
Method Article
Internalización y Observación de fluorescente Biomoléculas en microorganismos que viven a través de electroporación
En este artículo
Resumen
Studies of biomolecules in vivo are crucial for understanding molecular function in a biological context. Here we describe a novel method allowing the internalization of fluorescent biomolecules, such as DNA or proteins, into living microorganisms. Analysis of in vivo data recorded by fluorescence microscopy is also presented and discussed.
Resumen
The ability to study biomolecules in vivo is crucial for understanding their function in a biological context. One powerful approach involves fusing molecules of interest to fluorescent proteins such as GFP to study their expression, localization and function. However, GFP and its derivatives are significantly larger and less photostable than organic fluorophores generally used for in vitro experiments, and this can limit the scope of investigation.
We recently introduced a straightforward, versatile and high-throughput method based on electroporation, allowing the internalization of biomolecules labeled with organic fluorophores into living microorganisms. Here we describe how to use electroporation to internalize labeled DNA fragments or proteins into Escherichia coli and Saccharomyces cerevisiæ, how to quantify the number of internalized molecules using fluorescence microscopy, and how to quantify the viability of electroporated cells. Data can be acquired at the single-cell or single-molecule level using fluorescence or FRET. The possibility of internalizing non-labeled molecules that trigger a physiological observable response in vivo is also presented. Finally, strategies of optimization of the protocol for specific biological systems are discussed.
Introducción
La mayoría de los estudios de fluorescencia dentro de las células vivas dependen de fusiones de proteína con proteínas fluorescentes (fps), tales como GFP 1. Estas etiquetas fluorescentes permiten estudios del número de copia, patrón de difusión o la localización de las proteínas implicadas en procesos tales como la expresión de genes o de transporte de membrana 2-7. MF ofrecen una alta especificidad etiquetado, aplicación fácil, y están disponibles en un gran inventario de variantes con diferentes fotofísico y propiedades químicas 1. Sin embargo, fluoróforos orgánicos siguen siendo la elección principal para los experimentos in vitro debido a su mayor fotoestabilidad (hasta 100 veces más estable que los MF) 8,9, pequeño tamaño (hasta 100 veces menor volumen que los MF) y la facilidad de etiquetado intramolecular (principalmente a través de la utilización de residuos de cisteína). Todos estos factores son particularmente importantes para una sola molécula fluorescente y FRET estudios de 10.
Varios métodos de internalización combining las ventajas del etiquetado orgánico y en la detección in vivo se han introducido en la última década; sin embargo, tales métodos ya sea emplean polipéptidos relativamente grandes etiquetas (tags por ejemplo, TMP, halo, o 20 kDa SNAP) 11-14, requiere el uso de aminoácidos no naturales 15, o se limita a las células eucariotas grandes, de una sola membrana (por ejemplo. , carga por legrado, jeringa de carga, microinyección) 16-19.
Este protocolo describe un novedoso, sencillo y de alto rendimiento método de internalización que las parejas las ventajas de fluoróforos orgánicos con la observación in vivo. Para desarrollar esta técnica, se adaptó el procedimiento de electroporación comúnmente utilizado para transformar las células con el plásmido de ADN 20,21 para cargar microorganismos, tales como E. coli o S. cerevisiae con biomoléculas marcadas orgánicamente. El protocolo consiste en 4 sencillos pasos: la incubación de las células con biomoléculas marcadas,la electroporación, la recuperación celular, y lavado de células para eliminar biomoléculas no internalizadas. A continuación, presentamos este protocolo de electroporación, así como los procesos de formación de imágenes de células y análisis de datos para el estudio de la fluorescencia basada en células y de una sola molécula y FRET señales.
La electroporación se basa en la descarga de un campo eléctrico de alta tensión a través de una suspensión de células de baja fuerza iónica para formar poros de la membrana transitorios a través del cual biomoléculas pueden entrar en las células (Figura 1) 20,21. Así como con la transformación de bacterias o levadura con ADN de plásmido, las células tienen que estar preparados antes de la electroporación para asegurar su electrocompetency. Este procedimiento, que consta de varias etapas de lavado con agua, aumenta la permeabilidad de la membrana y disminuye la fuerza iónica de la solución de células para evitar la formación de arco en la cubeta de electroporación. En este protocolo, las células se pueden preparar como se describe a continuación (véase el protocolo: 1,1) o comprados de proveedor comercials.

Figura 1: Representación esquemática del protocolo de internalización De izquierda a derecha:. Añadir unos pocos microlitros de biomoléculas marcadas a la alícuota de células electrocompetentes (fragmentos de ADN doblemente marcada y bacterias en este ejemplo); incubar 1 a 10 min en hielo y transferir a una cubeta de electroporación previamente enfriada; electroporar y luego añadir 0,5 a 1 ml de medio rico a las células inmediatamente después; incubar a 37 ° C (o la temperatura requerida por el organismo, por ejemplo, 29 ° C para la levadura) para permitir que las células se recuperan; realizar 5 etapas de lavado para eliminar cualquier exceso de moléculas que no son internalizados etiquetados; resuspender el sedimento final en 100-200 l de tampón PBS y pipeta de 10 l en una almohadilla de agarosa; cubrir la almohadilla con un cubreobjetos limpio y la imagen en un microscopio de fluorescencia (en el modo de campo amplio o el modo de HILO).
células electrocompetentes se incuban con las biomoléculas marcadas justo antes de la electroporación, las cuales se pueden realizar utilizando electroporators estándar que se encuentran en la mayoría de los laboratorios de bioquímica. Inmediatamente después de la electroporación, las células se incuban en un medio rico que permite su recuperación antes del lavado (Figura 1). El exceso de biomoléculas marcadas no internalizados se retira primero por lavado en un tampón que contiene una concentración bastante alta de sal y un poco de detergente (véase el protocolo: 3,3). La presencia de sal interrumpe interacciones electrostáticas no específicas formados por biomoléculas marcadas no internalizados que de otro modo pueden pegar en la membrana externa. Del mismo modo, la presencia de detergente en el tampón de lavado interrumpe interacciones hidrófobas no específicas.
Mientras que la internalización de ADN es directa (Figura 2), las precauciones deben tomarse cuando internalizar proteínas marcadas mediante electroporación. En primer lugar, la muestra de stock de proteína marcada orgánicamente todavía puede contener un pequeño porcentaje de colorante libre. Moléculas de colorante libres son mucho más pequeñas que las proteínas y por lo tanto pueden ser internalizados preferentemente. Para asegurarse de que la gran mayoría de las moléculas fluorescentes internalizadas observadas corresponden a la proteína de interés, la muestra inicial de proteína debe contener menos de ~ 2% de colorante libre (Figura 5) 22. El exceso de proteínas marcadas no internalizados también puede adherirse a la membrana celular externa después de la electroporación; este fenómeno es específico de la proteína y necesita ser comprobada para cada nueva proteína. Proponemos varias opciones que permiten la eliminación de las proteínas no internalizadas de la muestra celular cargado (Ver PROTOCOLO: 3.3.3).
Finalmente, las células se resuspenden en un pequeño volumen de tampón de fosfato y se pipetearon en una almohadilla de agarosa, lo que permite su formación de imágenes en un microscopio de fluorescencia. La inmovilización en las almohadillas de agarosa es un simplE y forma eficiente de imágenes de células sobre un cubreobjetos sin dañar su integridad. La almohadilla debe contener un medio de cultivo bajo fluorescencia.
Imágenes de células se puede realizar tanto en campo amplio, el total de fluorescencia de reflexión interna (TIRF) o utilizando HILO (muy inclinadas y laminado Hoja óptica) microscopía. En la configuración HILO, el rayo láser penetra más profundamente en la muestra que en TIRF, sin embargo, no se ilumina toda la muestra como para campo amplio, lo que permite una mayor relación señal-ruido 23. Dependiendo de la resolución de energía y el tiempo de láser utilizado, biomoléculas internalizados pueden ser contados (usando análisis por pasos-photobleaching, Figura 3), localizados, o seguido 24-28. Internalización de los constructos doblemente marcada con un par de FRET de fluoróforos permite la cuantificación de FRET tanto a una sola célula o los niveles de una sola molécula (Figura 6).
Diferentes parámetros se pueden variardependiendo de la salida deseada y el sistema biológico estudiado. En primer lugar, la cantidad de material interiorizado por célula se puede ajustar mediante la alteración de la concentración de biomoléculas marcadas añadió a las células antes de electroporación (Figura 2). Intensidad de campo electroporación también influir tanto en la eficiencia de carga y la viabilidad celular; como se esperaba, mientras que los aumentos de la eficiencia de carga con el aumento de la intensidad de campo, la viabilidad de las células electroporadas disminuye (Figura 4A). Ambos parámetros pueden ser cuantificados mediante el registro del porcentaje de carga y división de las células después de la electroporación. Este ensayo de viabilidad junto con imágenes de fluorescencia también verifica la observación de biomoléculas internalizados en las células vivas y permite la observación continua durante varias generaciones (Figura 4B).
En resumen, este protocolo permite la internalización de la etiqueta fluorescente de ADN y proteínas en moléculasE. coli o S. cerevisiae 26. Las moléculas individuales marcadas con fluoróforos orgánicos pueden ser rastreados con alta resolución espacial y temporal para escalas de tiempo de un orden de magnitud más que los MF. Finalmente, este método es compatible con widefield, TIRF y detección confocal, así como esquemas de excitación pulsada, como ALEX (alternando excitación láser 28,29).
Protocolo
1. Preparación de la célula
- Preparación de bacterias electrocompetentes de laboratorio hechas
- Preparar un precultivo 5-10 ml durante la noche de una sola colonia de la cepa de E. coli de interés en un medio de baja fluorescencia tales como M9 o EZ Rich medio definido.
- Por la mañana, inocular una nueva cultura 400 ml con el precultivo durante la noche tal que OD 600 nm comienza en 0,02. Añadir a los 400 ml bajo fluorescencia 2,5 ml de 1 M MgSO 4 y 2,5 ml de 1 M MgCl 2 medianas.
- Crecer a 37 ° C y 250 rpm hasta que OD 600 nm alcanza 0,4 a 0,6.
- Detener el crecimiento por el frío de la cultura en un baño de agua helada durante 10 a 15 min.
Nota: A partir de ahora, llevar a cabo todos los pasos a 4 ° C (en hielo). - Centrifugar la cultura 15 min a 1000 x g. Desechar el sobrenadante y resuspender el botón celular en 250 ml refrigerados y estériles destilada H 2 O.
- Repetir la centrifugación y resuspensión pasos tWICE, disminuyendo el volumen de agua a 100 ml y luego 50 ml.
- Centrifugar el cultivo de 10 min a 1000 x g. Desechar el sobrenadante y resuspender el botón celular en 25 ml de H destilada fría y estéril 2 O + 10% de glicerol.
- Repetir la centrifugación y resuspensión tres veces los pasos, disminuyendo el volumen de solución de glicerol al 10% a 10 ml, 5 ml, y finalmente a 500 l.
- Alícuota de las células en alícuotas de 20 l cada uno, el flash de congelación en nitrógeno líquido y se almacena a -80 ° C.
- Células bacterianas electrocompetentes comerciales
- Diluir una alícuota de células comercial 1: 1 con agua destilada enfriada estéril. Hacer alícuotas 20 l y se almacena a -80 ° C.
- Preparación de la levadura electrocompetente
Nota: electrocompetentes S. células cerevisiae se preparan antes de cada experimento de electroporación y no pueden ser almacenadas a -80 ° C como para E. coli.- Para empezar, inocular 5Medio YPD 0 ml con una sola colonia de la cepa deseada de interés.
- Incubar a 30 ° C y 250 rpm hasta que la DO 600 nm alcanza 0,6 a 0,8.
- Centrifugar las células a 1000 xg durante 5 min a 4 ° C.
- Resuspender el precipitado en 25 ml refrigerados y estériles destilada H 2 O.
- Repetir las etapas de lavado resuspender dos veces en 25 ml de agua y resuspendiendo dos veces en 2 ml de una solución enfriada de sorbitol a 1 M.
- Resuspender las células en 250 l de 1 M sorbitol y dividir las células en 50 ml de alícuotas.
2. agarosa preparación pad
- Para eliminar las partículas fluorescentes de fondo, grabar un cubreobjetos en un horno a 500 ° C durante 1 hora. "Clean quemado" cubreobjetos se pueden almacenar durante semanas a temperatura ambiente cubierto de papel de aluminio.
Nota: Otros métodos de limpieza comunes tales como la limpieza de plasma o solución Piranha podrían ser utilizados siempre y cuando el fondofluorescencia de las diapositivas limpiado permanece casi nulo. - Preparar una solución de agarosa de baja fluorescencia por fusión en un horno de microondas una agarosa al 2% - solución de agua destilada (70 ° C). Inmediatamente añadir 500 l de la solución de agarosa al 2% claro para 500 l de 2X medio de cultivo de baja fluorescencia y mezclar suavemente.
- Antes de que se enfríe y se endurezca, pipetear con prontitud a esta agarosa - solución del medio en un cubreobjetos de microscopio (No 1.5 grosor) con el fin de formar un lecho de aproximadamente 2 cm de diámetro y una altura de unos pocos milímetros. Evite las burbujas y el pop con puntas de pipeta si es necesario.
- Acoplar la almohadilla con un segundo cubreobjetos "quemado" (No 1.5 espesor, ver Figura 1).
Nota: Esta cubreobjetos superior ayuda a formar una plataforma homogénea plana y proteger del polvo y secado mientras que las células se están preparando. Medio mínimo como M9 o medio rico como EZ Rich Media Definido han sido probados por su baja fluorescencia.
3. Electroporation
- Incubación
- Agregue hasta 5 l de moléculas marcadas almacenados en una memoria intermedia baja en sal ( Nota: La concentración de moléculas marcadas con fluorescencia en las soluciones madre y por lo tanto la cantidad de moléculas marcadas añadidas a la célula antes de la electroporación se correlaciona directamente con la eficiencia de carga (Figura 3, y Discusión). Como algunas proteínas son menos compatibles con la condición de baja sal, la concentración de sal en el tampón de almacenamiento podría ser aumentada pero el volumen de moléculas marcadas añadió a las células antes de la electroporación a continuación, necesita ser disminuido.
- Transferir la mezcla de células y biomoléculas marcadas en una cubeta de electroporación previamente enfriada (0,1 y 0,2 cm de espacio para las bacterias y la levadura, respectivamente). Golpee suavemente la cubeta en el banco para eliminar las burbujas potenciales de la solución.
- Coloque el cuvette en el electroporador y aplicar un pulso eléctrico de alta tensión a la solución (0,9 a 1,8 kV / cm, véase la discusión para más detalles sobre la elección de la tensión). Un pulso de tales formas de poros transitorios en las membranas celulares que permiten biomoléculas marcadas se difundan en las células.
- Compruebe que la constante de tiempo que aparece en la electroporador es entre 4 a 6 ms. Constantes de tiempo inferiores son a menudo debido a la muy alta concentración de sal y / o la presencia de burbujas en la cubeta, y dará lugar a muy bajo costo o sin carga de las células.
- Recuperación
- Inmediatamente después de la electroporación, añadir 500 l de medio rico como SOC, Medium EZ Rich Definido, YPD o cualquier medio rico a las células.
- Incubar la muestra a 37 ° C para las bacterias y 29 ° C para la levadura durante 2 a 10 min. Para las mediciones de viabilidad, donde el usuario quiere evaluar el porcentaje de células que crecen y se dividen después de la electroporación, utilizar un tiempo de recuperación más largo (hasta 1 hora) como we observar tales tiempos de retraso antes de la primera división celular.
- Las etapas de lavado
- Se lavan las células para eliminar cualquier biomoléculas no internalizadas desacelerándose las células durante 1 min a 3.300 xg y 4 ° C. Desechar el sobrenadante y resuspender las células en 500 l de PBS.
Nota: Para cada muestra, preparar un control negativo de las células incubadas con la misma cantidad de biomoléculas marcadas pero no electroporadas y lavados exactamente de la misma manera que la muestra principal. - Repita los pasos anteriores 3 veces.
- En el caso de la internalización de proteínas, optimizar el procedimiento de lavado en función de las propiedades y el comportamiento de la proteína marcada de interés. Los pasos siguientes son ejemplo de posibles optimizaciones:
- Realice los 3 primeros ciclos de lavado utilizando PBS que contenía NaCl 100 mM y 0,005% de Triton X100 para eliminar las proteínas no internalizadas que podría pegarse a la membrana celular externa 22, 26.
- Filtrar las células electroporadas con un filtro de 0,22 micras de diámetro de poro instalado en el interior de un tubo de microcentrífuga de 1,5 ml pipeteando las células electroporadas en el filtro. Centrifugar durante 3 minutos a 800 xg y 4 ° C. Deseche el flujo directo. Añadir 500 l nueva PBS sobre las células y hacerlas girar una vez más como antes y repita estos pasos una vez 22.
- Añadir una pequeña cantidad de proteasa K (10 ng en 500 l de PBS) durante el primer ciclo de lavado para permitir la digestión de cualquier proteína no internalizados.
- Realice los 3 primeros ciclos de lavado utilizando PBS que contenía NaCl 100 mM y 0,005% de Triton X100 para eliminar las proteínas no internalizadas que podría pegarse a la membrana celular externa 22, 26.
- Centrifugar las células durante 1 minuto a 3300 xg y 4 ° C. Desechar el sobrenadante y resuspender las células en 150 l de PBS.
- Corre la solución de células cargado en la plataforma de agarosa mediante la eliminación del cubreobjetos superior y la difusión de 10 l de la gotita de suspensión de células por gotita. Reemplazar un cubreobjetos limpio y sin usar quemada (No 1.5 espesor, que coincide con el microscopio especificación objetiva) en tque la parte superior de la almohadilla y presione suavemente en la diapositiva.
- Protege las células electroporadas de la luz mediante el almacenamiento de las pastillas en una caja opaca, mientras que imágenes de diferentes muestras.
- Se lavan las células para eliminar cualquier biomoléculas no internalizadas desacelerándose las células durante 1 min a 3.300 xg y 4 ° C. Desechar el sobrenadante y resuspender las células en 500 l de PBS.
Adquisición de datos 4. Microscopio
Nota: Una sola célula y microscopía de fluorescencia de una sola molécula en microorganismos vivos se pueden realizar en cualquier microscopio de fluorescencia apropiado (o comercial hecha a la medida).
- Ajustes
- Widefield o HILO iluminación
- Muestras de imagen con cualquier microscopio TIRF / sola molécula.
Nota: A modo de ejemplo, podemos utilizar en el laboratorio un microscopio invertido personalizado con una TIRF puesta a punto. Vigas de un 532-nm y un láser de diodo de 637 nm se combinan y colimados antes de centrarse en el plano focal posterior del objetivo. La fluorescencia de la muestra se recoge mediante el mismo objetivo, separada de la luz de excitación utilizando un paso largo y un filtro de muesca, y se dividió en red canales y verdes utilizando un espejo dicroico. Los dos canales son imágenes en mitades separadas del chip de un (EM-CCD) de la cámara de electrones multiplicando dispositivo de carga acoplada. Los vídeos se graban utilizando la modalidad cinética. Imágenes de luz blanca se obtienen usando una lámpara de luz blanca y un condensador unido al microscopio como una fuente de iluminación. - Por observación general de una sola molécula, establezca el modo de iluminación del microscopio para TIRF o HILO 23 (véase la discusión para más detalles acerca de TIRF frente de imágenes HILO). Para establecer un modo de HILO en un microscopio TIRF, disminuir ligeramente el ángulo de incidencia de la luz de excitación para cambiar el enfoque ligeramente más alto que la superficie de cubreobjetos (la imagen de la celda interior en lugar de su membrana inferior en contacto con el cubreobjetos, ver 4.5.4 ).
- Para el análisis a nivel celular, el seguimiento a largo sola molécula experimentos o análisis por etapas fotoblanqueo, establezca el modo de iluminación del microscopio a una ensuri modo de campo amplioendo la observación continua de todo el volumen de la célula y, por tanto, de todas las moléculas marcadas internalizados.
- Muestras de imagen con cualquier microscopio TIRF / sola molécula.
- Por lo general, los poderes uso de excitación alrededor 0,5-3 MW (~ 50 a 400 W / cm 2).
Nota: Las potencias de láser más bajos son útiles para lograr una larga vida observación de fluorescencia y el seguimiento (durante 1 minuto), mientras que potencias de láser superiores podrían ser necesarias para una mayor resolución espacial y temporal o análisis fotoblanqueo paso a paso. - Utilice tiempos de exposición que van desde 15 ms para el seguimiento de los experimentos a 100 ms para la observación más general y cuantificación intensidad. Nota: Otros velocidades y modos se pueden utilizar como iluminación estroboscópica, sobre todo para el estudio de especie que se difunde rápidamente 30.
- En el microscopio TIRF, grabar el canal de fluorescencia en un CCD de electrones multiplicando (EMCCD) cámara con un aumento resultante en una longitud de pixel de ~ 100 nm / pixel. La configuración TIRF es describir con mayor detalle en la referencia 26.
- Widefield o HILO iluminación
- La adquisición de datos
- Apagar o bloquear la iluminación láser hasta el comienzo del experimento. Cambie la ganancia de la cámara EMCCD apagado para evitar daños en la cámara debido a la sobreexposición.
- Colocar el sándwich de la almohadilla de agarosa al revés sobre la platina del microscopio, con el lado de la célula cubierta orientada hacia abajo, a fin de que la celda cerca de la objetiva. Ajuste el enfoque en las células en el modo de microscopía de luz transmitida 28. Grabar una imagen de cada célula de vista bajo la imagen de luz blanca con el fin de localizar el celular esboza antes de apagar la luz blanca.
- Evita que la muestra de la luz ambiente de laboratorio.
- Para el modo de excitación Hilo, ajustar el ángulo del haz de excitación a cada relación máxima de señal a ruido mediante la iluminación de sólo la sección de la muestra cerca de la superficie cubreobjetos.
- Para lograr la iluminación Hilo, enfocar el haz láser en el plano focal posterior de un 100x 1,4 NA objetivo 28 (mayor numeRICAL aberturas tales como 1,45 o 1,49 son también adecuados). Al desplazar la lente de enfoque perpendicular a la viga, el foco se mueve aparte del centro de objetivo de forma que la salida del haz del objetivo con un ángulo.
- Ajustar la posición de la lente a fin de maximizar la relación señal-ruido, la intensidad de fluorescencia intracelular en comparación con la señal de fondo extracelular.
- Cambie la ganancia de la cámara y comenzar la adquisición de datos antes de encender el láser.
- Durante la grabación de datos, adquirir una imagen de luz blanca de cada FOV antes o después de la grabación de los datos de fluorescencia; esto ayuda a identificar límites de las celdas en los canales de fluorescencia.
- Para la estimación de la viabilidad
- Utilice un medio rico bajo fluorescencia en la almohadilla de agarosa para permitir que las células crecen después de la electroporación.
- Equilibrar el microscopio a la temperatura óptima para el microorganismo estudiado (37 ° C para E. coli, 29 ° C para S. cerevisiae) Con un sistema de calentador de objetivo.
- Debe registrar tanto la luz y fluorescencia imágenes en blanco cada 30 minutos, asegurándose de permanecer exactamente en el mismo campo de visión durante toda la grabación de datos. Un retraso de ≈1 horas se observa normalmente antes de las células comienzan a dividirse.
- Contar el número de biomoléculas internalizadas por célula
- Ajuste la potencia del láser para valores altos (3.2 MW) y el tiempo de exposición a largo (100 ms).
- Ajuste la iluminación a modo de gran campo con el fin de iluminar toda la célula.
- Grabar películas como se describe en el paso 4.2 asegurándose de grabar imágenes adicionales (50-100 frames) después de la finalización de la fotoblanqueo fluorescencia.
5. Análisis de Datos
- Análisis general
- Analizar las imágenes grabadas y películas, tanto en blancos de luz y fluorescencia excitaciones, utilizando un software de imagen, como el ImageJ software libre.
- En ImageJ, abra las imágenes o películas grabadas en el microscopio (formato TIF) en Archivo> Abrir> Su ubicación del archivo.
- Para comparar cualitativamente la intensidad de fluorescencia en una pantalla de ordenador, asegúrese de que todas las imágenes de fluorescencia se muestran con los mismos ajustes de brillo y contraste en Imagen> Ajustar> Brillo / Contraste. Ajuste manual o automáticamente la configuración de una imagen seleccionada, pulse el botón "Set" y seleccionar la opción "Propagar a todas las otras imágenes".
- Establecer el tipo de información para extraer: Analizar> Medidas Set y seleccione (al menos) "Área", "desviación estándar", "Min y valor de gris max" y "Valor promedio gris".
- Para comparar las intensidades de fluorescencia celular, seleccione el área s de interés utilizando el botón de selección a mano alzada de ImageJ y extraer las intensidades de células en Analizar> Medida. La tabla resultante contiene los valores de medición ySe pueden guardar y / o copiado a otro software. El valor "media" corresponde a la intensidad promedio por píxel en el área seleccionada y se puede comparar directamente entre células o entre una célula y el fondo.
- En una muestra de células a electroporación, una célula se considera cargado si su intensidad medio por pixel es mayor que la intensidad media por píxel del control negativo plus 3 veces su desviación estándar (Av (I cargado celular)> Av (I -EP) + 3 * DesviaciónEstándar (I -EP)).
- Construir imágenes y películas de superposición falsa fluorescencia de color con el fin de evaluar la calidad y la carga de las muestras.
- En ImageJ, superposición de imágenes, como la de la imagen de fluorescencia correspondiente al mismo campo de visión en imágenes> imagen Colores y luz blanca> Combinar canales. Seleccione un color para cada imagen (C4 (gris) para la luz blanca, C1 (rojo) para el canal rojo, C2 (verde para el canal verde ... etc.).
- Comprobaren la superposición de imágenes que la fluorescencia se encuentra dentro de los límites de las celdas (imagen de luz blanca) y que la fluorescencia de fondo es bajo y homogéneo (no hay puntos brillantes fuera de los límites de las celdas).
- Antes de analizar un gran número de células, comprobar cualitativamente las imágenes correspondientes a la muestra negativa son similares a las imágenes de células vacías y mostrar intensidades mucho más bajas que las células electroporadas.
- Para los experimentos de viabilidad, contar manualmente los porcentajes de división de las células dañadas (muertos), no se dividen, pero visiblemente intacta (idéntico) y desde el mismo campo de visión que crece con el tiempo (Ver 4.2.6).
- Evaluar la viabilidad de al menos 200 células por muestra (electroporación, control negativo y celdas vacías) con el fin de reunir suficientes estadísticas.
- Analizar las imágenes grabadas y películas, tanto en blancos de luz y fluorescencia excitaciones, utilizando un software de imagen, como el ImageJ software libre.
- Análisis basado en células
- Contar el número de biomoléculas internalizadas por célula mediante análisis fotoblanqueo paso a paso
- Células segmentados según selecting del área de interés con el botón de selección a mano alzada de ImageJ y dibujar una forma que rodea precisamente la celda (equivalente a la membrana celular).
- Extracto de las intensidades de células a través del tiempo en Imagen> Pilas> El eje Z Parcela Perfil. El gráfico resultante representa la intensidad media por píxel para el área dentro de la frontera de células frente a cada fotograma de la película resulta en una curva de photobleaching para esa celda específica. Contiene una disminución exponencial de la intensidad inicial de células alcanzar una asíntota inferior (fluorescencia de fondo). Los valores de medición y se pueden guardar y / o copiar a otro software, haga clic en "Guardar" o "Copiar".
- Copiar y pegar los valores de blanqueo en una columna de la hoja de cálculo (I crudo).
- Calcular la autofluorescencia promedio por píxel que queda después de photobleaching (I automático) promediando los valores brutos I obtenidos durante los últimos 50 a 100 fotogramas (menor asíntota).
- Subtract la autofluorescencia promedio por píxel que queda después de photobleaching para esa celda de la curva inicial fotoblanqueo: Yo blanquear = I cruda - Me auto.
- Utilice la línea de base-resta fotoblanqueo TimeTraces (I blanquear contra el marco) que muestra menos de 10 pasos cuantificados para evaluar el tamaño de paso media (intensidad fluoróforo unitario) debido al blanqueo de un solo fluoróforo 26.
- Evaluar el número de moléculas internalizadas por célula dividiendo la intensidad de células de línea de base-resta inicial (I blanqueadora a t = 0) por la intensidad fluoróforo unitario.
- Eficiencia de FRET de una sola célula
- Medir la intensidad de células promedio por píxel en ambos canales donante y aceptor de emisión (tras la excitación del donante) y dentro del límite de la celda para cada canal como explicar en 5.1.1.4.
- Medir la intensidad media de píxeles para el fondo en cada cadannel de un área en blanco de la diapositiva.
- Restar esta intensidad de fondo de la intensidad media por píxel. Utilice estas intensidades de fondo-resta de fluorescencia para calcular FRET para cada celda mediante el cálculo de la intensidad aceptor de fondo-resta dividido por el total de (+ aceptor donante) intensidad de fondo-resta tras la excitación del donante: aceptor I / (I + I aceptor donante)
- Contar el número de biomoléculas internalizadas por célula mediante análisis fotoblanqueo paso a paso
- Análisis de una sola molécula
- Seguimiento de una sola molécula y el análisis de difusión
Nota: El protocolo para el seguimiento de la difusión de moléculas fluorescentes en las células vivas y para evaluar su coeficiente de difusión aparente se ha descrito 26, 28.- En pocas palabras, adaptarse a las imágenes de los fluoróforos individuales en cada cuadro por una gaussiana elíptica 2D. Enlace localizada moléculas a una pista si aparecían en tramas consecutivas dentro de una ventana de 5-7 pixeles (desde 0,48 hasta 0,67 m). Use un memory el parámetro de 1 marco para dar cuenta de la desaparición transitoria de un fluoróforo debido a parpadear o localización perdido.
- Análisis in vivo smFRET
- Identificar manualmente moléculas localizadas dentro de las células de las películas por ir a través de una película en ImageJ e identificar moléculas inmóviles (o bastante inmóviles) en el (aceptor) canal de FRET.
- Para extraer las intensidades en el canal aceptor y donante correspondiente a una molécula inmóvil, seleccionar el área alrededor de la molécula en cada canal utilizando el botón de selección "Oval" de ImageJ (círculo alrededor de cada solo fluoróforo, utilizando un radio ~ 3-pixel) y extraer las intensidades de moléculas en Analizar> Medida. La tabla resultante contiene los valores de medición y se puede guardar y / o copia en otro software.
- Calcular los valores de fondo por canal de la intensidad media de píxeles de un círculo del mismo tamaño en un área en blanco de la diapositiva sobre todos los marcos analizados.
- Utilice los valores de fluorescencia de fondo restado en los canales de donante y aceptor (tras la excitación del donante) para la fluorescencia y FRET trazas de tiempo, como en el caso de una sola célula FRET (véase 5.2.1.7).
Nota: automatizada y robusto análisis y algoritmos han sido descritos en las referencias 26-28, 31.
- Seguimiento de una sola molécula y el análisis de difusión
Resultados
Preparación de la muestra
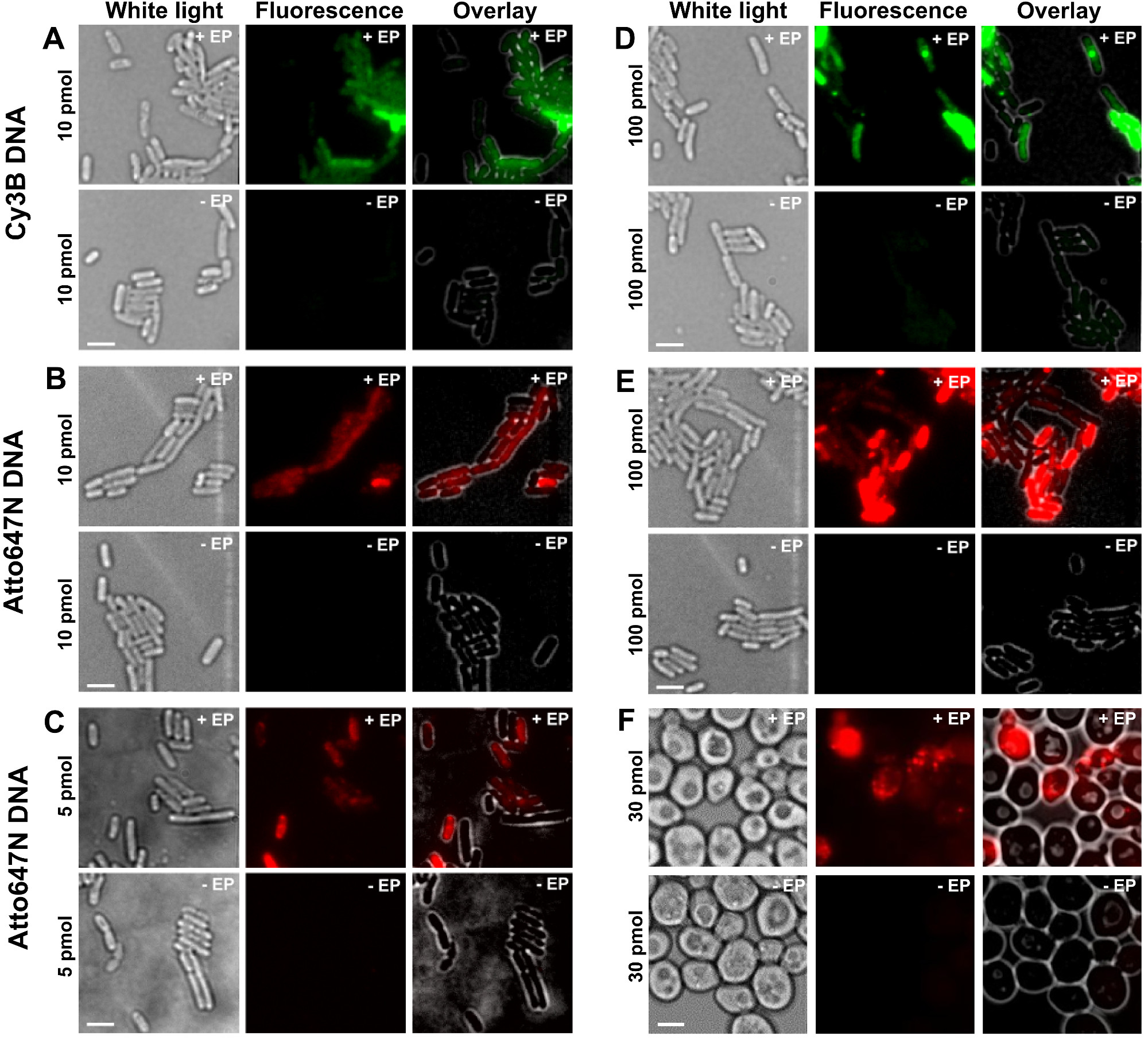
Las diferentes etapas del protocolo se presentan como esquemas de la figura 1. A modo de ejemplo, representamos a la carga de bacterias con doblemente marcada (donante y tintes aceptores) fragmentos de ADN. Los resultados representativos para la internalización de ADN se muestran en la Figura 2. Para cada muestra de electroporación, los datos para las celdas vacías y células no electroporadas También se registraron (Figura 2). "Las celdas vacías" corresponden a electrocompetent células no incubadas con biomoléculas fluorescentes ni electroporación; su intensidad en el canal de fluorescencia refleja el nivel de autofluorescencia en idénticas condiciones experimentales (potencia del láser, la resolución de tiempo, temperatura, etc.). "células para no electroporación" (también llamado -EP, es decir, menos EP) corresponden a un control negativo en el que electrocompetentes células han sido incubadas con el biomolec fluorescentemó- pero no electroporación. Estas células no electroporación deben exhibir un nivel de fluorescencia similar a la autofluorescencia de las células vacías y significativamente más baja que la intensidad de la fluorescencia mostrada por, células electroporadas cargados. Esto confirma la supresión de cualquier biomoléculas marcadas no internalizados que podrían se han adherido a la membrana celular externa.

Figura 2: Los resultados representativos para la internalización de ADN de doble cadena marcado con diferentes fluoróforos a diferentes concentraciones de bacterias (AE) y levadura (F) De izquierda a derecha:. De luz blanca, de fluorescencia y superposición de imágenes. - / + EP denota incubación sin / con electroporación. Las barras de escala: 3 m. A. Cy3B dsDNA, 10 pmol, E. coli. B. ATTO647N dsDNA, 10 pmol, E. coli. C. Alexa647 dsDNA, 5 pmol, E. coli. D. Cy3B dsDNA, 100 pmol, E. coli. E. ds ATTO647NADN, 100 pmol, E. coli. F. ATTO647N dsDNA, 30 pmol, de levadura. Esta cifra ha sido modificado de referencia 26. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Contar el número de biomoléculas internalizadas por célula
El procedimiento para estimar el número de biomoléculas marcadas internalizadas por célula mediante el análisis de photobleaching se presenta en la Figura 3 y complementario de la película 2, junto con los resultados representativos obtenidos con diferentes concentraciones de ADN marcado. Carga celular eficiencia aumenta con la cantidad inicial de ADN marcado se incuba, que permite al usuario ajustar el número de moléculas marcadas por célula de un nivel "de una sola molécula" (<10, complementario Movie 2B) a un nivel "ensemble" (> 10 , complementaria 2A Película). Una manera robusta de estimar el porcentaje de células cargadas es to contar el número de células electroporadas que muestran una intensidad de células por encima de la intensidad promedio de células de células no electroporación plus 3 veces su desviación estándar (es decir, Av (I-EP) + 3 * DesviaciónEstándar (I-EP), donde Av = promedio, I = intensidad por píxel, Std. Dev. = desviación estándar y -EP = no electroporación) como se muestra en la Figura 3.

Figura 3:. Contar el número de moléculas internalizados mediante el análisis de photobleaching (A) de celda única fotoblanqueo análisis. Ejemplos de TimeTraces de intensidad de fluorescencia (azules: datos brutos; rojo: ajuste; inserciones: WL y imágenes de fluorescencia de E. coli cargado con ATTO647N marcado dsDNA antes y después del blanqueo). Top: evento de blanqueamiento de un solo paso. Secundaria: célula que contiene ± 3 moléculas que muestran blanqueoy parpadeando. Abajo: célula que contiene> 10 pasos correspondientes a por lo menos 10 moléculas (B) histograma de intensidades de altura de un solo paso de un algoritmo automatizado paso de ajuste a partir de células que contienen menos de 57 6 pasos distinguibles.. Ajuste Single-Gaussian está centrado en 11 ± 3 au, que corresponde a una intensidad de fluoróforo unitaria de 8.100 fotones por segundo. El asterisco marca la recopilación de todas las alturas de escalón superior o igual 50 au (C) Histograma de moléculas internalizadas por célula electroporadas con diferentes cantidades de ATTO647N dsDNA, calculadas después de la división de la intensidad inicial de fluorescencia por la intensidad fluoróforo unitaria bin. De arriba a abajo: celdas vacías (es decir, no incubadas con moléculas fluorescentes y no electroporadas), no electroporación (pero incubadas con moléculas fluorescentes, llamado -EP), y las células electroporadas se incubaron con 10 y 100 pmol dsDNA (llamado + EP). Las celdas vacías y no corresponden a electroporacióna la autofluorescencia, células electroporadas mientras que muestran una amplia distribución de moléculas internalizados, con una mayor proporción de células altamente cargados en 100 pmol (≥ 4 moléculas, ver asterisco bin-marcado). La eficiencia de la internalización (fracción de células con Int.> Significa + 3x Std. Dev. De muestra no -EP) para los 10 y 100 pmol muestras fue de 94% y 90%, respectivamente. 121 ± 106 moléculas de 10 pmol ADN de doble cadena, y 176 ± 187 moléculas de 100 pmol dsDNA: número de moléculas internalizadas por célula media. Ajustes: 100 ms de exposición, iluminación de campo amplio. Las barras de escala: 1 m. Esta cifra ha sido modificado de referencia 26. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Carga y la viabilidad celular
Además de cambiar la cantidad de biomoléculas marcadas añadido a las células antes de la electroporación, lael usuario puede ajustar la cantidad de moléculas internalizados por la elección de diferentes intensidades de campo durante la electroporación (Figura 4, Película 1 complementario). Intensidades de campo más altas conducen a una mayor eficiencia de internalización, pero conducen a una ligera disminución de la viabilidad celular. Para la internalización de proteínas, el uso de una etapa de filtración puede ayudar a eliminar las proteínas no internalizados marcados (véase 3.3.3.1.1). En tales casos, la filtración celular asegura que las proteínas fluorescentes observados son de hecho internalizados en el interior del citoplasma bacteriano; Observamos, sin embargo, que la filtración también tiene un impacto negativo sobre la viabilidad celular (para más detalles, véase REF 22).

Figura 4: Influencia de la tensión de la electroporación durante la carga y la viabilidad celular (A) Diagrama de barras que representa el efecto de la intensidad de campo electroporación en carga efic.iency (barras rojas) y la viabilidad de las células (barras verdes). 84 ± 8% de la célula no-electroporación (0 kV / cm) se dividen después de 1 h en una almohadilla de agarosa a 37 ° C. En las mismas condiciones, 78 ± 3% y 49 ± 3% de las células electroporadas a 0,9 kV / cm y 1,8 kV / cm, respectivamente, se dividen después de 1 hr. Para la eficiencia de carga, 73 ± 8% de las células se cargan a 0,9 kV / cm, mientras que el 92 ± 6% de las células se cargan a 1,8 kV / cm. Las barras de error representan la desviación estándar calculada a partir de tres mediciones independientes; Se analizaron más de 200 células para cada muestra y cada repetición. En general los aumentos de eficiencia de carga con voltaje de electroporación a la ligera en detrimento de la viabilidad celular. (B) medidas de fluorescencia basados en células durante varias generaciones muestran que la intensidad de fluorescencia en general se comparte por igual entre ambas células hijas. Cell 1 y 2 se refieren al número de células en la imagen de luz blanca (izquierda) y la imagen de fluorescencia a t = 0. Escalabar: 1 m). Esta cifra ha sido modificado de referencia 26.
Internalización de proteínas
Los resultados representativos para la internalización de proteínas están en las figuras 5A & B. Es particularmente importante para eliminar la mayor cantidad de la libre (sin reaccionar) tinte restante de la muestra de proteína como sea posible antes de la electroporación. En los ejemplos de las figuras 5A y B, la muestra marcada con Cy3b Klenow Fragment (Cy3b-KF, donde KF es el fragmento Klenow de la ADN de E. coli polimerasa I, 66 kDa) sólo contiene 1% de colorante libre; dicha contribución tinte a la carga global de la célula es insignificante. Las comparaciones de la muestra a electroporación de interés con tanto las células no electroporación (incubadas con la misma cantidad de proteína marcada), así como células electroporadas con la cantidad equivalente de colorante libre constituyen dos controles necesarios para asegurar que las moléculas fluorescentes observadosson de hecho interiorizado proteínas marcadas.

Figura 5: internalización de proteínas en bacterias vivas (A) Representante campo de superposición de fluorescencia de puntos de vista.. Las células a electroporación a la tensión 1,4 kV con 50 pmol RNAP ω subunidad de una solución de proteína de valores que sólo contenía 1% libre de tinte Cy3b. Celdas vacías para no electroporación (no -EP) y se definen como anteriormente. Colorante libre se internaliza en la misma concentración como en el RNAP ω muestra de electroporación. Imaging en el modo de campo amplio, de excitación de 532 nm a 1 mW, 50 ms de exposición. (B) Distribución de intensidades celulares promediada no corregidas para las muestras en (A), dada en proporción del total de células. Fueron segmentados más de 400 células por muestra. Esta cifra ha sido modificado de referencia 22. (C) La internalización de unlaBeled ARN polimerasa de T7 (T7 RNAP, 98 kDa) en electrocompetentes DH5a que lleva el plásmido que codifica GFP esmeralda pRSET-EmGFP (EmGFP) bajo el control de un promotor T7. Izquierda: Esquema del ensayo. Secundaria: superposición de fluorescencia. Derecha: histogramas de la intensidad de fluorescencia basados en células para la muestra no electroporación (parte superior) y las células se incubaron y electroporación con T7 RNAP (parte inferior); aproximadamente el 11% de las células electroporadas muestran una alta intensidad de fluorescencia (fracción de células con Int.> significa + 3x Std. Dev. de muestra no -EP) que indica la expresión de EmGFP. Los asteriscos indican contenedores reuniendo todas las intensidades por encima o igual Barra de escala 1100 au: 3 micras. Esta cifra ha sido modificado de referencia 26.
La Figura 5C presenta otra aplicación de electroporación proteína. Aquí, la proteína electroporación es sin marcar, pero su internalización desencadena una respuesta fluorescente observable. Este experimento verifica la preSENCE y la funcionalidad de las proteínas electroporadas en el citoplasma de la célula. T7 RNA polimerasa sin rotular (98 kDa) se internaliza en E. coli DH5a cepa que contiene un plásmido que codifica para la proteína fluorescente EmGFP bajo el control de un promotor T7 26. Como el gen de la T7 RNA polimerasa está ausente en DH5a, la expresión EmGFP en nuestros experimentos requiere que funcional ARN polimerasa de T7 se introduce en las células a través de electroporación (Figura 5C). Después de la electroporación con 1 pmol T7 RNAP,> 11% de las células (azul bar, Figura 5C) exhibió fluorescencia mayor que el control negativo (incubadas con la misma cantidad de T7 RNAP, pero no a electroporación). Este resultado establece que una proporción de las moléculas de T7 RNAP internalizados por electroporación retener su integridad in vivo y puede realizar las funciones previstas en el citoplasma de la célula.
In vivo FRET en la molécula solay los niveles de una sola célula
Por último, la internalización y el análisis de las especies doblemente marcada en las bacterias que viven se presenta en la Figura 6 y Movie complementaria 3. fusiones de proteínas fluorescentes Como no son ideales para estudios in vivo smFRET, la capacidad de entregar biomoléculas doblemente marcados en las células vivas mediante electroporación es uno de los grandes activos de este método. Figura 6A presenta unicelular FRET análisis de bacterias cargadas con diferentes patrones de ADN FRET (utilizando Cy3B y fluoróforos ATTO647N como donador-aceptor par FRET). Las células se sometieron a electroporación con 20 pmol de tres estándares dsDNA FRET cortos doblemente marcada con eficiencia de FRET aparente (E *) de 0,17, 0,48, y 0,86 in vitro (anteriormente determinaron 26). Todos los ADNs entran en las células de manera eficiente (Figura 6A, izquierda) y el pico principal de cada una sola célula de E * distribución concuerda bien con los resultados in vitro (Figura 6A, A la derecha). En las muestras de media y alta de FRET, las poblaciones celulares con menor E * se observan, presumiblemente debido a una combinación de blanqueo aceptor y la inactividad fotofísico, carga de las células variable de lo esperado (por lo tanto, de relación variable de señal a ruido) y la degradación del ADN .

Figura 6:.. Los resultados representativos para una sola célula y de una sola molécula FRET observación en las bacterias que viven estudios Ensemble y smFRET en bacterias individuales (A) Análisis de células cargadas con 20 pmol de cada uno de tres patrones de ADN de FRET que exhiben baja (~ 0,17), intermedio (~ 0,48), y alto (~ 0,86) FRET (según se mide utilizando mediciones de una sola molécula in vitro; véase la ref 26). Izquierda: la luz blanca y las imágenes verde / rojo (FRET) superposición de fluorescencia (Escala de barras: 3 micras). Ejemplos de valores de FRET de diferentes células se indican (blanco). Aparejo ht (de arriba a abajo):. FRET basado en células sin corregir (E *) histogramas de donante sólo (verde oscuro), baja (verde claro), intermedio (amarillo), y alto (rojo) FRET estándares de ADN (B-D) En smFRET vivo. Las células se cargan con FRET intermedio-ADN 0,25 pmol (panel B), 0,25 pmol de alta FRET ADN (panel C), y 5 pmol doblemente etiquetadas KF (panel D). Columna de la izquierda: verde / superposición fluorescencia roja de un solo cuadro, antes y después de photobleaching aceptor. Columna central: rastros de tiempo correspondientes a la molécula en el círculo amarillo. Eficiencia de FRET, intensidades de emisión de los donantes, y las intensidades de emisión aceptor se muestran en azul, verde y rojo, respectivamente. Columna derecha: histogramas traste de donante sólo moléculas (verde) y las moléculas donantes aceptor (amarillo, rojo y gris) de 20 huellas de tiempo para cada muestra. Las barras de escala: 3 m para A, 1 m para B-D. Esta cifra ha sido modificado de referencia 26.208fig6large.jpg "target =" _ blank "> Haga clic aquí para ver una versión más grande de esta figura.
Observar smFRET in vivo para ADN o de proteína muestras, cantidades bajas (0,25 pmol) de patrones de ADN intermedio y alto FRET (Figura 6B, C) o 5 pmol de fluoróforos doble etiquetado KF (Alexa647 / Cy3B como FRET par, figura 6D) están a electroporación en E. coli. Tales concentraciones llevaron a muchas células cargadas con pocos (n = 1-10) moléculas marcadas, lo que permite la localización directa, el seguimiento, la vigilancia y Preocúpate por moléculas individuales. Algunas moléculas se difunden libremente, mientras que otros parecen inmóviles o se difunden lentamente (Película Complementario 3). TimeTraces de biomoléculas inmovilizadas doblemente marcada (Figura 6), medio duran de 1 a 30 s y mostrar las características de smFRET: cambios anticorrelated en el donante y aceptor de fluorescencia sobre blanqueo aceptor (por ejemplo, t ~ 16 seg; Figura 6B, medio ), seguido por solablanqueo -paso donante (por ejemplo, t ~; 19 seg; Figura 6B). Distribuciones FRET generados a partir de tales TimeTraces (Figura 6, derecha) dan como resultado un valor medio que se encuentra en excelente acuerdo con los publicados en estudios in vitro 26,31,32. Estos resultados establecen la capacidad para los estudios smFRET cuantitativos sobre los ADN y proteínas interiorizadas, y sugieren que las proteínas estén intactos y estructura a la electroporación y la internalización (como el apoyo de los experimentos T7 RNAP internalización).
Película complementaria 1:. La viabilidad celular Izquierda: imágenes de luz blanca. Imágenes de fluorescencia: Derecho. Animated GIF animado que muestra la división después de la electroporación (1,8 kV / cm) de las bacterias cargadas con ADN marcado Atto647 10 pmol. La aparente disminución global de la fluorescencia se debe a la dilución de ADN marcado en la división celular y también en parte a la photobleaching que se produce durante cadamedición.
Película complementaria 2: estudios fotoblanqueo basados en células A.. Ejemplo representativo de una célula muy cargado (que contiene> 100 moléculas de ADN ATTO647N marcado). Arriba a la izquierda, imagen de luz blanca de la célula de interés (rectángulo rojo). Arriba a la derecha, de la película de las células cargadas mostrando su decaimiento de fluorescencia durante varios minutos. Abajo, vestigios tiempo de la decadencia general de la intensidad de fluorescencia de la célula de interés. Fluoróforos orgánicos pueden exhibir fotoblanqueo vidas 2 órdenes de magnitud mayor que los MF (aquí, ~ 41 seg para ATTO647N). B. ejemplo representativo de una célula cargada con menos de 10 moléculas marcadas (3 en este caso). Top, mismo que el panel A. Bottom, marca temporal de la intensidad de fluorescencia en general de la célula de interés que muestra única etapa de blanqueo y / o parpadeando correspondiente a fluoróforos orgánicos individuales. La altura media de estas medidas corresponde a la intensidad unitaria de una sola molécula (aquí ~12 au) utilizados para estimar el número inicial de moléculas internalizadas por célula. Películas bajo continua excitación láser rojo de 300 mW de potencia y 100 milisegundos por trama.
Película Complementario 3: E n vivo sola molécula FRET Top:. Las células cargadas con 0,25 pmol de ADN de alto FRET (como en la figura 6C) vigilará continuamente en 50 ms por fotograma bajo iluminación nTIRF usando 1 mW verde (532 nm) láser. Cada marco es un / rojo de superposición (FRET) fluorescencia verde de cada canal. Difundir y rojo inmóvil (intacta) y verde (etiqueta activa individual) moléculas de ADN pueden ser observados. Rastro tiempo correspondiente a la molécula en el círculo amarillo: Bottom. Eficiencia de FRET, intensidades de emisión de los donantes, y las intensidades de emisión aceptor se muestran en azul, verde y rojo, respectivamente. Evento anti-blanqueo correlacionado aceptor (transiciones de rojo a verde) se corresponde con la firma de una sola molécula FRET.
Discusión
Muchos parámetros se pueden variar durante la electroporación de células y adquisición de datos en función del sistema biológico de interés y la naturaleza precisa del experimento (de nivel de celda o el análisis de una sola molécula). Por ejemplo, cuando la electroporación de ADN en bacterias de 0,25 a 5 pmol de fragmentos de dsDNA marcados conduce a una baja eficiencia de la internalización, lo que permite la detección directa de una sola molécula (es decir., Sin la necesidad de photobleaching de antemano). Por encima de 5 pmol ADN de doble cadena, las células tienden a ser muy cargado, un régimen más adecuado para el análisis de una sola célula. Todos los ADNs de la etiqueta también debe ser previamente purificado en gel con el fin de eliminar cualquier rastro de colorante libre (fluoróforo que no ha reaccionado) de la solución de ADN de valores. Además, los posibles problemas con la degradación del ADN, en particular para los experimentos smFRET, pueden abordarse mediante el uso de los ADN con los ácidos nucleicos no naturales, o motivos que protegen termini-exonucleasa accesible como bucles en horquilla.
Otra adjustable parámetro en la electroporación es la intensidad de campo aplicada durante la electroporación. La fuerza del campo baja (~ 1 kV / cm) dará lugar a una eficiencia de carga baja adecuada para los estudios de una sola molécula. Intensidades de campo superiores (hasta 1,8 kV / cm) se incrementará la eficiencia de carga; sin embargo, existe una correlación inversa entre la intensidad de campo y la viabilidad celular después de la electroporación (véase la Figura 4). Para referencia, una intensidad de campo normal utilizado para las bacterias y la levadura electroporación es ~ 1,5 kV / cm. La constante de tiempo, que representa la longitud de este decaimiento, es un parámetro conveniente seguir, ya que las gotas constantes de tiempo tan pronto como cualquier fenómeno de formación de arco se produce en la cubeta. En virtud de la configuración normal, la constante de tiempo debe ser mayor que 4 ms; valores más bajos conducirán a una baja eficiencia de carga o células dañadas aún no cargados. La mayoría de electroporators ofrecen otros grados de libertad (tales como "truncamiento pulso" o "forma de impulso") que puede ser modificado para sintonizar tantocarga de las células y la viabilidad. Hemos aplicado este método para ambas bacterias y levaduras, sin embargo procedimientos similares también deben permitir la internalización de las biomoléculas marcadas en células de mamífero usando los ajustes apropiados electroporador desde su membrana es en realidad menos complejo (una sola bicapa lipídica) y desde la electroporación ya se ha utilizado con tales células 21.
Cuando internalizar proteínas marcadas, todo tinte libre necesita ser removido de la solución de proteína de stock etiquetada electroporación antes. Moléculas de colorante gratuitos, debido a su menor tamaño, pueden ser internalizados preferentemente sobre las proteínas de interés, y son difíciles de discriminar durante el análisis de datos (a pesar de su difusión más rápida esperado). Como guía, para una muestra de proteína marcada orgánicamente sea adecuado para la electroporación, la cantidad de colorante libre restante debe ser inferior a 2% (detectada usando el escaneo fluorescente de un SDS-PAGE) 22. Este proceso es particularmente importante,como algunas moléculas podrían pegarse a las membranas externas de bacterias o levaduras electroporadas. A este respecto, la muestra de control negativo debe mostrar la intensidad de fluorescencia por célula claramente inferior a las células electroporadas, idealmente tan bajo como el nivel de autofluorescencia de las células vacías (células que no han sido incubadas con cualquier biomoléculas marcadas con fluorescencia ni electroporadas, Figura 2).
Como con dsDNA, la eficiencia de la internalización de proteínas marcadas está vinculada a la cantidad de biomoléculas añadió a las células antes de la electroporación. Sin embargo, otros parámetros, tales como el tamaño y la carga, juegan un papel en la internalización. Las proteínas pequeñas exhiben altas eficiencias de internalización, mientras que las proteínas más grandes (hasta 98 kDa) puede ser internalizado con éxito pero con una eficiencia más baja (Figura 5) 26. El punto isoeléctrico de la proteína, interacciones potenciales con la membrana celular y otros parámetros fisicoquímicos tambiéncarga de las células influencia durante la electroporación. Como resultado, los usuarios necesitan para optimizar los experimentos de su propio sistema, a sabiendas de que una alta concentración inicial de proteína marcada (> 50 micras) y darle la mejor oportunidad para la carga exitosa. La electroporación también ofrece una nueva herramienta para perturbar y analizar la función celular mediante la introducción de proteínas y otras biomoléculas en células (ya sean etiquetados o sin etiqueta). Los experimentos de ARN polimerasa T7 (Figura 5C) presenta un ejemplo de un experimento en el que podemos introducir una biomolécula que puede cambiar la expresión génica in vivo usando electroporación tal.
Cuando la realización de experimentos de fluorescencia de una sola molécula, la iluminación TIR generalmente se favorece sobre otros modos de iluminación, ya que ofrece la mejor relación señal-ruido por emocionantes sólo fluoróforos dentro de una sección delgada encima de la superficie cubreobjetos (~ 100 nm). Sin embargo de imagen etiquetada biomoléculas que se difunden dentro de microorganismos vivos podrían volvercuadernillo iluminación más profunda (hasta 0,8 m para E. coli). Iluminación más profunda se consigue en el modo de Hilo, preservando al mismo tiempo una alta relación señal-ruido. Por otro lado, las imágenes de campo amplio es particularmente importante para el análisis photobleaching paso a paso, donde el usuario es la estimación del número de moléculas internalizados por photobleaching una célula cargada entero con alto poder de láser y dividiendo la intensidad inicial de fluorescencia celular por la intensidad unitario producido por una sola molécula (paso photobleaching sola, la Figura 3). También se requiere formación de imágenes de campo amplio para el seguimiento molécula de largo plazo con el fin de localizar las moléculas de difusión de interés incluso si sus trayectorias cubren todo el volumen de la celda.
En este protocolo, se presenta cómo electroporación, una técnica estándar para biólogos y bioquímicos para la entrega de ácidos nucleicos en las células, constituye un método simple para la entrega de las biomoléculas fluorescentes en diversos tipos de células. Thes novedoso, técnica de alto rendimiento ofrece una herramienta única de observar moléculas marcadas en su ambiente nativo. En adición biomoléculas marcadas con fluoróforos que cubren una amplia gama de longitudes de onda, la electroporación puede entregar moléculas modificadas con muchos grupos químicos, tales como nucleótidos y aminoácidos no naturales, quelantes de metales, agentes de reticulación, y grupos introducción en jaula. Si el sistema biológico de interés no es esencial para el desarrollo de las células, el gen que codifica para la proteína diana también puede ser borrado (o derribado), asegurando que las proteínas observadas después de la internalización representan todos (o la mayoría) de la piscina intracelular de proteínas . En esencia, la electroporación puede "trasplante" la flexibilidad de bioconjugados in vitro en células vivas y, por tanto, beneficiar los esfuerzos de la biología sintética, la biología de sistemas, y vivo de detección de moléculas individuales en.
Divulgaciones
The authors have nothing to disclose.
Agradecimientos
We thank Stephan Uphoff for discussions.
R.C. was supported by Linacre College, Oxford University. A.P. was supported by the German Academic Exchange Service (DAAD), the German National Academic Foundation and EPSRC. M.S. was supported by the Wellcome Trust. A.N.K. was supported by a UK BBSRC grant (BB/H01795X/1), and a European Research Council Starter grant (261227).
Materiales
| Name | Company | Catalog Number | Comments |
| ElectroMax DH5-alpha Comptent cells | Invitrogen | 11319-019 | or any other commercial or lab-mage electrocompetant bacteria or yeast. |
| EZ Rich Defined Madia | Teknova | M2105 | low fluorescence rich media |
| MicroPulser Electroporation Apparatus | Biorad | 165-2100 | or any classical electroporator for microorganism transformation |
| Certified Molecular Biology agarose | Biorad | 161-3100 | low fluorescence agarose for agarose pad |
| Microscope coverslips No 1.5 thickness | Menzel | BB024060SC | remove background particles by heating slides in furnace at 500 °C for 1h |
| Single-molecule fluorescence microscope | Home-built | described in REFs | |
| Localization software | Custom-written, available online | MATLAB and C++ software package that can be adapted for localization analysis. | |
| Tracking software | Available online | MATLAB implementation by Blair and Dufresne. |
Referencias
- Tsien, R. Y. The green fluorescent protein. Annu Rev Biochem. 67, 509-544 (1998).
- Leake, M. C., et al. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature. 443, 355-358 (2006).
- Taniguchi, Y., Kawakami, M. Application of HaloTag protein to covalent immobilization of recombinant proteins for single molecule force spectroscopy. Langmuir. 26, 10433-10436 (2010).
- Xie, X. S., Choi, P. J., Li, G. W., Lee, N. K., Lia, G. Single-molecule approach to molecular biology in living bacterial cells. Annual review of biophysics. 37, 417-444 (2008).
- Lee, J. H., et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 343, 1360-1363 (2014).
- Miesenbock, G., De Angelis, D. A., Rothman, J. E. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 394, 192-195 (1998).
- Sauer, M. Localization microscopy coming of age: from concepts to biological impact. J Cell Sci. 126, 3505-3513 (2013).
- Dempsey, G. T., Vaughan, J. C., Hao Chen, K., Zhuang, X. Evaluation of fluorophores for optimal performance in localizationbased super-resolution imaging. Nat Meth. 8, 1027-1041 (2011).
- Shaner, N. C., Steinbach, P. A., Tsien, R. Y. A guide to choosing fluorescent proteins. Nat Meth. 2, 905-909 (2005).
- Landgraf, D., Okumus, B., Chien, P., Baker, T. A., Paulsson, J. Segregation of molecules at cell division reveals native protein localization. Nat. Methods. 9, 480-482 (2012).
- Jaitin, D. A., et al. Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science. 343, 776-779 (2014).
- Aldridge, S., et al. AHT-ChIP-seq: a completely automated robotic protocol for high-throughput chromatin immunoprecipitation. Genome Biol. 14, R124 (2013).
- Keppler, A., et al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 21, 86-89 (2003).
- Wombacher, R., et al. Live-cell super-resolution imaging with trimethoprim conjugates. Nat. Methods. 7, 717-719 (2010).
- Zhang, Z., et al. A new strategy for the site-specific modification of proteins in vivo. Biochemistry. 42, 6735-6746 (2003).
- McNeil, P. L., Murphy, R. F., Lanni, F., Taylor, D. L. A method for incorporating macromolecules into adherent cells. J Cell Biol. 98, 1556-1564 (1984).
- Clarke, M. S., McNeil, P. L. Syringe loading introduces macromolecules into living mammalian cell cytosol. J Cell Sci. 102, 533-541 (1992).
- Sakon, J. J., Weninger, K. R. Detecting the conformation of individual proteins in live cells. Nat. Methods. 7, 203-205 (2010).
- Taylor, L. S. Electromagnetic syringe. IEEE Trans. Biomed. Eng. 25, 303-304 (1978).
- Dower, W. J., Miller, J. F., Ragsdale, C. W. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 16, 6127-6145 (1988).
- Neumann, E., Schaefer-Ridder, M., Wang, Y., Hofschneider, P. H. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J. 1, 841-845 (1982).
- Sustarsic, M., et al. Optimized delivery of fluorescently labeled proteins in live bacteria using electroporation. Histochem Cell Biol. , (2014).
- Tokunaga, M., Imamoto, N., Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods. 5, 159-161 (2008).
- Sinha, A., et al. A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature. 000, 1-5 (2014).
- English, B. P., et al. Single-molecule investigations of the stringent response machinery in living bacterial cells. Proc Natl Acad Sci U S A. 108, E365-E373 (2011).
- Crawford, R., et al. Long-lived intracellular single-molecule fluorescence using electroporated molecules. Biophys J. 105, 2439-2450 (2013).
- Uphoff, S., Reyes-Lamothe, R., Garza de Leon, F., Sherratt, D. J., Kapanidis, A. N. Single-molecule DNA repair in live bacteria. Proc Natl Acad Sci U S A. 110, 8063-8068 (2013).
- Uphoff, S., Sherratt, D. J., Kapanidis, A. N. Visualizing Protein-DNA Interactions in Live Bacterial Cells Using Photoactivated Single-molecule Tracking. J Vis Exp. , (2014).
- Hohlbein, J., Gryte, K., Heilemann, M., Kapanidis, A. N. Surfing on a new wave of single-molecule fluorescence methods. Phys Biol. 7, 031001 (2010).
- Xie, X. S., Yu, J., Yang, W. Y. Perspective - Living cells as test tubes. Science. 312, 228-230 (2006).
- Santoso, Y., et al. Conformational transitions in DNA polymerase I revealed by single-molecule FRET. Proc Natl Acad Sci U S A. 107, 715-720 (2010).
- Hohlbein, J., et al. Conformational landscapes of DNA polymerase I and mutator derivatives establish fidelity checkpoints for nucleotide insertion. Nature communications. 4, 2131 (2013).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados