
Method Article
Mapping Mammalian 3D Genome Interactions with Micro-C-XL
In This Article
Summary
A protocol for mapping the three-dimensional genome organization with nucleosome resolution using the genome-wide chromosome conformation capture method Micro-C-XL is presented here.
Abstract
Three-dimensional (3D) chromosome organization is a major factor in genome regulation and cell-type specification. For example, cis-regulatory elements, known as enhancers, are thought to regulate the activity of distal promoters via interaction in 3D space. Genome-wide chromosome conformation capture (3C)-technologies, such as Hi-C, have transformed our understanding of how genomes are organized in cells. The current understanding of 3D genome organization is limited by the resolution with which the topological organization of chromosomes in 3D space can be resolved. Micro-C-XL measures chromosome folding with resolution at the level of the nucleosome, the basic unit of chromatin, by utilizing micrococcal nuclease (MNase) to fragment genomes during the chromosome conformation capture protocol. This results in an improved signal-to-noise ratio in the measurements, thus facilitating the better detection of insulation sites and chromosome loops compared to other genome-wide 3D technologies. A visually supported, detailed, step-by-step protocol for preparing high-quality Micro-C-XL samples from mammalian cells is presented in this article.
Introduction
Micro-C-XL is a genome-wide technique to measure 3D genome conformation with nucleosome resolution. Micro-C-XL builds on the widely used proximity ligation-based Hi-C technology, which has transformed our understanding of how 3D genomes are organized1. Micro-C-XL and its first iteration, Micro-C, were initially developed in Saccharomyces cerevisiae2,3 and later adapted to mammalian cell systems, for which the protocol has demonstrated its full potential in detecting short-range features of the 3D genome, such as chromosome loops and insulation sites. This version is based on recent mammalian Micro-C-XL publications4,5. As Micro-C-XL supersedes Micro-C, Micro-C-XL is henceforth referred to as Micro-C in the manuscript.
The major differences between Micro-C and Hi-C6 are as follows: 1) genome fragmentation with micrococcal nuclease (MNase) compared to restriction enzymes and 2) additional crosslinkers with larger atomic spacing between the reactive groups compared to only formaldehyde. Both steps contribute significantly to the improved signal-to-noise ratio of Micro-C compared to conventional Hi-C. The fragmentation size limits the resolution to which the 3D genome organization can be resolved during the proximity ligation protocol. MNase is a nuclease that preferentially digests accessible DNA and leaves nucleosomal-protected DNA intact. Nucleosome footprinting using MNase-sequencing has shown that nucleosomes fully cover most eukaryotic genomes7. As nucleosomes are distributed throughout the genome with an average spacing of 160-220 bp, depending on species and cell type, MNase is the ideal enzyme for high-resolution mapping of the genome architecture.
The use of an additional crosslinker in combination with formaldehyde (FA) in the Micro-C method additionally improves the signal-to-noise ratio2,8. Amine-specific crosslinkers with longer atomic spacers between the reactive groups facilitate protein-protein crosslinks. These are typically disuccinimidyl glutarate (DSG) or ethylene glycol bis-succinimidyl succinate (EGS) with 7.7 Å and 16.1 Å spacers, respectively. The reduction of noise through EGS or DSG is particularly apparent in experiments with high fragmentation rates, such as Micro-C, and presumably occurs due to a reduction in the rate of random ligation events8.
A recently developed Hi-C 3.0 protocol that utilizes ESG/DSG crosslinking and multiple combinations of restriction enzymes reduces noise in Hi-C experiments and significantly improves the detection of chromosome loops and insulation sites8,9. Still, a site-by-site comparison of various interaction data features found that Micro-C had superior detection of short-range features, such as chromosome loops and insulation sites, compared to both Hi-C 3.0 and conventional Hi-C8. However, Hi-C 3.0 does improve the detection of short-range features and maintains strong detection of genome compartmentalization compared to conventional Hi-C. In summary, the choice of a chromosome conformation capture method should be determined by the objective and biological question.
Here, we provide a step-by-step protocol for successful Micro-C experiments that can unravel 3D genome organization.
Protocol
1. Cell culture and crosslinking
- Culture cells according to the experimental needs to obtain a minimum of 1 x 107 cells. Here, the cells were grown at 37 °C with 5% CO2 in E14 medium (DMEM with pyruvate and L-glutamine, 15% FBS, 1x LIF, 1x NEAA, 1% pen-strep, 0.1 mM β-mercaptoethanol [see Table of Materials]) and passaged every second day.
NOTE: This protocol has been successfully applied to various cell types from multiple species, such as Homo sapiens and Mus musculus. In this example, mouse embryonic stem (ES) cells were used. - Harvest the cells at 70%-80% confluency by aspirating the medium. Wash once with 5 mL of DPBS, and incubate the cells with 3 mL of prewarmed 0.25% trypsin per 10 cm dish for 2-3 min at 37 °C.
- Quench the trypsin with 7 mL of prewarmed E14 medium and transfer the detached cells to a 50 mL tube.
- Pellet the cells by centrifugation at 300 x g for 5 min at room temperature (RT). Discard the supernatant and resuspend the cell pellet in the medium. Count the cells with a cell counter.
NOTE The protocol is robust to some variation in cell concentrations, and various cell counters have been used to quantify the cell numbers; however, be aware of the dynamic range of the cell counter used. This is typically between 1 x 105-1 x 107 cells/mL. While the centrifugation speed and duration have been tested for this cell type, different, smaller cells may require a higher centrifugation speed or longer spin times, and the centrifugation should be adjusted accordingly. - Collect the cells by centrifugation at 300 x g for 5 min at RT and resuspend the cells in DPBS at a final concentration of 1 x 106 cells/mL. For example, if the yield is 1 x 107 cells, resuspend the cells in 10 mL of DPBS.
- For the first crosslinking step, add 37% formaldehyde (FA) to a final concentration of 1% to the cell suspension, and incubate the cell suspension for 10 min at RT with rotation (15-20 rpm); as an example, if the yield is 1 x 107 cells, add 270 µL of FA to the 10 mL cell suspension.
NOTE: The FA solution is typically stable at RT for up to 3 months after opening. - Quench the reaction by adding 2.5 M glycine to a final concentration of 0.25 M and incubate for 5 min at RT with rotation (15-20 rpm). For example, if the yield is 1 x 107 cells, add 1.027 mL of 2.5 M glycine to the cell suspension.
- Collect the cells by centrifugation at 300 x g for 5 min at RT. Discard the supernatant and resuspend the cells in 5 mL of DPBS. Repeat the centrifugation step once and resuspend the pelleted cells in DPBS to 4 x 106 cells/mL. For example, if the yield is 1 x 107 cells, resuspend the cells in 2.5 mL of DPBS.
- For the second crosslinking step, prepare a 0.3 M stock solution of ethylene glycol bis-succinimidyl succinate (EGS) in DMSO (13.6 mg of EGS in 100 µL of DMSO). Add EGS at a final concentration of 3 mM to the cell suspension, and incubate at RT for 40 min with rotation (15-20 rpm). For example, if the yield is 1 x 107 cells, add 25 µL of 0.3 M EGS stock solution to 2.5 mL of the cell suspension.
NOTE: Equilibrate the EGS to RT for at least 20 min before weighing for preparing the stock solution. - Quench the reaction by adding 2.5 M glycine to a final concentration of 0.4 M, and incubate for 5 min at RT with rotation (15-20 rpm). For example, if the yield is 1 x 107 cells, add 400 µL of 2.5 M glycine to the cell suspension.
- Collect the cells by centrifugation at 1,000 x g for 5 min at RT, and resuspend the pelleted cells in DPBS to 5 x 106 cells/mL. Distribute 5 x 106 cells/tube for preparative libraries and 1 x 106 cells/tube for titration for MNase digestion.
NOTE: It is suggested to titrate the optimal digestion degree for each cross-linked cell batch. Ideally, collect two to three aliquots of 1 x 106 cells for MNase titration experiments (step 2) and two to four aliquots of 5 x 106 cells for preparative experiments (step 3). - Collect the cells by centrifugation at 1,000 x g for 5 min at RT and remove the supernatant. Snap-freeze the cell pellets in liquid nitrogen, and store them at −80 °C.
NOTE: Successful Micro-C libraries can be prepared from samples stored for up to 3 months.
2. MNase titration
NOTE: Performing an MNase titration is necessary to determine the optimal concentration of MNase before processing the double-crosslinked cells' preparative library.
- To perform the MNase titration, thaw one pellet of 1 x 106 cells on ice for 10 min and resuspend the cells in 500 µL of DPBS (add 1x BSA if the cells stick to the wall). Incubate the cell suspension on ice for 20 min.
- Collect the cells by centrifugation at 10,000 x g for 5 min at RT, and remove the supernatant. Resuspend the pelleted cells in 500 µL of MB#1 buffer (50 mM NaCl, 10 mM Tris-HCl, 5 mM MgCl2, 1 mM CaCl2, 0.2% NP-40, 1x protease inhibitor [see Table of Materials], pH 7.4).
- Collect the cells by centrifugation at 10,000 x g for 5 min, and remove the supernatant. Resuspend the cells in 200 µL of MB#1 buffer, and split the sample into four tubes.
- Thaw one vial of MNase (20 U/µL), and dilute with 10 mM Tris, pH 7.4, in a series of 1:2, 1:4, 1:4, and 1:4 to achieve concentrations of 10 U/µL, 2.5 U/µL, 0.625 U/µL, and 0.1256 U/µL respectively (one for each digestion condition). With appropriate time intervals (10-20 s), add 1 µL of MNase solution to one of the four samples, vortex, and incubate on a thermomixer at 37 °C for 10 min (800 rpm shaking). Continue with adding 1 µL from the remaining MNase dilutions to the remaining cell aliquots.
- Stop the Mnase digestion by adding 200 µL of freshly prepared STOP buffer (150 µL of 10 mM Tris, pH 7.4, 25 µL of 10 % SDS, 25 µL of 20 mg/mL proteinase K, 2 µL of 0.5 M EGTA) to each tube in the same order and with the same time interval that the MNase was added. Incubate at 65 °C for 2 h.
- Add 500 µL of phenol-chloroform-isoamyl alcohol (PCI) to each sample, and mix thoroughly by vortexing. Centrifuge at 19,800 x g for 5 min at RT to separate the phases, and transfer the aqueous phase to new tubes (approximately 200 µL/sample).
CAUTION: PCI contains numerous toxic components and should only be handled in a chemical safety hood. Please consult the manufacturer for more details. - Purify the DNA using a commercial DNA purification kit (see Table of Materials) according to the manufacturer's instructions and elute the samples in 12 µL of elution buffer.
NOTE: The SDS concentration from the deproteinization step (step 2.5) is inhibitory for some DNA purification kits. The DNA purification kit used here performs comparably to ethanol precipitation. - Add 2-5 µL of loading dye, and run the samples on a 1.5% agarose gel at 120 V for 30-50 min (until properly separated; Figure 1A).
- Choose the best degree of digestion for the experiment, and continue with the preparative MNase digestion. An optimal digestion degree displays little to no subnucleosomal fragments and a 70%-90 % mono- to di-nucleosome ratio.
NOTE: In this experiment, the digestion degree of Lane 4 in Figure 1A was determined to be the optimal digestion obtained with 0.625-1.25 U of Mnase for 2.5 x 105 cells (= 2.5-5 U of Mnase for 1 x 106 cells). For a detailed discussion, see the representative results.
3. Preparative MNase digestion
- To fragment chromatin to the mono-nucleosomes via MNase digestion, thaw previously double cross-linked 5 x 106 cell aliquots, and resuspend in 1 mL of DPBS. Incubate on ice for 20 min (add 1x BSA if the cells stick to the tube wall).
- Collect the cells by centrifugation at 10,000 x g for 5 min at RT, and discard the supernatant. Resuspend the pellet in 500 µL of MB#1 buffer. Repeat the centrifugation step once. Resuspend the pellet in 1 mL of MB#1 buffer, and make 200 µL aliquots (1 x 106 cells per aliquot).
- Based on the MNase titration (described in step 2), digest the chromatin by adding the appropriate amount of MNase (usually 2.5-10 U/µL per 1 x 106 cells) to each aliquot. Mix well (vortex and spin quickly), and incubate on a thermomixer at 37 °C for 10 min with 800 rpm shaking.
- Stop the MNase digestion by adding 1.6 µL of 0.5 M EGTA (final 4 mM) to each aliquot, and incubate on a thermomixer at 65 °C for 10 min with 800 rpm shaking.
- Collect the sample by centrifugation at 10,000 x g for 5 min at RT, and discard the supernatant. Resuspend the cell pellet in 500 µL of 1x NEBuffer 2.1.
- Pool samples equivalent to an input of 5 x 106 cells or less for further processing.
NOTE: If more than 5 x 106 cells are to be processed, process these samples in parallel, as the enzymatic conditions are optimized for 5 x 106 cells. - Before proceeding to the proximity ligation steps, transfer 10% of the sample as an input control to control for the MNase digestion level. Add 150 µL of 10 mM Tris, pH 7.4, 25 µL of 10% SDS, and 25 µL of 20 mg/mL proteinase K to this sample, and incubate overnight at 65 °C.
4. DNA end processing and proximity ligation
- Collect the remaining sample by centrifugation at 10,000 x g for 5 min at 4 °C, and discard the supernatant. Resuspend the pellet in 90 µL of freshly prepared Micro-C master mix 1 (Table 1), and incubate on a thermomixer for 15 min at 37 °C with 800 rpm shaking.
- Add 10 µL of 5 U/µL Klenow Fragment, and incubate on a thermomixer for 15 min at 37 °C with 800 rpm shaking.
- Add 100 µL of freshly prepared Micro-C master mix 2 (Table 2), and incubate on a thermomixer for 45 min at 25 °C with 800 rpm shaking. After incubation, quench the enzymatic reaction by adding EDTA to a final concentration of 30 mM. Incubate on a thermomixer for 20 min at 65 °C with 800 rpm shaking.
- Collect the sample by centrifugation at 10,000 x g for 5 min at 4 °C, and discard the supernatant. Resuspend the sample in 500 µL of freshly prepared Micro-C master mix 3 (Table 3), and incubate for 2.5 h at RT with rotation (15-20 rpm).
- Collect the sample by centrifugation at 10,000 x g for 5 min at 4 °C, and discard the supernatant. Resuspend the sample in 200 µL of freshly prepared Micro-C master mix 4 (Table 4), and incubate on a thermomixer for 15 min at 37 °C with 800 rpm shaking.
- For reverse crosslinking and deproteination, add 25 µL of 20 mg/mL proteinase K and 25 µL of 10% SDS to the sample, and incubate at 65 °C overnight with intermittent mixing.
5. Di-nucleosomal DNA purification and size selection
- Add 500 µL of PCI to the samples and input control, and mix by vortexing. Separate the phases by centrifugation at 19,800 x g for 5 min, and transfer the upper aqueous phase to a fresh tube.
- Concentrate the DNA either using a DNA purification kit or by ethanol precipitation. Elute the samples in 30 µL (step 5.3) and the input controls (step 3.11) in 15 µL, and run a 1.5 % agarose gel to separate the mono-nucleosomes and di-nucleosomes (Figure 1B).
NOTE: The SDS concentration from the deproteinization step is inhibitory for some DNA purification kits. The DNA purification kit used here (see Table of Materials) performs comparably to ethanol precipitation. Depending on the number of cells used, the input can range from 100 ng to 10 µg. Typically, 1-5 µg of DNA is extracted from 5 x 106 cells. - Excise the DNA fragments that have a di-nucleosomal size (approximately 300 bp). Use a commercially available DNA gel elution kit (see Table of Materials) to extract the DNA from the agarose gel, and elute in 150 µL.
6. Preparation of the streptavidin beads
- Transfer 10 µL of streptavidin beads (see Table of Materials) per sample into a reaction tube.
- Place it into an appropriate magnet for 1.5 mL tubes (see Table of Materials). After the solution clears (1-2 min), remove the supernatant, and resuspend the beads in 300 µL of 1x TBW (5 mM Tris-HCl, pH 7.5, 0.5 mM EDTA, 1 M NaCl, 0.05% Tween 20) per sample. Repeat this step once.
- Resuspend the beads in 150 µL of 2x B&W buffer (10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 2 M NaCl) per sample processed in step 7.
7. Streptavidin pull-down and on-bead library preparation
- Add 150 µL of pre-prepared beads (step 6.3) to 150 µL of the sample (step 5.3). Incubate for 20 min at RT with rotation (15-20 rpm).
- Place the tubes into an appropriate magnet, and wait until the solution clears (1-2 min). Remove the supernatant and resuspend the beads in 300 µL of 1x TBW. Repeat this step.
- Place the tubes into an appropriate magnet and wait until the solution clears (1-2 min). Remove the supernatant and resuspend the beads in 100 µL of 0.1x TE (1 mM Tris, 0.1 mM EDTA, pH 8.0).
- Place the tubes into an appropriate magnet and wait until the solution clears (1-2 min). Remove the supernatant, resuspend the beads in 50 µL of 0.1x TE, and transfer them to PCR tubes.
NOTE: The volume of 0.1x TE used (50 µL) corresponds to the input volume of the DNA sequencing library preparation kit (see Table of Materials) used in this protocol. If a different kit or strategy is used, adjust the volume accordingly. - Perform the DNA manipulation steps of the sequencing library preparation kit according to the manufacturer's protocol. These steps typically include DNA blunting, A-tailing, adaptor ligation, and U excision. The last step (U excision) is specific to the kit used in this study. If using that kit, follow the manufacturer's instructions from step 1 to step 2.6 in the kit protocol using non-diluted adaptors, and do not consider −20 °C storage after step 2.6.
NOTE: The buffer changes required by the sequencing kits must be exchanged by the binding of the beads to a magnet and washes (steps 7.3 to 7.4), as DNA is still bound to the streptavidin beads. Additionally, because the DNA is bound to the magnetic beads, ignore any purification and size selection steps after adapter ligation from the kit protocol. Continue to step 7.6 (this protocol). - After adapter ligation is completed, wash the sample as described in step 7.2. Discard the supernatant and resuspend the beads in 20 µL of 0.1x TE.
8. Estimation of the required PCR cycles
NOTE: Estimating the required PCR cycles for library amplification is advisable. Typically, a Micro-C library requires 8-15 cycles of PCR. Although the step is not essential, it helps avoid over-amplification and reduces the risk of PCR duplicates.
- To define the minimum number of PCR cycles required, perform PCR with 1 µL of streptavidin-biotin-DNA sample (step 7.6). To do this, add PCR master mix (3.2 µL of H2O, 0.4 µL of i5 primer, 0.4 µL of i7 primer, and 5 µL of Q5 high-fidelity DNA polymerase) to the sample. Perform PCR with 16 cycles according to the manufacturer's instructions for the DNA library preparation kit used.
NOTE: The primers used here are purchased in a separate kit (see Table of Materials) that needs to be compatible with the library preparation kit used. - After the PCR, collect the beads with an appropriate magnet, and measure the DNA concentration of 1 µL of the supernatant using a high-sensitivity DNA quantification instrument (see Table of Materials). To obtain the total DNA concentration, multiply this value by 10 (total volume of the PCR reaction). Estimate the required number of PCR cycles as discussed in the representative results.
- Add 2 µL of 6x loading buffer to the remaining 9 µL of PCR mixture. Resolve the PCR amplified library on a 1% agarose gel to determine successful adaptor ligation, resulting in approximately 450 bp size (Figure 1C).
NOTE: The gel should show a distinct band of approximately 420 bp (Micro-C library plus adaptors). Bands of 120 bp represent adapter dimers and are indicative of low-complexity libraries. Bands of lower molecular weights may appear (less than 100 bp), and these are unused primers from the PCR reaction.
9. Sequencing library amplification
- Add PCR master mix (65 µL of H2O, 100 µL of Q5 high-fidelity DNA polymerase, 8 µL of i5 primer, 8 µL of i7 primer) to the remaining 19 µL of the streptavidin-biotin-DNA sample. Split the reaction mixture into 50-100 µL aliquots.
NOTE: The optimal volume for the PCR depends on the PCR machine; typically, 50 µL offers the most reproducible amplification in common PCR machines. - Perform PCR according to the manufacturer's instructions of the DNA library preparation kit with the number of cycles determined in step 8. If step 8 was omitted, 14 cycles are recommended for PCR.
- Purify the DNA with paramagnetic beads (see Table of Materials) at a ratio of 1:0.9 according to the manufacturer's protocol. Elute in 20 µL of 0.1% TE.
NOTE: If adapter dimers are detected in step 8.5, perform purification twice using the same ratio. - Determine the DNA concentration and run the samples on a quality control system (see Table of Materials). Ensure good quality of the Micro-C library by the presence of a single precise band (Figure 1D).
NOTE: Adaptor dimers are uncommon in on-bead library preparations due to the sample washes before the PCR. Thus, the appearance of adaptor dimers indicates low library complexity. If adaptor dimers are observed, it is highly recommended to quality control the sample with low input sequencing.
10. DNA sequencing and data processing
- Sequence the Micro-C library with paired-end sequencing according to the sequencing provider's requirements.
NOTE: Ideally, the samples are sequenced on a platform in paired-end mode with 50 bp per read. Older platforms that offer shorter read lengths, such as 2 x 35 bp, have also been used successfully. Importantly, if repetitive genomic regions are studied, it might be advisable to sequence with a longer read length. - To assess the quality of the Micro-C library, perform low input sequencing with 5 x 106 to 1 x 107 reads per sample.
- Process the sequencing files (fastq files) with Distiller10. Map the reads against the appropriate reference genome, here mm10.
NOTE: The sequencing files can be processed using various pipelines on local computers or computing clusters. For samples with low sequencing depths, larger bin sizes, such as 10,000 bp, 50,000 bp, 100,000 bp, and 500,000 bp, can reduce computing demands and file sizes. Distiller (used in this study) generates all the required file types to assess the quality of the Micro-C library. The generated *.stats file contains the information on the map rate, cis-trans ratios, and read orientation stratified by the distance between the read pairs. These parameters are visualized in Figure 2, and the assessment of the Micro-C library quality is discussed in the representative results. The processing software also generates mcool files that can be directly loaded into HiGlass (https://docs.higlass.io/) to generate interaction matrices9.
Results
The successful preparation of Micro-C libraries can be evaluated in several steps of the protocol. The most important step is the choice of a proper MNase digestion degree. Therefore, the MNase concentration must be titrated to consistently yield 70%-90% mono-nucleosomes over di-nucleosomes for every sample. It is important to note that chromatin digestion is different for eu- and hetero-chromatin, with MNase digesting heterochromatin less efficiently. Thus, the optimal digestion degree depends on the chromatin region of interest and the cell type studied since the relative proportion of eu- and hetero-chromatin is cell type-specific. Therefore, it is advisable to carefully titrate the MNase concentration required and to first evaluate the Micro-C experiment's success by low-input sequencing.
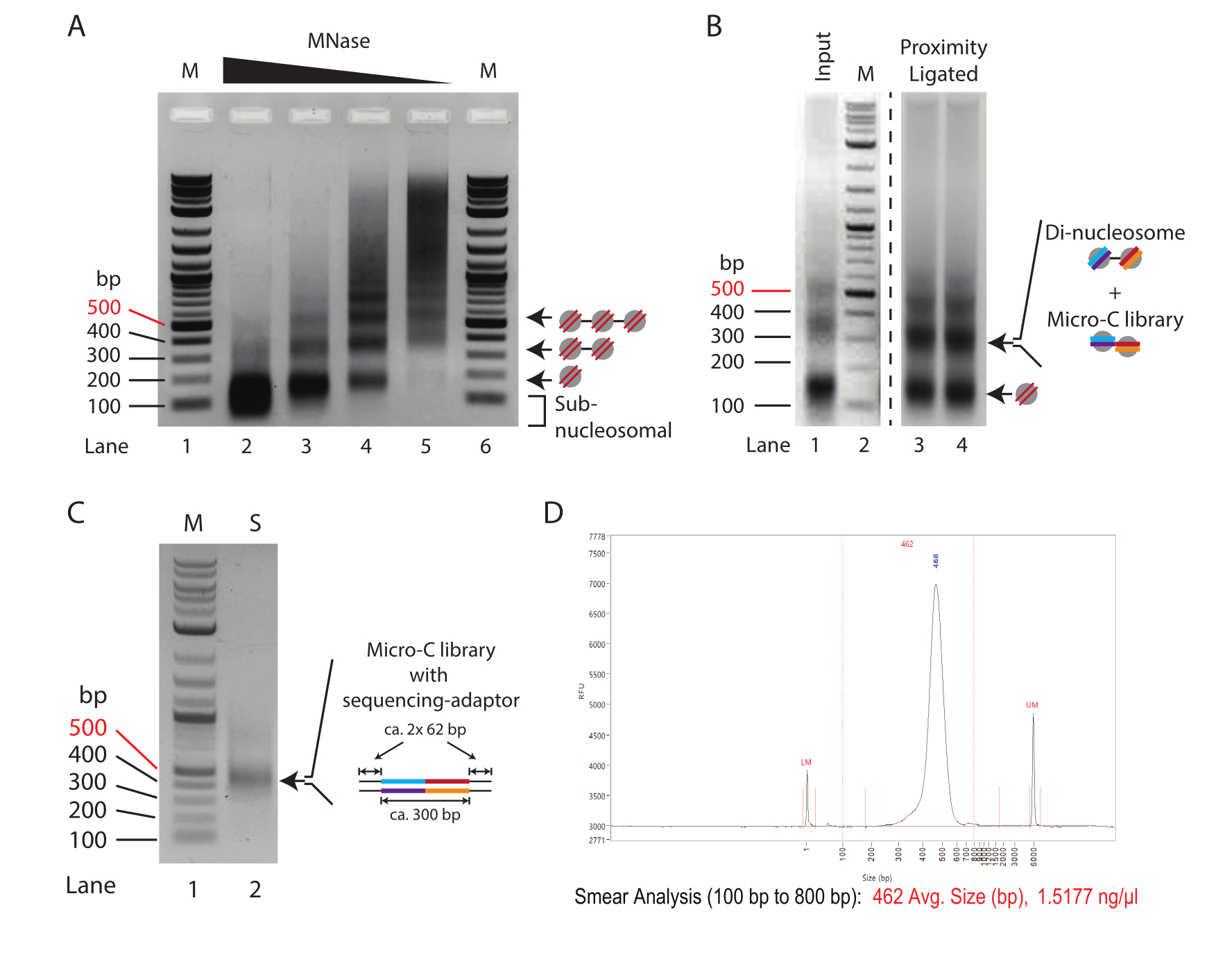
A typical MNase titration pattern of chromatin treated with decreasing amounts of MNase is shown in Figure 1A. Here, chromatin from 250,000 cells per reaction is digested with a four-fold dilution of MNase. The highest concentration (10 U of MNase, Lane 2) shows over-digested chromatin almost exclusively consisting of mono-nucleosomal DNA (~150 bp). Notably, the center of the mono-nucleosomal band runs lower in the agarose gel compared to the corresponding bands in the samples with reduced MNase concentrations, indicating an over-digestion of nucleosomal DNA. Over-digested nucleosomes are inefficiently ligated in the proximity ligation reaction; therefore, the sample in Lane 2 is suboptimal for Micro-C experiments. Lane 3 (2.5 U of MNase) displays an almost appropriate digestion degree for Micro-C experiments. Here, the mono-nucleosomal band is the dominant species, and the subnucleosomal smear, indicative of over-digested nucleosomes, is reduced; however, it is still present. The digestion degree in Lane 4 (0.635 U of MNase) is an ideal condition for a Micro-C experiment in this titration example. A clear mono-nucleosomal band without sub-nucleosomal DNA is present. The band intensity for the mono- and di-nucleosome DNA is almost equal, indicating a mono-nucleosome yield of 66% or higher. It is worth noting that the di-nucleosomal DNA is approximately twice the size of the mono-nucleosomal DNA (~320 bp vs. ~150 bp), so its band intensity per mole of DNA is twice as high compared to its mono-nucleosomal counterpart. The digestion degree in Lane 5 (0.156 U of MNase) shows under-digested chromatin with almost no nucleosomal DNA, and this, therefore, represents a suboptimal sample.
In conclusion, in this example, the digestion of 2.5 x 105 mouse ES cells with 0.625 U of MNase (corresponding to 2.5 U of MNase for 1 x 106 cells in 200 µL) offers the most promising starting point for preparative digestions in Micro-C experiments. However, an intermediate MNase concentration between the conditions used for the samples in Lane 3 and Lane 4 (corresponding to 5 U of MNase for 1 x 106 cells in 200 µL) should also be considered. Importantly, chromatin digestion with MNase cannot be scaled linearly, and it is not recommended to upscale the preparative digestion more than 4x. To prepare Micro-C libraries from more than 1 x 106 cells, it is advisable to digest the chromatin in aliquots of 1 x 106 cells and pool them after MNase inactivation.
To assess the success of the proximity ligation protocol, the input control, which is MNase-digested and not proximity ligated (step 3.8), should be compared to the proximity-ligated sample (step 5.3) by 1.5% agarose gel electrophoresis (Figure 1B). The proximity-ligated mono-nucleosome band has an approximate size of 300 bp, similar to that of di-nucleosomes. Therefore, the mono- to di- nucleosomal band signal ratio should shift from predominantly mono-nucleosomes (Lane 1) toward di-nucleosomes (Lane 3 and Lane 4). As the agarose gel in this step is the di-nucleosomal DNA that is excised and purified, splitting the samples into multiple lanes is advisable to avoid over-loading.
Assessing the quality and quantity of the prepared sequencing library by minimal PCR is recommended. Here, DNA from 1 µL of beads (1/20 of the total sample) is amplified for 16 cycles in 10 µL of PCR reaction. The total concentration of the minimal PCR library typically ranges from 50-500 ng after 16 PCR cycles. In theory, this corresponds to a 1-10 µg library from the remaining 19 µL sample if it were also amplified for 16 cycles. It is recommended to use the minimum number of PCR cycles needed to generate a library of approximately 100 ng from the total DNA. Assuming logarithmic amplification in the PCR, the theoretical concentration of the DNA obtained from the 19 µL input at 16 cycles can be divided successively by two to calculate the number of PCR cycles required to generate a 100 ng library. For example, a 100 ng yield from 1 µL after 16 cycles corresponds to a 1,900 ng yield amplified from 19 µL. In this scenario, 12 cycles should ideally generate a 118 ng sequencing library from the total DNA (1,900 ng/[2 × 2 × 2 × 2] = 118 ng). The remaining 9 µL sample from the minimal PCR can then be used to assess the quality of the library by agarose gel electrophoresis (Figure 1C). Visualization should show one distinct band at 420 bp and no bands for adaptor dimers (120 bp). Smaller fragments may also appear, and these correspond to unused PCR primers.
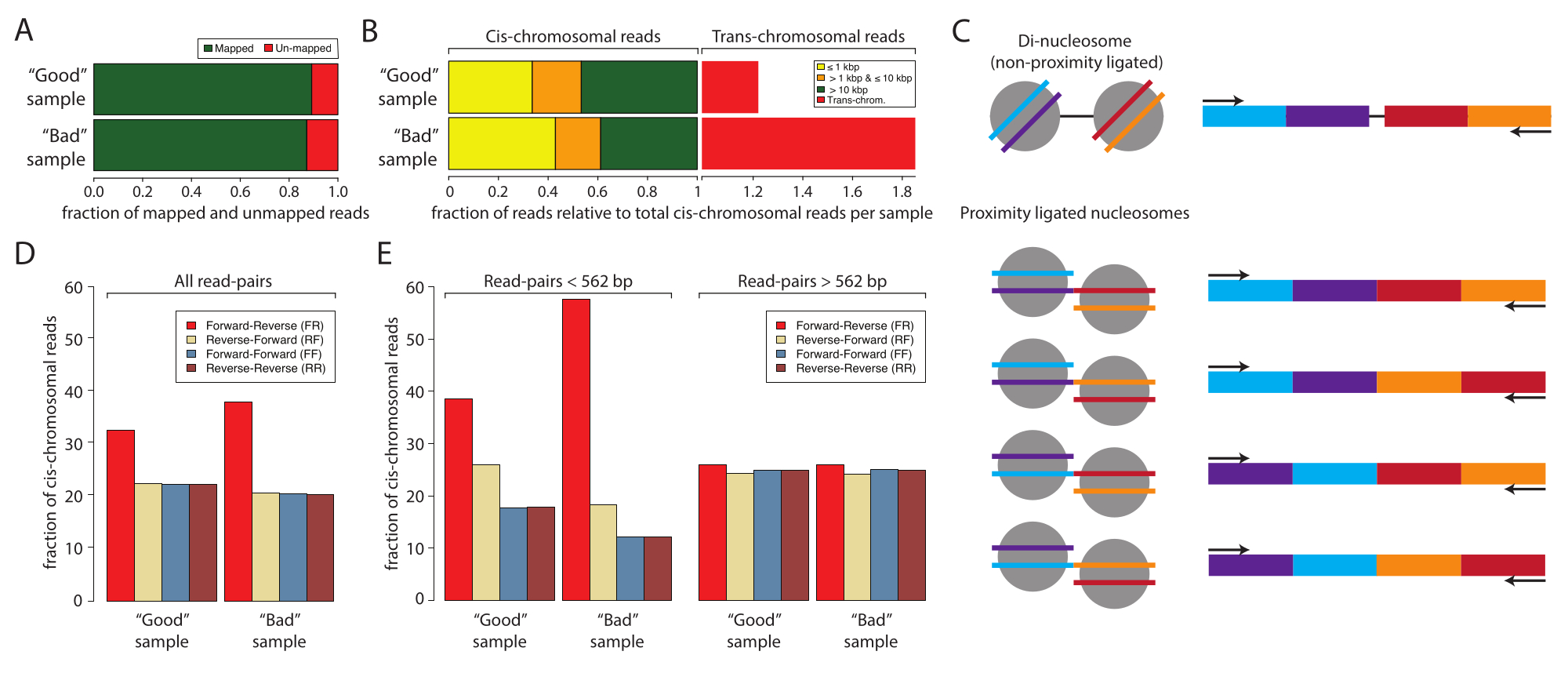
Next, analyzing and confirming successful Micro-C sample preparation by low-input sequencing is recommended before committing to resource-intensive deep sequencing. Typically, libraries are sequenced to a read depth of 5 x 106 to 1 x 107 and evaluated based on the following criteria: the sequencing read duplication rate, the cis- versus trans-chromosomal interaction rate, and the sequencing read orientation frequency. The Micro-C libraries are processed with Distiller, a full-service pipeline that processes the data from sequencing read files (Fastq format) to read-pairs files (Bedpe format) and scalable interaction matrices (Cool and Mcool formats) using cooler, pairtools, and cooltools10,11,12. The pipeline also generates a summary file that is ideal for assessing the quality of the Micro-C libraries10 (https://github.com/open2c/distiller-nf). The PCR duplication rate provides information on the sequencing library complexity and can be extracted from the *.stats file generated. High-quality Micro-C libraries have less than 5%-10% PCR duplication rates when generated from 5 million or more cells. Notably, some sequencing platforms generate PCR duplicates during cluster formation independent of the sequencing library complexity. Figure 2A shows the relative duplication rates of two experiments: one that we consider a good sample and one bad sample. In this example, both samples displayed acceptable map rates. The next criterias to assess the quality of Micro-C libraries is the cis versus trans ratio and read orientation frequencies. Within the nucleus, chromosomes inhabit individual chromosome territories and, thus, rarely interact with other chromosomes. A high rate of detected trans-chromosomal interactions indicates a high rate of random ligations. It should be noted that at this level of analysis, the bad sample showed a high rate of trans-chromosomal interactions compared to the good sample (Figure 2B). For Micro-C, a 70% or higher cis-chromosomal interaction rate is desirable.
A Micro-C library has a fragment size similar to the di-nucleosomal DNA band, which can co-purify with the proximity ligated sample and contaminate the experiment. These contaminates are always cis-chromosomal interactions. Therefore, it is important to also evaluate the read orientation rates. The rate of di-nucleosomal contamination can be estimated by low-input sequencing. Di-nucleosomal DNA stems from two neighboring nucleosomes that have not been cleaved by MNase. Thus, the resulting sequencing reads will always display a forward-reverse read orientation (F and R), and the distance between the read pairs will be around 320 bp. Proximity-ligated fragments, in comparison, can be ligated in four orientations, yielding read-pairs with F-R, R-R, R-F, and F-F, ideally with equal abundance (Figure 2C). In addition, they display various distances between the two read pairs. To estimate the quantity of di-nucleosomal contaminates, the frequency of read orientations can be calculated from the *stats files generated by distiller (Figure 2D). Notably, in this work, the fraction of F-R reads (red) was higher in the bad sample compared to the good sample, and this became more apparent when the read orientations were stratified by distance (Figure 2E). The F-R fraction is dominated by di-nucleosomal fragments compared to Micro-C libraries when the read pairs are stratified into reads with distances <562 bp or ≥562 bp. Here, the fraction of reads with distance <562 bp are dominated by F-R reads, whereas the fraction with distances ≥562 bp displays an even distribution between the four possible orientations, indicating that the global over-representation of F-R reads stems from di-nucleosomal contaminants. The choice of 562 bp as the threshold for subsetting is defined by the binning in the *stats file generated. Although not necessary for this quality control, more defined subsetting can be achieved by extracting the distances from the *pairs file, which is also generated by distiller. It is important to note that di-nucleosomal reads do not decrease the quality of the Micro-C sample as they can be identified and ignored during data processing. However, they do not contain valuable information about 3D interactions, and they dilute the informative reads.
Thus, careful MNase titration and thorough quality control with low-input sequencing are the best tools to optimize the quality of Micro-C experiments.

Figure 1: Intermediate stages of the Micro-C protocol. (A) Agarose gel electrophoresis of chromatin from 2.5 x 105 mouse ES cells digested with varying MNase concentrations. The mono-, di-, and tri-nucleosomal bands are indicated by arrows. M: DNA ladder (Lane 1/6); 10 U of MNase per 250,000 cells (Lane 2); 2.5 U of MNase per 250,000 cells (Lane 3); 0.625 U of MNase per 250,000 cells (Lane 4); 0.156 U of MNase per 250,000 cells (Lane 2). (B) The 1.0% agarose gel electrophoresis of the Micro-C prepared samples (Lane 3 and Lane 4) and the MNase digested input control (Lane 1). Lane 1 and Lane 2 (M: DNA ladder) are enhanced to emphasize the relative change in mono- to di-nucleosomal fragment intensity. The mono- and di-nucleosomal bands are indicated by arrows. The di-nucleosomal band in the proximity-ligated sample combines di-nucleosomal and Micro-C library DNA. (C) The 1.0% agarose gel electrophoresis of the Micro-C sequencing libraries amplified from 1 µL sample to evaluate the quality. Lane 1 (M): DNA ladder; Lane 2 (S): Mirco-C library. (D) Fragment Analyzer trace of the final Micro-C library. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Sample statistics for the low-input sequencing of a good sample and a bad sample. (A) Bar graph of the percentage mapped (green) and unmapped (red) reads. (B) Normalized fraction of reads mapping cis and trans-chromosomal interactions. The data sets were normalized to the cis-mapping read. The cis-mapping reads were stratified by the distance between the first and the second reads of the paired-end sequenced samples: ≤1 kbp (yellow), >1 kbp and ≤10 kbp (orange), and >10 kbp (red). (C) Schematic of the potential molecular species with the di-nucleosomal sizes. (D) Percentages of read-pair orientations of all the reads of the good sample and the bad sample. (E) Same as panel (D) but stratified by distances (left, <562 bp and right, ≥562 bp). Please click here to view a larger version of this figure.
{kind=link}
| Components | 1x | 4.4x |
| 10x NEBuffer 2.1, | 10 µL | 44 µL |
| 2 µL 100 mM ATP | 2 µL | 8.8 µL |
| 100 mM DTT | 5 µL | 22 µL |
| H2O | 68 µL | 299.2 µL |
| 10 U/µL T4 PNK | 5 µL | 22 µL |
| Total | 90 µL | 396 µL |
Table 1: Micro-C master mix 1. Composition of the master mix for the end chewing reaction.
| Components | 1x | 4.4x |
| 1 mM Biotin-dATP | 10 µL | 44 µL |
| 1 mM Biotin-dCTP | 10 µL | 44 µL |
| 10 mM mix of dTTP and dGTP | 1 µL | 4.4 µL |
| 10x T4 DNA Ligase Buffer | 5 µL | 22 µL |
| 200x BSA | 0.25 µL | 1.1 µL |
| H2O | 23.75 µL | 104.5 µL |
Table 2: Micro-C master mix 2. Composition of the master mix for the end labeling reaction.
| Components | 1x | 4.4x |
| 10x NEB T4 Ligase reaction buffer | 50 µL | 220 µL |
| H2O | 422,5 µL | 1859 µL |
| T4 DNA Ligase | 25 µL | 110µL |
Table 3: Micro-C master mix 3. Composition of the master mix for the proximity ligation reaction.
| Components | 1x | 4.4x |
| 10x NEBuffer 1.1 | 20 µL | 88 µL |
| H2O | 180 µL | 792 µL |
| ExoIII nuclease | 10 µL | 44 µL |
Table 4: Micro-C master mix 4. Composition of the master mix for the biotin removal reaction.
Discussion
The success of a Micro-C experiment depends on a few critical steps in the protocol that need to be carefully executed. First, crosslinking with the additional crosslinker DSG or EGS can lead to the aggregation of cells, depending on the cell type. Adding 0.1%-0.5% BSA to the crosslinking reaction significantly reduces the aggregation without affecting the crosslinking efficiency. Inefficient crosslinking can result in increased rates of trans-chromosomal interactions that are indicative of random ligations. The second, but most crucial, step in this protocol is the digestion of chromatin with MNase. Suboptimal chromatin digestion leads to inefficient proximity ligation (over-digestion) or increased rates of non-proximity-ligated di-nucleosomes (under-digestion). The efficiency of the ligation reaction can be evaluated by agarose gel electrophoresis (Figure 1B) and is additionally best estimated by low-input sequencing. If the low-input sequencing reveals either a high duplication rate (inefficient ligation) or increased di-nucleosome rates, the MNase digestion degree should be re-evaluated. Notably, the loss of sample when executing the protocol can lead to reduced library complexity. The concentration of a sample is best evaluated after DNA purification (step 5.3) or by minimal PCR (step 8). The total yield of DNA from 5 x 106 mammalian cells after DNA purification is typically >2 µg. The DNA concentration should be controlled after the MNase digestion, ExoIII digestion, and DNA purification. Endogenous nucleases, the abundance of which is cell type-specific and species-specific, can be a source of DNA degradation. In addition, column-based DNA purification can lead to sample loss due to incompatibility with the SDS from deproteination reactions. Ethanol precipitation can be considered if the DNA concentration is low at this step.
As Micro-C requires sample-specific MNase titration, it is challenging to apply Micro-C to small cell populations, such as with small organs from various model organisms, embryos and single cells, organoids, or patient biopsies. Here, Hi-C 3.0 offers a well-established alternative using an endpoint reaction by sequence-specific restriction endonucleases8,9.
Micro-C is a widely applicable high-resolution chromosome conformation technology with a high dynamic range and a low signal-to-noise ratio, which makes it particularly suitable for investigating short-range chromosome features4,5,8, such as chromosome loops. The resolution of Micro-C allows for capturing promoter-enhancer loops, which are beyond the detection limit of Hi-C, efficiently enabling a more detailed analysis of the relationship between genome organization and regulation13,14,15. Furthermore, DNA capture strategies have recently been combined with Micro-C to increase the locus-specific resolution of the targeted genomic loci to unprecedented levels, revealing novel insights into the ultrastructure of the 3D genome16,17,18. In summary, we envision that Micro-C and its derivates will be a key technology for dissecting the role of the 3D genome in transcriptional regulation and, consequently, cell type differentiation and maintenance.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We thank Christl Gaubitz and Kathleen Stewart-Morgan for their critical reading of the manuscript. We thank Anja Groth and the Groth lab for their support in establishing our lab. We thank the staff of the CPR/reNEW Genomics Platform for support: H. Wollmann, M. Michaut, and A. Kalvisa. The Novo Nordisk Foundation Center for Stem Cell Medicine (reNEW) is supported by the Novo Nordisk Foundation grant number NNF21CC0073729. The Novo Nordisk Foundation Center for Protein Research (CPR) is supported by the Novo Nordisk Foundation grant number NNF14CC0001. We thank the Brickman lab at the Novo Nordisk Center for Stem Cell Medicine, reNEW Copenhagen, for the mouse ES cells.
Materials
| Name | Company | Catalog Number | Comments |
| 1 mM Biotin dATP | Jenna Bioscience | NU-835-Bio14-S | |
| 1 mM Biotin dCTP | Jenna Bioscience | NU-809-BioX-S | |
| 10 mM dGTP | NEB | N0442S | |
| 10 mM dTTP | NEB | N0443S | |
| 10 U/ml T4 PNK | NEB | M0201L | |
| 100 U/L Exonuclease III | NEB | M0206L | |
| 10x NEBuffer 1.1 | NEB | B7001S | |
| 10x NEBuffer 2.1 | NEB | B7202S | |
| 10x T4 DNA Ligase buffer | NEB | B0202A | |
| 1x DPBS w/o Mg2+ and Ca2+ | ThermoFisher | 14190144 | |
| 1x LIF | |||
| 2_Mercaptoethanol 50 mM | Gibco | 31350010 | 0.1 mM b-mercaptoethanol |
| 37% Formaldehyde | Sigma Aldrich | 252549-500ML | Caution. See manufactures MSDS |
| 400 U/ml T4 DNA Ligase | NEB | M0202L | |
| 5 U/ml Klenow Fragment | NEB | M0210L | |
| Agarose | BIO-RAD | 1613102 | Caution. See manufactures MSDS |
| BSA 20mg/ml | NEB | B9000S | |
| CaCl2 | |||
| cell counter | |||
| Dimethyl Sulfoxide (DMSO) | Sigma Aldrich | D8418-100ML | Caution. See manufactures MSDS |
| Dynabeads MyOne Streptavidin C1 | Invitrogen | 65001 | |
| DynaMag-2 Magnet | Invitrogen | 12321D | refered to as: magnet magnet for 1.5 ml tubes |
| DynaMag-PCR Magnet | Invitrogen | 492025 | refered to as: magnet magnet for PCR tubes |
| EDTA Ultrapure 0.5M pH 8.0 | Invitrogen | 15575-038 | |
| EGTA Ultrapure 0.5M pH 8.0 | BioWorld | 40121266-1 | |
| Ethanol 96% | VWR Chemicals | 20824365 | quality control system |
| Ethidium Bromide | Invitrogen | 15585-011 | |
| Ethylene glycol bis(succinimidyl succinate) (EGS) | ThermoFisher | 21565 | |
| Fetl Bovin Serum | Sigma Aldrich | F7524 | 15% FBS |
| Gel Loading dye purple (6X) | NEB | B7024S | |
| Glycine | PanReac AppliChem | A1067.0500 | |
| Halt Proteinase inhibitor (100x) | ThermoFisher | 78430 | Caution. See manufactures MSDS |
| IGEPAL CA-630 (NP-40) | Sigma Aldrich | 18896-50ML | |
| MgCl 1 M | Invitrogen | AM9530G | |
| Micrococcal Nuclease (MNase) | Worthington | LS004798 | |
| mouse embryonic stem cells | |||
| NaCl | Sigma Aldrich | S9888-1KG | |
| NEBNext Multiplex Oligos for Illumina (Dual Index primers) | NEB | E7600S | amplification primers for sequencing libraries |
| NEBNext Ultra II DNA library prep kit for Illumina | NEB | E7645L | sequencing library preparation kit |
| NEBNext Ultra II Q5 Master mix | NEB | M0544S | Caution. See manufactures MSDS |
| Non-Essential Amino Acids Solution | Gibco | 11140050 | 1x NEAA |
| Penicillin-Streptomycin (10,000 U/mL) | Gibco | 15140148 | 1% Pen-Strep |
| Proteinase K (40 mg/ml) | GoldBio | P-480-1 | Caution. See manufactures MSDS |
| QIAquick Gel extraction kit | QIAgen | 28706 | refered to as: DNA gel elution kit |
| QIAquick PCR purification kit | QIAgen | 28106 | refered to as: commercial DNA purification kit |
| Qubit dsDNA HS Assay kit | Invitrogen | Q32854 | high sensitivity DNA quantification instrument |
| Quick load purple 1kb plus DNA Ladder | NEB | N0550S | |
| SPRIselect size selection beads | Beckman Coulter | B23319 | paramagnetic beads |
| ThermoMixer C | Eppendorf | 5382000015 | refered to as: thermomixer |
| Tris | Merck | 10708976001 | |
| Trypsin | |||
| Tween20 | Sigma Aldrich | P7949-100ML | |
| Ultrapure 10% SDS | Invitrogen | 15553-035 | |
| Ultrapure Phenol Chloroform Isoamyl Alcohol (PCI) | Invitrogen | 15593-031 | |
| Fragment Analyzer |
References
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Hsieh, T. -. H. S., Fudenberg, G., Goloborodko, A., Rando, O. J. Micro-C XL: Assaying chromosome conformation from the nucleosome to the entire genome. Nature Methods. 13 (12), 1009-1011 (2016).
- Hsieh, T. -. H. S., et al. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell. 162 (1), 108-119 (2015).
- Krietenstein, N., et al. Ultrastructural details of mammalian chromosome architecture. Molecular Cell. 78 (3), 554-565 (2020).
- Hsieh, T. -. H. S., et al. Resolving the 3D landscape of transcription-linked mammalian chromatin folding. Molecular Cell. 78 (3), 539-553 (2020).
- Belaghzal, H., Dekker, J., Gibcus, J. H. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods. 123, 56-65 (2017).
- van Holde, K. E. . Chromatin. , (1989).
- Oksuz, B. A. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18, 1046-1055 (2021).
- Lafontaine, D. L., Yang, L., Dekker, J., Gibcus, J. H. Hi-C 3.0: Improved protocol for genome-wide chromosome conformation capture. Current Protocols. 1 (7), 198 (2021).
- Goloborodko, A., Venev, S., Abdennur, N., Di Tommaso, P. Mirnylab/distiller-nf; v033. Zenodo. , (2019).
- Venev, S., et al. Open2c/cooltools: v0.4.1. Zenodo. , (2021).
- Abdennur, N., Mirny, L. A. Cooler: Scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 36 (1), 311-316 (2019).
- Zhang, S., Übelmesser, N., Barbieri, M., Papantonis, A. Enhancer-promoter contact formation requires RNAPII and antagonizes loop extrusion. BioRxiv. , (2022).
- Barshad, G., et al. RNA polymerase II and PARP1 shape enhancer-promoter contacts. BioRxiv. , (2022).
- Hansen, A. S., et al. Distinct classes of chromatin loops revealed by deletion of an RNA-binding region in CTCF. Molecular Cell. 76 (3), 395-411 (2019).
- Hua, P., et al. Defining genome architecture at base-pair resolution. Nature. 595 (7865), 125-129 (2021).
- Downes, D. J. High-resolution targeted 3C interrogation of cis-regulatory element organization at genome-wide scale. Nature Communications. 12, 531 (2021).
- Goel, V. Y., Huseyin, M. K., Hansen, A. S. Region capture Micro-C reveals coalescence of enhancers and promoters into nested microcompartments. BioRxiv. , (2022).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved