
Method Article
Dreidimensionale Motornerve Organoid Generation
In diesem Artikel
Zusammenfassung
Dieses Protokoll bietet ein umfassendes Verfahren zur Herstellung des menschlichen iPS-Zell-abgeleiteten motorischen Nervenorganoids durch spontane Montage eines robusten Bündels von Axonen, das von einem Sphäroid in einem Gewebekulturchip erweitert wird.
Zusammenfassung
Eine Faszikel von Axonen ist eines der wichtigsten strukturellen Motive im Nervensystem beobachtet. Eine Störung der Axonfaszien könnte entwicklungs- und neurodegenerative Erkrankungen verursachen. Obwohl zahlreiche Studien mit Axonen durchgeführt wurden, ist unser Verständnis der Bildung und Dysfunktion von Axonfascicles aufgrund des Mangels an robusten dreidimensionalen In-vitro-Modellen immer noch begrenzt. Hier beschreiben wir ein Schritt-für-Schritt-Protokoll zur schnellen Generierung eines Motornervenorganoids (MNO) aus humaninduzierten pluripotenten Stammzellen (iPS) in einem mikrofluidischen Gewebekulturchip. Zunächst wird die Herstellung von Chips beschrieben, die für das Verfahren verwendet werden. Aus menschlichen iPS-Zellen wird ein motorischer Neuronsphäroid (MNS) gebildet. Als nächstes wird das differenzierte MNS in den Chip übertragen. Danach wachsen Axone spontan aus dem Sphäroid heraus und fügen sich in einem Mikrokanal, der im Chip ausgestattet ist, zu einem Faszikel zusammen, der ein MNO-Gewebe erzeugt, das ein Bündel von Axonen trägt, das aus dem Sphäroid ausgestreckt wird. Für die nachgelagerte Analyse können MNOs aus dem Chip entnommen werden, um für morphologische Analysen fixiert oder für biochemische Analysen sowie Kalzium-Bildgebung und Multi-Elektroden-Array-Aufnahmen seziert zu werden. Mit diesem Protokoll generierte MNOs können Arzneimitteltests und -screenings erleichtern und zum Verständnis der Mechanismen beitragen, die der Entwicklung und Erkrankungen von Axonfaszikeln zugrunde liegen.
Einleitung
Spinale motorische Neuronen (MN) erweitern Axone auf Skelettmuskeln, um die Körperbewegung zu steuern. Ihre axonalen Bahnen sind im Entwicklungsprozess hoch organisiert und reguliert. Trotz vieler Studien über Axonverlängerung und Anleitung1werden die Mechanismen für die organisierte Axonbündelbildung noch untersucht. Axone von motorischen Neuronen werden oft durch neurodegenerative Erkrankungen wie amyotrophe Lateralsklerose (ALS)2geschädigt, aber pathophysiologische Mechanismen der Schäden an Axonfascicles sind schlecht verstanden. Daher ist ein physiologisches und pathologisches Modell zur Rekapitulation der Axonbündelbildung und Regression im Feld erforderlich.
Ein menschliches, von Stammzellen abgeleitetes motores Neuron ist eine vielversprechende Plattform, um die Entwicklung und Krankheiten wie ALS3zu verstehen. Humaninduzierte pluripotente Stammzellen (iPS-Zellen) können verwendet werden, um Krankheiten mit Patienten-abgeleiteten Zellen zu modellieren. Bis heute wurden verschiedene Differenzierungsmethoden von pluripotenten Stammzellen in MN4,5,6berichtet. Axone von Neuronen in zweidimensionaler Kultur sind jedoch zufällig orientiert und rekapitulieren nicht in vivo Mikroumgebung innerhalb sich entwickelnder Nerven, in denen Axone durch dichte axo-axonale Wechselwirkungen unidirektional zusammengesetzt werden7. Um dieses Problem zu überwinden, haben wir eine Technik entwickelt, um ein dreidimensionales Gewebe zu erzeugen, das dem motorischen Nerv aus den menschlichen iPS-Zellen8ähnelt, und das Gewebe als Motornervenorganoid (MNO) bezeichnet. Das MNO besteht aus Zellkörpern, die sich in einem motorischen Neuronensphäroid (MNS) und einem axonalen Faszikel befinden, das aus dem Sphäroid ausgestreckt ist. Die Axone im Faszikel sind unidirektional ausgerichtet, was Axonen bei der Entwicklung von Motornerven ähnelt. Daher bieten MNOs auf einzigartige Weise eine physiologische axonale Mikroumgebung, was von keiner anderen zuvor entwickelten neuronalen Kulturmethode durchgeführt wurde.
In diesem Protokoll beschreiben wir Methoden für die Gewebekultur-Chips-Herstellung, schnelle motorische Neurondifferenzierung und motorische Nervenorganoidbildung in entwickelten Chips. Unser Gewebekultur-Chip ist sehr einfach, und er enthält nur ein Fach zum Akzeptieren eines Sphäroids, einen Mikrokanal zur Bildung eines Axonbündels und ein Fach für das Gehäuse von Axonklemmen. Das Gerät enthält keine komplexen Strukturen, einschließlich Mikrogrooves oder Mikroporenfilter, die häufig verwendet werden, um Axone und Zellkörper nach Größe9,10zu trennen. Daher können unsere Geräte einfach hergestellt werden, indem Sie die in diesem Protokoll beschriebenen Schritte befolgen, wenn ein Photolithographie-Setup verfügbar ist.
Eine schnelle Differenzierung menschlicher iPS-Zellen wird durch eine optimierte Kombination von Induzierenden und Musterfaktoren (SB431542, LDN-193189, Retinoinsäure (RA) und geglätteten Agonisten (SAG)) und Beschleunigungsfaktoren (SU5402 und DAPT) erreicht. Es wurde berichtet, dass die Kombination von SU5402 und DAPT die Differenzierung von peripheren Neuronen und neuronalen Kammzellenbeschleunigt 11. In diesem Protokoll bieten wir drei verschiedene Methoden zur Generierung von MNOs an, damit Leser sich für eine Methode entscheiden können, die ihren Bedürfnissen am besten entspricht. Wir empfehlen die Differenzierung menschlicher iPS-Zellen nach der Bildung eines Sphäroids (die 3D-Methode), da das differenzierte MNS direkt in einen Gewebekulturchip übertragen werden kann. Alternativ können menschliche iPS-Zellen in monolayer (2D)-Kultur in motorische Neuronen unterschieden und dann zu dreidimensionalen motorischen Neuronsphäroiden erzeugt werden, wie wir zuvor berichteten8. Wir haben das Protokoll aktualisiert, und mit der in diesem Protokoll beschriebenen dreidimensionalen Differenzierungsmethode kann der Übergang von 2D zu 3D vermieden werden und MNOs können mit kürzerer Differenzierungszeit, weniger Schritten und reduzierten technischen Risiken ohne den Dissoziationsschritt erhalten werden. Kommerziell erhältliche Neuronen können auch verwendet werden, um MNS zu generieren, um die Zeit für differenzierung zu reduzieren.
Um ein MNO zu generieren, haben wir ein MNS im Gewebekulturchip kultiviert. Die Axone verlängern sich aus dem Sphäroid und erstrecken sich in den Mikrokanal, in dem sich Axone unidirektional sammeln und ausrichten. Dies erleichtert die axo-axonale Interaktion und spontane Bildung eines eng zusammengesetzten unidirektionalen Bündelgewebes von Axonen im Mikrokanal, das durch dieses Protokoll einzigartig erreicht wird, während entweder eine spontane Bündelbildung oder eine geführte axonale Orientierung allein durch andere Protokolle12,13,14erreicht werden kann. In einem typischen Experiment wandern nur wenige Zellen aus Sphäroiden zum Mikrokanal und die meisten Zellen bleiben in der Nähe von Sphäroiden. Diese Methode ermöglicht es, Axone spontan von den Sphäroiden zu trennen, ohne größenabhängige physikalische Barrieren (z. B. Mikronuten oder Mikroporenfilter) zu verwenden, um Axone von Zellkörpern zu trennen.
Das resultierende MNO kann verschiedenen Untersuchungen unterzogen werden, einschließlich morphologischer, biochemischer und physikalischer Analysen. Der Zellkörper und das verlängerte Axonbündel können durch Schneiden physikalisch isoliert und für nachgelagerte Experimente, z.B. biochemische Assays, separat analysiert werden. Biologische Materialien wie RNA und Protein können aus wenigen Axonbündeln für regelmäßige biochemische Tests wie RT-PCR und Western Blotting isoliert werden. Hier beschreiben wir ein Protokoll zur Erzeugung von Motornervenorganoid aus iPS-Zellen, das ein attraktives physiologisches und pathologisches Modell bietet, um den Mechanismus zu untersuchen, der der Entwicklung und Krankheit von Axonfascicles zugrunde liegt.
Protokoll
1. SU-8 Formherstellung durch Photolithographie
HINWEIS: Bei diesem Verfahren handelt es sich um gefährliche Chemikalien. Verwenden Sie Rauchhaube und PSA durchgängig.
- Den Siliziumwafer (4 Zoll Durchmesser, 1 mm Dicke, poliert) mit Aceton reinigen und mit Stickstoffgas blasen. Dann bei 180 °C für 3 min zum Trocknen backen.
- Geben Sie 3 ml SU-8 2100 auf einen gereinigten Wafer auf.
- Die SU-8 gleichmäßig auf dem Wafer mit einem Spincoater bei 500 U/min für 10 s beschichten und dann nacheinander bei 1500 U/min für 30 s mit einer Beschleunigung von 300 U/m/s drehen, um eine 150 m dicke Schicht SU-8 zu erhalten.
HINWEIS: Stellen Sie sicher, dass der Siliziumwafer in der Mitte des Spincoaters positioniert und durch Vakuum korrekt fixiert ist. - Den Wafer auf der Kochplatte bei 50 °C für 10 min, bei 65 °C 7 min und bei 95 °C für 45 min weich backen.
- Stellen Sie die Fotomaske (Abbildung 1) auf den Maskenausrichter ein und setzen Sie UV-Licht (365 nm) für 60 s aus.
HINWEIS: Die Belichtungszeit muss durch eine angemessene Dosis UV-Licht optimiert werden. - Nach der Belichtung den Wafer bei 65 °C für 6 min und bei 95 °C für 13 min auf der Kochplatte backen.
- Entwickeln Sie den Wafer für 10-20 min in SU-8 Entwickler mit Agitation mit einem Orbital-Shaker, die Entwicklung der Lösung einmal während des Prozesses ändern.
HINWEIS: Verlängern Sie die Entwicklungszeit, wenn die Trümmer von SU-8 verbleiben. - Spülen Sie den Wafer in Isopropanol und trocknen Sie den Wafer vorsichtig mit Stickstoffgas.
- Messen Sie die Höhe des abgelagerten SU-8 mit einem Messmikroskop und stellen Sie sicher, dass er ca. 150 m dick ist. Es kann auf unbestimmte Zeit bei Raumtemperatur gelagert werden.
2. PDMS mikrofluidische Gewebekultur Chip-Fertigung
- Befestigen Sie den SU-8-abgelagerten Wafer mit doppeltem Seitenband in einem Behälter (z. B. 15 cm Kunststoff Petrischale).
- Blasen Sie den Staub mit Stickstoffgas vom Wafer.
- Zum Silanisieren den SU-8-abgelagerten Wafer zusammen mit einem kleinen Behälter (z.B. 35 mm Schale) in eine Vakuumkammer geben. Lassen Sie 10 l (Tridecafluor-1,1,2,2-Tetrahydrooctyl)-1-Trichlorsilan in den kleinen Behälter fallen. Das (Tridecafluor-1,1,2,2-Tetrahydrooctyl)-1-Trichlorsilan nicht direkt auf SU-8-Wafer auftragen.
- Schließen Sie die Vakuumkammer fest und schalten Sie eine Vakuumpumpe für mindestens 2 h ein.
- Nehmen Sie einen Plastikbecher und gießen Sie das Silikonelastomer (z.B. Silpot 184 oder entsprechend Sylgard 184) und das Härtungsmittel mit einem Gewichtsverhältnis von 10:1. Dann gut mit einem Spachtel und Degas in der Vakuumkammer mischen, bis Blasen vollständig entfernt werden.
- Gießen Sie das PDMS-Gemisch mit dem SU-8-Wafer auf die gewünschte Dicke (3-4 mm) in den Behälter und entgasen Sie erneut, um Blasen zu entfernen.
- Backen Sie das PDMS im Ofen bei 60 °C für mindestens 3 h, um das PDMS vollständig auszuhärten.
- Nach dem Abkühlen das ausgehärtete PDMS mit einem Skalpell oder einer Rasierklinge vom Wafer abschneiden.
- Um die beiden Kammern des Gewebekulturchips zu erstellen, schlagen Sie zwei Löcher, in denen sich die beiden Fächer befinden, indem Sie einen Biopsie-Punch mit einem Durchmesser von 1,5 mm verwenden.
- Um ein mittleres Reservoir zu erstellen, bereiten Sie eine weitere PDMS-Mischung (Silikonelastomer und das Härtungsmittel bei einem Gewichtsverhältnis von 10:1) vor und gießen Sie es in eine neue 10 cm Petrischale. Stellen Sie das Gießvolumen auf 5 mm Dicke von PDMS ein.
- Backen Sie das PDMS im Ofen bei 60 °C für mindestens 3 h, um das PDMS vollständig auszuhärten.
- Nach dem Abkühlen des PDMS schneiden Sie das ausgehärtete PDMS mit einem Skalpell ab, um einen rechteckigen Ring zu erhalten.
- Verkleben Sie die untere Schicht mit dem medium Reservoir, indem Sie ungehärtetes PDMS zwischen ihnen auftragen und die montierten PDMS-Schichten backen. Diese gebundene Struktur führt zum PDMS-Gewebekulturchip.
- Reinigen Sie den PDMS-Gewebekulturchip mit einem Scotch-Tape, um Staub und kleine Partikel von der Oberfläche zu entfernen. PDMS Gewebekulturchips können bei Raumtemperatur gelagert werden, wenn sie vor Staub und UV geschützt sind.
3. Vorbereitung der Kultur
- Kulturmedium
HINWEIS: Alle unten aufgeführten Medien sollten für die Sterilisation gefiltert werden, sofern nicht anders angegeben. Vorbereitete Medien können bei 4 °C gelagert und innerhalb eines Monats verwendet werden.- Zur Zubereitung von mTeSR Plus Medium: Kombinieren Sie eine Flasche 100 ml mTeSR Plus 5x Ergänzung mit einer Flasche 400 ml mTeSR Plus Basal Medium.
- Zur Vorbereitung von 100 ml KSR-Medium: In 85 ml DMEM/F12 15 ml Knockout-Serumersatz (KSR, 15%), 1 ml kommerzielleglutamin-Ergänzung (1%) hinzufügen und 1 ml nicht essentielle Aminosäure (NEAA, 1%).
- Zur Vorbereitung 100 ml N2 Medium: In 100 ml neurobasalen Mediums 1 ml N2 (100x), 1 ml kommerzielle Glutaminergänzung und 1 ml NEAA hinzufügen.
- Zur Vorbereitung von 250 ml Reifungsmedium: In 250 ml neurobasalen Mediums 5 ml B27 (2%), 2,5 ml kommerzielleGlutaminergänzung (1%) und 2,5 ml Penicillin/Streptomycin (1%) hinzufügen.
- Setzen Sie die Verbindungen (Retinosäure (RA), SB431542, LDN-193189, SU5402, DAPT, SAG, Y-27632) in der Zellkultur-Grade-DMSO auf die gewünschte Konzentration aus. Aliquots vorbereiten und bis zu 6 Monate bei -20 °C lagern. Folgende Lagerlösungen kommen zum Einsatz: 1 mM RA, 10 mM SB431542, 100 m LDN-193189, 10 mM SU5402, 10 mM DAPT, 1 mM SAG, 10 mM Y-27632.
- Beschichtung
HINWEIS: Um eine Polymerisation der Kellermembranmatrix durch Hitze zu verhindern, vermeiden Sie sich wiederholende Gefrier-Tau-Zyklen. Behandeln Sie alle Beschichtungsverfahren mit vorgekühlten Pipettenspitzen und Rohren, wenn möglich. Die Kellermembranmatrix sollte über Nacht bei 4 °C aufgetaut und mit vorgekühlten Pipettenspitzen und -rohren aliquotediert werden. Die Aliquots können bei -20 °C oder -80 °C eingefroren werden.- Das gefrorene Aliquot bei 4°C auf Eis auftauen. Das Aliquot sollte während des Beschichtungsvorgangs kalt gehalten werden. Mit einer vorgekühlten Pipettenspitze die Kellermembranmatrix mit eiskaltem DMEM/F12 im Verhältnis 1:40 verdünnen. Nicht verwendete verdünnte Kellermembranmatrix kann bei 4 °C für 2-3 Tage gelagert werden, da keine Polymerisation aufgetreten ist.
- Fügen Sie 1 ml der Kellermembranmatrix/DMEM-F12 Lösung hinzu, um einen Brunnen der 6 Wellplatte zu beschichten.
- Inkubieren Sie die Platte bei Raumtemperatur für mindestens eine Stunde oder 4 °C über Nacht. Beschichtete Platten können maximal eine Woche bei 4 °C gelagert werden.
4. Wartung von iPS-Zellen
HINWEIS: Undifferenzierte iPS-Zellen werden in mTeSR Plus-Medium und Subkulturbeibehalten beibehalten, wenn die Konfluenz von ≥ 90 % in einer 6-Well-Platte in diesem Protokoll beobachtet wird. Für iPS-Zellen, die in anderen Medien kultiviert werden, sind möglicherweise geringfügige Anpassungen erforderlich.
- Bereiten Sie Kellermembran-Matrix-beschichtete Gerichte vor, wie bereits in Schritt 3.2 erwähnt.
- Das mTeSR Plus Medium vollständig aspirieren. Waschen Sie den Brunnen einmal mit PBS und fügen Sie 0,5 ml Passaging-Reagenz hinzu (siehe Tabelle der Materialien). Warten Sie einige Sekunden und saugen Sie die Lösung an.
- Inkubieren Sie die Platte bei 37 °C im Inkubator für 5 min oder bis die Zellen rund werden.
HINWEIS: Die Inkubationszeit kann zwischen verschiedenen iPS-Zelllinien und der Konfluenz variieren. Bitte überprüfen Sie regelmäßig unter dem Mikroskop, um die Dissoziationszeit während der Inkubation zu bestimmen. - Fügen Sie 1 ml mTeSR Plus Medium hinzu und tippen Sie auf die Platte für 30-60 s, um die Kolonien zu lösen.
- 1 ml der Zellsuspensionslösung mit 7 ml frischem mTeSR plus Medium vorsichtig mischen. Pipette nicht mehr als 5 Mal.
- Platte im Verhältnis 1:8. In der Regel 1 ml der Suspension aus Schritt 4.5 hinzufügen und 1 ml mTeSR plus Medien hinzufügen, ergänzt durch 5-10 'M y-27632 (ROCK-Hemmer). Das Verdünnungsverhältnis der Passage hängt von der iPSC-Leitung ab.
- Legen Sie die Zelle in einen 5% CO2/37 °C Inkubator. Entfernen Sie am nächsten Tag Y-27632, indem Sie ein frisches mTeSR Plus-Medium hinzufügen. Danach wechseln Sie die Medien zunächst jeden zweiten Tag und jeden Tag, wenn die Zelle eine höhere Konfluenz erreicht.
5. Differenzierung von iPS-Zellen in motorische Neuronen
HINWEIS: Alle optionen unten (5.2, 5.3 und 5.4) erzeugen MNOs mit > 90% Effizienz.
- Passaging iPSC zur motorischen Neurondifferenzierung
HINWEIS: Die Differenzierung kann entweder in 3D-Protokollen (5.2) oder 2D (5.3) erfolgreich durchgeführt werden.- Lassen Sie undifferenzierte iPS-Zellen wachsen, bis sie eine Konfluenz von ca. 80% in mTeSR Plus Medium in einer 6-Well-Platte erreicht.
- Das Medium vollständig ansaugen. Waschen Sie den Brunnen sofort einmal mit sterilem PBS und fügen Sie 0,5 ml Zelldissoziationslösung in die Zellen.
- Inkubieren Sie die Platte bei 37 °C im Inkubator für ca. 2-3 min, oder bis die Zellen abgetrennt und rund werden, aber am Brunnen befestigt bleiben.
- Mit einer 5 ml serologischen Pipette 1 ml Medium und sanfte Pipette ein paar Mal nach oben und unten geben. Übertragen Sie die Zellsuspension in ein 15 ml Rohr, das aus 4 ml des Mediums besteht.
- Zentrifuge bei 200 x g für 3 min.
- Den Überstand vorsichtig ansaugen, das Pellet ungestört lassen und die Zellen in 1 ml des Mediums, ergänzt durch 10 M Y-27632, wieder aussetzen.
- Zählen Sie die Zellen mit einem Hämozytometer und fahren Sie entweder zu 5,2 (3D-Differenzierung) oder 5,3 (2D-Differenzierung).
- Bildung von Motorneuronsphäroid (MNS) in 3D-Differenzierung ("3D-Protokoll")
HINWEIS: Eine vollständige mittlere Änderung erfolgt täglich ab den Tagen 0-12 der Differenzierung (Abbildung 2).- Säen Sie die iPS-Zellen von Schritt 5.1.7 bis zu einer 96 Well U Bodenplatte bei 40.000 Zellen/Well in 100 l mTeSR Plus, ergänzt um 10 m Y-27632.
- Am nächsten Tag jeden Brunnen durch 100 l des frischen Mediums ersetzen.
- An den Tagen 0 und 1: Aspirieren Sie das Kulturmedium und ersetzen Sie es durch 100 l KSR-Medium (3.1.2), ergänzt durch 10 m SB431542 und 100 nM LDN-193189.
- An den Tagen 2 und 3: Saugen Sie das Kulturmedium und ersetzen Sie es durch 100 l KSR-Medium, ergänzt durch 10 M SB431542, 100 nM LDN-193189, 5 'M DAPT, 5 'M SU5402, 1'M RA und 1 'M SAG.
- An den Tagen 4 und 5: Bereiten Sie ein Mischmedium vor, das aus 75% KSR medium und 25% N2 medium (3.1.3) besteht. Dann saugen Sie das Kulturmedium an und ersetzen Sie es durch 100 l Mischmedium, ergänzt durch 10 M SB431542, 100 nM LDN-193189, 5 'M DAPT, 5'M SU5402, 1'M RA und 1'M SAG.
- An den Tagen 6 und 7: Bereiten Sie ein Mischmedium vor, das aus 50% KSR medium und 50% N2 medium besteht. Dann, Aspirat-Kultur-Medium und ersetzen Sie es durch 100 l gemischtes Medium ergänzt mit 5 M DAPT, 5 M SU5402, 1 M Retinsäure und 1 M SAG.
- An den Tagen 8 und 9: Bereiten Sie ein Mischmedium vor, das aus 25% KSR medium und 75% N2 medium besteht. Dann, Aspirat-Kultur-Medium und ersetzen sie mit 100 l gemischtes Medium ergänzt mit 5 M DAPT, 5 M SU5402, 1 M RA und 1 M SAG.
- An den Tagen 10 und 11: Ersetzen Sie das Medium durch 100 l N2-Medium, ergänzt durch 5 M DAPT, 5 M SU5402, 1 M RA und 1 M SAG.
- Am 12. Tag: Übertragen Sie die MNs in den Gewebekulturchip (Schritt 6) oder ersetzen Sie das Medium durch 100 l des Reifungsmediums (3.1.4), ergänzt durch den 20 ng/mL-neurotrophen Faktor (BDNF).
HINWEIS: Das MNS kann ab Tag 12 bis 19 auf den Gewebekulturchip übertragen werden. Sphäroide, die nicht übertragen werden, sollten bis zur Übertragung in 96 Well U Bodenplatten im Reifungsmedium kultiviert gehalten werden, ergänzt mit 20 ng/mL BDNF.
- (Alternative Option) 2D-Differenzierung und Übergang zu 3D-MNS ("2D-Protokoll")
- Saugen Sie die Beschichtungslösung aus einer vorbeschichteten 12-Well-Platte.
- Säen Sie die iPS-Zellen aus Schritt 5.1.7 mit einer Dichte von 100.000 – 200.000 Zellen pro Brunnen in mTeSR Plus Medium mit 10 m Y-27632.
HINWEIS: Die undifferenzierte iPS-Zellen in mTeSR Plus-Medium ohne Y-27632 weiter kultivieren, bis die Zellen 80 % Konfluenz erreichen, wenn die Zellen für den nächsten Schritt zu spärlich sind (5.3.3). - An den Tagen 0 und 1: Saugkulturmedium und ersetzen durch 1 ml KSR-Medium (3.1.2), ergänzt durch 10 M SB431542 und 100 nM LDN-193189.
- An den Tagen 2 und 3: Saugkulturmedium und ersetzen Sie es durch 1 ml KSR-Medium, ergänzt durch 10 M SB431542, 100 nM LDN-193189, 5 'M DAPT, 5'M SU5402, 1'M RA und 1 'M SAG.
- An den Tagen 4 und 5: Bereiten Sie ein Mischmedium vor, das aus 75% KSR medium und 25% N2 medium (3.1.3) besteht. Aspiratkulturmedium und ersetzen durch 1 ml Mischmedium, ergänzt durch 10 M SB431542, 100 nM LDN-193189, 5 m DAPT, 5 m SU5402, 1 M RA und 1 M SAG.
- An den Tagen 6 und 7: Bereiten Sie ein Mischmedium vor, das aus 50% KSR medium und 50% N2 medium besteht. Aspirieren Sie das Kulturmedium und ersetzen Sie es durch 1 ml Mischmedium, ergänzt durch 5 M DAPT, 5 M SU5402, 1 M Retinsäure und 1 m SAG.
- An den Tagen 8 und 9: Bereiten Sie ein Mischmedium vor, das aus 25% KSR medium und 75% N2 medium besteht. Aspirieren Sie das Kulturmedium und ersetzen Sie es durch 1 ml des Mischmediums, ergänzt durch 5 M DAPT, 5 M SU5402, 1 M RA und 1 M SAG.
- An den Tagen 10 und 11: Ersetzen Sie das Medium durch 1 ml N2-Medium, ergänzt durch 5 M DAPT, 5 M SU5402, 1 M RA und 1 M SAG.
- An Tag 12: Das Differenzierungsmedium ansaugen, einmal mit PBS gut waschen und 0,5 ml des Zellablösungsmediums hinzufügen. Legen Sie die Platte in einen 37 °C-Inkubator für 1-3 min (z. B. bei Verwendung von TrypLE Express) oder 20-30 min (z. B. bei Anwendung von Accutase).
- Mit einer P1000 Pipette die Zellen vorsichtig sammeln und die Zellen in ein 15 ml konisches Rohr mit frischem Reifungsmedium und Zentrifuge bei 200 x g für 3 min übertragen. Wenn Zellen klumpig sind, pipette ein paar Mal sanft nach oben und unten. Pipette nicht zu viel, da dies Schäden an den Zellen verursachen kann.
- Aspirieren Sie den Überstand und setzen Sie das Pellet in 1 ml Reifungsmedium (3.1.4) mit 20 ng/mL BDNF ergänzt auf.
- Zählen Sie die Zelle mit einem Hämozytometer. Die Zellen bei 10.000-40.000 Zellen pro Brunnen in einer 96 well U Bodenplatte im Reifungsmedium mit 20 ng/ml BDNF ergänzt. Die anfängliche Saatdichte sollte in Abhängigkeit von der iPS-Zelllinie und dem Zustand der Zellen optimiert werden, so dass der Durchmesser des Sphäroids bei der Einweinung in den Gewebekulturchip 800-900 m beträgt. In den meisten Fällen beginnen Sie zunächst bei 20.000 Zellen pro Brunnen und erhöhen oder verringern sie dann die Anzahl der Zellen entsprechend der Größe.
- Kultur für weitere 3-10 Tage, bis die Zellen ein Sphäroid mit einer glatten Kante bilden.
- (Alternative Option): MNS-Bildung aus motorischen Neuronen
HINWEIS: Kommerziell erhältliche menschliche iPS-Zell-abgeleitete motorische Neuronen (siehe Materialtabelle) können verwendet werden, um MNOs zu generieren, anstatt sich von menschlichen iPS-Zellen zu unterscheiden.- Nach dem Auftauen der Kryovial der motorischen Neuronen, schnell die Zellen mit 9 ml des motorischen Neuronen Medium sausen. Drehen Sie bei 400 x g für 5 min bei Raumtemperatur.
- Aspirieren Sie den Überstand und setzen Sie das Pellet mit dem Motorneuronmedium wieder auf.
- Führen Sie die gleichen Schritte wie oben (5.3.12- 5.3.13) aus, um MNSs zu erstellen.
6. Vorbereitung des Gewebekulturchips zur Motornervenorganoidbildung (MNO)
- Sterilisieren Sie das vorbereitete PDMS (ab Schritt 2.13), indem Sie es mindestens 1 h in 70% Ethanol in die Petrischale eintauchen.
HINWEIS: Alle folgenden Schritte sollten in einem Biosicherheitsschrank manipuliert werden. - Sterilisieren Sie das Mikroskopglas (76 x 52 mm), indem Sie es in 70% Ethanol in die Petrischale eintauchen.
- Während des Trocknungsprozesses des Mikroskopglases das PDMS-Gerät auf das halb nasses Mikroskopglas legen und durch Warten über Nacht vollständig trocknen lassen. Nach dem vollständigen Trocknen sollte das PDMS-Gerät am Glas haften.
HINWEIS: Diese Verklebung ist nicht dauerhaft, um das Lösen der PDMS-Geräte aus dem Mikroskopglas nach der Kultur für die Gewebesammlung zu ermöglichen. Die permanente Verklebung durch Sauerstoffplasma kann verwendet werden, um die Haftung zwischen PDMS und Glas zu maximieren, aber es würde die Demontage der Chips und der Gewebesammlung verbieten. - Beschichten Sie die Oberfläche des Mikrokanals in PDMS-Gerät und Mikroskopglas mit 30 l verdünnter Kellermembranmatrix in DMEM/F12 (1:40), indem Sie einen Tröpfchen auf einer Seite des Kanaleinlasses anlegen und dann die Lösung von der anderen Seite des Einlasses mit einer Pipette oder Saugpumpe ansaugen (Abbildung 3A). Nicht zu viel Lösungsvolumen ansaugen, um Blasenkontamination zu vermeiden.
- Dann inkubieren Sie das PDMS-Gerät für 1 Stunde bei Raumtemperatur oder über Nacht bei 4 °C in einem Sekundärbehälter (z.B. Petrischale).
7. Motornervenorganoid (MNO) Bildung
- Ersetzen Sie die Beschichtungslösung durch vorgewärmte 150 l Reifungsmedium, ergänzt durch 20 ng/ml BDNF kurz vor Gebrauch.
- Legen Sie dann das MNS von Schritt 5.2.9 oder 5.3.13 mit einer Mikropipette mit einer Breitbohrspitze in den Einlass des Mikrokanals. MNS kann sich durch Schwerkraft spontan am Boden des Geräts niederlassen. Wenden Sie beim Einspritzen des MNS nicht zu viel Druck auf (Abbildung 3B).
HINWEIS: Wenn das MNS an der Seitenwand eines Lochs in einem Gewebekulturchip klebt, saugen Sie die Lösung vorsichtig von einer anderen Seite des Einlasses an. - Füllen Sie ein kleines Reservoir (z. B. eine Kappe von 15 ml Rohr) mit sterilem Wasser und legen Sie es in der Nähe des Gewebekulturchips in den Sekundärbehälter, um eine mittlere Verdunstung zu verhindern. Dann legen Sie es in einen 5% CO2/37 °C Inkubator.
- Für eine mittlere Veränderung, aspirieren Sie das erschöpfte Kulturmedium aus der Mitte des mittleren Reservoirs (Abbildung 3C). Besaugen Sie nicht das gesamte Medium und das Gewebe.
- Fügen Sie vorsichtig frisches Reifungsmedium (mit BDNF) hinzu. Das Medium sollte alle 2-3 Tage gewechselt werden. Trocknen Sie das Kulturmedium zu keinem Zeitpunkt während der Kultur. Axone wachsen von einem MNS in den Kanal und fügen sich spontan in 2-3 Wochen zu einem einzigen Bündel zusammen, was zur Bildung eines MNO führt. MNO kann für mehr als 1 Monat im Gerät kultiviert werden.
8. Nachgelagerte Analyse von MNO
- Ganzkörper-Immunostainierung
- Bringen Sie das Gerät oder Geschirr zu einer chemischen Rauchhaube. Fixieren Sie MNO, indem Sie dem Medium ungefähr ein ungefähres Volumen von 8 % Paraformaldehyd (PFA) hinzufügen, um eine Endkonzentration von 4 % PFA zu erreichen. Das PDMS-Gerät aus dem Glas abziehen und 15-20 min bei Raumtemperatur brüten.
- Waschen Sie den MNO mit PBS und wiederholen Sie dann PBS Waschen 2x.
- Permeabilisieren Sie die MNO mit 0,2% Triton X-100 in PBS und inkubieren Sie für 5 min bei Raumtemperatur.
- Waschen Sie den MNO mit PBS und wiederholen Sie dann PBS Waschen 2x. Blockieren Sie dann das MNO mit PBS, das 1% BSA enthält, und brüten sie 1 h bei Raumtemperatur.
- Primäre Antikörper in PBS mit 0,1% BSA verdünnen und MNO mit der primären Antikörperlösung über Nacht bei 4 °C inkubieren. Waschen Sie den MNO dreimal mit PBS.
- Sekundärantikörper in PBS mit 0,1% BSA verdünnen und MNO mit der Sekundärantikörperlösung für 2 h bei Raumtemperatur inkubieren. Dann waschen Sie die MNO dreimal mit PBS.
- Beflecken Sie das MNO mit Hoechst in PBS mit 0,2% Triton-X und brüten sie 5 min bei Raumtemperatur.
- Waschen Sie den MNO dreimal mit PBS. Das immunbefleckte MNO ist bereit für die Bildgebung mit fluoreszierenden oder konfokalen Lasermikroskopen.
- Gewebesammlung und Isolierung des Axonbündels
- 10 ml HBSS-Lösung in eine 10 cm Petrischale gießen. Tauchen Sie dann das gesamte PDMS-Gerät in die HBSS-Lösung ein.
- Lösen Sie das PDMS vorsichtig vom Mikroskopglas unter einem Stereomikroskop. Wenn das Gewebe am PDMS-Gerät klebt, tragen Sie vorsichtig 1 ml HBSS-Lösung von der Oberseite des Lochs mit einer Pipette auf.
- Sobald das Gewebe vom PDMS abkommt, tragen Sie vorsichtig 1 ml HBSS auf das Mikroskopglas auf, um das PDMS vollständig vom Mikroskopglas zu lösen.
- Um ein Axonbündel von einem Sphäroid zu isolieren, schneiden Sie das Axonbündel mit einem chirurgischen Messer oder einer Pinzette unter das Mikroskop. Schneiden Sie das Axonbündel etwas weit vom Sphäroid ab, um eine Kontamination der migrierten Zellen zu vermeiden.
- Isolierte Axonbündel und Sphäroide können durch verschiedene nachgeschaltete Analysen wie RT-PCR, RNA-seq und Western Blotting weiter analysiert werden.
- Der PDMS-Kulturchip kann wiederverwendet werden. Um den Kulturchip zu reinigen, beschallen Sie den Kulturchip in destilliertem Wasser für 15 min. Dann beschallen Sie den Chip in destilliertem Wasser, ergänzt mit 1% Reinigungsmittel. Waschen Sie den Kulturchip mit destilliertem Wasser 5 mal.
- Calcium-Bildgebung
HINWEIS: Die neuronale Aktivität kann mit Kalziumindikator sowohl vor als auch nach der Entnahme von Gewebe aus dem Gewebekulturchip (ab 8.2) gemessen werden. Das Protokoll basiert auf einem bestimmten kommerziellen Kit (siehe Tabelle der Materialien). Alternativ können auch andere Calcium-Bildgebungskits oder gleichwertige Methoden verwendet werden.- Waschen Sie den MNO dreimal mit PBS (ohne Ca2+ und Mg2+).
- Inkubieren Sie das Gewebe mit 5 m Fluo-4AM im Aufnahmemedium (20 mM HEPES, 115 mM NaCl, 5,4 mM KCl, 0,8 mM MgCl2, 1,8 mM, CaCl2, 13,8 mM Glukose) für 30-60 min bei 37 °C. Eine Zugabe von 0,01-0,02 % von Pluronic F-127 kann die Aufnahme von Fluo-4 AM in die Zellen unterstützen.
- Dann waschen Sie das Gewebe mit PBS (ohne Ca2+ und Mg2+)und ersetzen Sie es durch Aufnahmemedium.
- Erfassen Sie Zeitrafferbilder mit fluoreszierender Mikroskopie mit einem GFP/Cy2-Filtersatz. Stellen Sie die Belichtungszeit weniger als 20 ms pro Frame ein.
- Öffnen Sie die erfasste Filmdatei als Bildsequenz mit Bild J.
- Wenn Sie eine Farb-CCD- oder CMOS-Kamera verwenden, konvertieren Sie RGB-Bilder in 16-Bit-Monobilder. Öffnen "Analysieren | Werkzeuge | ROI Manager", Zeichnen Sie die Region von Interesse und klicken Sie auf "Hinzufügen". Klicken Sie auf "Multi Measure". Der Mittelwert der Intensität in mehreren ROI wird im Ergebnis angezeigt.
- Zeichnen Sie die Änderung der Signalintensität mit einer allgemeinen Datenanalysesoftware.
- Messung der neuronalen Aktivität durch Multielektroden-Array
HINWEIS: Die Aktivität von motorischen Neuronen kann durch ein Multi-Elektroden-Array (MEA) erfasst werden.- Nach der Erstellung einer MNO, übertragen Sie es auf eine Kellermembran Matrix beschichtet MEA Sonde. Positionieren Sie das Gewebe auf Elektroden.
- Fügen Sie 200 L Reifungsmedium hinzu und brüten Sie 1-2 h bei 37 °C, damit sich das MNO an der Oberfläche anheften kann.
- Ersetzen Sie das Medium durch aufnahmemittel (8.3.2). Legen Sie Gitter und Gewicht optional auf ein MNO, um den Kontrakt mit den Elektroden zu erhöhen.
- Stellen Sie die MEA-Sonde auf die Aufnahmekopfstufe ein. Wischen Sie den Kontakt zwischen der MEA-Sonde und der Kopfstufe mit 70% Ethanol ab.
- Beginnen Sie mit der Aufzeichnung neuronaler Aktivität, indem Sie den Anweisungen der Hersteller des MEA folgen.
Ergebnisse
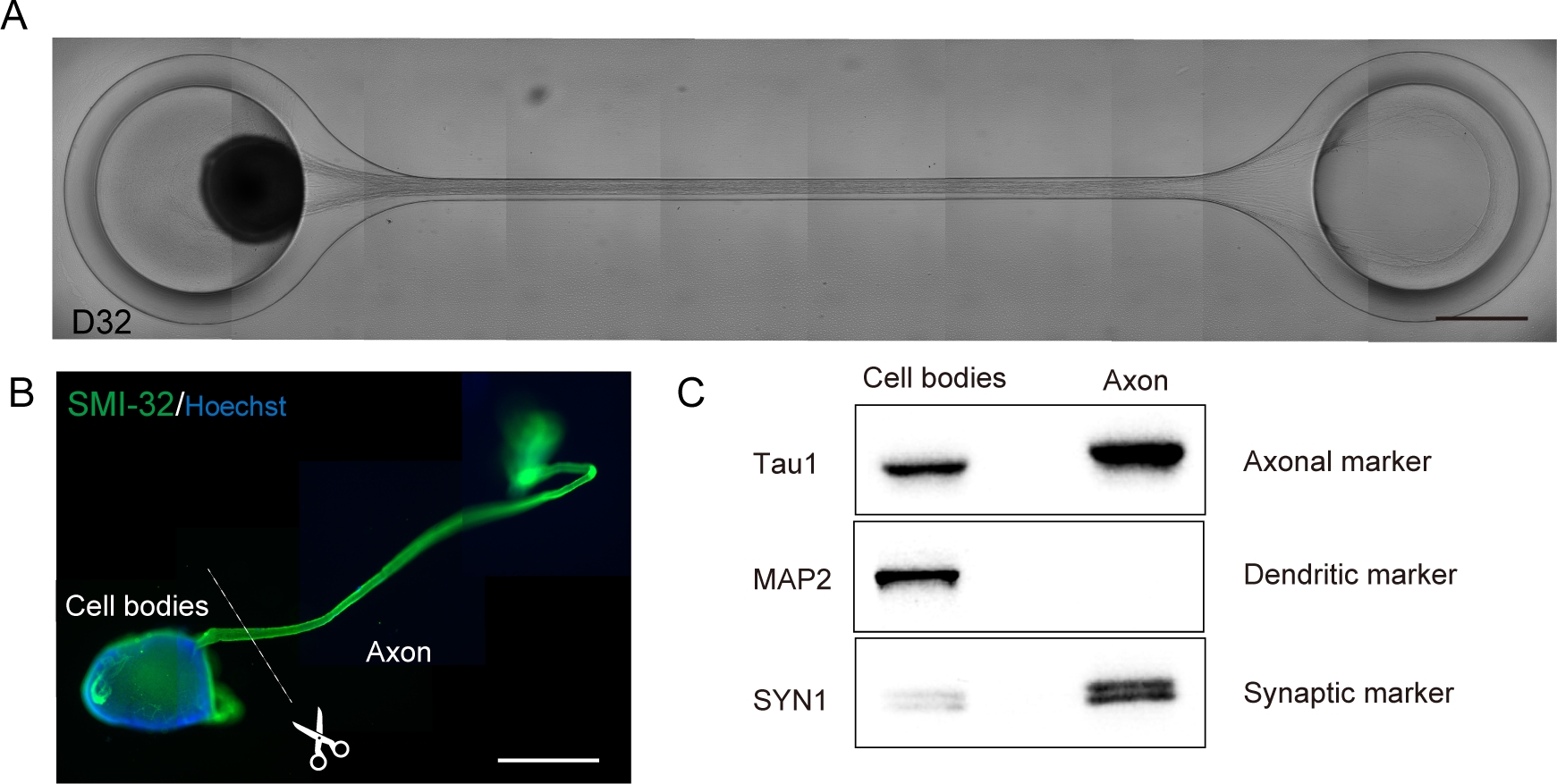
Motorneuronen wurden innerhalb von 12-14 Tagen in 3D-Differenzierungsverfahren unterschieden (Abbildung 4 und Abbildung 5). Wichtig ist, dass mehr als 60% der Zellen während der Differenzierung den motorischen Neuronenmarker HB9 exprimiert haben. Die Immunhistochemie ergab, dass etwa 80% der Zellen im MNS SMI32-positive motorische Neuronen waren. HB9 und SMI32 sind die etablierten motorischen Neuronmarker im Frühstadium15,16. Die Expression von HB9 und SMI32 sind die wichtigsten Parameter, die bestätigt werden müssen, um die zelluläre Identität der motorischen Neuronen zu gewährleisten. Nach der Einführung eines MNS in den Kulturchip erstrecken sich Axone in den Kanal und ein Axonbündel bildet sich. Durch Mikrokanäle, die als physikalische Leitfäden dienen, erlängen sich Axone aus dem MNS und bilden ein Bündel durch axo-axonale Wechselwirkung (Abbildung 6A). Es ist wichtig, die Bildung des Axonbündels durch mikroskopische Beobachtung zu bestätigen, um die Erzeugung eines MNO zu bestätigen. Ein erfolgreiches MNO trägt ein Axonbündel, das breiter als 50 m ist, und wenige isolierte Axone aus dem Bündel im Kanal. Die anfängliche Dehnung der Axone kann 24 Stunden nach der Einführung des Sphäroids beobachtet werden. Innerhalb der nächsten 3-4 Tage erreichten die Axone die Mitte des Mikrokanals und erreichten dann innerhalb von weiteren 10 Tagen das andere Ende(Abbildung 6A). Folglich sammelten sich die Axone und bildeten ein gerades und unidirektionales Bündel in 2-3 Wochen in einem Chip, und danach wurden neuronale Aktivitäten beobachtet.
Motornervenorganoide können aus dem Chip gesammelt werden, indem das PDMS aus dem Mikroskopglas für die biologische Analyse abgelöst wird (Abbildung 6B). Axonbündel und Zellkörper können durch Schneiden mit einem chirurgischen Messer oder einer Pinzette unter dem Mikroskop seziert und isoliert werden (Abbildung 6B). Diese biologischen Materialien einschließlich RNA und Protein können für regelmäßige biochemische Tests wie RT-PCR und Western Blotting verwendet werden. In Axonbündeln von MNOs werden nukleare oder dendritische Herstellerproteine bei der westlichen Blotting nicht nachgewiesen (Abbildung 6C).
In Kombination mit einem Kalziumindikator (Fluo-4 AM) kann die neuronale Aktivität im Gewebekulturchip erfasst werden. Spontane Aktivität von motorischen Neuronen im Sphäroid und im Axonbündel wurde innerhalb des MNO beobachtet. Auch wurden die neuronalen Aktivitäten mit Hilfe eines Multi-Elektroden-Array-Systems beobachtet.

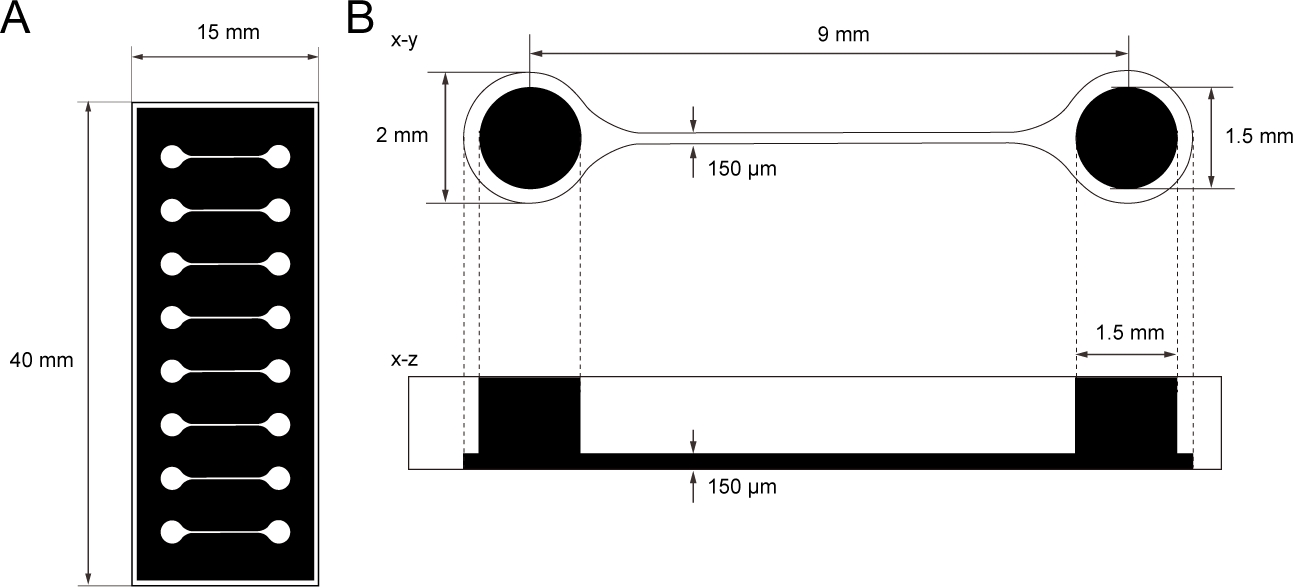
Abbildung 1: Die Dimension des PDMS-Gewebekulturchips.
(A) Fotomaske des Gewebekulturchips. (B) Abmessungen des Mikrokanals im Gewebekulturchip. Der Durchmesser der Basiskammer zum Halten des motorischen Neuronsphäroids beträgt 2 mm und das Loch von PDMS über der Kammer 1,5 mm. Die Breite und Höhe eines Mikrokanals, der zwei Kammern überbrückt, sind beide 150 m. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

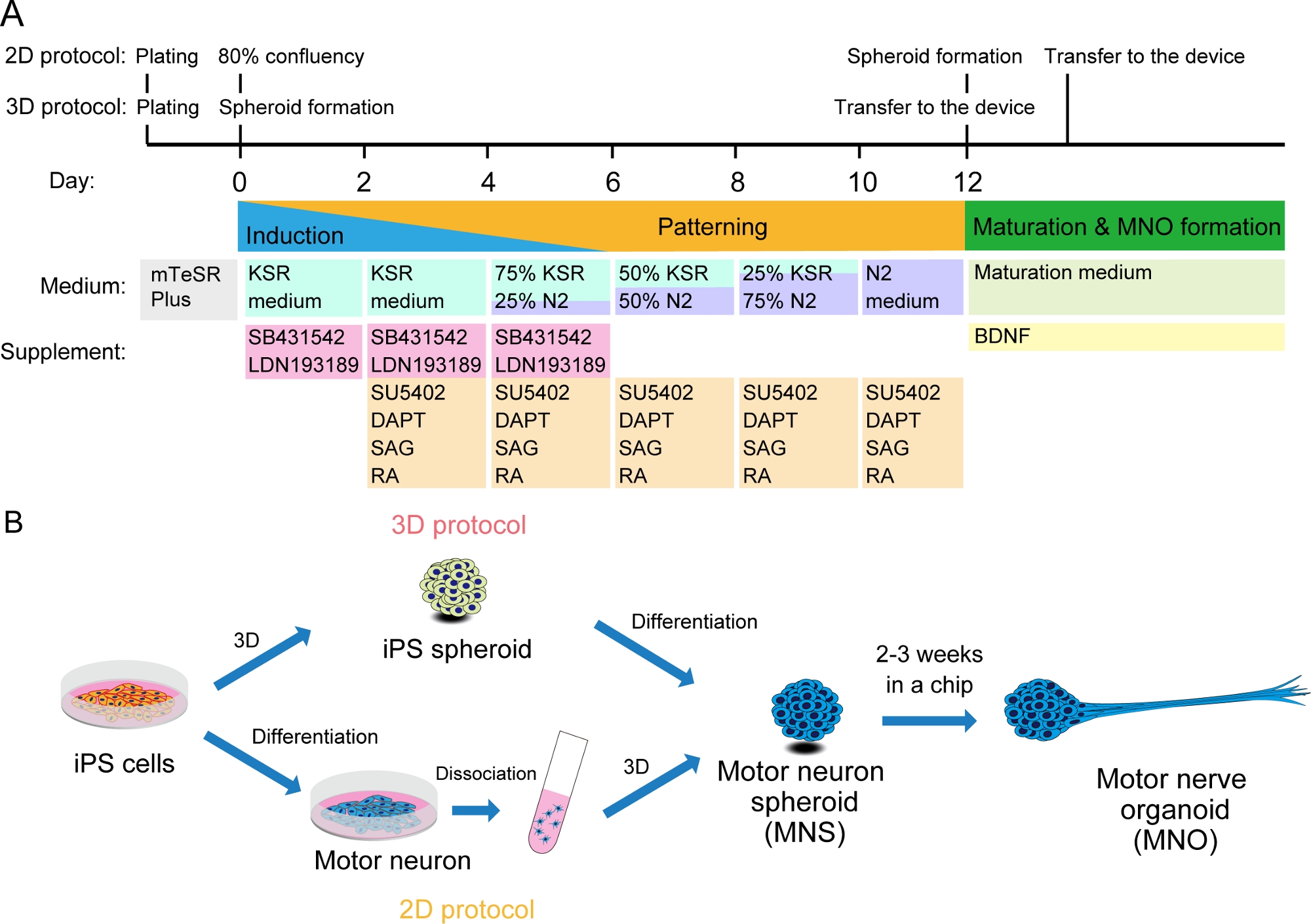
Abbildung 2: Schematische Darstellung der motorischen Neuronendifferenzierung.
(A) Die Differenzierungsschritte beinhalteten die neuronale Induktion, die Musterung in die Motorneuronenlinie und die Reifung der motorischen Neuronen. (B) Zwei Optionen zum Erstellen von Motorneuronsphäroid (MNS) aus iPS-Zellen: 3D-Protokoll und ein 2D-Protokoll mit Dissoziationsschritt von motorischen Neuronen. Motornervenorganoid (MNO) kann durch beide Protokolle erhalten werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

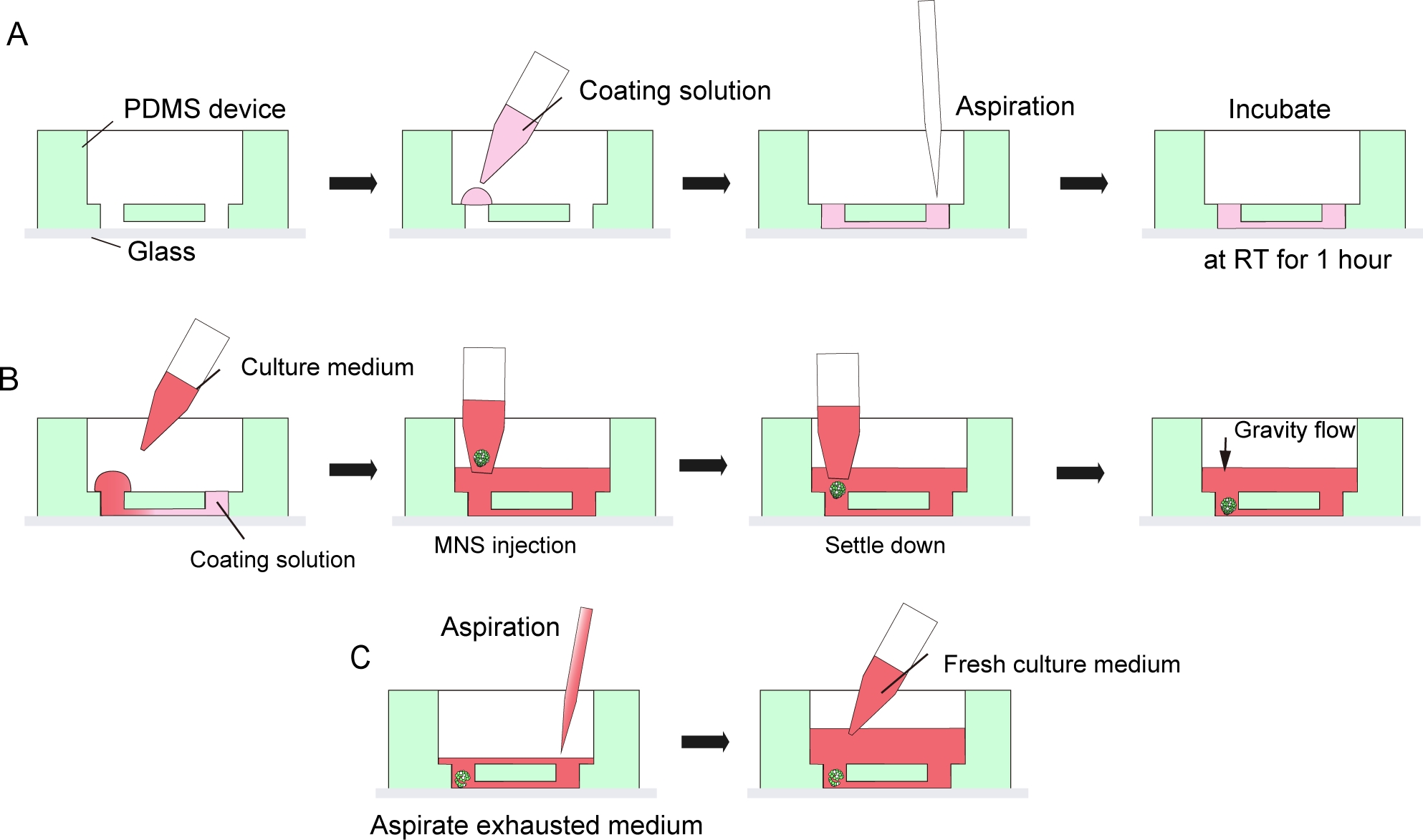
Abbildung 3: Schritt-für-Schritt-Protokoll für Keller-Membran-Matrix-Beschichtung und Motorneuron-Sphäroid-Einführung.
(A) Kellermembran-Matrixbeschichtung im Kanal des Gewebekulturchips. (B) MNS-Einführung in das Loch des Chips. (C) Kulturmedium veränderung durch Streben nach erschöpftem Medium. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

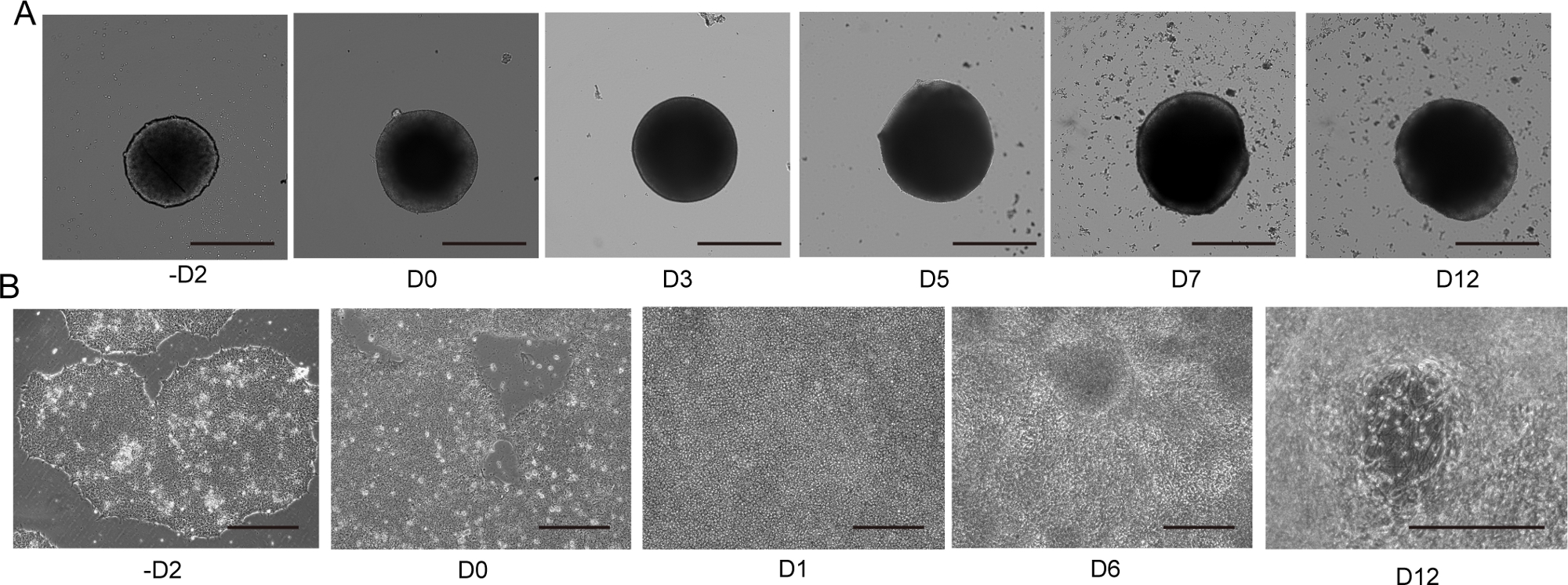
Abbildung 4: Differenzierung von 2D- und 3D-Motorneuronen.
(A) Zeitverlauf der repräsentativen 3D-MNS-Differenzierung (3D-Protokoll). Die Größe des MNS nahm im Laufe der Zeit allmählich zu. Maßstabsleiste: 500 m. (B) Zeitverlauf der 2D-Differenzierung auf -D2, D0, D1, D6, D12 (2D-Protokoll). Maßstabsleiste: 500 m. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

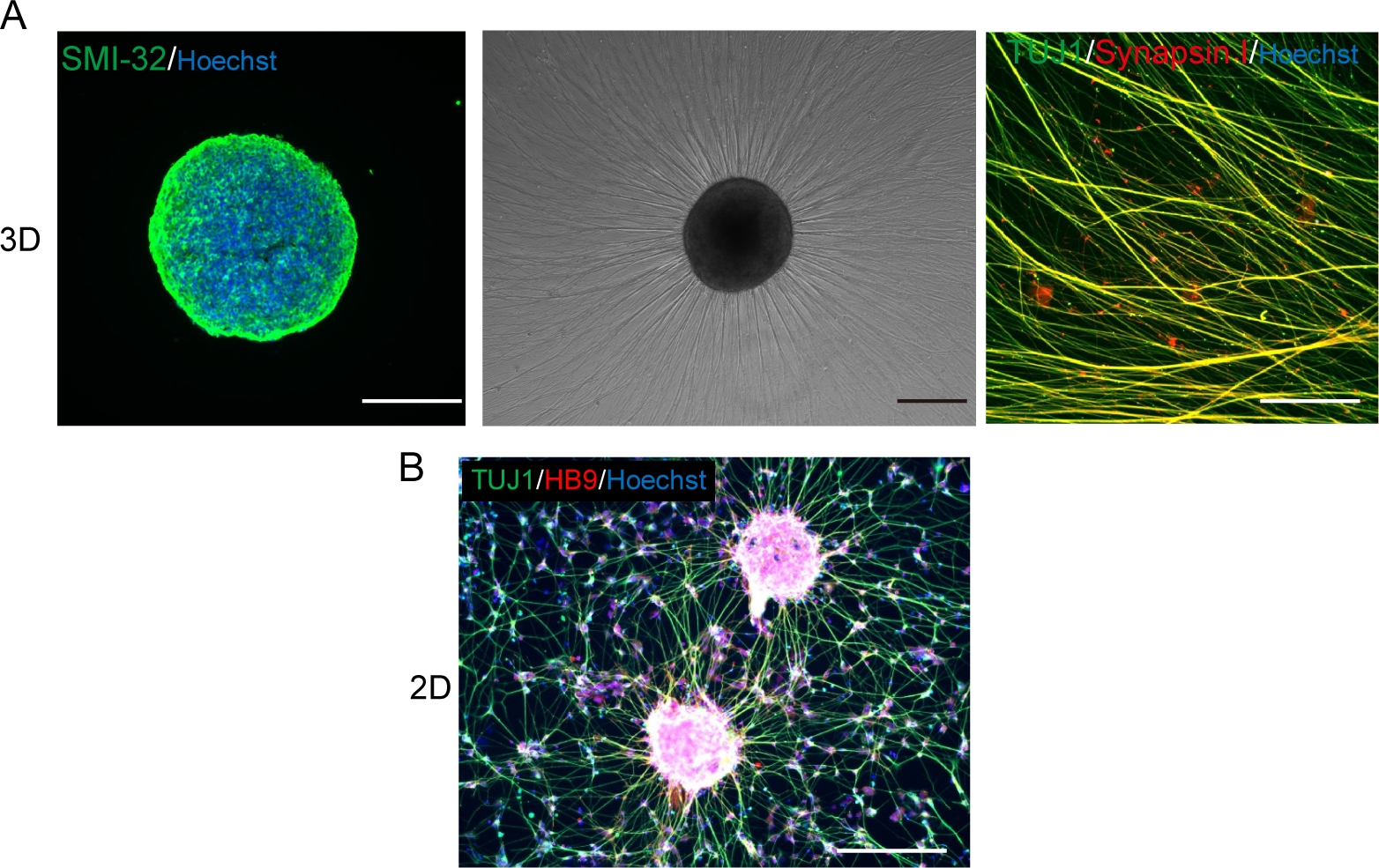
Abbildung 5: Charakterisierung der motorischen Neuronen.
(A) (Links) Eine Kryosektion eines MNS, der mit SMI-32-Antikörper und DAPI gefärbt ist. (Mitte) Ein Phasenkontrastbild von neu beschichtetem MNS auf einer mit der Kellermembran beschichteten Oberfläche. Axonale Dehnung wurde beobachtet. (Rechts) Axone von replated MNS mit Synapsin I und Tuj1 Antikörpern gefärbt. Skalenbalken: 500 m (Links und Mitte) und 50 m (rechts). (B) Ein repräsentatives Bild von 2D-Motorneuronen, die mit Tuj- und HB9-Antikörpern immunostainiert sind. Maßstabsleiste: 200 m. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 6: Charakterisierung eines Motornervenorganoids (MNO), das in einem Kulturchip erzeugt wird.
(A) Repräsentative Bilder der Axondehnung und der dicken Axonbündelbildung auf D32. Maßstabsleiste: 500 m. (B) Immunostainierung des Motornervenorganoids (MNO) durch SMI-32 und DAPI. Axone und Zellkörper können durch physikalisch schneiden isoliert werden. Scale bar: 1 mm. (C) Reinheit des Proteins aus Axonen und Zellkörpern, quantifiziert durch western Blotting. MAP2, ein dendritischer Marker, wurde im axonalen Protein nicht nachgewiesen, während der axonale Marker Tau1 im axonalen Protein angereichert ist. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Diskussion
Dieses Protokoll beschreibt die Bildung eines Motornervenorganoids (MNO), das ein Axonbündel hat, das aus einem motorischen Neuronensphäroid aus menschlichen iPS-Zellen ausgefahren wird. Das geformte Axonbündel ist dick, flexibel und in unidirektionalen Strukturen gut organisiert. Durch die Sezieren des Axonbündels können hochreines axonales Protein und RNA ausreichend für biochemische Analysen gewonnen werden. Die neuronale Aktivität kann in Axonbündeln und Sphäroiden mit Kalziumbildgebung gemessen werden. Eine Kontamination von kerntechnischen und dendritischen Proteinen im axonalen Lysat wurde durch westliches Blotting nicht nachgewiesen, was zeigt, dass unsere Methode Axone effizient von Zellkörpern und Dendriten trennte.
Einer der Vorteile dieses Protokolls ist die schnelle Differenzierung und Generierung von MNO mit einem Axonbündel, in dem alle Prozesse in 4 Wochen mit dem 3D-Protokoll und 5-6 Wochen mit dem 2D-Protokoll durchgeführt werden können. Dies ist kurz im Vergleich zu anderen Protokollen, die in der Regel 3-4 Wochen dauern, um einfach in MN von embryonalen Stammzellen und iPS-Zellen17 zu unterscheiden, und es dauert zusätzliche 2-4 Wochen, um axonale Dehnung zu erhalten. Das 3D-Protokoll wird aufgrund der kürzeren Differenzierungszeit, weniger Schritten und reduzierten technischen Risiken ohne den Dissoziationsschritt im Vergleich zum 2D-Protokoll in der Regel gegenüber dem 2D-Protokoll bevorzugt. Der mikrofluidische Gewebekulturchip wurde so konzipiert, dass sich die Axone von MNS durch den Mikrokanal zum anderen Fach hin ausziehen können, was die Bildung eines Bündels von Axonen erleichtert, indem axo-axonale Wechselwirkungen und Affinität zwischen Axonen induzieren. Aufgrund des einfachen Versuchsaufbaus können alle hier beschriebenen Protokolle nicht nur von Bioingenieuren durchgeführt werden, die mit der Manipulation eines Gewebekulturchips vertraut sind, sondern auch von Biologen und Neurowissenschaftlern, die mit Mikrofluidik und Mikrofabrikationstechniken nicht vertraut sind. Es sollte beachtet werden, dass die Schritte 1 und 2 auch mithilfe eines externen Fertigungsdienstes ausgeführt werden können.
Einer der entscheidenden Schritte zum Ausführen des Protokolls ist ein sequenzieller Wechsel des Kulturmediums. Es wird empfohlen, das Kulturmedium bei jedem Schritt während der Differenzierung vollständig zu ändern, damit Faktoren im verbrauchten Medium die MN-Differenzierung nicht stören. Ein weiterer kritischer Punkt dieses Protokolls ist die Aufrechterhaltung undifferenzierter iPS-Zellen in guter Qualität. Die Qualität der anfänglichen iPS-Zellkultur wirkt sich signifikant auf die Effizienz bei der Gewinnung von motorischen Neuronen und MNO aus. Ein weiterer Punkt ist, dass der Durchmesser von MNS kleiner sein sollte als die Größe der Bohrung des Chips (1,5 mm). Größere Sphäroide können nicht in die Kammer gelangen und können im Mittelteil eine schwere hypoxische Nekrose erleben. Die Größe von MNSs kann gesteuert werden, indem eine anfängliche Aussaatanzahl von iPS-Zellen (im 3D-Protokoll) oder motorischen Neuronen (im 2D-Protokoll) geändert wird. Die Sädichte der Zellen sollte für jede iPS-Zelllinie optimiert werden.
Unterteilte mikrofluidische Geräte mit Mikrorillen und kleinen Porenfiltern wurden häufig verwendet, um Axone von Zellkörpern und Dendriten zu trennen. Diese Technik kann auch Axone von Zellkörpern und Dendriten trennen, mit überlegener Fülle von Axonen in gebündeltem Gewebe. Im Vergleich zu den anderen Methoden besteht eine wesentliche Einschränkung dieser Methode darin, dass sie nicht zwei verschiedene Kulturmedien in der aktuellen Gestaltung des Gewebekulturchips trennen kann, was die Fähigkeit der Kokultur zweier unterschiedlicher Zellen behindert, die zwei unterschiedliche Medien erfordern. Eine weitere Einschränkung ist, dass der PDMS-Chip eine vorbestimmte Beschränkung der Größe des Gewebes vorgegeben hat. Ein Sphäroid, das größer als das Loch ist, kann nicht in die Kammer gelangen, und das Axonbündel kann nicht dicker als die Breite des mikrofluidischen Kanals werden.
Diese Methode kann auf andere Arten von Neuronen angewendet werden. Unsere Gruppe hat gezeigt, dass sie in der Lage ist, einen Zerebralparesetrakt mit einer modifizierten Methode in Kombination mit zerebralen Organoidtechniken zu modellieren18. Kortikale Sphäroide wurden in beide Fächer eingeführt und die Axone spontan wechselseitig zu jedem Sphäroid gestreckt, und in der Folge bildete sich spontan ein Axonbündel. Dadurch können zwei kortikale Sphäroide durch ein Axonbündel verbunden werden, und das Gewebe konnte als ein Stück erhalten werden. Dies zeigt, dass der Ansatz sehr vielseitig ist, um Axon-Bündelgewebe unabhängig von neuronalen Zelltypen zu bilden. In diesem Protokoll wurden menschliche iPS-Zellen verwendet, jedoch können andere Stammzellen, einschließlich menschlicher ES-Zellen und menschlicher neuronaler Stammzellen, mit Änderungen am vorgestellten Protokoll verwendet werden. 3D-Sphäroide von Neuronen können durch verschiedene Protokolle19,20erzeugt werden. Diese Methode der Herstellung von Geweben mit einem Axon-Bundle kann möglicherweise in Der Zukunft mit den anderen Differenzierungsprotokollen für die Herstellung von 3D MN Sphäroid kombiniert werden. Darüber hinaus können die Dicke und Länge des Axonbündels durch einfache Änderung der Breite und Höhe der Mikrokanäle des Gewebekulturchips für zukünftige Entwicklungen gesteuert werden.
Wir glauben, dass dieses Protokoll für Arzneimitteltests und -screenings verwendet werden kann und zum Verständnis der Mechanismen beitragen kann, die der Entwicklung und Denkkrankheiten von Axonfaszien zugrunde liegen.
Offenlegungen
In einem Teil dieses Protokolls wurde Jiksak Bioengineering, Inc., das von Jiro Kawada gegründet wurde, ein Patent lizenziert.
Danksagungen
Diese Studie wurde von der Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for Scientific Research 17H05661 und 18K19903, Core-2-Core-Programm und Beyond AI Institute unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| (Tridecafluoro-1,1,2,2-tetrahydrooctyl)-1-trichlorosilane | Sigma | 440302 | |
| 16% Paraformaldehyde (formaldehyde) aqueous solution | Electron Microscopy Sciences | 15710 | |
| 200µl Wide Bore Pipet Tips | BMBio | BMT-200WRS | |
| 6-well plates | Violamo | 2-8588-01 | |
| Accutase | ICT | AT104 | |
| B-27 Supplement (50X) | Gibco | 17504044 | |
| Bovine serum albumin | Sigma | A6003 | |
| Brain-derived neurotrophic factor (BDNF) | Wako | 020-12913 | |
| CO2 incubator | Panasonic | MCO-18AIC | |
| Cryostor CS10 | Stem Cell Technologies | 07959 | |
| DAPT | Sigma | D5942 | |
| DMEM/F12 | Sigma | D8437 | |
| Fluo-4 AM | Dojindo Laboratories | CS22 | |
| GlutaMAX Supplement | Gibco | 35050-061 | |
| Growth factor reduced Matrigel (basement membrane matrix) | Corning | 354230 | |
| HB9 Antibody | Santa Cruz | sc-22542 | |
| HBSS | Wako | 085-09355 | |
| Hoechst 33342 | Sigma | 14533 | |
| iCell motor neuron (commercially available human iPS cell-derived motor neurons) | Cellular Dynamics | R1051 | |
| Isopropyl alcohol (IPA) | Wako | 166-04836 | |
| Knock Out Serum Replacement | Gibco | 10828028 | |
| LDN193189 | Sigma | SML0559 | |
| MEA probe | Alpha MED Scientific inc | MED-P5004A | |
| MEM Non-essential Amino Acid Solution (100x) (NEAA) | Sigma | M7145 | |
| Microscope Glass | Matsunami | S9111 | |
| mTeSR Plus | Stem Cell Technologies | 05825 | |
| N2 supplement | Wako | 141-08941 | |
| Neurobasal medium | Gibco | 21103049 | |
| Penicillin-streptomycin | Gibco | 15140122 | |
| Photoresist SU-8 2100 | Microchem | #SU-8 2100 | |
| Prime surface 96U | Sumitomo Bakelite | MS-9096U | |
| ReLeSR (passaging reagent) | Stem Cell Technologies | 05872 | |
| Retinoic acid | Wako | 186-01114 | |
| SAG | Sigma | SML1314 | |
| SB431542 | Wako | 192-16541 | |
| Silicon wafer | SUMCO | PW-100-100 | |
| Silpot 184 w/c kit | Dow Toray | Silpot 184 w/c kit | |
| Smi32 Antibody | Biolegend | 801701 | |
| SU5402 | Sigma | SML0443 | |
| SU-8 Developer | Microchem | Y020100 | |
| Synapsin I Antibody | Millipore | Ab1543 | |
| TrypLE Express liquid without phenol red (dissociation solution) | Gibco | 12604-021 | |
| Tuj1 Antibody | Biolegend | 801202 | |
| Y-27632 | Wako | 030-24021 |
Referenzen
- Raper, J., Mason, C. Cellular strategies of axonal pathfinding. Cold Spring Harbor Perspective Biology. 2 (9), 001933 (2010).
- Ito, Y., et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 353 (6299), 603-608 (2016).
- Fujimori, K., et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nature Medicine. 24 (10), 1579-1589 (2018).
- Chen, H., et al. Modeling ALS with iPSCs Reveals that Mutant SOD1 Misregulates Neurofilament Balance in Motor Neurons. Cell Stem Cell. 14 (6), 796-809 (2014).
- Osaki, T., Uzel, S. G. M., Kamm, R. D. Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Science Advances. 4 (10), (2018).
- Imamura, K., et al. The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Science Translational Medicine. 9 (391), (2017).
- Wang, L., Marquardt, T. What axons tell each other: axon-axon signaling in nerve and circuit assembly. Current Opinion in Neurobiology. 23 (6), 974-982 (2013).
- Kawada, J., et al. Generation of a Motor Nerve Organoid with Human Stem Cell-Derived Neurons. Stem Cell Reports. 9 (5), 1441-1449 (2017).
- Nagendran, T., Poole, V., Harris, J., Taylor, A. M. Use of Pre-Assembled Plastic Microfluidic Chips for Compartmentalizing Primary Murine Neurons. JoVE. (141), e58421 (2018).
- Paranjape, S. R., Nagendran, T., Poole, V., Harris, J., Taylor, A. M. Compartmentalization of Human Stem Cell-Derived Neurons within Pre-Assembled Plastic Microfluidic Chips. JoVE. (147), e59250 (2019).
- Chambers, S. M., et al. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nature Biotechnology. 30 (7), 715-720 (2012).
- Rimington, R. P., Fleming, J. W., Capel, A. J., Wheeler, P. C., Lewis, M. P. Bioengineered model of the human motor unit with physiologically functional neuromuscular junctions. bioRxiv. , (2020).
- Cullen, D. K., et al. Bundled Three-Dimensional Human Axon Tracts Derived from Brain Organoids. iScience. 21, 57-67 (2019).
- Giandomenico, S. L., et al. Cerebral organoids at the air-liquid interface generate diverse nerve tracts with functional output. Nature Neurosciences. 22 (4), 669-679 (2019).
- Egawa, N., et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Science Translational Medicine. 4 (145), (2012).
- Sances, S., et al. Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nature Neurosciences. 19 (4), 542-553 (2016).
- Qu, Q., et al. High-efficiency motor neuron differentiation from human pluripotent stem cells and the function of Islet-1. Nature Communications. 5 (1), 3449 (2014).
- Kirihara, T., et al. A Human Induced Pluripotent Stem Cell-Derived Tissue Model of a Cerebral Tract Connecting Two Cortical Regions. iScience. 14, 301-311 (2019).
- Rigamonti, A., et al. Large-Scale Production of Mature Neurons from Human Pluripotent Stem Cells in a Three-Dimensional Suspension Culture System. Stem Cell Reports. 6 (6), 993-1008 (2016).
- Yan, Y., Song, L., Madinya, J., Ma, T., Li, Y. Derivation of Cortical Spheroids from Human Induced Pluripotent Stem Cells in a Suspension Bioreactor. Tissue Engineering Part A. 24 (5-6), 418-431 (2017).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten