
Method Article
Mapeo de las interacciones del genoma 3D de los mamíferos con Micro-C-XL
En este artículo
Resumen
Aquí se presenta un protocolo para mapear la organización tridimensional del genoma con resolución de nucleosomas utilizando el método de captura de conformación cromosómica de todo el genoma Micro-C-XL.
Resumen
La organización cromosómica tridimensional (3D) es un factor importante en la regulación del genoma y la especificación del tipo de célula. Por ejemplo, se cree que los elementos cis-reguladores, conocidos como potenciadores, regulan la actividad de los promotores distales a través de la interacción en el espacio 3D. Las tecnologías de captura de la conformación cromosómica (3C) de todo el genoma, como Hi-C, han transformado nuestra comprensión de cómo se organizan los genomas en las células. La comprensión actual de la organización del genoma 3D está limitada por la resolución con la que se puede resolver la organización topológica de los cromosomas en el espacio 3D. Micro-C-XL mide el plegamiento cromosómico con resolución a nivel del nucleosoma, la unidad básica de la cromatina, mediante la utilización de la nucleasa microcócica (MNasa) para fragmentar los genomas durante el protocolo de captura de la conformación cromosómica. Esto da como resultado una mejor relación señal-ruido en las mediciones, lo que facilita una mejor detección de sitios de aislamiento y bucles cromosómicos en comparación con otras tecnologías 3D de todo el genoma. En este artículo se presenta un protocolo paso a paso, detallado y con soporte visual para preparar muestras de Micro-C-XL de alta calidad a partir de células de mamíferos.
Introducción
Micro-C-XL es una técnica de todo el genoma para medir la conformación del genoma en 3D con resolución de nucleosomas. Micro-C-XL se basa en la tecnología Hi-C basada en la ligadura de proximidad ampliamente utilizada, que ha transformado nuestra comprensión de cómose organizan los genomas 3D. Micro-C-XL y su primera iteración, Micro-C, se desarrollaron inicialmente en Saccharomyces cerevisiae2,3 y posteriormente se adaptaron a sistemas celulares de mamíferos, para los que el protocolo ha demostrado todo su potencial en la detección de características de corto alcance del genoma 3D, como bucles cromosómicos y sitios de aislamiento. Esta versión se basa en publicaciones recientes de Micro-C-XL en mamíferos 4,5. Como Micro-C-XL reemplaza a Micro-C, Micro-C-XL se denominará Micro-C en el manuscrito.
Las principales diferencias entre Micro-C y Hi-C6 son las siguientes: 1) fragmentación del genoma con nucleasa microcócica (MNasa) en comparación con enzimas de restricción y 2) reticulantes adicionales con mayor espaciamiento atómico entre los grupos reactivos en comparación con solo formaldehído. Ambos pasos contribuyen significativamente a la mejora de la relación señal-ruido de Micro-C en comparación con Hi-C convencional. El tamaño de la fragmentación limita la resolución a la que se puede resolver la organización del genoma 3D durante el protocolo de ligadura de proximidad. La MNasa es una nucleasa que digiere preferentemente el ADN accesible y deja intacto el ADN protegido por nucleosomas. La huella de nucleosomas mediante secuenciación de MNasa ha demostrado que los nucleosomas cubren completamente la mayoría de los genomas eucariotas7. Como los nucleosomas se distribuyen por todo el genoma con un espaciamiento medio de 160-220 pb, dependiendo de la especie y el tipo de célula, la MNasa es la enzima ideal para el mapeo de alta resolución de la arquitectura del genoma.
El uso de un reticulante adicional en combinación con formaldehído (FA) en el método Micro-C mejora adicionalmente la relación señal-ruido 2,8. Los reticulantes específicos de aminas con espaciadores atómicos más largos entre los grupos reactivos facilitan los reticulantes proteína-proteína. Por lo general, se trata de glutarato de disuccinimidil (DSG) o succinimidato de etilenglicol bis-succinimidil (EGS) con espaciadores de 7,7 Å y 16,1 Å, respectivamente. La reducción del ruido a través de EGS o DSG es particularmente evidente en experimentos con altas tasas de fragmentación, como Micro-C, y presumiblemente ocurre debido a una reducción en la tasa de eventos de ligadura aleatoria8.
Un protocolo Hi-C 3.0 recientemente desarrollado que utiliza la reticulación ESG/DSG y múltiples combinaciones de enzimas de restricción reduce el ruido en los experimentos Hi-C y mejora significativamente la detección de bucles cromosómicos y sitios de aislamiento 8,9. Aun así, una comparación sitio por sitio de varias características de datos de interacción encontró que Micro-C tenía una detección superior de características de corto alcance, como bucles cromosómicos y sitios de aislamiento, en comparación con Hi-C 3.0 y Hi-C8 convencional. Sin embargo, Hi-C 3.0 mejora la detección de características de corto alcance y mantiene una fuerte detección de la compartimentación del genoma en comparación con Hi-C convencional. En resumen, la elección de un método de captura de la conformación cromosómica debe estar determinada por la cuestión objetiva y biológica.
Aquí, proporcionamos un protocolo paso a paso para experimentos exitosos de Micro-C que pueden desentrañar la organización del genoma 3D.
Protocolo
1. Cultivo celular y reticulación
- Cultivar células de acuerdo a las necesidades experimentales para obtener un mínimo de 1 x 107 células. Aquí, las células se cultivaron a 37 °C con 5% de CO2 en medio E14 (DMEM con piruvato y L-glutamina, 15% FBS, 1x LIF, 1x NEAA, 1% pen-strep, 0,1 mM β-mercaptoetanol [ver Tabla de Materiales]) y se dejaron pasar cada dos días.
NOTA: Este protocolo se ha aplicado con éxito a varios tipos de células de múltiples especies, como Homo sapiensy Mus musculus. En este ejemplo, se utilizaron células madre embrionarias (ES) de ratón. - Coseche las células al 70%-80% de confluencia aspirando el medio. Lavar una vez con 5 ml de DPBS e incubar las células con 3 ml de tripsina precalentada al 0,25% por plato de 10 cm durante 2-3 min a 37 °C.
- Apague la tripsina con 7 ml de medio E14 precalentado y transfiera las células desprendidas a un tubo de 50 ml.
- Granular las células por centrifugación a 300 x g durante 5 min a temperatura ambiente (RT). Deseche el sobrenadante y vuelva a suspender el gránulo celular en el medio. Cuente las celdas con un contador de celdas.
NOTA El protocolo es robusto a algunas variaciones en las concentraciones celulares, y se han utilizado varios contadores celulares para cuantificar el número de células; Sin embargo, tenga en cuenta el rango dinámico del contador de celdas utilizado. Por lo general, esto está entre 1 x 105-1 x 107 células/ml. Si bien la velocidad y la duración de la centrifugación se han probado para este tipo de celda, las celdas diferentes y más pequeñas pueden requerir una velocidad de centrifugación más alta o tiempos de centrifugado más largos, y la centrifugación debe ajustarse en consecuencia. - Recoger las células por centrifugación a 300 x g durante 5 min a RT y resuspender las células en DPBS a una concentración final de 1 x 106 células/mL. Por ejemplo, si el rendimiento es de 1 x 107 células, vuelva a suspender las células en 10 ml de DPBS.
- Para el primer paso de reticulación, agregue un 37% de formaldehído (FA) a una concentración final del 1% a la suspensión celular e incube la suspensión celular durante 10 min a RT con rotación (15-20 rpm); por ejemplo, si el rendimiento es de 1 x 107 células, agregue 270 μL de AG a la suspensión celular de 10 mL.
NOTA: La solución FA suele ser estable en RT hasta 3 meses después de la apertura. - Apagar la reacción añadiendo 2,5 M de glicina a una concentración final de 0,25 M e incubar durante 5 min a RT con rotación (15-20 rpm). Por ejemplo, si el rendimiento es de 1 x 107 células, agregue 1,027 ml de glicina 2,5 M a la suspensión celular.
- Recoja las células por centrifugación a 300 x g durante 5 min en RT. Deseche el sobrenadante y vuelva a suspender las células en 5 mL de DPBS. Repita el paso de centrifugación una vez y vuelva a suspender las células peletizadas en DPBS a 4 x 106 células/ml. Por ejemplo, si el rendimiento es de 1 x 107 células, vuelva a suspender las células en 2,5 ml de DPBS.
- Para el segundo paso de reticulación, prepare una solución madre 0,3 M de succinimidil de etilenglicol bis-succinimidil (EGS) en DMSO (13,6 mg de EGS en 100 μL de DMSO). Añadir EGS a una concentración final de 3 mM a la suspensión celular, e incubar a RT durante 40 min con rotación (15-20 rpm). Por ejemplo, si el rendimiento es de 1 x 107 células, añadir 25 μL de solución madre de 0,3 M de EGS a 2,5 mL de la suspensión celular.
NOTA: Equilibre el EGS a RT durante al menos 20 minutos antes de pesar para preparar la solución madre. - Apagar la reacción añadiendo 2,5 M de glicina a una concentración final de 0,4 M, e incubar durante 5 min a RT con rotación (15-20 rpm). Por ejemplo, si el rendimiento es de 1 x 107 células, agregue 400 μL de glicina 2,5 M a la suspensión celular.
- Recoja las células por centrifugación a 1.000 x g durante 5 min a RT y vuelva a suspender las células peletizadas en DPBS a 5 x 106 células/mL. Distribuya 5 x 10 6 células/tubo para bibliotecas preparativas y 1 x 106 células/tubo para la valoración para la digestión de la MNasa.
NOTA: Se sugiere valorar el grado óptimo de digestión para cada lote de células reticuladas. Idealmente, recolectar de dos a tres alícuotas de 1 x 10 6 células para los experimentos de valoración de MNasa (paso 2) y de dos a cuatro alícuotas de 5 x 106 células para los experimentos preparativos (paso 3). - Recoger las células por centrifugación a 1.000 x g durante 5 min a RT y eliminar el sobrenadante. Congele rápidamente los gránulos de celda en nitrógeno líquido y guárdelos a -80 °C.
NOTA: Las bibliotecas Micro-C exitosas se pueden preparar a partir de muestras almacenadas durante un máximo de 3 meses.
2. Valoración de MNasa
NOTA: Es necesario realizar una valoración de MNasa para determinar la concentración óptima de MNasa antes de procesar la biblioteca preparativa de las células doblemente reticuladas.
- Para realizar la valoración de MNasa, descongele un gránulo de 1 x 106 células en hielo durante 10 min y vuelva a suspender las células en 500 μL de DPBS (añadir 1x BSA si las células se adhieren a la pared). Incubar la suspensión celular en hielo durante 20 min.
- Recoger las células por centrifugación a 10.000 x g durante 5 min a RT, y eliminar el sobrenadante. Resuspender las células peletizadas en 500 μL de tampón MB#1 (50 mM de NaCl, 10 mM de Tris-HCl, 5 mM de MgCl2, 1 mM de CaCl 2,0,2% de NP-40, 1x inhibidor de la proteasa [ver Tabla de Materiales], pH 7.4).
- Recoger las células por centrifugación a 10.000 x g durante 5 min, y retirar el sobrenadante. Vuelva a suspender las células en 200 μL de tampón MB#1 y divida la muestra en cuatro tubos.
- Descongele un vial de MNasa (20 U/μL) y diluya con 10 mM de Tris, pH 7,4, en una serie de 1:2, 1:4, 1:4 y 1:4 para alcanzar concentraciones de 10 U/μL, 2,5 U/μL, 0,625 U/μL y 0,1256 U/μL respectivamente (una para cada condición de digestión). Con intervalos de tiempo apropiados (10-20 s), añadir 1 μL de solución de MNasa a una de las cuatro muestras, vórtice, e incubar en una termomezcladora a 37 °C durante 10 min (agitación a 800 rpm). Continúe con la adición de 1 μL de las diluciones restantes de MNasa a las alícuotas celulares restantes.
- Detenga la digestión de la Mnasa añadiendo 200 μL de tampón STOP recién preparado (150 μL de 10 mM de Tris, pH 7,4, 25 μL de SDS al 10 %, 25 μL de 20 mg/ml de proteinasa K, 2 μL de 0,5 M de EGTA) a cada tubo en el mismo orden y con el mismo intervalo de tiempo en que se añadió la MNasa. Incubar a 65 °C durante 2 h.
- Agregue 500 μL de alcohol fenol-cloroformo-isoamílico (PCI) a cada muestra y mezcle bien mediante vórtice. Centrifugar a 19.800 x g durante 5 min a RT para separar las fases, y transferir la fase acuosa a nuevos tubos (aproximadamente 200 μL/muestra).
PRECAUCIÓN: El PCI contiene numerosos componentes tóxicos y solo debe manipularse en una campana de seguridad química. Consulte al fabricante para obtener más detalles. - Purifique el ADN utilizando un kit comercial de purificación de ADN (consulte la Tabla de materiales) de acuerdo con las instrucciones del fabricante y eluya las muestras en 12 μL de tampón de elución.
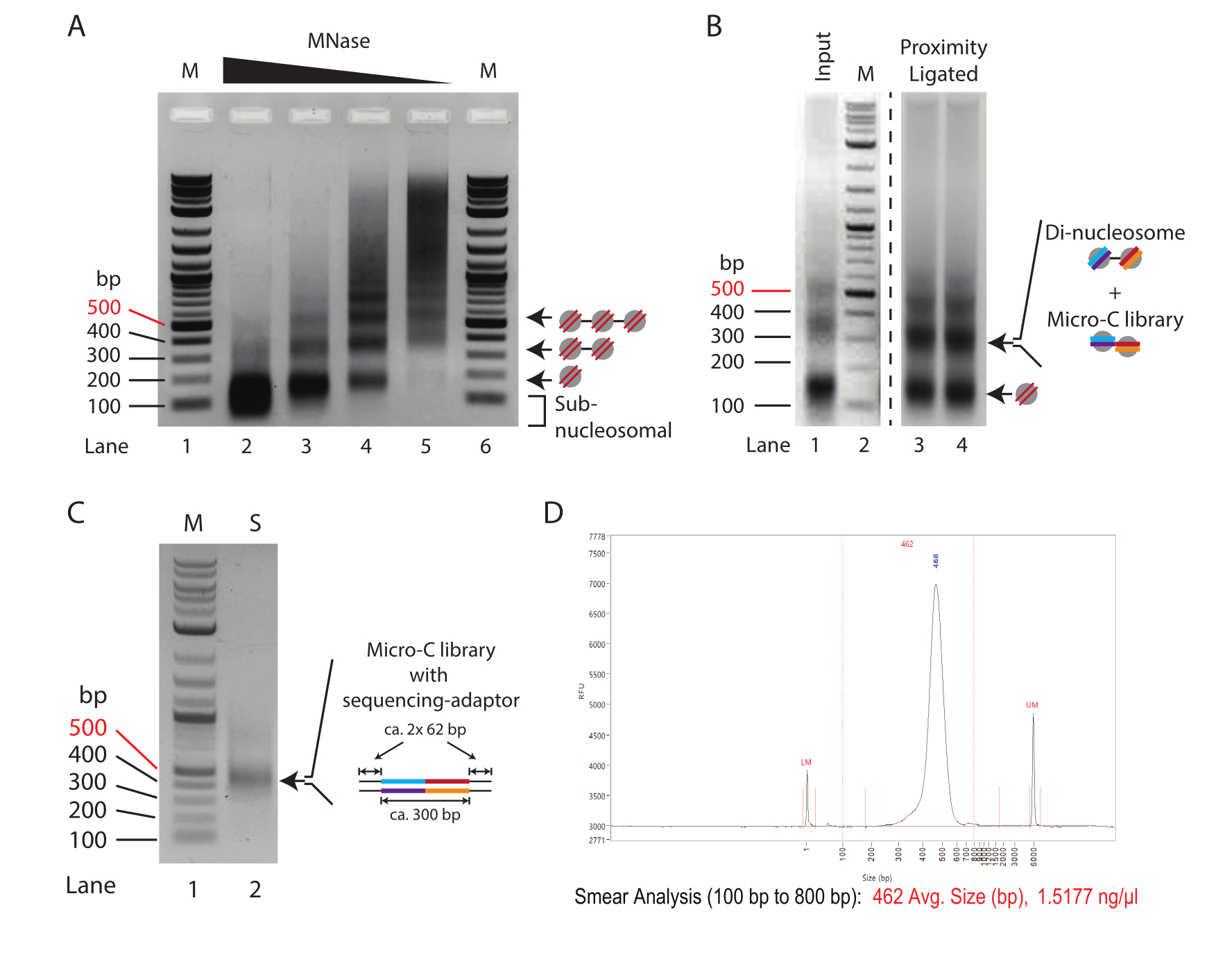
NOTA: La concentración de SDS de la etapa de desproteinización (paso 2.5) es inhibidora para algunos kits de purificación de ADN. El kit de purificación de ADN utilizado aquí tiene un rendimiento comparable al de la precipitación de etanol. - Agregue 2-5 μL de colorante de carga y ejecute las muestras en un gel de agarosa al 1,5% a 120 V durante 30-50 min (hasta que se separen correctamente; Figura 1A).
- Elija el mejor grado de digestión para el experimento y continúe con la digestión preparativa de MNasa. Un grado óptimo de digestión muestra poco o ningún fragmento subnucleosomal y una proporción de mononucleosoma a dinucleosoma del 70 % al 90 %.
NOTA: En este experimento, se determinó que el grado de digestión del carril 4 en la Figura 1A es la digestión óptima obtenida con 0,625-1,25 U de Mnase para 2,5 x 10 5 células (= 2,5-5 U de Mnase para 1 x 106 células). Para una discusión detallada, consulte los resultados representativos.
3. Digestión preparativa de la MNasa
- Para fragmentar la cromatina en los mononucleosomas a través de la digestión de MNasa, descongelar alícuotas de 5 x 106 células previamente reticuladas previamente y resuspender en 1 ml de DPBS. Incubar en hielo durante 20 minutos (añadir 1x BSA si las células se adhieren a la pared del tubo).
- Recoger las células por centrifugación a 10.000 x g durante 5 min a RT, y desechar el sobrenadante. Vuelva a suspender el gránulo en 500 μL de tampón MB#1. Repita el paso de centrifugación una vez. Vuelva a suspender el gránulo en 1 ml de tampón MB#1 y haga 200 μL de alícuotas (1 x 106 células por alícuota).
- Con base en la valoración de MNasa (descrita en el paso 2), digiera la cromatina agregando la cantidad adecuada de MNasa (generalmente 2,5-10 U/μL por 1 x 106 células) a cada alícuota. Mezclar bien (vórtice y centrifugar rápidamente) e incubar en una termobatidora a 37 °C durante 10 min agitando a 800 rpm.
- Detenga la digestión de MNasa añadiendo 1,6 μL de 0,5 M EGTA (4 mM finales) a cada alícuota, e incube en una termomezcladora a 65 °C durante 10 min con agitación a 800 rpm.
- Recoger la muestra por centrifugación a 10.000 x g durante 5 min a RT, y desechar el sobrenadante. Resuspender el gránulo celular en 500 μL de 1x NEBuffer 2.1.
- Agrupar muestras equivalentes a una entrada de 5 x 106 celdas o menos para su posterior procesamiento.
NOTA: Si se van a procesar más de 5 x 10 6 células, procese estas muestras en paralelo, ya que las condiciones enzimáticas están optimizadas para 5 x 106 células. - Antes de continuar con los pasos de ligadura de proximidad, transfiera el 10% de la muestra como control de entrada para controlar el nivel de digestión de MNasa. Añadir a esta muestra 150 μL de Tris 10 mM, pH 7,4, 25 μL de SDS al 10% y 25 μL de 20 mg/mL de proteinasa K e incubar durante la noche a 65 °C.
4. Procesamiento de extremos de ADN y ligadura de proximidad
- Recoger la muestra restante por centrifugación a 10.000 x g durante 5 min a 4 °C y desechar el sobrenadante. Vuelva a suspender el gránulo en 90 μL de mezcla maestra Micro-C 1 recién preparada (Tabla 1) e incube en una termomezcladora durante 15 min a 37 °C con agitación a 800 rpm.
- Añadir 10 μL de 5 U/μL de Klenow Fragment e incubar en una termomezcladora durante 15 min a 37 °C agitando a 800 rpm.
- Añadir 100 μL de mezcla maestra Micro-C 2 recién preparada (Tabla 2) e incubar en una termomezcladora durante 45 min a 25 °C con agitación a 800 rpm. Después de la incubación, apagar la reacción enzimática añadiendo EDTA a una concentración final de 30 mM. Incubar en una termomezcladora durante 20 minutos a 65 °C agitando a 800 rpm.
- Recoger la muestra por centrifugación a 10.000 x g durante 5 min a 4 °C, y desechar el sobrenadante. Resuspender la muestra en 500 μL de mezcla maestra Micro-C 3 recién preparada (Tabla 3) e incubar durante 2,5 h a RT con rotación (15-20 rpm).
- Recoger la muestra por centrifugación a 10.000 x g durante 5 min a 4 °C, y desechar el sobrenadante. Volver a suspender la muestra en 200 μL de mezcla maestra Micro-C 4 recién preparada (Tabla 4) e incubar en una termomezcladora durante 15 min a 37 °C con agitación a 800 rpm.
- Para la reticulación inversa y la desproteinación, añadir 25 μL de 20 mg/ml de proteinasa K y 25 μL de SDS al 10% a la muestra, e incubar a 65 °C durante la noche con mezcla intermitente.
5. Purificación de ADN dinucleosomal y selección de tamaño
- Agregue 500 μL de PCI a las muestras y al control de entrada, y mezcle por vórtice. Separar las fases por centrifugación a 19.800 x g durante 5 min, y transferir la fase acuosa superior a un tubo nuevo.
- Concentrar el ADN utilizando un kit de purificación de ADN o mediante precipitación de etanol. Eluir las muestras en 30 μL (paso 5.3) y los controles de entrada (paso 3.11) en 15 μL, y ejecutar un gel de agarosa al 1,5 % para separar los mononucleosomas y dinucleosomas (Figura 1B).
NOTA: La concentración de SDS de la etapa de desproteinización es inhibidora para algunos kits de purificación de ADN. El kit de purificación de ADN utilizado aquí (ver Tabla de Materiales) tiene un rendimiento comparable al de la precipitación de etanol. Dependiendo del número de células utilizadas, la entrada puede oscilar entre 100 ng y 10 μg. Normalmente, se extraen de 1 a 5 μg de ADN de 5 x 106 células. - Extirpar los fragmentos de ADN que tienen un tamaño dinucleosomal (aproximadamente 300 pb). Utilice un kit de elución de gel de ADN disponible en el mercado (consulte la Tabla de materiales) para extraer el ADN del gel de agarosa y eluir en 150 μL.
6. Preparación de las perlas de estreptavidina
- Transfiera 10 μL de perlas de estreptavidina (ver Tabla de materiales) por muestra a un tubo de reacción.
- Colóquelo en un imán apropiado para tubos de 1,5 ml (consulte la tabla de materiales). Después de que la solución se aclare (1-2 min), retire el sobrenadante y vuelva a suspender las perlas en 300 μL de 1x TBW (5 mM de Tris-HCl, pH 7,5, 0,5 mM EDTA, 1 M de NaCl, 0,05% Tween 20) por muestra. Repita este paso una vez.
- Vuelva a suspender las perlas en 150 μL de tampón 2x B&W (10 mM de Tris-HCl, pH 7,5, 1 mM EDTA, 2 M de NaCl) por muestra procesada en el paso 7.
7. Preparación de la biblioteca desplegable y en el cordón de estreptavidina
- Añadir 150 μL de perlas preparadas previamente (paso 6.3) a 150 μL de la muestra (paso 5.3). Incubar durante 20 min a RT con rotación (15-20 rpm).
- Coloque los tubos en un imán apropiado y espere hasta que la solución se aclare (1-2 min). Retire el sobrenadante y vuelva a suspender las perlas en 300 μL de 1x TBW. Repita este paso.
- Coloque los tubos en un imán apropiado y espere hasta que la solución se aclare (1-2 min). Retirar el sobrenadante y volver a suspender las perlas en 100 μL de 0,1x TE (1 mM de Tris, 0,1 mM de EDTA, pH 8,0).
- Coloque los tubos en un imán apropiado y espere hasta que la solución se aclare (1-2 min). Retirar el sobrenadante, resuspender las perlas en 50 μL de TE 0,1x y transferirlas a tubos de PCR.
NOTA: El volumen de 0,1x TE utilizado (50 μL) corresponde al volumen de entrada del kit de preparación de la biblioteca de secuenciación de ADN (véase la Tabla de materiales) utilizado en este protocolo. Si se utiliza un kit o una estrategia diferente, ajuste el volumen en consecuencia. - Realice los pasos de manipulación del ADN del kit de preparación de la biblioteca de secuenciación de acuerdo con el protocolo del fabricante. Estos pasos suelen incluir el embotamiento del ADN, la cola A, la ligadura del adaptador y la escisión en U. El último paso (escisión en U) es específico del kit utilizado en este estudio. Si utiliza ese kit, siga las instrucciones del fabricante desde el paso 1 hasta el paso 2.6 del protocolo del kit utilizando adaptadores no diluidos, y no considere el almacenamiento a -20 °C después del paso 2.6.
NOTA: Los cambios de tampón requeridos por los kits de secuenciación deben intercambiarse mediante la unión de las perlas a un imán y lavados (pasos 7.3 a 7.4), ya que el ADN todavía está unido a las perlas de estreptavidina. Además, debido a que el ADN está unido a las perlas magnéticas, ignore cualquier paso de purificación y selección de tamaño después de la ligadura del adaptador del protocolo del kit. Continúe con el paso 7.6 (este protocolo). - Una vez completada la ligadura del adaptador, lave la muestra como se describe en el paso 7.2. Deseche el sobrenadante y vuelva a suspender las perlas en 20 μL de 0,1x TE.
8. Estimación de los ciclos de PCR requeridos
NOTA: Se recomienda estimar los ciclos de PCR necesarios para la amplificación de la biblioteca. Por lo general, una biblioteca Micro-C requiere de 8 a 15 ciclos de PCR. Aunque el paso no es esencial, ayuda a evitar la sobreamplificación y reduce el riesgo de duplicados de PCR.
- Para definir el número mínimo de ciclos de PCR necesarios, realice la PCR con 1 μL de muestra de estreptavidina-biotina-ADN (paso 7.6). Para ello, añada a la muestra la mezcla maestra de PCR (3,2 μL deH2O, 0,4 μL de cebador i5, 0,4 μL de cebador i7 y 5 μL de ADN polimerasa Q5 de alta fidelidad). Realice la PCR con 16 ciclos de acuerdo con las instrucciones del fabricante para el kit de preparación de la biblioteca de ADN utilizado.
NOTA: Los cebadores utilizados aquí se compran en un kit separado (consulte la Tabla de materiales) que debe ser compatible con el kit de preparación de la biblioteca utilizado. - Después de la PCR, recoja las perlas con un imán adecuado y mida la concentración de ADN de 1 μL del sobrenadante utilizando un instrumento de cuantificación de ADN de alta sensibilidad (ver Tabla de materiales). Para obtener la concentración total de ADN, multiplique este valor por 10 (volumen total de la reacción de PCR). Calcule el número requerido de ciclos de PCR como se discute en los resultados representativos.
- Agregue 2 μL de tampón de carga 6x a los 9 μL restantes de mezcla de PCR. Resuelva la biblioteca amplificada de PCR en un gel de agarosa al 1% para determinar el éxito de la ligadura del adaptador, lo que da como resultado un tamaño de aproximadamente 450 pb (Figura 1C).
NOTA: El gel debe mostrar una banda distintiva de aproximadamente 420 pb (biblioteca Micro-C más adaptadores). Las bandas de 120 pb representan dímeros adaptadores y son indicativas de bibliotecas de baja complejidad. Pueden aparecer bandas de menor peso molecular (menos de 100 pb), y se trata de cebadores no utilizados de la reacción de PCR.
9. Amplificación de la biblioteca de secuenciación
- Añadir mezcla maestra de PCR (65 μL deH2O, 100 μL de ADN polimerasa de alta fidelidad Q5, 8 μL de cebador i5, 8 μL de cebador i7) a los 19 μL restantes de la muestra de estreptavidina-biotina-ADN. Divida la mezcla de reacción en alícuotas de 50-100 μL.
NOTA: El volumen óptimo para la PCR depende de la máquina de PCR; Por lo general, 50 μL ofrecen la amplificación más reproducible en las máquinas de PCR comunes. - Realice la PCR de acuerdo con las instrucciones del fabricante del kit de preparación de la biblioteca de ADN con el número de ciclos determinado en el paso 8. Si se omitió el paso 8, se recomiendan 14 ciclos para la PCR.
- Purifique el ADN con perlas paramagnéticas (consulte la Tabla de materiales) en una proporción de 1:0.9 de acuerdo con el protocolo del fabricante. Eluir en 20 μL de TE al 0,1%.
NOTA: Si se detectan dímeros adaptadores en el paso 8.5, realice la purificación dos veces utilizando la misma proporción. - Determine la concentración de ADN y ejecute las muestras en un sistema de control de calidad (consulte la tabla de materiales). Asegure una buena calidad de la biblioteca Micro-C mediante la presencia de una sola banda precisa (Figura 1D).
NOTA: Los dímeros adaptadores son poco comunes en las preparaciones de bibliotecas en cordones debido a los lavados de muestras antes de la PCR. Por lo tanto, la aparición de dímeros adaptadores indica una baja complejidad de la biblioteca. Si se observan dímeros adaptadores, se recomienda encarecidamente controlar la calidad de la muestra con una secuenciación de entrada baja.
10. Secuenciación del ADN y procesamiento de datos
- Secuenciar la biblioteca Micro-C con secuenciación de extremos emparejados de acuerdo con los requisitos del proveedor de secuenciación.
NOTA: Idealmente, las muestras se secuencian en una plataforma en modo de extremo emparejado con 50 pb por lectura. Las plataformas más antiguas que ofrecen longitudes de lectura más cortas, como 2 x 35 pb, también se han utilizado con éxito. Es importante destacar que, si se estudian regiones genómicas repetitivas, podría ser aconsejable secuenciar con una longitud de lectura más larga. - Para evaluar la calidad de la biblioteca Micro-C, realice una secuenciación de entrada baja con 5 x 106 a 1 x 107 lecturas por muestra.
- Procese los archivos de secuenciación (archivos fastq) con Distiller10. Asigne las lecturas con el genoma de referencia apropiado, aquí mm10.
NOTA: Los archivos de secuenciación se pueden procesar mediante varias canalizaciones en equipos locales o clústeres informáticos. En el caso de las muestras con profundidades de secuenciación bajas, los tamaños de ubicación más grandes, como 10.000 pb, 50.000 pb, 100.000 pb y 500.000 pb, pueden reducir las demandas informáticas y el tamaño de los archivos. Distiller (utilizado en este estudio) genera todos los tipos de archivos necesarios para evaluar la calidad de la biblioteca Micro-C. El archivo *.stats generado contiene la información sobre la velocidad del mapa, las relaciones cis-trans y la orientación de lectura estratificada por la distancia entre los pares de lectura. Estos parámetros se visualizan en la Figura 2, y la evaluación de la calidad de la biblioteca Micro-C se discute en los resultados representativos. El software de procesamiento también genera archivos mcool que se pueden cargar directamente en HiGlass (https://docs.higlass.io/) para generar matrices de interacción9.
Resultados
La preparación exitosa de las bibliotecas Micro-C se puede evaluar en varios pasos del protocolo. El paso más importante es la elección de un grado de digestión de MNasa adecuado. Por lo tanto, la concentración de MNasa debe valorarse para producir de manera consistente un 70%-90% de mononucleosomas sobre dinucleosomas para cada muestra. Es importante tener en cuenta que la digestión de la cromatina es diferente para la eu- y la heterocromatina, ya que la MNasa digiere la heterocromatina de manera menos eficiente. Por lo tanto, el grado óptimo de digestión depende de la región de cromatina de interés y del tipo de célula estudiado, ya que la proporción relativa de eu- y heterocromatina es específica del tipo de célula. Por lo tanto, es aconsejable valorar cuidadosamente la concentración de MNasa requerida y evaluar primero el éxito del experimento Micro-C mediante secuenciación de baja entrada.
En la Figura 1A se muestra un patrón típico de titulación de MNasa de cromatina tratada con cantidades decrecientes de MNasa. Aquí, la cromatina de 250.000 células por reacción se digiere con una dilución cuádruple de MNasa. La concentración más alta (10 U de MNasa, Carril 2) muestra cromatina sobredigerida que consiste casi exclusivamente en ADN mononucleosómico (~150 pb). En particular, el centro de la banda mononucleosomal es más bajo en el gel de agarosa en comparación con las bandas correspondientes en las muestras con concentraciones reducidas de MNasa, lo que indica una digestión excesiva del ADN nucleosomal. Los nucleosomas sobredigeridos se ligan de manera ineficiente en la reacción de ligadura de proximidad; por lo tanto, la muestra en el carril 2 no es óptima para experimentos de Micro-C. El carril 3 (2,5 U de MNasa) muestra un grado de digestión casi apropiado para experimentos de Micro-C. Aquí, la banda mononucleosomal es la especie dominante, y el frotis subnucleosomal, indicativo de nucleosomas sobredigeridos, se reduce; sin embargo, todavía está presente. El grado de digestión en el carril 4 (0,635 U de MNasa) es una condición ideal para un experimento de Micro-C en este ejemplo de valoración. Está presente una banda mononucleosómica clara sin ADN subnucleosomal. La intensidad de la banda para el ADN mononucleosoma y dinucleosoma es casi igual, lo que indica un rendimiento de mononucleosoma del 66% o más. Vale la pena señalar que el ADN dinucleosomal es aproximadamente el doble del tamaño del ADN mononucleosomal (~ 320 pb frente a ~ 150 pb), por lo que su intensidad de banda por mol de ADN es dos veces mayor en comparación con su contraparte mononucleosomal. El grado de digestión en el carril 5 (0,156 U de MNasa) muestra cromatina subdigerida con casi ningún ADN nucleosomal, y esto, por lo tanto, representa una muestra subóptima.
En conclusión, en este ejemplo, la digestión de 2,5 x 10 5 células madre embrionarias de ratón con 0,625 U de MNasa (correspondiente a2,5 U de MNasa para 1 x 106 células en 200 μL) ofrece el punto de partida más prometedor para las digestiones preparativas en experimentos de Micro-C. Sin embargo, también debe considerarse una concentración intermedia de MNasa entre las condiciones utilizadas para las muestras en el carril 3 y el carril 4 (correspondiente a 5 U de MNasa para 1 x 106 células en 200 μL). Es importante destacar que la digestión de la cromatina con MNasa no se puede escalar linealmente y no se recomienda aumentar la digestión preparativa más de 4 veces. Para preparar bibliotecas de Micro-C a partir de más de 1 x 10 6 células, es aconsejable digerir la cromatina en alícuotas de 1 x 106 células y agruparlas después de la inactivación de la MNasa.
Para evaluar el éxito del protocolo de ligadura de proximidad, el control de entrada, que es digerido por MNasa y no ligado por proximidad (paso 3.8), debe compararse con la muestra ligada por proximidad (paso 5.3) mediante electroforesis en gel de agarosa al 1,5% (Figura 1B). La banda mononucleosoma ligada por proximidad tiene un tamaño aproximado de 300 pb, similar al de los dinucleosomas. Por lo tanto, la relación de señal de banda mononucleosoma a dinucleosoma debe cambiar de predominantemente mononucleosomas (carril 1) a dinucleosomas (carril 3 y carril 4). Como el gel de agarosa en este paso es el ADN dinucleosomal que se extirpa y purifica, es aconsejable dividir las muestras en varios carriles para evitar la sobrecarga.
Se recomienda evaluar la calidad y cantidad de la biblioteca de secuenciación preparada mediante una PCR mínima. Aquí, el ADN de 1 μL de perlas (1/20 de la muestra total) se amplifica durante 16 ciclos en 10 μL de reacción de PCR. La concentración total de la biblioteca mínima de PCR suele oscilar entre 50 y 500 ng después de 16 ciclos de PCR. En teoría, esto corresponde a una biblioteca de 1-10 μg de la muestra restante de 19 μL si también se amplificara durante 16 ciclos. Se recomienda utilizar el número mínimo de ciclos de PCR necesarios para generar una biblioteca de aproximadamente 100 ng a partir del ADN total. Suponiendo una amplificación logarítmica en la PCR, la concentración teórica del ADN obtenida a partir de la entrada de 19 μL a 16 ciclos se puede dividir sucesivamente por dos para calcular el número de ciclos de PCR necesarios para generar una biblioteca de 100 ng. Por ejemplo, un rendimiento de 100 ng de 1 μL después de 16 ciclos corresponde a un rendimiento de 1.900 ng amplificado de 19 μL. En este escenario, lo ideal es que 12 ciclos generen una biblioteca de secuenciación de 118 ng a partir del ADN total (1.900 ng/[2 × 2 × 2 × 2] = 118 ng). La muestra restante de 9 μL de la PCR mínima se puede utilizar para evaluar la calidad de la biblioteca mediante electroforesis en gel de agarosa (Figura 1C). La visualización debe mostrar una banda distinta a 420 pb y ninguna banda para los dímeros adaptadores (120 pb). También pueden aparecer fragmentos más pequeños, que corresponden a cebadores de PCR no utilizados.
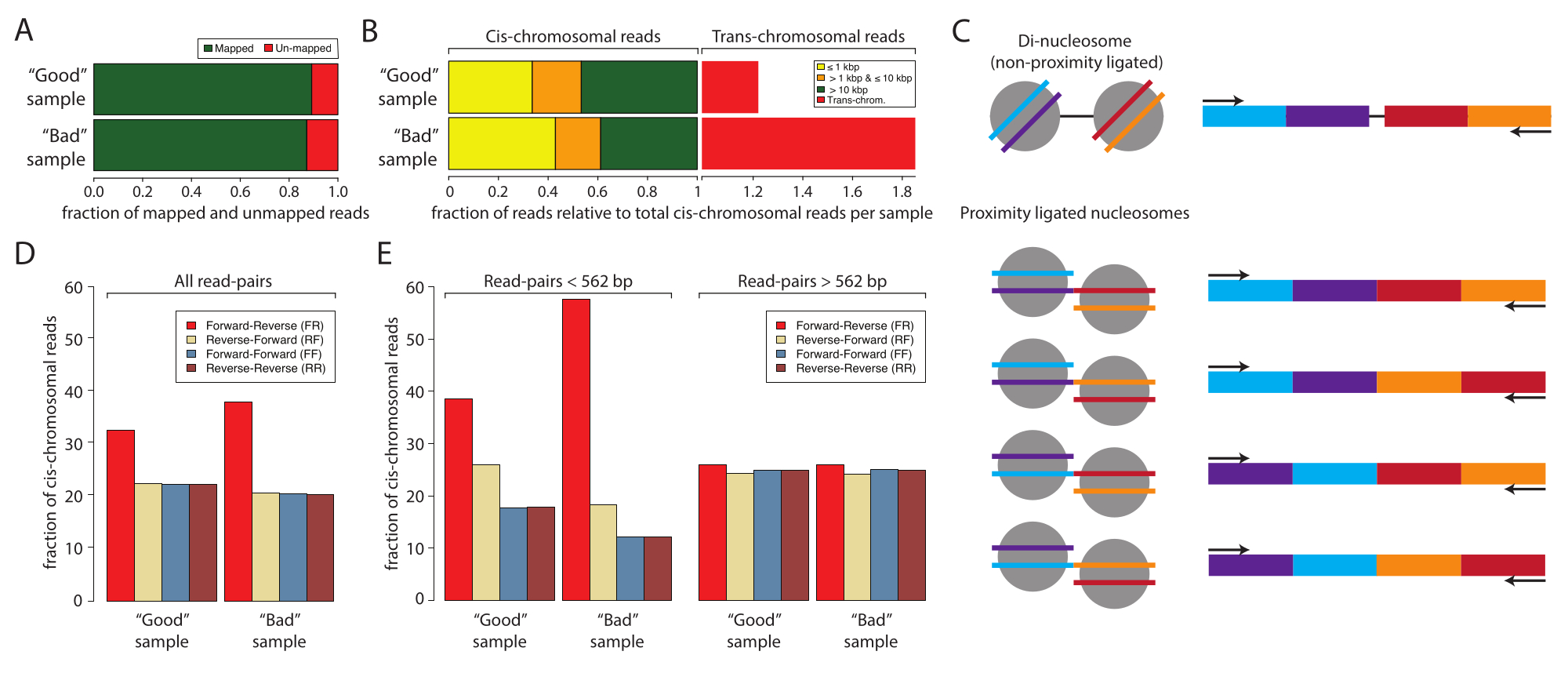
A continuación, se recomienda analizar y confirmar el éxito de la preparación de la muestra de Micro-C mediante la secuenciación de baja entrada antes de comprometerse con la secuenciación profunda que consume muchos recursos. Normalmente, las bibliotecas se secuencian a una profundidad de lectura de 5 x 106 a 1 x 107 y se evalúan en función de los siguientes criterios: la tasa de duplicación de lectura de secuenciación, la tasa de interacción cisómica frente a la transcromosómica y la frecuencia de orientación de lectura de secuenciación. Las bibliotecas Micro-C se procesan con Distiller, una canalización de servicio completo que procesa los datos desde la secuenciación de archivos de lectura (formato Fastq) hasta archivos de pares de lectura (formato Bedpe) y matrices de interacción escalables (formatos Cool y Mcool) utilizando cooler, pairtools y cooltools10,11,12. La canalización también genera un archivo de resumen que es ideal para evaluar la calidad de las bibliotecas de Micro-C10 (https://github.com/open2c/distiller-nf). La tasa de duplicación de PCR proporciona información sobre la complejidad de la biblioteca de secuenciación y se puede extraer del archivo *.stats generado. Las bibliotecas Micro-C de alta calidad tienen tasas de duplicación de PCR inferiores al 5%-10% cuando se generan a partir de 5 millones o más de células. En particular, algunas plataformas de secuenciación generan duplicados de PCR durante la formación del clúster, independientemente de la complejidad de la biblioteca de secuenciación. La Figura 2A muestra las tasas de duplicación relativas de dos experimentos: uno que consideramos una muestra buena y otro que una muestra mala. En este ejemplo, ambas muestras mostraron velocidades de mapa aceptables. El siguiente criterio para evaluar la calidad de las bibliotecas Micro-C es la relación cis frente a trans y las frecuencias de orientación de lectura. Dentro del núcleo, los cromosomas habitan territorios cromosómicos individuales y, por lo tanto, rara vez interactúan con otros cromosomas. Una alta tasa de interacciones transcromosómicas detectadas indica una alta tasa de ligaduras aleatorias. Cabe destacar que en este nivel de análisis, la muestra mala mostró una alta tasa de interacciones transcromosómicas en comparación con la muestra buena (Figura 2B). Para Micro-C, es deseable una tasa de interacción ciscromosómica del 70% o más.
Una biblioteca Micro-C tiene un tamaño de fragmento similar a la banda de ADN dinucleosomal, que puede copurificarse con la muestra ligada por proximidad y contaminar el experimento. Estos contaminantes son siempre interacciones ciscromosómicas. Por lo tanto, es importante evaluar también las tasas de orientación de lectura. La tasa de contaminación dinucleosómica puede estimarse mediante secuenciación de bajos insumos. El ADN dinucleosomal proviene de dos nucleosomas vecinos que no han sido escindidos por la MNasa. Por lo tanto, las lecturas de secuenciación resultantes siempre mostrarán una orientación de lectura hacia adelante-hacia atrás (F y R), y la distancia entre los pares de lectura será de alrededor de 320 pb. Los fragmentos ligados por proximidad, en comparación, se pueden ligar en cuatro orientaciones, lo que produce pares de lectura con F-R, R-R, R-F y F-F, idealmente con la misma abundancia (Figura 2C). Además, muestran varias distancias entre los dos pares de lectura. Para estimar la cantidad de contaminantes dinucleosómicos, la frecuencia de las orientaciones de lectura se puede calcular a partir de los archivos *stats generados por el destilador (Figura 2D). En particular, en este trabajo, la fracción de lecturas F-R (rojo) fue mayor en la muestra mala en comparación con la muestra buena, y esto se hizo más evidente cuando las orientaciones de lectura se estratificaron por distancia (Figura 2E). La fracción F-R está dominada por fragmentos dinucleosomales en comparación con las bibliotecas Micro-C cuando los pares de lectura se estratifican en lecturas con distancias <562 pb o ≥562 pb. Aquí, la fracción de lecturas con distancia <562 pb está dominada por lecturas F-R, mientras que la fracción con distancias ≥562 pb muestra una distribución uniforme entre las cuatro orientaciones posibles, lo que indica que la sobrerrepresentación global de lecturas F-R se debe a contaminantes dinucleosómicos. La elección de 562 pb como umbral para el subconjunto se define mediante la discretización en el archivo *stats generado. Aunque no es necesario para este control de calidad, se puede lograr un subconjunto más definido extrayendo las distancias del archivo *pairs, que también es generado por distiller. Es importante tener en cuenta que las lecturas dinucleosomales no disminuyen la calidad de la muestra de Micro-C, ya que pueden identificarse e ignorarse durante el procesamiento de datos. Sin embargo, no contienen información valiosa sobre las interacciones 3D y diluyen las lecturas informativas.
Por lo tanto, la valoración cuidadosa de MNasa y el control de calidad exhaustivo con secuenciación de baja entrada son las mejores herramientas para optimizar la calidad de los experimentos de Micro-C.

Figura 1: Etapas intermedias del protocolo Micro-C . (A) Electroforesis en gel de agarosa de cromatina de 2,5 x 105 células madre embrionarias de ratón digeridas con concentraciones variables de MNasa. Las bandas mononucleosomales, dinucleosómicas y trinucleosómicas se indican con flechas. M: Escalera de ADN (Carril 1/6); 10 U de MNasa por cada 250.000 células (carril 2); 2,5 U de MNasa por cada 250.000 células (carril 3); 0,625 U de MNasa por cada 250.000 células (carril 4); 0,156 U de MNasa por cada 250.000 células (carril 2). (B) La electroforesis en gel de agarosa al 1,0% de las muestras preparadas con Micro-C (Carril 3 y Carril 4) y el control de entrada digerido por MNasa (Carril 1). El carril 1 y el carril 2 (M: escalera de ADN) se mejoran para enfatizar el cambio relativo en la intensidad de los fragmentos mononucleosomales a dinucleosomales. Las bandas mono y dinucleosomales se indican con flechas. La banda dinucleosomal en la muestra ligada por proximidad combina ADN dinucleosomal y de la biblioteca Micro-C. (C) La electroforesis en gel de agarosa al 1,0% de las bibliotecas de secuenciación Micro-C amplificada a partir de una muestra de 1 μL para evaluar la calidad. Carril 1 (M): escalera de ADN; Carril 2 (S): Biblioteca Mirco-C. (D) Traza del analizador de fragmentos de la biblioteca Micro-C final. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Estadísticas de muestra para la secuenciación de entrada baja de una muestra buena y una muestra mala . (A) Gráfico de barras del porcentaje mapeado (verde) y no mapeado (rojo) lecturas. (B) Fracción normalizada de lecturas que mapea las interacciones cis y transcromosómicas. Los conjuntos de datos se normalizaron a la lectura de mapeo cis. Las lecturas de mapeo cis se estratificaron por la distancia entre la primera y la segunda lectura de las muestras secuenciadas de extremos pareados: ≤1 kbp (amarillo), >1 kbp y ≤10 kbp (naranja) y >10 kbp (rojo). (C) Esquema de las especies moleculares potenciales con los tamaños dinucleosomales. (D) Porcentajes de orientaciones de pares de lectura de todas las lecturas de la muestra buena y de la muestra mala. (E) Igual que el panel (D) pero estratificado por distancias (izquierda, <562 pb y derecha, ≥562 pb). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Componentes | 1 vez | 4.4x |
| 10x NEBuffer 2.1, | 10 μL | 44 μL |
| 2 μL 100 mM ATP | 2 μL | 8,8 μL |
| TDT de 100 mM | 5 μL | 22 μL |
| H2O | 68 μL | 299,2 μL |
| 10 U/μL T4 PNK | 5 μL | 22 μL |
| Total | 90 μL | 396 μL |
Tabla 1: Mezcla maestra Micro-C 1. Composición de la mezcla maestra para la reacción de masticación final.
| Componentes | 1 vez | 4.4x |
| 1 mM de biotina-dATP | 10 μL | 44 μL |
| 1 mM de biotina-dCTP | 10 μL | 44 μL |
| Mezcla de 10 mM de dTTP y dGTP | 1 μL | 4,4 μL |
| 10x Tampón de ADN ligasa T4 | 5 μL | 22 μL |
| 200x BSA | 0,25 μL | 1,1 μL |
| H2O | 23,75 μL | 104,5 μL |
Tabla 2: Mezcla maestra Micro-C 2. Composición de la mezcla maestra para la reacción de etiquetado final.
| Componentes | 1 vez | 4.4x |
| 10x Tampón de reacción de ligasa NEB T4 | 50 μL | 220 μL |
| H2O | 422,5 μL | 1859 μL |
| ADN ligasa T4 | 25 μL | 110μL |
Tabla 3: Mezcla maestra Micro-C 3. Composición de la mezcla maestra para la reacción de ligadura de proximidad.
| Componentes | 1 vez | 4.4x |
| 10x NEBuffer 1.1 | 20 μL | 88 μL |
| H2O | 180 μL | 792 μL |
| Nucleasa ExoIII | 10 μL | 44 μL |
Tabla 4: Mezcla maestra Micro-C 4. Composición de la mezcla maestra para la reacción de eliminación de biotina.
Discusión
El éxito de un experimento Micro-C depende de unos pocos pasos críticos en el protocolo que deben ejecutarse cuidadosamente. En primer lugar, la reticulación con el reticulante adicional DSG o EGS puede conducir a la agregación de células, dependiendo del tipo de célula. La adición de 0,1%-0,5% de BSA a la reacción de reticulación reduce significativamente la agregación sin afectar la eficiencia de la reticulación. La reticulación ineficiente puede dar lugar a un aumento de las tasas de interacciones transcromosómicas que son indicativas de ligaduras aleatorias. El segundo paso, pero el más importante, de este protocolo es la digestión de la cromatina con MNasa. La digestión subóptima de la cromatina conduce a una ligadura de proximidad ineficiente (digestión excesiva) o a un aumento de las tasas de dinucleosomas no ligados a la proximidad (digestión insuficiente). La eficiencia de la reacción de ligadura puede evaluarse mediante electroforesis en gel de agarosa (Figura 1B) y, además, se estima mejor mediante secuenciación de baja entrada. Si la secuenciación de baja entrada revela una alta tasa de duplicación (ligadura ineficiente) o un aumento de las tasas de dinucleosomas, se debe volver a evaluar el grado de digestión de la MNasa. En particular, la pérdida de muestra al ejecutar el protocolo puede conducir a una reducción de la complejidad de la biblioteca. La concentración de una muestra se evalúa mejor después de la purificación del ADN (paso 5.3) o mediante una PCR mínima (paso 8). El rendimiento total de ADN de 5 x 106 células de mamíferos después de la purificación del ADN suele ser de >2 μg. La concentración de ADN debe controlarse después de la digestión de MNasa, la digestión de ExoIII y la purificación del ADN. Las nucleasas endógenas, cuya abundancia es específica del tipo de célula y de la especie, pueden ser una fuente de degradación del ADN. Además, la purificación de ADN basada en columnas puede provocar la pérdida de muestras debido a la incompatibilidad con la SDS de las reacciones de desproteinación. Se puede considerar la precipitación de etanol si la concentración de ADN es baja en este paso.
Dado que Micro-C requiere una valoración de MNasa específica de la muestra, es un reto aplicar Micro-C a poblaciones de células pequeñas, como órganos pequeños de varios organismos modelo, embriones y células individuales, organoides o biopsias de pacientes. En este caso, Hi-C 3.0 ofrece una alternativa bien establecida que utiliza una reacción de punto final mediante endonucleasas de restricción específica de secuencia 8,9.
Micro-C es una tecnología de conformación cromosómica de alta resolución ampliamente aplicable con un alto rango dinámico y una baja relación señal-ruido, lo que la hace particularmente adecuada para investigar características cromosómicas de corto alcance 4,5,8, como bucles cromosómicos. La resolución de Micro-C permite capturar bucles promotor-potenciador, que están más allá del límite de detección de Hi-C, lo que permite un análisis más detallado de la relación entre la organización del genoma y la regulación13,14,15. Además, las estrategias de captura de ADN se han combinado recientemente con Micro-C para aumentar la resolución específica del locus de los loci genómicos objetivo a niveles sin precedentes, revelando nuevos conocimientos sobre la ultraestructura del genoma 3D16,17,18. En resumen, prevemos que Micro-C y sus derivados serán una tecnología clave para diseccionar el papel del genoma 3D en la regulación transcripcional y, en consecuencia, en la diferenciación y mantenimiento de los tipos celulares.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Agradecemos a Christl Gaubitz y Kathleen Stewart-Morgan por su lectura crítica del manuscrito. Agradecemos a Anja Groth y al laboratorio de Groth por su apoyo en el establecimiento de nuestro laboratorio. Agradecemos al personal de la Plataforma de Genómica CPR/reNEW por su apoyo: H. Wollmann, M. Michaut y A. Kalvisa. El Centro de Medicina de Células Madre de la Fundación Novo Nordisk (reNEW) cuenta con el apoyo de la subvención número NNF21CC0073729 de la Fundación Novo Nordisk. El Centro de Investigación de Proteínas (CPR) de la Fundación Novo Nordisk cuenta con el apoyo de la subvención número NNF14CC0001 de la Fundación Novo Nordisk. Agradecemos al laboratorio Brickman del Centro Novo Nordisk de Medicina de Células Madre, reNEW Copenhague, por las células madre embrionarias de ratón.
Materiales
| Name | Company | Catalog Number | Comments |
| 1 mM Biotin dATP | Jenna Bioscience | NU-835-Bio14-S | |
| 1 mM Biotin dCTP | Jenna Bioscience | NU-809-BioX-S | |
| 10 mM dGTP | NEB | N0442S | |
| 10 mM dTTP | NEB | N0443S | |
| 10 U/ml T4 PNK | NEB | M0201L | |
| 100 U/L Exonuclease III | NEB | M0206L | |
| 10x NEBuffer 1.1 | NEB | B7001S | |
| 10x NEBuffer 2.1 | NEB | B7202S | |
| 10x T4 DNA Ligase buffer | NEB | B0202A | |
| 1x DPBS w/o Mg2+ and Ca2+ | ThermoFisher | 14190144 | |
| 1x LIF | |||
| 2_Mercaptoethanol 50 mM | Gibco | 31350010 | 0.1 mM b-mercaptoethanol |
| 37% Formaldehyde | Sigma Aldrich | 252549-500ML | Caution. See manufactures MSDS |
| 400 U/ml T4 DNA Ligase | NEB | M0202L | |
| 5 U/ml Klenow Fragment | NEB | M0210L | |
| Agarose | BIO-RAD | 1613102 | Caution. See manufactures MSDS |
| BSA 20mg/ml | NEB | B9000S | |
| CaCl2 | |||
| cell counter | |||
| Dimethyl Sulfoxide (DMSO) | Sigma Aldrich | D8418-100ML | Caution. See manufactures MSDS |
| Dynabeads MyOne Streptavidin C1 | Invitrogen | 65001 | |
| DynaMag-2 Magnet | Invitrogen | 12321D | refered to as: magnet magnet for 1.5 ml tubes |
| DynaMag-PCR Magnet | Invitrogen | 492025 | refered to as: magnet magnet for PCR tubes |
| EDTA Ultrapure 0.5M pH 8.0 | Invitrogen | 15575-038 | |
| EGTA Ultrapure 0.5M pH 8.0 | BioWorld | 40121266-1 | |
| Ethanol 96% | VWR Chemicals | 20824365 | quality control system |
| Ethidium Bromide | Invitrogen | 15585-011 | |
| Ethylene glycol bis(succinimidyl succinate) (EGS) | ThermoFisher | 21565 | |
| Fetl Bovin Serum | Sigma Aldrich | F7524 | 15% FBS |
| Gel Loading dye purple (6X) | NEB | B7024S | |
| Glycine | PanReac AppliChem | A1067.0500 | |
| Halt Proteinase inhibitor (100x) | ThermoFisher | 78430 | Caution. See manufactures MSDS |
| IGEPAL CA-630 (NP-40) | Sigma Aldrich | 18896-50ML | |
| MgCl 1 M | Invitrogen | AM9530G | |
| Micrococcal Nuclease (MNase) | Worthington | LS004798 | |
| mouse embryonic stem cells | |||
| NaCl | Sigma Aldrich | S9888-1KG | |
| NEBNext Multiplex Oligos for Illumina (Dual Index primers) | NEB | E7600S | amplification primers for sequencing libraries |
| NEBNext Ultra II DNA library prep kit for Illumina | NEB | E7645L | sequencing library preparation kit |
| NEBNext Ultra II Q5 Master mix | NEB | M0544S | Caution. See manufactures MSDS |
| Non-Essential Amino Acids Solution | Gibco | 11140050 | 1x NEAA |
| Penicillin-Streptomycin (10,000 U/mL) | Gibco | 15140148 | 1% Pen-Strep |
| Proteinase K (40 mg/ml) | GoldBio | P-480-1 | Caution. See manufactures MSDS |
| QIAquick Gel extraction kit | QIAgen | 28706 | refered to as: DNA gel elution kit |
| QIAquick PCR purification kit | QIAgen | 28106 | refered to as: commercial DNA purification kit |
| Qubit dsDNA HS Assay kit | Invitrogen | Q32854 | high sensitivity DNA quantification instrument |
| Quick load purple 1kb plus DNA Ladder | NEB | N0550S | |
| SPRIselect size selection beads | Beckman Coulter | B23319 | paramagnetic beads |
| ThermoMixer C | Eppendorf | 5382000015 | refered to as: thermomixer |
| Tris | Merck | 10708976001 | |
| Trypsin | |||
| Tween20 | Sigma Aldrich | P7949-100ML | |
| Ultrapure 10% SDS | Invitrogen | 15553-035 | |
| Ultrapure Phenol Chloroform Isoamyl Alcohol (PCI) | Invitrogen | 15593-031 | |
| Fragment Analyzer |
Referencias
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Hsieh, T. -. H. S., Fudenberg, G., Goloborodko, A., Rando, O. J. Micro-C XL: Assaying chromosome conformation from the nucleosome to the entire genome. Nature Methods. 13 (12), 1009-1011 (2016).
- Hsieh, T. -. H. S., et al. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell. 162 (1), 108-119 (2015).
- Krietenstein, N., et al. Ultrastructural details of mammalian chromosome architecture. Molecular Cell. 78 (3), 554-565 (2020).
- Hsieh, T. -. H. S., et al. Resolving the 3D landscape of transcription-linked mammalian chromatin folding. Molecular Cell. 78 (3), 539-553 (2020).
- Belaghzal, H., Dekker, J., Gibcus, J. H. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods. 123, 56-65 (2017).
- van Holde, K. E. . Chromatin. , (1989).
- Oksuz, B. A. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18, 1046-1055 (2021).
- Lafontaine, D. L., Yang, L., Dekker, J., Gibcus, J. H. Hi-C 3.0: Improved protocol for genome-wide chromosome conformation capture. Current Protocols. 1 (7), 198 (2021).
- Goloborodko, A., Venev, S., Abdennur, N., Di Tommaso, P. Mirnylab/distiller-nf; v033. Zenodo. , (2019).
- Venev, S., et al. Open2c/cooltools: v0.4.1. Zenodo. , (2021).
- Abdennur, N., Mirny, L. A. Cooler: Scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 36 (1), 311-316 (2019).
- Zhang, S., Übelmesser, N., Barbieri, M., Papantonis, A. Enhancer-promoter contact formation requires RNAPII and antagonizes loop extrusion. BioRxiv. , (2022).
- Barshad, G., et al. RNA polymerase II and PARP1 shape enhancer-promoter contacts. BioRxiv. , (2022).
- Hansen, A. S., et al. Distinct classes of chromatin loops revealed by deletion of an RNA-binding region in CTCF. Molecular Cell. 76 (3), 395-411 (2019).
- Hua, P., et al. Defining genome architecture at base-pair resolution. Nature. 595 (7865), 125-129 (2021).
- Downes, D. J. High-resolution targeted 3C interrogation of cis-regulatory element organization at genome-wide scale. Nature Communications. 12, 531 (2021).
- Goel, V. Y., Huseyin, M. K., Hansen, A. S. Region capture Micro-C reveals coalescence of enhancers and promoters into nested microcompartments. BioRxiv. , (2022).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados