
Method Article
Kartierung von 3D-Genominteraktionen von Säugetieren mit Micro-C-XL
In diesem Artikel
Zusammenfassung
Ein Protokoll zur Kartierung der dreidimensionalen Genomorganisation mit Nukleosomenauflösung unter Verwendung der genomweiten Chromosomenkonformationserfassungsmethode Micro-C-XL wird hier vorgestellt.
Zusammenfassung
Die dreidimensionale (3D) Chromosomenorganisation ist ein wichtiger Faktor bei der Genomregulation und der Zelltypspezifikation. Zum Beispiel wird angenommen, dass cis-regulatorische Elemente, die als Enhancer bekannt sind, die Aktivität distaler Promotoren durch Interaktion im 3D-Raum regulieren. Genomweite Chromosomenkonformationserfassungstechnologien (3C) wie Hi-C haben unser Verständnis der Organisation von Genomen in Zellen verändert. Das derzeitige Verständnis der 3D-Genomorganisation ist begrenzt durch die Auflösung, mit der die topologische Organisation von Chromosomen im 3D-Raum aufgelöst werden kann. Micro-C-XL misst die Chromosomenfaltung mit Auflösung auf der Ebene des Nukleosoms, der Grundeinheit des Chromatins, indem es die Mikrokokken-Nuklease (MNase) verwendet, um Genome während des Chromosomenkonformationserfassungsprotokolls zu fragmentieren. Dies führt zu einem verbesserten Signal-Rausch-Verhältnis in den Messungen und ermöglicht so eine bessere Detektion von Isolationsstellen und Chromosomenschleifen im Vergleich zu anderen genomweiten 3D-Technologien. In diesem Artikel wird ein visuell unterstütztes, detailliertes Schritt-für-Schritt-Protokoll zur Herstellung hochwertiger Micro-C-XL-Proben aus Säugetierzellen vorgestellt.
Einleitung
Micro-C-XL ist eine genomweite Technik zur Messung der 3D-Genomkonformation mit Nukleosomenauflösung. Micro-C-XL baut auf der weit verbreiteten Proximity-Ligations-basierten Hi-C-Technologie auf, die unser Verständnis der Organisation von 3D-Genomen verändert hat1. Micro-C-XL und seine erste Iteration, Micro-C, wurden ursprünglich in Saccharomyces cerevisiae2,3 entwickelt und später an Säugetierzellsysteme angepasst, für die das Protokoll sein volles Potenzial bei der Erkennung von kurzreichweitigen Merkmalen des 3D-Genoms, wie z. B. Chromosomenschleifen und Isolationsstellen, unter Beweis gestellt hat. Diese Version basiert auf neueren Micro-C-XL-Veröffentlichungen von Säugetieren 4,5. Da Micro-C-XL Micro-C ersetzt, wird Micro-C-XL im Manuskript fortan als Micro-C bezeichnet.
Die Hauptunterschiede zwischen Micro-C und Hi-C6 sind wie folgt: 1) Genomfragmentierung mit Mikrokokken-Nuklease (MNase) im Vergleich zu Restriktionsenzymen und 2) zusätzliche Vernetzer mit größerem Atomabstand zwischen den reaktiven Gruppen im Vergleich zu nur Formaldehyd. Beide Schritte tragen maßgeblich zum verbesserten Signal-Rausch-Verhältnis von Micro-C im Vergleich zu herkömmlichem Hi-C bei. Die Fragmentierungsgröße begrenzt die Auflösung, mit der die 3D-Genomorganisation während des Proximity-Ligationsprotokolls aufgelöst werden kann. MNase ist eine Nuklease, die bevorzugt zugängliche DNA verdaut und nukleosomal-geschützte DNA intakt lässt. Das Nukleosomen-Footprinting mittels MNase-Sequenzierung hat gezeigt, dass Nukleosomen die meisten eukaryotischen Genome vollständig abdecken7. Da Nukleosomen je nach Spezies und Zelltyp mit einem durchschnittlichen Abstand von 160-220 bp über das Genom verteilt sind, ist MNase das ideale Enzym für die hochauflösende Kartierung der Genomarchitektur.
Die Verwendung eines zusätzlichen Vernetzers in Kombination mit Formaldehyd (FA) im Micro-C-Verfahren verbessert zusätzlich das Signal-Rausch-Verhältnis 2,8. Aminspezifische Vernetzer mit längeren atomaren Spacern zwischen den reaktiven Gruppen erleichtern Protein-Protein-Vernetzungen. Dabei handelt es sich typischerweise um Disuccinimidylglutarat (DSG) oder Ethylenglykolbisuccinimidylsuccinat (EGS) mit 7,7 Å bzw. 16,1 Å Abstandshaltern. Die Reduktion des Rauschens durch EGS oder DSG zeigt sich besonders in Experimenten mit hohen Fragmentierungsraten, wie z.B. Micro-C, und tritt vermutlich aufgrund einer Verringerung der Rate zufälliger Ligationsereignisseauf 8.
Ein kürzlich entwickeltes Hi-C 3.0-Protokoll, das ESG/DSG-Vernetzung und mehrere Kombinationen von Restriktionsenzymen verwendet, reduziert das Rauschen in Hi-C-Experimenten und verbessert die Detektion von Chromosomenschleifen und Isolationsstellen erheblich 8,9. Ein Standort-für-Standort-Vergleich verschiedener Interaktionsdatenmerkmale ergab jedoch, dass Micro-C im Vergleich zu Hi-C 3.0 und konventionellem Hi-C8 eine überlegene Erkennung von kurzreichweitigen Merkmalen wie Chromosomenschleifen und Isolationsstellen aufwies. Hi-C 3.0 verbessert jedoch die Detektion von kurzreichweitigen Merkmalen und behält im Vergleich zu herkömmlichem Hi-C eine starke Detektion der Genomkompartimentierung bei. Zusammenfassend lässt sich sagen, dass die Wahl einer Methode zur Erfassung der Chromosomenkonformation durch die objektive und biologische Fragestellung bestimmt werden sollte.
Hier stellen wir ein Schritt-für-Schritt-Protokoll für erfolgreiche Micro-C-Experimente zur Verfügung, die die Organisation des 3D-Genoms entschlüsseln können.
Protokoll
1. Zellkultur und Vernetzung
- Kultur von Zellen entsprechend den experimentellen Erfordernissen, um mindestens 1 x 107 Zellen zu erhalten. Hier wurden die Zellen bei 37 °C mit 5 %CO2 in E14-Medium (DMEM mit Pyruvat und L-Glutamin, 15 % FBS, 1x LIF, 1x NEAA, 1 % Pen-Streptokokken, 0,1 mM β-Mercaptoethanol [siehe Materialtabelle]) gezüchtet und jeden zweiten Tag durchgelassen.
HINWEIS: Dieses Protokoll wurde erfolgreich auf verschiedene Zelltypen verschiedener Spezies angewendet, wie z. B. Homo sapiensund Mus musculus. In diesem Beispiel wurden embryonale Stammzellen (ES-Zellen) von Mäusen verwendet. - Ernten Sie die Zellen bei 70%-80% Konfluenz durch Absaugen des Mediums. Einmal mit 5 ml DPBS waschen und die Zellen mit 3 ml vorgewärmtem 0,25%igem Trypsin pro 10-cm-Schale für 2-3 Minuten bei 37 °C inkubieren.
- Das Trypsin wird mit 7 ml vorgewärmtem E14-Medium abgeschreckt und die abgelösten Zellen in ein 50-ml-Röhrchen überführt.
- Die Zellen werden durch Zentrifugation bei 300 x g für 5 Minuten bei Raumtemperatur (RT) pelletiert. Verwerfen Sie den Überstand und resuspendieren Sie das Zellpellet im Medium. Zählen Sie die Zellen mit einem Zellzähler.
HINWEIS Das Protokoll ist robust gegenüber einigen Schwankungen der Zellkonzentrationen, und verschiedene Zellzähler wurden verwendet, um die Zellzahlen zu quantifizieren. Beachten Sie jedoch den Dynamikumfang des verwendeten Zellzählers. Dies liegt in der Regel zwischen 1 x 105-1 x 107 Zellen/ ml. Während die Zentrifugationsgeschwindigkeit und -dauer für diesen Zelltyp getestet wurden, können andere, kleinere Zellen eine höhere Zentrifugationsgeschwindigkeit oder längere Schleuderzeiten erfordern, und die Zentrifugation sollte entsprechend angepasst werden. - Die Zellen werden durch Zentrifugation bei 300 x g für 5 min bei RT gesammelt und die Zellen in DPBS bei einer Endkonzentration von 1 x 106 Zellen/ml resuspendiert. Wenn die Ausbeute z. B. 1 x 107 Zellen beträgt, resuspendieren Sie die Zellen in 10 ml DPBS.
- Für den ersten Vernetzungsschritt werden 37 % Formaldehyd (FA) bis zu einer Endkonzentration von 1 % zur Zellsuspension gegeben und die Zellsuspension 10 Minuten lang bei RT mit Rotation (15-20 U/min) inkubiert. Wenn die Ausbeute beispielsweise 1 x 107 Zellen beträgt, werden 270 μl FA zu der 10-ml-Zellsuspension hinzugefügt.
HINWEIS: Die FA-Lösung ist in der Regel bis zu 3 Monate nach dem Öffnen bei RT stabil. - Die Reaktion wird durch Zugabe von 2,5 M Glycin zu einer Endkonzentration von 0,25 M gelöscht und 5 min bei RT mit Rotation (15-20 U/min) inkubiert. Wenn die Ausbeute beispielsweise 1 x 107 Zellen beträgt, fügen Sie der Zellsuspension 1,027 ml 2,5 M Glycin hinzu.
- Die Zellen werden durch Zentrifugation bei 300 x g für 5 min bei RT gesammelt. Verwerfen Sie den Überstand und resuspendieren Sie die Zellen in 5 ml DPBS. Wiederholen Sie den Zentrifugationsschritt einmal und resuspendieren Sie die pelletierten Zellen in DPBS auf 4 x 106 Zellen/ml. Wenn die Ausbeute z. B. 1 x 107 Zellen beträgt, resuspendieren Sie die Zellen in 2,5 ml DPBS.
- Für den zweiten Vernetzungsschritt wird eine 0,3 M Stammlösung von Ethylenglykolbisuccinimidylsuccinat (EGS) in DMSO (13,6 mg EGS in 100 μl DMSO) hergestellt. EGS in einer Endkonzentration von 3 mM zur Zellsuspension geben und bei RT für 40 min mit Rotation (15-20 U/min) inkubieren. Wenn die Ausbeute z. B. 1 x 107 Zellen beträgt, werden 25 μl 0,3 M EGS-Stammlösung zu 2,5 ml der Zellsuspension hinzugefügt.
HINWEIS: Äquilibrieren Sie das EGS für RT für mindestens 20 Minuten vor dem Wiegen zur Herstellung der Stammlösung. - Die Reaktion wird durch Zugabe von 2,5 M Glycin zu einer Endkonzentration von 0,4 M gelöscht und 5 min bei RT mit Rotation (15-20 U/min) inkubiert. Wenn die Ausbeute beispielsweise 1 x 107 Zellen beträgt, fügen Sie der Zellsuspension 400 μl 2,5 M Glycin hinzu.
- Die Zellen werden durch Zentrifugation bei 1.000 x g für 5 min bei RT gesammelt und die pelletierten Zellen in DPBS auf 5 x 106 Zellen/ml resuspendiert. Verteilen Sie 5 x 10 6 Zellen/Röhrchen für präparative Bibliotheken und 1 x 106 Zellen/Röhrchen für die Titration für den MNase-Verdau.
HINWEIS: Es wird empfohlen, den optimalen Aufschlussgrad für jede vernetzte Zellcharge zu titrieren. Im Idealfall werden zwei bis drei Aliquots von 1 x 10 6 Zellen für MNase-Titrationsexperimente (Schritt 2) und zwei bis vier Aliquoten von 5 x 106 Zellen für präparative Experimente (Schritt 3) gesammelt. - Die Zellen werden durch Zentrifugation bei 1.000 x g für 5 min bei RT gesammelt und der Überstand entfernt. Die Zellpellets werden in flüssigem Stickstoff eingefroren und bei −80 °C gelagert.
HINWEIS: Erfolgreiche Micro-C-Bibliotheken können aus Proben hergestellt werden, die bis zu 3 Monate gelagert werden.
2. MNase-Titration
HINWEIS: Die Durchführung einer MNase-Titration ist notwendig, um die optimale Konzentration von MNase zu bestimmen, bevor die präparative Bibliothek der doppelt vernetzten Zellen verarbeitet wird.
- Um die MNase-Titration durchzuführen, tauen Sie ein Pellet mit 1 x 10 6 Zellen 10 Minuten lang auf Eis auf und resuspendieren Sie die Zellen in 500 μl DPBS (fügen Sie 1x BSA hinzu, wenndie Zellen an der Wand kleben). Die Zellsuspension 20 Minuten lang auf Eis inkubieren.
- Die Zellen werden durch Zentrifugation bei 10.000 x g für 5 Minuten bei RT gesammelt und der Überstand entfernt. Resuspendieren Sie die pelletierten Zellen in 500 μl MB#1-Puffer (50 mM NaCl, 10 mM Tris-HCl, 5 mM MgCl 2, 1 mM CaCl 2,0,2 % NP-40, 1x Protease-Inhibitor [siehe Materialtabelle], pH 7,4).
- Die Zellen werden durch Zentrifugation bei 10.000 x g für 5 Minuten gesammelt und der Überstand entfernt. Resuspendieren Sie die Zellen in 200 μl MB#1-Puffer und teilen Sie die Probe in vier Röhrchen auf.
- Eine Durchstechflasche MNase (20 U/μl) auftauen und mit 10 mM Tris, pH 7,4, in einer Reihe von 1:2, 1:4, 1:4 und 1:4 verdünnen, um Konzentrationen von 10 U/μl, 2,5 U/μl, 0,625 U/μl bzw. 0,1256 U/μl (eine für jede Aufschlussbedingung) zu erreichen. In angemessenen Zeitabständen (10-20 s) 1 μl MNase-Lösung zu einer der vier Proben geben, vortexen und auf einem Thermomixer bei 37 °C für 10 Minuten inkubieren (800 U/min schütteln). Fahren Sie mit der Zugabe von 1 μl aus den verbleibenden MNase-Verdünnungen zu den verbleibenden Zellaliquoten fort.
- Stoppen Sie den Mnase-Aufschluss, indem Sie 200 μl frisch zubereiteten STOP-Puffer (150 μl 10 mM Tris, pH 7,4, 25 μl 10 % SDS, 25 μl 20 mg/ml Proteinase K, 2 μl 0,5 M EGTA) in die gleiche Reihenfolge und mit dem gleichen Zeitintervall geben, in dem die MNase zugegeben wurde. Bei 65 °C 2 h inkubieren.
- Zu jeder Probe werden 500 μl Phenol-Chloroform-Isoamylalkohol (PCI) gegeben und durch Wirbeln gründlich vermischt. Zentrifugieren Sie bei 19.800 x g für 5 Minuten bei RT, um die Phasen zu trennen, und überführen Sie die wässrige Phase in neue Röhrchen (ca. 200 μl/Probe).
ACHTUNG: PCI enthält zahlreiche toxische Bestandteile und sollte nur in einer Chemikalienschutzhaube gehandhabt werden. Bitte wenden Sie sich an den Hersteller für weitere Details. - Reinigen Sie die DNA mit einem handelsüblichen DNA-Aufreinigungskit (siehe Materialtabelle) gemäß den Anweisungen des Herstellers und eluieren Sie die Proben in 12 μl Elutionspuffer.
HINWEIS: Die SDS-Konzentration aus dem Deproteinisierungsschritt (Schritt 2.5) ist für einige DNA-Aufreinigungskits hemmend. Das hier verwendete DNA-Aufreinigungskit ist vergleichbar mit der Ethanolfällung. - Fügen Sie 2-5 μl Beladungsfarbstoff hinzu und lassen Sie die Proben 30-50 Minuten lang bei 120 V auf einem 1,5%igen Agarose-Gel laufen (bis sie ordnungsgemäß getrennt sind; Abbildung 1A).
- Wählen Sie den besten Verdausgrad für das Experiment und fahren Sie mit dem präparativen MNase-Aufschluss fort. Ein optimaler Verdauungsgrad weist wenig bis gar keine subnukleosomalen Fragmente und ein Verhältnis von 70%-90 % Mono- zu Dinukleosomen auf.
ANMERKUNG: In diesem Experiment wurde der Aufschlussgrad von Spur 4 in Abbildung 1A als optimaler Aufschluss mit 0,625-1,25 U Mnase für 2,5 x 10 5 Zellen (= 2,5-5 U Mnase für 1 x 106 Zellen) bestimmt. Eine ausführliche Diskussion finden Sie in den repräsentativen Ergebnissen.
3. Präparativer MNase-Verdau
- Um das Chromatin über MNase-Verdau in die Mononukleosomen zu fragmentieren, werden zuvor doppelt vernetzte 5 x 10 6-Zell-Aliquots aufgetaut und in 1 ml DPBS resuspendiert. 20 min auf Eis inkubieren (1x BSA zugeben, wenn die Zellen an der Röhrchenwand kleben).
- Die Zellen werden durch Zentrifugation bei 10.000 x g für 5 Minuten bei RT gesammelt und der Überstand verworfen. Resuspendieren Sie das Pellet in 500 μl MB#1-Puffer. Wiederholen Sie den Zentrifugationsschritt einmal. Das Pellet wird in 1 ml MB#1-Puffer resuspendiert und 200 μl Aliquots (1 x 106 Zellen pro Aliquot) hergestellt.
- Basierend auf der MNase-Titration (beschrieben in Schritt 2) wird das Chromatin verdaut, indem die entsprechende Menge MNase (normalerweise 2,5-10 U/μl pro 1 x 106 Zellen) zu jedem Aliquot hinzugefügt wird. Gut mischen (Wirbel und schnell schleudern) und auf einem Thermomixer bei 37 °C 10 min bei 800 U/min schütteln.
- Stoppen Sie den MNase-Aufschluss, indem Sie 1,6 μl 0,5 M EGTA (letzte 4 mM) zu jedem Aliquot hinzufügen und auf einem Thermomixer bei 65 °C für 10 Minuten mit 800 U/min schütteln inkubieren.
- Die Probe wird durch Zentrifugation bei 10.000 x g für 5 Minuten bei RT entnommen und der Überstand verworfen. Resuspendieren Sie das Zellpellet in 500 μl 1x NEBuffer 2.1.
- Poolproben, die einem Input von 5 x 106 Zellen oder weniger entsprechen, zur weiteren Verarbeitung.
HINWEIS: Wenn mehr als 5 x 10 6 Zellen verarbeitet werden sollen, verarbeiten Sie diese Proben parallel, da die enzymatischen Bedingungen für 5 x 106 Zellen optimiert sind. - Bevor Sie mit den Schritten der Proximity-Ligatur fortfahren, übertragen Sie 10 % der Probe als Eingangskontrolle, um den MNase-Aufschluss zu kontrollieren. Zu dieser Probe werden 150 μl 10 mM Tris, pH 7,4, 25 μl 10 % SDS und 25 μl 20 mg/ml Proteinase K gegeben und über Nacht bei 65 °C inkubiert.
4. DNA-Endprozessierung und Proximity-Ligatur
- Die verbleibende Probe wird durch Zentrifugation bei 10.000 x g für 5 min bei 4 °C aufgefangen und der Überstand verworfen. Das Pellet wird in 90 μl frisch zubereitetem Micro-C-Mastermix 1 (Tabelle 1) resuspendiert und 15 Minuten lang auf einem Thermomixer bei 37 °C mit 800 U/min geschüttelt.
- Geben Sie 10 μl 5 U/μl Klenow-Fragment hinzu und inkubieren Sie 15 Minuten lang auf einem Thermomixer bei 37 °C mit 800 U/min Schütteln.
- 100 μl frisch zubereiteter Micro-C-Mastermix 2 (Tabelle 2) zugeben und 45 Minuten lang auf einem Thermomixer bei 25 °C mit 800 U/min schütteln inkubieren. Nach der Inkubation wird die enzymatische Reaktion durch Zugabe von EDTA zu einer Endkonzentration von 30 mM gelöscht. Auf einem Thermomixer 20 min bei 65 °C mit 800 U/min schütteln.
- Die Probe wird durch Zentrifugation bei 10.000 x g für 5 min bei 4 °C entnommen und der Überstand verworfen. Die Probe wird in 500 μl frisch zubereitetem Micro-C-Mastermix 3 (Tabelle 3) resuspendiert und 2,5 h bei RT mit Rotation (15-20 U/min) inkubiert.
- Die Probe wird durch Zentrifugation bei 10.000 x g für 5 min bei 4 °C entnommen und der Überstand verworfen. Die Probe wird in 200 μl frisch zubereitetem Micro-C-Mastermix 4 (Tabelle 4) resuspendiert und 15 Minuten lang auf einem Thermomixer bei 37 °C mit 800 U/min geschüttelt.
- Für die Rückwärtsvernetzung und Deproteinierung werden 25 μl 20 mg/ml Proteinase K und 25 μl 10 % SDS in die Probe gegeben und über Nacht bei 65 °C mit intermittierendem Mischen inkubiert.
5. Di-nukleosomale DNA-Reinigung und Größenauswahl
- Geben Sie 500 μl PCI zu den Proben und der Eingangskontrolle und mischen Sie sie durch Vortexing. Die Phasen werden durch Zentrifugation bei 19.800 x g für 5 min getrennt und die obere wässrige Phase in ein frisches Röhrchen überführt.
- Konzentrieren Sie die DNA entweder mit einem DNA-Reinigungskit oder durch Ethanolfällung. Die Proben in 30 μl (Schritt 5.3) und die Eingangskontrollen (Schritt 3.11) in 15 μl eluieren und ein 1,5 %iges Agarose-Gel verwenden, um die Mononkleosomen und Dinukleosomen zu trennen (Abbildung 1B).
HINWEIS: Die SDS-Konzentration aus dem Deproteinisierungsschritt ist für einige DNA-Aufreinigungskits hemmend. Das hier verwendete DNA-Aufreinigungskit (siehe Materialtabelle) ist vergleichbar mit der Ethanolfällung. Je nach Anzahl der verwendeten Zellen kann der Input zwischen 100 ng und 10 μg liegen. Typischerweise werden 1-5 μg DNA aus 5 x 106 Zellen extrahiert. - Entfernen Sie die DNA-Fragmente, die eine di-nukleosomale Größe (ca. 300 bp) haben. Verwenden Sie ein handelsübliches DNA-Gel-Elutionskit (siehe Materialtabelle), um die DNA aus dem Agarose-Gel zu extrahieren und in 150 μl zu eluieren.
6. Herstellung der Streptavidin-Kügelchen
- 10 μl Streptavidin-Kügelchen (siehe Materialtabelle) pro Probe in ein Reaktionsröhrchen überführen.
- Legen Sie es in einen geeigneten Magneten für 1,5-ml-Röhrchen (siehe Materialtabelle). Nachdem die Lösung geklärt ist (1-2 min), entfernen Sie den Überstand und resuspendieren Sie die Kügelchen in 300 μl 1x TBW (5 mM Tris-HCl, pH 7,5, 0,5 mM EDTA, 1 M NaCl, 0,05 % Tween 20) pro Probe. Wiederholen Sie diesen Schritt einmal.
- Resuspendieren Sie die Kügelchen in 150 μl 2x B&W-Puffer (10 mM Tris-HCl, pH 7,5, 1 mM EDTA, 2 M NaCl) pro in Schritt 7 verarbeiteter Probe.
7. Streptavidin-Pull-Down- und On-Bead-Bibliotheksvorbereitung
- 150 μl vorbereitete Kügelchen (Schritt 6.3) zu 150 μl der Probe (Schritt 5.3) geben. 20 Minuten bei RT mit Rotation (15-20 U/min) inkubieren.
- Legen Sie die Röhrchen in einen geeigneten Magneten und warten Sie, bis die Lösung geklärt ist (1-2 Minuten). Entfernen Sie den Überstand und resuspendieren Sie die Kügelchen in 300 μl 1x TBW. Wiederholen Sie diesen Schritt.
- Legen Sie die Röhrchen in einen geeigneten Magneten und warten Sie, bis die Lösung geklärt ist (1-2 min). Entfernen Sie den Überstand und resuspendieren Sie die Kügelchen in 100 μl 0,1x TE (1 mM Tris, 0,1 mM EDTA, pH 8,0).
- Legen Sie die Röhrchen in einen geeigneten Magneten und warten Sie, bis die Lösung geklärt ist (1-2 min). Entfernen Sie den Überstand, resuspendieren Sie die Kügelchen in 50 μl 0,1x TE und überführen Sie sie in PCR-Röhrchen.
HINWEIS: Das verwendete Volumen von 0,1x TE (50 μl) entspricht dem Eingangsvolumen des in diesem Protokoll verwendeten Vorbereitungskits für die DNA-Sequenzierungsbibliothek (siehe Materialtabelle). Wenn ein anderes Kit oder eine andere Strategie verwendet wird, passen Sie die Lautstärke entsprechend an. - Führen Sie die DNA-Manipulationsschritte des Vorbereitungskits für die Sequenzierungsbibliothek gemäß dem Protokoll des Herstellers durch. Zu diesen Schritten gehören in der Regel DNA-Abstumpfung, A-Tailing, Adaptor-Ligatur und U-Exzision. Der letzte Schritt (U-Exzision) ist spezifisch für das in dieser Studie verwendete Kit. Wenn Sie dieses Kit verwenden, befolgen Sie die Anweisungen des Herstellers von Schritt 1 bis Schritt 2.6 im Kit-Protokoll mit unverdünnten Adaptern und ziehen Sie nach Schritt 2.6 keine Lagerung bei −20 °C in Betracht.
ANMERKUNG: Die für die Sequenzierungskits erforderlichen Pufferwechsel müssen durch die Bindung der Kügelchen an einen Magneten und Waschungen (Schritte 7.3 bis 7.4) ausgetauscht werden, da die DNA immer noch an die Streptavidin-Kügelchen gebunden ist. Da die DNA an die magnetischen Kügelchen gebunden ist, ignorieren Sie außerdem alle Reinigungs- und Größenauswahlschritte nach der Adapterligatur aus dem Kit-Protokoll. Fahren Sie mit Schritt 7.6 (dieses Protokoll) fort. - Nachdem die Adapterligation abgeschlossen ist, waschen Sie die Probe wie in Schritt 7.2 beschrieben. Verwerfen Sie den Überstand und resuspendieren Sie die Kügelchen in 20 μl 0,1x TE.
8. Abschätzung der erforderlichen PCR-Zyklen
HINWEIS: Es ist ratsam, die erforderlichen PCR-Zyklen für die Bibliotheksamplifikation abzuschätzen. In der Regel erfordert eine Micro-C-Bibliothek 8-15 PCR-Zyklen. Obwohl dieser Schritt nicht unbedingt erforderlich ist, hilft er, eine Überamplifikation zu vermeiden und das Risiko von PCR-Duplikaten zu verringern.
- Um die erforderliche Mindestanzahl von PCR-Zyklen zu definieren, führen Sie eine PCR mit 1 μl Streptavidin-Biotin-DNA-Probe durch (Schritt 7.6). Dazu wird der Probe ein PCR-Mastermix (3,2 μlH2O, 0,4 μl i5-Primer, 0,4 μl i7-Primer und 5 μl Q5-High-Fidelity-DNA-Polymerase) zugegeben. Führen Sie eine PCR mit 16 Zyklen gemäß den Anweisungen des Herstellers für das verwendete DNA-Bibliotheksvorbereitungskit durch.
HINWEIS: Die hier verwendeten Primer werden in einem separaten Kit (siehe Materialtabelle) erworben, das mit dem verwendeten Bibliotheksvorbereitungskit kompatibel sein muss. - Nach der PCR werden die Kügelchen mit einem geeigneten Magneten gesammelt und die DNA-Konzentration von 1 μl des Überstandes mit einem hochempfindlichen DNA-Quantifizierungsinstrument gemessen (siehe Materialtabelle). Um die Gesamt-DNA-Konzentration zu erhalten, multiplizieren Sie diesen Wert mit 10 (Gesamtvolumen der PCR-Reaktion). Schätzen Sie die erforderliche Anzahl von PCR-Zyklen, wie in den repräsentativen Ergebnissen beschrieben.
- 2 μl 6x Ladepuffer zu den restlichen 9 μl PCR-Mischung hinzufügen. Lösen Sie die PCR-amplifizierte Bibliothek auf einem 1%igen Agarose-Gel auf, um die erfolgreiche Adaptorligatur zu bestimmen, was zu einer Größe von etwa 450 bp führt (Abbildung 1C).
HINWEIS: Das Gel sollte eine deutliche Bande von ca. 420 bp aufweisen (Micro-C-Bibliothek plus Adapter). Banden von 120 bp stellen Adapter-Dimere dar und weisen auf Bibliotheken mit geringer Komplexität hin. Banden mit niedrigerem Molekulargewicht können auftreten (weniger als 100 bp), und dies sind unbenutzte Primer aus der PCR-Reaktion.
9. Erweiterung der Sequenzierungsbibliothek
- Zu den restlichen 19 μl der Streptavidin-Biotin-DNA-Probe wird der PCR-Mastermix (65 μlH2O, 100 μl Q5-High-Fidelity-DNA-Polymerase, 8 μl i5-Primer, 8 μl i7-Primer) gegeben. Die Reaktionsmischung wird in 50-100 μl Aliquots aufgespalten.
HINWEIS: Das optimale Volumen für die PCR hängt vom PCR-Gerät ab. In der Regel bieten 50 μl die reproduzierbarste Amplifikation in gängigen PCR-Geräten. - Führen Sie die PCR gemäß den Anweisungen des Herstellers des DNA-Bibliotheksvorbereitungskits mit der in Schritt 8 festgelegten Anzahl von Zyklen durch. Wenn Schritt 8 weggelassen wurde, werden 14 Zyklen für die PCR empfohlen.
- Reinigen Sie die DNA mit paramagnetischen Kügelchen (siehe Materialtabelle) in einem Verhältnis von 1:0,9 gemäß dem Protokoll des Herstellers. Eluieren Sie in 20 μl 0,1 % TE.
HINWEIS: Wenn in Schritt 8.5 Adapterdimere erkannt werden, führen Sie die Aufreinigung zweimal mit demselben Verhältnis durch. - Bestimmen Sie die DNA-Konzentration und führen Sie die Proben auf ein Qualitätskontrollsystem (siehe Materialtabelle). Stellen Sie eine gute Qualität der Micro-C-Bibliothek sicher, indem Sie ein einzelnes präzises Band verwenden (Abbildung 1D).
HINWEIS: Adapterdimere sind bei der Vorbereitung von On-Bead-Bibliotheken aufgrund der Probenwäsche vor der PCR ungewöhnlich. Das Auftreten von Adapterdimeren deutet also auf eine geringe Bibliothekskomplexität hin. Wenn Adapterdimere beobachtet werden, wird dringend empfohlen, die Qualität der Probe mit einer Low-Input-Sequenzierung zu kontrollieren.
10. DNA-Sequenzierung und Datenverarbeitung
- Sequenzieren Sie die Micro-C-Bibliothek mit Paired-End-Sequenzierung gemäß den Anforderungen des Sequenzierungsanbieters.
HINWEIS: Im Idealfall werden die Proben auf einer Plattform im Paired-End-Modus mit 50 bp pro Lesevorgang sequenziert. Auch ältere Plattformen, die kürzere Leselängen bieten, wie z.B. 2 x 35 bp, wurden erfolgreich eingesetzt. Wichtig ist, dass es bei der Untersuchung repetitiver genomischer Regionen ratsam sein kann, mit einer längeren Leselänge zu sequenzieren. - Um die Qualität der Micro-C-Bibliothek zu beurteilen, führen Sie eine Low-Input-Sequenzierung mit 5 x 106 bis 1 x 107 Reads pro Probe durch.
- Verarbeiten Sie die Sequenzierungsdateien (fastq-Dateien) mit Distiller10. Mappen Sie die Reads mit dem entsprechenden Referenzgenom, hier mm10.
HINWEIS: Die Sequenzdateien können mithilfe verschiedener Pipelines auf lokalen Computern oder Computerclustern verarbeitet werden. Bei Proben mit geringer Sequenzierungstiefe können größere Behältergrößen wie 10.000 bp, 50.000 bp, 100.000 bp und 500.000 bp den Rechenaufwand und die Dateigröße reduzieren. Distiller (in dieser Studie verwendet) generiert alle erforderlichen Dateitypen, um die Qualität der Micro-C-Bibliothek zu bewerten. Die generierte *.stats-Datei enthält die Informationen über die Abbildungsrate, das cis-trans-Verhältnis und die Leseorientierung, geschichtet durch den Abstand zwischen den Lesepaaren. Diese Parameter sind in Abbildung 2 visualisiert, und die Bewertung der Qualität der Micro-C-Bibliothek wird in den repräsentativen Ergebnissen diskutiert. Die Verarbeitungssoftware erzeugt auch mcool-Dateien, die direkt in HiGlass (https://docs.higlass.io/) geladen werden können, um Interaktionsmatrizenzu generieren 9.
Ergebnisse
Die erfolgreiche Präparation von Micro-C-Bibliotheken kann in mehreren Schritten des Protokolls evaluiert werden. Der wichtigste Schritt ist die Wahl des richtigen MNase-Aufschlussgrades. Daher muss die MNase-Konzentration titriert werden, um für jede Probe konstant 70 % bis 90 % Mononukleosomen gegenüber Dinukleosomen zu erhalten. Es ist wichtig zu beachten, dass der Chromatinverdau für eu- und heterochromatin unterschiedlich ist, wobei MNase Heterochromatin weniger effizient verdaut. Der optimale Verdauungsgrad hängt also von der interessierenden Chromatinregion und dem untersuchten Zelltyp ab, da der relative Anteil von eu- und heterochromatin zelltypspezifisch ist. Daher ist es ratsam, die erforderliche MNase-Konzentration sorgfältig zu titrieren und den Erfolg des Micro-C-Experiments zunächst durch Low-Input-Sequenzierung zu evaluieren.
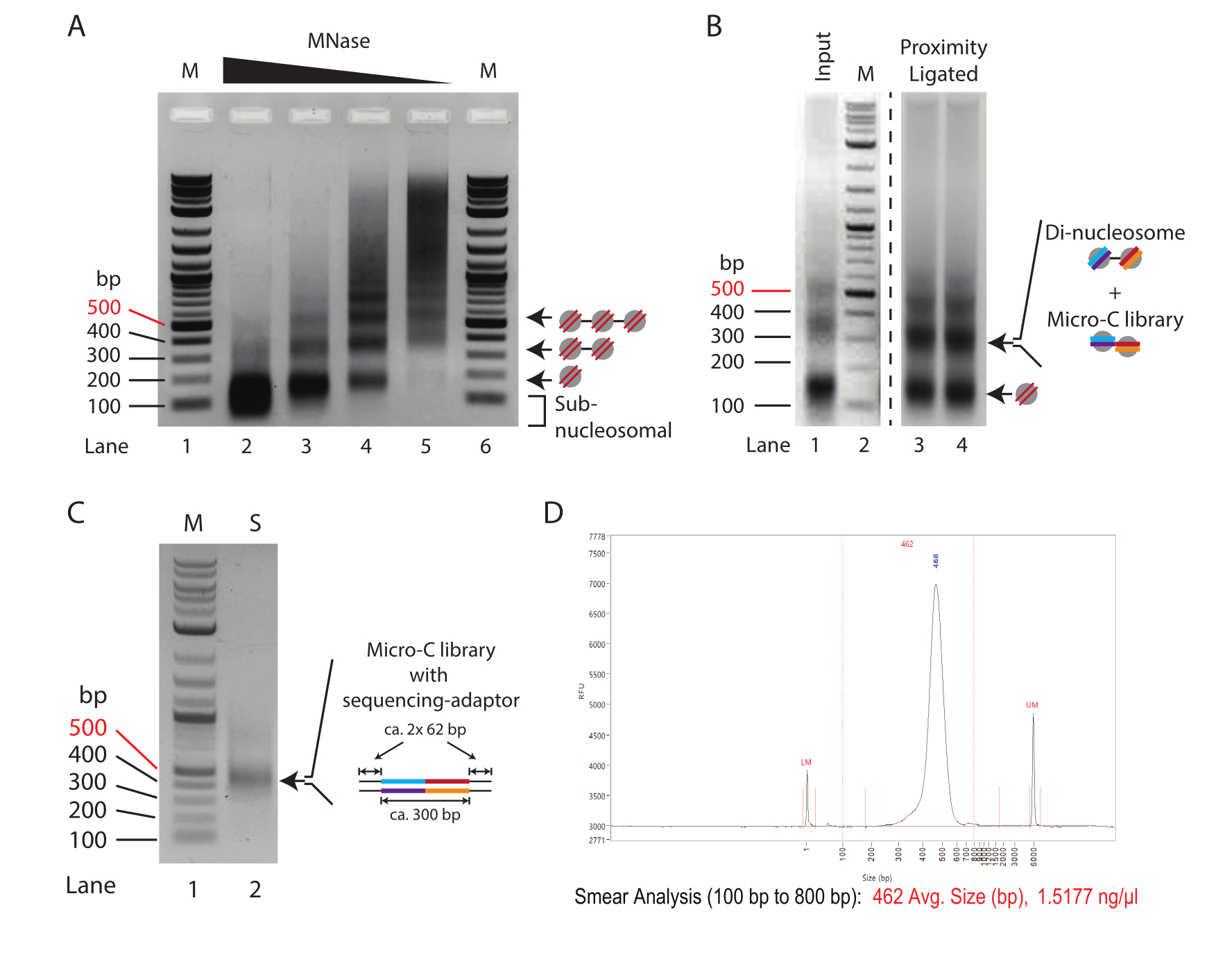
Ein typisches MNase-Titrationsmuster von Chromatin, das mit abnehmenden Mengen an MNase behandelt wurde, ist in Abbildung 1A dargestellt. Hier wird das Chromatin von 250.000 Zellen pro Reaktion mit einer vierfachen Verdünnung der MNase verdaut. Die höchste Konzentration (10 U MNase, Spur 2) zeigt ein überverdautes Chromatin, das fast ausschließlich aus mononukleosomaler DNA besteht (~150 bp). Bemerkenswert ist, dass die Mitte der mononukleosomalen Bande im Agarose-Gel im Vergleich zu den entsprechenden Banden in den Proben mit reduzierten MNase-Konzentrationen niedriger verläuft, was auf eine Überverdauung der nukleosomalen DNA hindeutet. Überverdaute Nukleosomen werden in der Proximity-Ligationsreaktion ineffizient ligiert; daher ist die Probe in Spur 2 für Micro-C-Experimente suboptimal. Spur 3 (2,5 U MNase) weist einen nahezu geeigneten Aufschlussgrad für Mikro-C-Experimente auf. Hier ist die mononukleosomale Bande die dominierende Spezies, und der subnukleosomale Abstrich, der auf überverdaute Nukleosomen hinweist, ist reduziert; Es ist jedoch immer noch vorhanden. Der Aufschlussgrad in Spur 4 (0,635 U MNase) ist eine ideale Bedingung für ein Micro-C-Experiment in diesem Titrationsbeispiel. Es ist eine klare mononukleosomale Bande ohne subnukleosomale DNA vorhanden. Die Bandenintensität für die Mono- und Dinukleosomen-DNA ist nahezu gleich, was auf eine Mononukleosomenausbeute von 66% oder mehr hindeutet. Es ist erwähnenswert, dass die di-nukleosomale DNA etwa doppelt so groß ist wie die mononukleosomale DNA (~320 bp vs. ~150 bp), so dass ihre Bandenintensität pro DNA-Mol im Vergleich zu ihrem mononukleosomalen Gegenstück doppelt so hoch ist. Der Aufschlussgrad in Spur 5 (0,156 U MNase) zeigt ein unterverdautes Chromatin mit fast keiner nukleosomalen DNA, was eine suboptimale Probe darstellt.
Zusammenfassend lässt sich sagen, dass in diesem Beispiel der Aufschluss von 2,5 x 10 5 Maus-ES-Zellen mit 0,625 U MNase (entsprechend2,5 U MNase für 1 x 106 Zellen in 200 μL) den vielversprechendsten Ausgangspunkt für präparative Aufschlüsse in Micro-C-Experimenten bietet. Es sollte jedoch auch eine mittlere MNase-Konzentration zwischen den für die Proben in Spur 3 und Spur 4 verwendeten Bedingungen (entsprechend 5 U MNase für 1 x 106 Zellen in 200 μl) berücksichtigt werden. Wichtig ist, dass der Chromatinverdau mit MNase nicht linear skaliert werden kann, und es wird nicht empfohlen, den präparativen Verdau mehr als 4x zu skalieren. Zur Herstellung von Micro-C-Bibliotheken aus mehr als 1 x 10 6 Zellen ist es ratsam, das Chromatin in Aliquoten von 1 x 106 Zellen zu verdauen und diese nach MNase-Inaktivierung zu poolen.
Um den Erfolg des Proximity-Ligationsprotokolls zu beurteilen, sollte die Eingangskontrolle, die MNase-verdaut und nicht Proximity-ligiert ist (Schritt 3.8), mit der Proximity-ligierten Probe (Schritt 5.3) durch 1,5%ige Agarose-Gelelektrophorese verglichen werden (Abbildung 1B). Die Proximity-ligierte Mononukleosomenbande hat eine ungefähre Größe von 300 bp, ähnlich der von Dinukleosomen. Daher sollte sich das mono- zu dikleosomale Bandsignalverhältnis von überwiegend Mononukleosomen (Spur 1) hin zu Dinukleosomen (Spur 3 und Spur 4) verschieben. Da es sich bei dem Agarose-Gel in diesem Schritt um die dinukleosomale DNA handelt, die herausgeschnitten und gereinigt wird, ist es ratsam, die Proben in mehrere Bahnen aufzuteilen, um eine Überlastung zu vermeiden.
Es wird empfohlen, die Qualität und Quantität der vorbereiteten Sequenzierungsbibliothek mittels minimaler PCR zu beurteilen. Hier wird DNA aus 1 μl Kügelchen (1/20 der Gesamtprobe) für 16 Zyklen in 10 μl PCR-Reaktion amplifiziert. Die Gesamtkonzentration der minimalen PCR-Bibliothek liegt typischerweise zwischen 50 und 500 ng nach 16 PCR-Zyklen. Theoretisch entspricht dies einer 1-10 μg-Bibliothek aus der verbleibenden 19 μL-Probe, wenn diese auch für 16 Zyklen amplifiziert würde. Es wird empfohlen, die Mindestanzahl von PCR-Zyklen zu verwenden, die erforderlich sind, um eine Bibliothek von etwa 100 ng aus der Gesamt-DNA zu generieren. Unter der Annahme einer logarithmischen Amplifikation in der PCR kann die theoretische Konzentration der DNA, die aus dem 19-μl-Input bei 16 Zyklen erhalten wurde, nacheinander durch zwei geteilt werden, um die Anzahl der PCR-Zyklen zu berechnen, die zur Erstellung einer 100-ng-Bibliothek erforderlich sind. Zum Beispiel entspricht eine Ausbeute von 100 ng aus 1 μl nach 16 Zyklen einer Ausbeute von 1.900 ng, die aus 19 μl amplifiziert wurde. In diesem Szenario sollten 12 Zyklen idealerweise eine 118 ng Sequenzierungsbibliothek aus der gesamten DNA generieren (1.900 ng/[2 × 2 × 2 × 2] = 118 ng). Die verbleibenden 9 μl Probe aus der minimalen PCR können dann verwendet werden, um die Qualität der Bibliothek durch Agarose-Gelelektrophorese zu beurteilen (Abbildung 1C). Die Visualisierung sollte eine deutliche Bande bei 420 bp und keine Banden für Adapterdimere (120 bp) zeigen. Es können auch kleinere Fragmente auftreten, die unbenutzten PCR-Primern entsprechen.
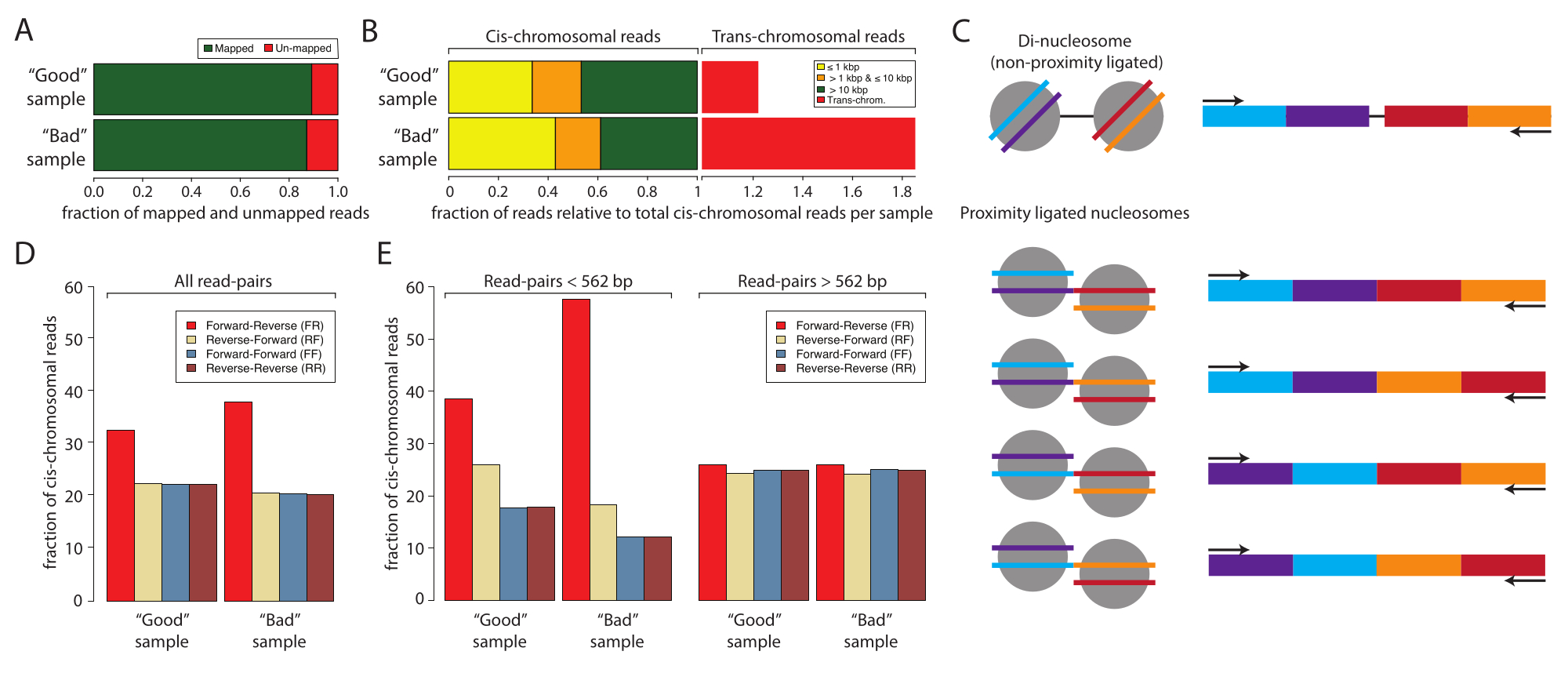
Als nächstes wird empfohlen, die erfolgreiche Micro-C-Probenvorbereitung durch Low-Input-Sequenzierung zu analysieren und zu bestätigen, bevor man sich für eine ressourcenintensive Tiefensequenzierung entscheidet. Typischerweise werden Bibliotheken auf eine Lesetiefe von 5 x 106 bis 1 x 107 sequenziert und anhand der folgenden Kriterien bewertet: die Sequenzierungs-Leseduplikationsrate, die cis- versus transchromosomale Interaktionsrate und die Sequenzierungs-Leseorientierungsfrequenz. Die Micro-C-Bibliotheken werden mit Distiller verarbeitet, einer Full-Service-Pipeline, die die Daten von der Sequenzierung gelesener Dateien (Fastq-Format) bis hin zu Lesepaardateien (Bedpe-Format) und skalierbaren Interaktionsmatrizen (Cool- und Mcool-Formate) unter Verwendung von cooler, pairtools und cooltools10,11,12 verarbeitet. Die Pipeline generiert auch eine Zusammenfassungsdatei, die sich ideal für die Bewertung der Qualität der Micro-C-Bibliotheken10 (https://github.com/open2c/distiller-nf) eignet. Die PCR-Duplikationsrate gibt Aufschluss über die Komplexität der Sequenzierungsbibliothek und kann aus der generierten *.stats-Datei extrahiert werden. Hochwertige Micro-C-Bibliotheken haben eine PCR-Duplikationsrate von weniger als 5 % bis 10 %, wenn sie aus 5 Millionen oder mehr Zellen hergestellt werden. Bemerkenswert ist, dass einige Sequenzierungsplattformen während der Clusterbildung PCR-Duplikate erzeugen, die unabhängig von der Komplexität der Sequenzierungsbibliothek sind. Abbildung 2A zeigt die relativen Duplikationsraten von zwei Experimenten: eines, das wir als gute Stichprobe betrachten, und eines, das wir als schlechte Stichprobe betrachten. In diesem Beispiel zeigten beide Stichproben akzeptable Kartenraten. Das nächste Kriterium zur Beurteilung der Qualität von Micro-C-Bibliotheken ist das Verhältnis von cis zu trans und die Häufigkeit der Leseorientierung. Innerhalb des Zellkerns besiedeln Chromosomen einzelne Chromosomenterritorien und interagieren daher selten mit anderen Chromosomen. Eine hohe Rate an detektierten transchromosomalen Interaktionen deutet auf eine hohe Rate an zufälligen Ligaturen hin. Es sollte beachtet werden, dass die schlechte Probe auf dieser Analyseebene eine hohe Rate an transchromosomalen Interaktionen im Vergleich zur guten Probe aufwies (Abbildung 2B). Für Mikro-C ist eine cis-chromosomale Interaktionsrate von 70 % oder mehr wünschenswert.
Eine Micro-C-Bibliothek hat eine Fragmentgröße, die der di-nukleosomalen DNA-Bande ähnelt, die mit der Proximity-ligierten Probe mitreinigen und das Experiment kontaminieren kann. Bei diesen Verunreinigungen handelt es sich immer um cis-chromosomale Interaktionen. Daher ist es wichtig, auch die Leseorientierungsraten auszuwerten. Die Rate der dinukleosomalen Kontamination kann durch Low-Input-Sequenzierung abgeschätzt werden. Die di-nukleosomale DNA stammt von zwei benachbarten Nukleosomen, die nicht von MNase gespalten wurden. Daher weisen die resultierenden sequenzierenden Lesevorgänge immer eine Vorwärts-Rückwärts-Leseorientierung (F und R) auf, und der Abstand zwischen den Lesepaaren beträgt etwa 320 bp. Im Vergleich dazu können Proximity-ligierte Fragmente in vier Orientierungen ligiert werden, was Lesepaare mit F-R, R-R, R-F und F-F ergibt, idealerweise mit gleicher Häufigkeit (Abbildung 2C). Darüber hinaus zeigen sie verschiedene Abstände zwischen den beiden Lesepaaren an. Um die Menge der dinukleosomalen Verunreinigungen abzuschätzen, kann die Häufigkeit der Leseorientierungen aus den vom Destillateur erzeugten *stats-Dateien berechnet werden (Abbildung 2D). Bemerkenswert ist, dass in dieser Arbeit der Anteil der F-R-Lesevorgänge (rot) in der schlechten Stichprobe höher war als in der guten Stichprobe, und dies wurde deutlicher, wenn die Leseorientierungen nach Entfernung geschichtet wurden (Abbildung 2E). Die F-R-Fraktion wird von di-nukleosomalen Fragmenten im Vergleich zu Micro-C-Bibliotheken dominiert, wenn die Read-Paare in Reads mit Abständen <562 bp oder ≥562 bp stratifiziert werden. Hier wird der Anteil der Reads mit einem Abstand von <562 bp von F-R-Reads dominiert, während der Anteil mit Abständen ≥562 bp eine gleichmäßige Verteilung zwischen den vier möglichen Orientierungen aufweist, was darauf hindeutet, dass die globale Überrepräsentation von F-R-Reads von di-nukleosomalen Verunreinigungen herrührt. Die Wahl von 562 bp als Schwellenwert für die Teilmenge wird durch das Binning in der generierten *stats-Datei definiert. Obwohl dies für diese Qualitätskontrolle nicht erforderlich ist, kann eine definiertere Teilmenge erreicht werden, indem die Abstände aus der *pairs-Datei extrahiert werden, die ebenfalls von distiller erzeugt wird. Es ist wichtig zu beachten, dass dinukleosomale Reads die Qualität der Micro-C-Probe nicht beeinträchtigen, da sie während der Datenverarbeitung identifiziert und ignoriert werden können. Sie enthalten jedoch keine wertvollen Informationen über 3D-Interaktionen und verwässern die informativen Lektüre.
Daher sind eine sorgfältige MNase-Titration und eine gründliche Qualitätskontrolle mit Low-Input-Sequenzierung die besten Werkzeuge, um die Qualität von Micro-C-Experimenten zu optimieren.

Abbildung 1: Zwischenstufen des Micro-C-Protokolls . (A) Agarose-Gelelektrophorese des Chromatins von 2,5 x 105 ES-Zellen der Maus, die mit unterschiedlichen MNase-Konzentrationen verdaut wurden. Die mono-, di- und trinukleosomalen Banden sind durch Pfeile gekennzeichnet. M: DNA-Leiter (Spur 1/6); 10 E MNase pro 250.000 Zellen (Spur 2); 2,5 U MNase pro 250.000 Zellen (Spur 3); 0,625 U MNase pro 250.000 Zellen (Spur 4); 0,156 E MNase pro 250.000 Zellen (Spur 2). (B) Die 1,0%ige Agarose-Gelelektrophorese der Mikro-C-präparierten Proben (Spur 3 und Spur 4) und die MNase-verdaute Eingangskontrolle (Spur 1). Spur 1 und Spur 2 (M: DNA-Leiter) werden verbessert, um die relative Änderung der Intensität mono- bis dinukleosomaler Fragmente zu betonen. Die mono- und dinukleosomalen Banden sind durch Pfeile gekennzeichnet. Die di-nukleosomale Bande in der Proximity-ligierten Probe kombiniert di-nukleosomale und Micro-C-Bibliotheks-DNA. (C) Die 1,0%ige Agarose-Gelelektrophorese der Micro-C-Sequenzierungsbibliotheken, die aus einer 1-μl-Probe amplifiziert wurden, um die Qualität zu bewerten. Bahn 1 (M): DNA-Leiter; Spur 2 (S): Mirco-C-Bibliothek. (D) Fragment Analyzer-Spur der endgültigen Micro-C-Bibliothek. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Stichprobenstatistik für die Low-Input-Sequenzierung einer guten und einer schlechten Stichprobe . (A) Balkendiagramm des Prozentsatzes der zugeordneten (grün) und nicht zugeordneten (rot) Lesevorgänge. (B) Normalisierter Anteil der Reads, der cis- und transchromosomale Interaktionen abbildet. Die Datensätze wurden auf das cis-Mapping normalisiert. Die cis-Mapping-Reads wurden nach dem Abstand zwischen dem ersten und dem zweiten Reads der sequenzierten Probenpaare geschichtet: ≤1 kbp (gelb), >1 kbp und ≤10 kbp (orange) und >10 kbp (rot). (C) Schematische Darstellung der potentiellen Molekülspezies mit den dinukleosomalen Größen. (D) Prozentsätze der Read-Pair-Orientierungen aller Reads der guten und der schlechten Sample. (E) Wie Tafel (D), jedoch geschichtet nach Entfernungen (links, <562 bp und rechts, ≥562 bp). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
| Komponenten | 1-fach | 4,4-fach |
| 10x NEBuffer 2.1, | 10 μl | 44 μl |
| 2 μl 100 mM ATP | 2 μl | 8,8 μl |
| 100 mM DVB-T | 5 μl | 22 μl |
| H2O | 68 μl | 299,2 μl |
| 10 U/μL T4 PNK | 5 μl | 22 μl |
| Gesamt | 90 μl | 396 μl |
Tabelle 1: Micro-C-Mastermix 1. Zusammensetzung der Mastermischung für die Endkaureaktion.
| Komponenten | 1-fach | 4,4-fach |
| 1 mM Biotin-dATP | 10 μl | 44 μl |
| 1 mM Biotin-dCTP | 10 μl | 44 μl |
| 10 mM Mix aus dTTP und dGTP | 1 μl | 4,4 μl |
| 10x T4 DNA-Ligase-Puffer | 5 μl | 22 μl |
| 200x BSA | 0,25 μl | 1,1 μl |
| H2O | 23,75 μl | 104,5 μl |
Tabelle 2: Micro-C-Mastermix 2. Zusammensetzung des Mastermixes für die Endmarkierungsreaktion.
| Komponenten | 1-fach | 4,4-fach |
| 10x NEB T4 Ligase Reaktionspuffer | 50 μl | 220 μl |
| H2O | 422,5 μl | 1859 μl |
| T4-DNA-Ligase | 25 μl | 110μL |
Tabelle 3: Micro-C-Mastermix 3. Zusammensetzung der Mastermischung für die Proximity-Ligationsreaktion.
| Komponenten | 1-fach | 4,4-fach |
| 10x NEBuffer 1.1 | 20 μl | 88 μl |
| H2O | 180 μl | 792 μl |
| ExoIII-Nuklease | 10 μl | 44 μl |
Tabelle 4: Micro-C-Mastermix 4. Zusammensetzung des Mastermixes für die Biotinentfernungsreaktion.
Diskussion
Der Erfolg eines Micro-C-Experiments hängt von einigen kritischen Schritten im Protokoll ab, die sorgfältig ausgeführt werden müssen. Zum einen kann die Vernetzung mit dem zusätzlichen Vernetzer DSG oder EGS je nach Zelltyp zur Aggregation von Zellen führen. Die Zugabe von 0,1 % bis 0,5 % BSA zur Vernetzungsreaktion reduziert die Aggregation erheblich, ohne die Vernetzungseffizienz zu beeinträchtigen. Ineffiziente Vernetzung kann zu erhöhten Raten von transchromosomalen Interaktionen führen, die auf zufällige Ligaturen hinweisen. Der zweite, aber wichtigste Schritt in diesem Protokoll ist der Verdau von Chromatin mit MNase. Ein suboptimaler Chromatinverdau führt zu einer ineffizienten Proximity-Ligatur (Überverdauung) oder zu erhöhten Raten von nicht-proximity-ligierten Dinukleosomen (Unterverdauung). Die Effizienz der Ligationsreaktion kann durch Agarose-Gelelektrophorese (Abbildung 1B) bewertet werden und lässt sich zusätzlich am besten durch Low-Input-Sequenzierung abschätzen. Wenn die Low-Input-Sequenzierung entweder eine hohe Duplikationsrate (ineffiziente Ligation) oder erhöhte Dinukleosomenraten zeigt, sollte der MNase-Verdausgrad neu bewertet werden. Insbesondere der Verlust von Proben beim Ausführen des Protokolls kann zu einer geringeren Komplexität der Bibliothek führen. Die Konzentration einer Probe wird am besten nach der DNA-Aufreinigung (Schritt 5.3) oder durch minimale PCR (Schritt 8) bestimmt. Die Gesamtausbeute an DNA aus 5 x 106 Säugetierzellen nach DNA-Aufreinigung beträgt typischerweise >2 μg. Die DNA-Konzentration sollte nach dem MNase-Verdau, dem ExoIII-Verdau und der DNA-Reinigung kontrolliert werden. Endogene Nukleasen, deren Häufigkeit zelltypspezifisch und speziesspezifisch ist, können eine Quelle des DNA-Abbaus sein. Darüber hinaus kann die säulenbasierte DNA-Aufreinigung aufgrund von Inkompatibilität mit dem SDS aufgrund von Deproteinierungsreaktionen zu Probenverlusten führen. Eine Ethanolfällung kann in Betracht gezogen werden, wenn die DNA-Konzentration in diesem Schritt niedrig ist.
Da Micro-C eine probenspezifische MNase-Titration erfordert, ist es schwierig, Micro-C auf kleine Zellpopulationen anzuwenden, z. B. mit kleinen Organen aus verschiedenen Modellorganismen, Embryonen und Einzelzellen, Organoiden oder Patientenbiopsien. Hier bietet Hi-C 3.0 eine etablierte Alternative, die eine Endpunktreaktion durch sequenzspezifische Restriktionsendonukleasenverwendet 8,9.
Micro-C ist eine weit verbreitete hochauflösende Chromosomenkonformationstechnologie mit einem hohen Dynamikbereich und einem niedrigen Signal-Rausch-Verhältnis, wodurch sie sich besonders für die Untersuchung von kurzreichweitigen Chromosomenmerkmalen 4,5,8, wie z. B. Chromosomenschleifen, eignet. Die Auflösung von Micro-C ermöglicht die Erfassung von Promotor-Enhancer-Schleifen, die jenseits der Nachweisgrenze von Hi-C liegen, was eine detailliertere Analyse der Beziehung zwischen Genomorganisation und Regulation ermöglicht13,14,15. Darüber hinaus wurden kürzlich DNA-Capture-Strategien mit Micro-C kombiniert, um die locus-spezifische Auflösung der anvisierten genomischen Loci auf ein noch nie dagewesenes Niveau zu erhöhen und neue Einblicke in die Ultrastruktur des 3D-Genoms zu ermöglichen16,17,18. Zusammenfassend stellen wir uns vor, dass Micro-C und seine Derivate eine Schlüsseltechnologie sein werden, um die Rolle des 3D-Genoms bei der Transkriptionsregulation und damit bei der Zelltypdifferenzierung und -erhaltung zu untersuchen.
Offenlegungen
Die Autoren haben nichts zu verraten.
Danksagungen
Wir danken Christl Gaubitz und Kathleen Stewart-Morgan für die kritische Lektüre des Manuskripts. Wir danken Anja Groth und dem Labor Groth für die Unterstützung beim Aufbau unseres Labors. Wir danken den Mitarbeitern der CPR/reNEW Genomics Plattform für die Unterstützung: H. Wollmann, M. Michaut und A. Kalvisa. Das Novo Nordisk Foundation Center for Stem Cell Medicine (reNEW) wird durch die Novo Nordisk Foundation Fördernummer NNF21CC0073729 unterstützt. Das Novo Nordisk Foundation Center for Protein Research (CPR) wird von der Novo Nordisk Foundation mit der Fördernummer NNF14CC0001 unterstützt. Wir danken dem Brickman-Labor am Novo Nordisk Center for Stem Cell Medicine, reNEW Copenhagen, für die ES-Zellen der Maus.
Materialien
| Name | Company | Catalog Number | Comments |
| 1 mM Biotin dATP | Jenna Bioscience | NU-835-Bio14-S | |
| 1 mM Biotin dCTP | Jenna Bioscience | NU-809-BioX-S | |
| 10 mM dGTP | NEB | N0442S | |
| 10 mM dTTP | NEB | N0443S | |
| 10 U/ml T4 PNK | NEB | M0201L | |
| 100 U/L Exonuclease III | NEB | M0206L | |
| 10x NEBuffer 1.1 | NEB | B7001S | |
| 10x NEBuffer 2.1 | NEB | B7202S | |
| 10x T4 DNA Ligase buffer | NEB | B0202A | |
| 1x DPBS w/o Mg2+ and Ca2+ | ThermoFisher | 14190144 | |
| 1x LIF | |||
| 2_Mercaptoethanol 50 mM | Gibco | 31350010 | 0.1 mM b-mercaptoethanol |
| 37% Formaldehyde | Sigma Aldrich | 252549-500ML | Caution. See manufactures MSDS |
| 400 U/ml T4 DNA Ligase | NEB | M0202L | |
| 5 U/ml Klenow Fragment | NEB | M0210L | |
| Agarose | BIO-RAD | 1613102 | Caution. See manufactures MSDS |
| BSA 20mg/ml | NEB | B9000S | |
| CaCl2 | |||
| cell counter | |||
| Dimethyl Sulfoxide (DMSO) | Sigma Aldrich | D8418-100ML | Caution. See manufactures MSDS |
| Dynabeads MyOne Streptavidin C1 | Invitrogen | 65001 | |
| DynaMag-2 Magnet | Invitrogen | 12321D | refered to as: magnet magnet for 1.5 ml tubes |
| DynaMag-PCR Magnet | Invitrogen | 492025 | refered to as: magnet magnet for PCR tubes |
| EDTA Ultrapure 0.5M pH 8.0 | Invitrogen | 15575-038 | |
| EGTA Ultrapure 0.5M pH 8.0 | BioWorld | 40121266-1 | |
| Ethanol 96% | VWR Chemicals | 20824365 | quality control system |
| Ethidium Bromide | Invitrogen | 15585-011 | |
| Ethylene glycol bis(succinimidyl succinate) (EGS) | ThermoFisher | 21565 | |
| Fetl Bovin Serum | Sigma Aldrich | F7524 | 15% FBS |
| Gel Loading dye purple (6X) | NEB | B7024S | |
| Glycine | PanReac AppliChem | A1067.0500 | |
| Halt Proteinase inhibitor (100x) | ThermoFisher | 78430 | Caution. See manufactures MSDS |
| IGEPAL CA-630 (NP-40) | Sigma Aldrich | 18896-50ML | |
| MgCl 1 M | Invitrogen | AM9530G | |
| Micrococcal Nuclease (MNase) | Worthington | LS004798 | |
| mouse embryonic stem cells | |||
| NaCl | Sigma Aldrich | S9888-1KG | |
| NEBNext Multiplex Oligos for Illumina (Dual Index primers) | NEB | E7600S | amplification primers for sequencing libraries |
| NEBNext Ultra II DNA library prep kit for Illumina | NEB | E7645L | sequencing library preparation kit |
| NEBNext Ultra II Q5 Master mix | NEB | M0544S | Caution. See manufactures MSDS |
| Non-Essential Amino Acids Solution | Gibco | 11140050 | 1x NEAA |
| Penicillin-Streptomycin (10,000 U/mL) | Gibco | 15140148 | 1% Pen-Strep |
| Proteinase K (40 mg/ml) | GoldBio | P-480-1 | Caution. See manufactures MSDS |
| QIAquick Gel extraction kit | QIAgen | 28706 | refered to as: DNA gel elution kit |
| QIAquick PCR purification kit | QIAgen | 28106 | refered to as: commercial DNA purification kit |
| Qubit dsDNA HS Assay kit | Invitrogen | Q32854 | high sensitivity DNA quantification instrument |
| Quick load purple 1kb plus DNA Ladder | NEB | N0550S | |
| SPRIselect size selection beads | Beckman Coulter | B23319 | paramagnetic beads |
| ThermoMixer C | Eppendorf | 5382000015 | refered to as: thermomixer |
| Tris | Merck | 10708976001 | |
| Trypsin | |||
| Tween20 | Sigma Aldrich | P7949-100ML | |
| Ultrapure 10% SDS | Invitrogen | 15553-035 | |
| Ultrapure Phenol Chloroform Isoamyl Alcohol (PCI) | Invitrogen | 15593-031 | |
| Fragment Analyzer |
Referenzen
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Hsieh, T. -. H. S., Fudenberg, G., Goloborodko, A., Rando, O. J. Micro-C XL: Assaying chromosome conformation from the nucleosome to the entire genome. Nature Methods. 13 (12), 1009-1011 (2016).
- Hsieh, T. -. H. S., et al. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell. 162 (1), 108-119 (2015).
- Krietenstein, N., et al. Ultrastructural details of mammalian chromosome architecture. Molecular Cell. 78 (3), 554-565 (2020).
- Hsieh, T. -. H. S., et al. Resolving the 3D landscape of transcription-linked mammalian chromatin folding. Molecular Cell. 78 (3), 539-553 (2020).
- Belaghzal, H., Dekker, J., Gibcus, J. H. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods. 123, 56-65 (2017).
- van Holde, K. E. . Chromatin. , (1989).
- Oksuz, B. A. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18, 1046-1055 (2021).
- Lafontaine, D. L., Yang, L., Dekker, J., Gibcus, J. H. Hi-C 3.0: Improved protocol for genome-wide chromosome conformation capture. Current Protocols. 1 (7), 198 (2021).
- Goloborodko, A., Venev, S., Abdennur, N., Di Tommaso, P. Mirnylab/distiller-nf; v033. Zenodo. , (2019).
- Venev, S., et al. Open2c/cooltools: v0.4.1. Zenodo. , (2021).
- Abdennur, N., Mirny, L. A. Cooler: Scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 36 (1), 311-316 (2019).
- Zhang, S., Übelmesser, N., Barbieri, M., Papantonis, A. Enhancer-promoter contact formation requires RNAPII and antagonizes loop extrusion. BioRxiv. , (2022).
- Barshad, G., et al. RNA polymerase II and PARP1 shape enhancer-promoter contacts. BioRxiv. , (2022).
- Hansen, A. S., et al. Distinct classes of chromatin loops revealed by deletion of an RNA-binding region in CTCF. Molecular Cell. 76 (3), 395-411 (2019).
- Hua, P., et al. Defining genome architecture at base-pair resolution. Nature. 595 (7865), 125-129 (2021).
- Downes, D. J. High-resolution targeted 3C interrogation of cis-regulatory element organization at genome-wide scale. Nature Communications. 12, 531 (2021).
- Goel, V. Y., Huseyin, M. K., Hansen, A. S. Region capture Micro-C reveals coalescence of enhancers and promoters into nested microcompartments. BioRxiv. , (2022).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten