)。在下面的函数中选择副因子=5,与之前在Cytosplore中应用的副因子5。
fcs_raw <- fs应用(fcs_raw,函数(x,副因子{5}}
共名(x) <- fcs_raw{1}@parameters@data$desc
expr <- exprs(x)
expr <- asinh(expr_,-c(1,2,4,5,6,17,21,24,25,34,35,37,38,51)*/共同因素)
expr(x) <- expr
返回(x)* - 使用FlowSOM函数对数据进行聚类。要比较FlowSOM和Cytosplore,请选择将数据集中到十个子集中,作为细胞分裂之前产生的输出。
fsom <- FlowSOM(fcs_raw,转换函数 = FALSE,缩放 = FALSE,
缩放.center = FALSE,缩放.scale = FALSE,静音 = FALSE,colsToUse = c(1:37),
nClus = 10,最大Meta = 10,重要性 = NULL,种子 = 123)
注:用户可以手动更改。 - 将每个单元格分配给其标识的子集和示例 ID。
subset_id <- 作为.因子(fsom$FlowSOM$map$映射{1})
级别(subset_id) <- fsom$元聚类
头(subset_id) - 在包含组分配的 R 中加载元数据文件(在补充文件 5中可用),并将其链接到 .fcs 文件。
示例 id <- read_excel("C:\用户\用户名\桌面\sample_id.xlsx")
示例 <- na.omit(示例)
示例$样本 ID <- 作为.因子(示例$示例ID)
示例$组 <- 作为.因子(样本$组)
样本$CSPLR_ST <- 作为.因子(样本$CSPLR_ST)
示例 id <- 作为.data.frame(示例id)
名称(示例)[3] <- "示例ID"
示例 ID <- l 应用(fsom$FlowSOM$metaData,函数(x)[rep(x=1],每个 = 长度(x{1}:x{2})))
attr(示例ID,"名称") <- NULL
示例 ID <- 作为因子(非列表(示例 ID))
示例 id <- 数据.帧(示例id)
级别(示例 ID)<- 粘贴("ID",1:dim(示例){1},sep="\""
df <- 数据.帧(subset_id、示例 ID、fsom$FlowSOM$data_,c(1:37)*)
重命名 <- 数据.帧(colpar_fcs_raw{1}@parameters@data$desc)
colnames(df)<-c("群集ID","示例ID",重命名$colpar_c(1:37)])
df$ 群集 ID <- 作为.因子(df$ 群集 ID)
df $sampleID <- 作为.因子(df$示例ID) - 通过运行以下脚本,基于从FlowSOM获得的数据帧创建 cfList:
cfData <- cfList(样本 = 样本,
expr = df) - 重新排序标记,使之与细胞分裂分析的输出类似。
cfData@expr <- cfData@expr],c(1,2,34,36,37,10,23,24,31,22,
38,15,8,3,27,9,11,28,35,26,
14,33,17,20,21,18,25,29,13,
30,12,16,32,4,5,6,7,19,39)]
注: FlowSOM的聚类现已完成。接下来,执行聚类输出的可视化。
- 通过运行以下命令在 R 中首次安装FlowSOM:
3. 可视化:后处理聚类分析
注意:此步骤是两种聚类方法共有的方法。因此,它可以在聚类后使用FlowSOM或Cytosplore执行。
- 在创建热图之前,请使用函数单元格Counts生成每个样本的计数表,如下代码所示。由于某些群集包含的单元格比其他群集少,因此通过在函数单元计数中指定"缩放 = TRUE"来缩放每个群集的数据,以便很容易看到样本之间的分散。
cfData <- 单元格计数(cfData、频率 = TRUE、刻度 = TRUE)
注:数据现在可以可视化。 - 使用热图可视化。
注:此包的主要功能之一是细胞热图,用于可视化所创建的聚类的表型及其相对于样本的异质性。
细胞热图(cfData,组="组",图例=真) - 使用框图进行可视化
注:可以通过调用函数细胞盒图以定量方式表示数据。此函数的输出表示每个群集的每个样本的比例。- 如在步骤 3.1 中所做的那样生成单元格计数,但不缩放数据以获取每个群集的频率。
cfData <- 单元格计数(cfData、频率 = TRUE、刻度 = FALSE)
细胞盒图(cfData,组 = "组")
- 如在步骤 3.1 中所做的那样生成单元格计数,但不缩放数据以获取每个群集的频率。
- 具有中值强度信号直方图的可视化
注:数据也可以通过表示中值强度信号直方图进行可视化。- 可视化三个标记的表达强度:CD45、CD11c 和 CD54。
- 通过调用以下行检查所需标记的名称。请注意标记名称并将其包含在 msiPlot 函数中。
名称(cfData@expr)
注:此处,重点关注 CD45、CD11c 和 CD54。检查标记的确切拼写,并根据需要进行调整:
msiPlot(cfData,标记 = c("89Y_CD45"、"167Er_CD11c"、"164Dy_CD54",按组"组")
结果
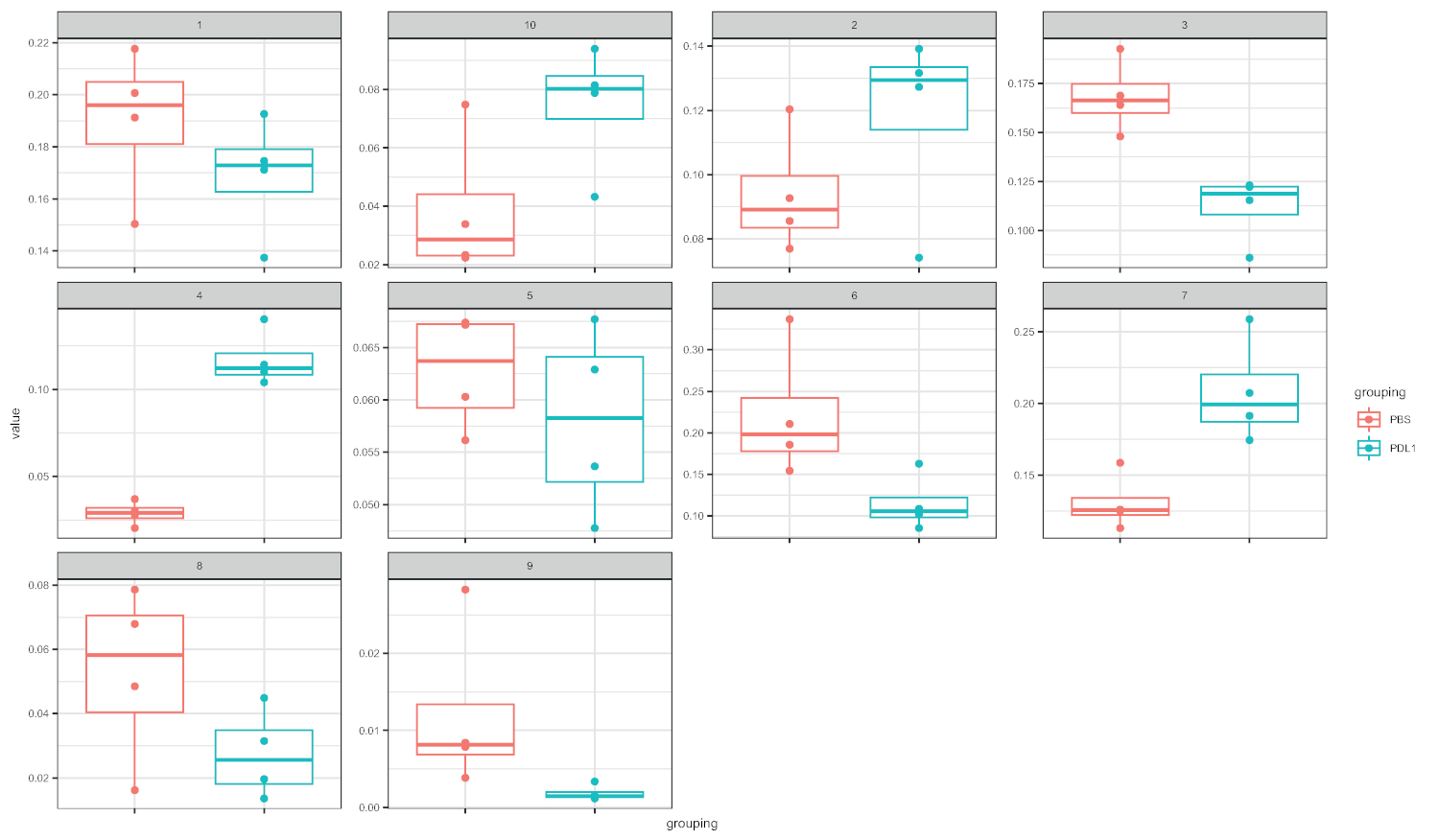
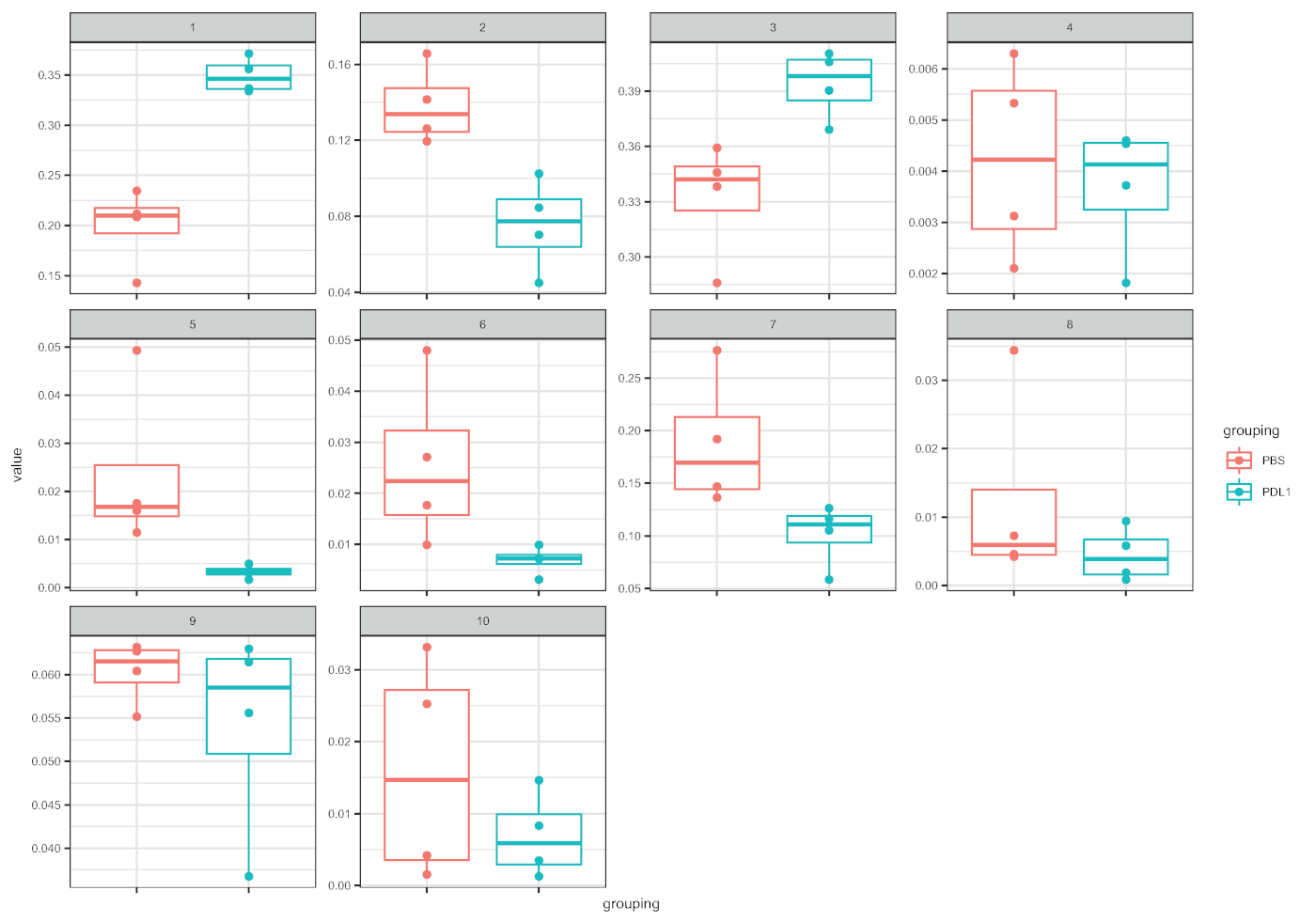
工作流Cytofast(图 1) 旨在对最初由分析软件(即FlowSOM或Cytosplore) 聚类的数据提供定量和定性概述。Cytofast运行多个可能的输出,包括分析中标识并基于标记表达式的所有聚类的热图(图 2和图 3)。顶部的树状图表示标识的群集之间的分层相似性。上部面板显示另一个热图,显示每个样本中相应子集的相对数量。右侧的树状图显示了样本之间的相似性,并且基于在欧氏体距离上执行的分层聚类。在图2中,为FlowSOM显示组合热图,后跟Cytofast,图3中为细胞快车,后为Cytofast。Cytofast还可用于定量显示数据,并在框图中显示结果(通过使用细胞因子图函数),如图4和图5所示。
在两种不同的方法之间发现了类似的聚类(例如,来自Cytosplore的簇8对应于FlowSOM中的簇10),并且PD-1和LAG-3等一些抑制标记的共表示在这两种方法中仍然可见。这两种聚类方法都允许对 PD-L1和之间进行区分。PBS治疗小鼠。相反,可以突出显示两种方法之间的一些差异。FlowSOM识别 2 个群集 (MHC-II=),而细胞群只显示一个群集 (MHC-II+dim)。这是由于最初的门控策略,其中NK细胞被手动封闭在CD161+细胞上,然后由FlowSOM进一步处理。然而,Cytosplore自动封闭来自CD45的细胞——第一个HSNE水平上的群,然后聚集在更高的层次层次。因此,Cytosplore比手动浇注在CD161上更精确地定义了NK细胞子集。然而,如右侧的树状图所示,保留了样本的分层聚类,这表明两个组(PD-L1 和 PBS)之间的隔离不依赖于所选的聚类方法。
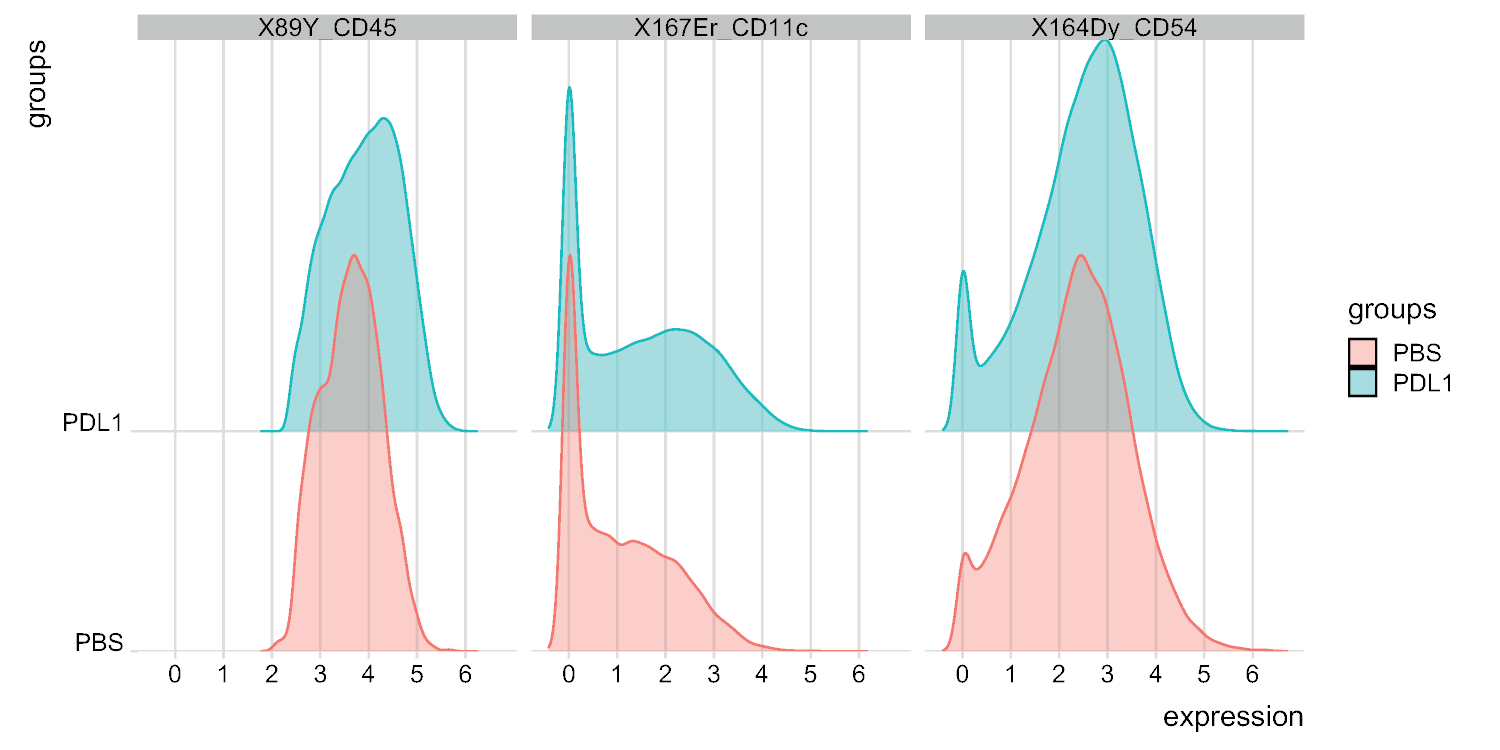
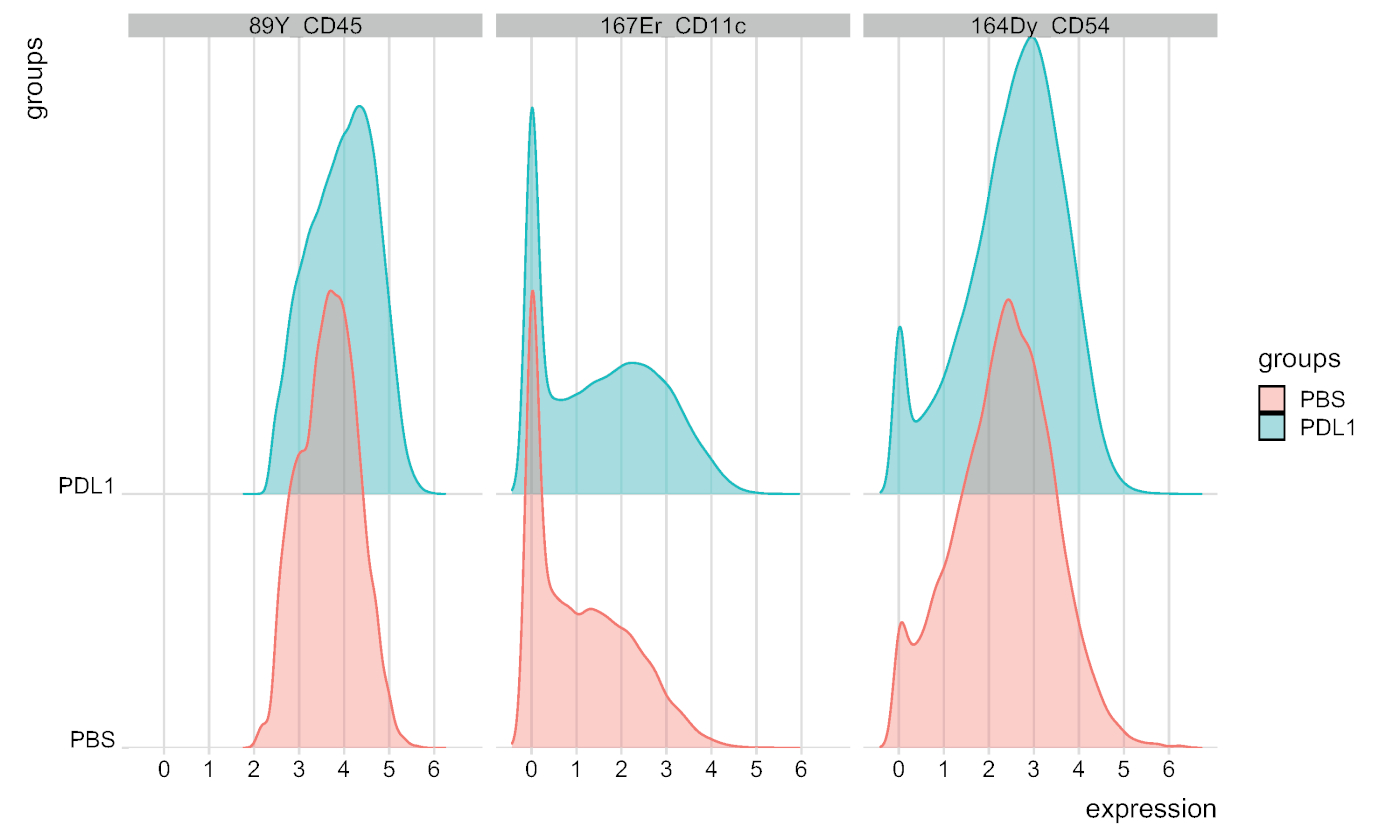
可以使用这两种方法手动定义群集数。Cytofast使用户能够评估其数据的异质性,并可以深入了解如何选择数据应划分的群集数。Cytofast封装中包括其他功能,如 msiPlot 函数(步骤 3.4.2),显示每个组每个标记的中值信号强度 (MSI) 图(图 6和图 7)。此功能允许检测全局变化,例如 PD-L1 处理组的 NK 细胞中 CD54 或 CD11c 表达的增加。可选功能可以集成在Cytofast封装中,例如以条形图和其他数据表示方法显示数据。后者需要添加 ggplot 工具,这些工具可以由 R 生成。

图1:Cytofast封装的工作流。这些数据是通过在免疫治疗治疗后3天从肿瘤中进行大量细胞测定产生的,或者未经治疗。比较了两种不同的聚类技术:细胞分裂和流水。Cytofast用于可视化这两种技术之间的差异。请点击此处查看此图的较大版本。
{kind=link}

图2:聚类概述和聚类丰度按Cytosore以下的Cytofast分析。所有NK细胞簇的热图(CD161+细胞由细胞分裂自动定义),在免疫治疗(PD-L1)后3天被识别。显示的数据基于细胞分裂,并汇集从未经处理和PD-L1处理的组。ArcSinh5 转换的表达式标记的级别以彩虹比例显示。在下面板上,每个样品的相对丰度由绿色到紫色的比例表示。右侧的树状图表示基于子集频率的样本之间的相似性。频率刻度表示平均值的色散。低频率或高频分别由绿色或紫色表示。请点击此处查看此图的较大版本。
{kind=link}

图3:按FlowSOM之后的Cytofast分析的每个组的聚类概述和聚类丰度。所有NK细胞簇的热图(CD161+事件预封闭),在免疫治疗(PD-L1)后3天确定。显示的数据基于FlowSOM聚类,并汇集自未经处理和 PD-L1 处理的组。ArcSinh5 转换的表达式标记的级别以彩虹比例显示。在下面板上,每个样品的相对丰度由绿色到紫色的比例表示。右侧的树状图表示基于子集频率的样本之间的相似性。频率刻度表示平均值的色散。低频率或高频分别由绿色或紫色表示。请点击此处查看此图的较大版本。
{kind=link}

图4:细胞快速表示,具有由细胞分裂定义的聚类的框图。每个群集的频率以箱线图表示,分为两个组(PBS 和 PD-L1)。单个点对应于一个鼠标。请点击此处查看此图的较大版本。
{kind=link}

图 5:由 FlowSOM定义的聚类的框图的Cytofast表示。每个群集的频率以箱线图表示,分为两个组(PBS 和 PD-L1)。单个点对应于一个鼠标。请点击此处查看此图的较大版本。
{kind=link}

图6:NK细胞信号强度图的分布由细胞自动封闭。信号强度的分布以直方图显示,用于三个特定标记:CD45、CD11c 和 CD54。请点击此处查看此图的较大版本。
{kind=link}

图7:由FlowSOM自动封闭NK细胞的信号强度图分布。信号强度的分布以直方图显示,三个特定标记:CD45、CD11c 和 CD54,按 PBS 和 PD-L1 组分隔。请点击此处查看此图的较大版本。
{kind=link}
补充文件 1.1_1.8。请点击此处查看此文件(右键单击下载)。
补充文件 2.1_2.10。请点击此处查看此文件(右键单击下载)。
补充文件3.请点击此处查看此文件(右键单击下载)。
补充文件 4.1_4.8。请点击此处查看此文件(右键单击下载)。
补充文件5.请点击此处查看此文件(右键单击下载)。
讨论
Cytofast是一种快速计算工具,通过突出显示和量化特定于治疗的细胞子集,提供对细胞测量数据的快速全球探索。所述协议旨在进一步使用细胞聚子或FlowSOM进行过程聚类分析。其他聚类分析工具适用于Cytofast,但这需要使用Cytofast将每个细胞分配给一个子集。然而,Cytofast不是一种聚类方法,因此在使用之前需要聚类过程.
分析表明,肿瘤微环境中的某些CD161+NK细胞子集对PD-L1阻断敏感。这一点从它们的表型和丰度的变化中得到证明,这些变化是使用Cytosplore和FlowSOM作为聚类方法观察到的。两种方法都区分了主要NK细胞簇(CD11b+ NKG2A+),频率略有不同(细胞孢子为15%-20%,FlowSOM为30%~40%)。 丰度差异和此近似值不会影响全局模式,因为图 2和图3右侧面板中显示的树状图都显示了类似的结果。通过使用Cytofast,因此可以(独立于选择的聚类方法)根据NK细胞簇状型和丰度分析分离PD-L1处理和未经治疗的小鼠。
根据记录的参数,需要修改协议。具体而言,在执行聚类分析时,必须删除某些参数(如时间和背景)。此外,将每个单元格分配给一个子集也很重要。cfData 函数只需将每个样本每个群集的原始单元计数添加到 cfList 中。从此步骤中,可以构建细胞热图,如第 3 节所述。
Cytofast已经成功地用作可视化和定量工具,比较不同的聚类方法13。此 R 包还与高级功能兼容,例如globaltest14,它可以使用临床变量测试群集组之间的关联。将来,全球测试工具和其他算法可以与Cytofast集成,以便进行更深入的可视化和量化。
披露声明
作者没有什么可透露的。
致谢
我们确认欧盟委员会根据提案号 675743 (ISPIC) 获得 H2020 MSCA 奖励。我们感谢特杰·范德·斯卢伊斯和艾里斯·帕迪克测试了协议。
材料
| Name | Company | Catalog Number | Comments |
| Computer | Dell | NA | NA |
参考文献
- Anchang, B., et al. Visualization and cellular hierarchy inference of single-cell data using SPADE. Nature Protocols. 11 (7), 1264-1279 (2016).
- Zunder, E. R., Lujan, E., Goltsev, Y., Wernig, M., Nolan, G. P. A continuous molecular roadmap to iPSC reprogramming through progression analysis of single-cell mass cytometry. Cell Stem Cell. 16 (3), 323-337 (2015).
- Van Gassen, S., et al. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A. 87 (7), 636-645 (2015).
- Levine, J. H., et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell. 162 (1), 184-197 (2015).
- Samusik, N., Good, Z., Spitzer, M. H., Davis, K. L., Nolan, G. P. Automated mapping of phenotype space with single-cell data. Nature Methods. 13 (6), 493-496 (2016).
- Spitzer, M. H., et al. An interactive reference framework for modeling a dynamic immune system. Science. 349 (6244), 1259425 (2015).
- Hotelling, H. Analysis of a complex of statistical variables into principal components. Journal of Educational Psychology. 24 (6), 417-441 (1933).
- van der Maaten, L., Hinton, G. Visualizing Data using t-SNE. Journal of Machine Learning Research. , (2008).
- Pezzotti, N., Hollt, T., Lelieveldt, B., Eisemann, E., Vilanova, A. Hierarchical Stochastic Neighbor Embedding. Computer Graphics Forum. 35 (3), 21-30 (2016).
- Becht, E., et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nature Biotechnology. 37, 38 (2018).
- Haghverdi, L., Buettner, F., Theis, F. J. Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics. 31 (18), 2989-2998 (2015).
- Beyrend, G., Stam, K., Höllt, T., Ossendorp, F., Arens, R. Cytofast: A workflow for visual and quantitative analysis of flow and mass cytometry data to discover immune signatures and correlations. Computational and Structural Biotechnology Journal. 16, 435-442 (2018).
- Beyrend, G., et al. PD-L1 blockade engages tumor-infiltrating lymphocytes to co-express targetable activating and inhibitory receptors. Journal for ImmunoTherapy of Cancer. 7 (1), 217 (2019).
- Goeman, J. J., van de Geer, S. A., de Kort, F., van Houwelingen, H. C. A global test for groups of genes: testing association with a clinical outcome. Bioinformatics. 20 (1), 93-99 (2004).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。