
Method Article
일렉트로을 통해 생활 미생물의 국제화 및 형광 생체 분자의 관측
요약
Studies of biomolecules in vivo are crucial for understanding molecular function in a biological context. Here we describe a novel method allowing the internalization of fluorescent biomolecules, such as DNA or proteins, into living microorganisms. Analysis of in vivo data recorded by fluorescence microscopy is also presented and discussed.
초록
The ability to study biomolecules in vivo is crucial for understanding their function in a biological context. One powerful approach involves fusing molecules of interest to fluorescent proteins such as GFP to study their expression, localization and function. However, GFP and its derivatives are significantly larger and less photostable than organic fluorophores generally used for in vitro experiments, and this can limit the scope of investigation.
We recently introduced a straightforward, versatile and high-throughput method based on electroporation, allowing the internalization of biomolecules labeled with organic fluorophores into living microorganisms. Here we describe how to use electroporation to internalize labeled DNA fragments or proteins into Escherichia coli and Saccharomyces cerevisiæ, how to quantify the number of internalized molecules using fluorescence microscopy, and how to quantify the viability of electroporated cells. Data can be acquired at the single-cell or single-molecule level using fluorescence or FRET. The possibility of internalizing non-labeled molecules that trigger a physiological observable response in vivo is also presented. Finally, strategies of optimization of the protocol for specific biological systems are discussed.
서문
살아있는 세포 내부의 대부분의 형광 연구는 GFP 1 형광 단백질 (FPS)와 단백질의 융합에 따라 달라집니다. 이러한 형광 태그는 유전자 발현 또는 2-7 수송 막 등의 처리에 관여하는 단백질의 카피 수, 확산 패턴 또는 지역화 연구를 허용한다. FPS는 높은 라벨 특이성, 쉬운 구현을 제공하며, 다양한 광 물리, 화학적 특성 1 변종의 큰 목록에서 사용할 수 있습니다. 그러나, 유기 형광가 큰 광 안정성에 의한 시험 관내 실험에 대한 주요 선택을 유지 (최대 FPS보다 안정적 100 배) 8, 9, 작은 크기 (더 작은 FPS보다 볼륨 100 배에) 및 분자 표지의 용이성 (주로 시스테인 잔기의 사용을 통해). 이러한 모든 요소는 단일 분자 형광에 특히 중요하며, FRET (10)을 연구합니다.
여러 국제화 메소드 COMBIN유기 라벨링 및 생체 검출 장점을 보내고 것은 지난 10 년 동안 소개되었다; 그러나, 이러한 방법은 비 천연 아미노산 (15)의 사용을 필요로하거나, 예를 들면 (대형 단일 막 진핵 세포로 제한되어, 비교적 큰 폴리펩티드 태그 (예를 들어, TMP, 할로, 또는 20 kDa의 SNAP 태그) 11-14을 사용 하나. 로드, 주사기 로딩, 미세 주입) 16 ~ 19를 긁어.
이 프로토콜은 그 생체 내 관찰과 커플 유기 형광의 새로운 장점을 간단하고 높은 처리량 내재화 방법을 설명한다. 이 기술을 개발하기 위해, 우리는 일반적으로 예컨대 E. 같은 미생물을로드하기 위해 플라스미드 DNA (20, 21)와 함께 세포를 형질 전환하는 데 사용되는 전기 천공 절차를 채택 대장균 또는 S. 유기적 표시 생체 분자와 사카로 마이 세스 세레 비시 애. 이 프로토콜은 4 간단한 단계로 구성 세포의 배양을 표시 생체 분자와,전기, 세포 복구, 세포 세척이 아닌 내면화 된 생체 분자를 제거합니다. 여기서, 우리는이 전기 프로토콜뿐만 아니라, 셀 기반 및 단일 분자의 형광을 연구하고 신호를 안달 세포 이미징 데이터 분석 및 처리를 제시한다.
전기 천공은 생체 분자 (도 1) 셀 (20, 21)를 입력 할 수있는 과도 멤브레인 기공을 형성하기 위해 낮은 이온 강도 세포 현탁액을 가로 질러 고전압 전기장 방전에 의존한다. 다만 플라스미드 DNA로 형질 전환 박테리아 또는 효모와 같은 세포는 electrocompetency을 보장하기 위해 전기 천공 전에 제조되어야한다. 이 절차는, 물과 여러 세척 단계로 이루어지는, 막 투과성을 증가시키고, 전기 천공 큐벳 내의 아킹을 피하기 위해 세포 용액의 이온 강도를 낮춘다. 이 프로토콜에서, 세포 (PROTOCOL 참조 : 1.1)을 후술하는 바와 같이 제조 될 수 있거나 또는 상업적 공급자로부터 구입의.

그림 1 : 국제화 프로토콜의 도식 표현 왼쪽에서 오른쪽으로 :. electrocompetent 세포 (이중 표지 DNA 단편이 예에서 박테리아)의 나누어지는 레이블 된 생체 분자의 몇 마이크로 리터를 추가; 얼음에 1 분 10 품어과 미리 냉장 전기의 베트로 전송; electroporate 다음 직후 세포에 0.5 ml의 다양한 매체를 추가; 세포가 복구 할 수 있도록 37 ° C (또는 유기체에 필요한 온도, 예를 들어, 효모 29 ° C)에서 배양; 임의의 과량의 비 표지 된 내면화 분자를 제거하는 세척 단계 5를 수행; PBS 버퍼와 피펫 아가로 오스 패드에 10 μL의 100 ~ 200 μL의 최종 펠렛을 재현 탁; (와이드 필드 모드 또는 힐로 모드) 형광 현미경 커버 슬립 세정 및 이미지 패드 커버.
Electrocompetent 세포는 대부분의 생화학 적 실험실에서 발견 electroporators 표준을 사용하여 수행 될 수있다 바로 전에 전기 생체 분자 표지와 함께 인큐베이션 하였다. 즉시 전기 후, 세포를 세척하기 전에 복구 (그림 1)을 허용하는 풍부한 매체에서 배양된다. 비 표지 된 생체 분자의 내재화 과량 제 염의 비교적 높은 농도의 일부를 함유하는 세제 세척 버퍼에 의해 제거된다 (3.3 PROTOCOL 참조). 염의 존재는 그렇게 외막에 붙어있다 내재화 비 표지 된 생체 분자에 의해 형성된 비 - 특이 정전 기적 상호 작용을 방해. 마찬가지로, 세척 완충액 세제의 존재하에 비특이적 소수성 상호 작용을 방해.
DNA 내재화 (그림 2) 간단하지만,주의 사항은 전기를 사용하여 표지 단백질을 내면화 할 때 수행해야. 첫째, 유기적 표지 단백질의 주식 샘플은 여전히 무료 염료의 작은 비율을 포함 할 수 있습니다. 무료 염료 분자는 단백질보다 훨씬 작고, 따라서 우선적으로 내면화 될 수 있습니다. 관찰 된 내면화 형광 분자의 대다수가 관심의 단백질에 대응되도록하려면 초기 단백질 샘플은 적은 2 ~ % 이상에게 무료 염료 (그림 5) (22)를 포함해야합니다. 비 표지 된 단백질의 내재화 과량 또한 일렉트로 후 세포 외막에 찌를 수있다; 이러한 현상은 특정 단백질이며, 각 새로운 단백질에 대해 확인을받을 필요가있다. 우리는 (: PROTOCOL 3.3.3 참조)로드 셀의 샘플 비 단백질의 내재화를 제거 할 수있는 여러 옵션을 제시한다.
마지막으로, 세포를 인산 완충액의 작은 부피에 재현 탁하고 형광 현미경에 자신의 이미징을 허용 아가 패드 상으로 피펫 팅한다. 아가로 오스 패드에 고정화는 간체입니다전자 및 무결성을 해치지 않고 커버 슬립에 세포 이미징의 효율적인 방법. 패드는 낮은 형광 문화 매체를 포함해야합니다.
셀 이미징은 하나 위드의 총 내부 반사 형광 (TIRF) 또는 HILO를 사용하여 (높은 경사와 적층 광학 시트) 현미경을 수행 할 수 있습니다. 힐로 구성에서, 레이저 빔은 큰 신호 대 잡음비 (23)을 허용 위드로서는 전체 샘플을 아직 조명하지 않는,보다 TIRF 시험편 깊숙히. 사용되는 레이저 파워 및 시간 해상도에 따라, 바이오 분자 내재화는 (도 3을 단계적-photobleaching에 분석을 사용하여) 계산 국소 또는 24-28를 추적 할 수있다. 형광의 FRET 쌍으로 이중 라벨 구조의 내면화는 단일 셀 또는 단일 분자 수준 (그림 6)에서 FRET 모두의 정량화 할 수 있습니다.
다른 파라미터는 변경 될 수있다원하는 출력과 연구 생물학적 시스템에 따라 다릅니다. 첫째, 셀당 내재화 물질의 양은 종래 전기 세포 (도 2)에 첨가 표지 된 생체 분자의 농도를 변경하여 조정될 수있다. 전기 천공 전계 강도는 적재 효율 및 세포 생존 둘 다에 영향을 미친다; 예상대로 증가 전계 강도 적재 효율이 증가하면서, 일렉트로 세포의 생존율 (도 4A)을 감소시킨다. 두 매개 변수는로드의 비율을 기록 및 일렉트로 후 세포 분할함으로써 정량화 할 수있다. 형광 이미징과 함께이 생존 분석은 또한 살아있는 세포에 내재화 된 생체 분자의 관찰을 확인하고 여러 세대 (그림 4B)를 통해 지속적으로 관찰 할 수 있습니다.
요약하면,이 프로토콜은 형광 표지 된 DNA와 단백질 분자의 내면화로 할 수 있습니다대장균 또는 S. 사카로 마이 세스 세레 비시 애 (26). 유기 형광으로 표지 개별 분자 크기의 순서 이상 FPS보다 시간 척도에 대한 높은 시공간 해상도로 추적 할 수 있습니다. 마지막으로,이 방법은 위드, TIRF 및 공 초점 검출뿐만 아니라 ALEX (레이저 여기 교번 28,29)와 같은 펄스 음원 방식과 호환 가능하다.
프로토콜
1. 셀 준비
- 실험실에서 만든 electrocompetent 박테리아의 준비
- 중간 정의 같은 M9 또는 EZ 리치와 같은 낮은 형광 매체에 관심있는 대장균 균주의 단일 콜로니에서 5 ~ 10 ml의 하룻밤 예비 배양을 준비합니다.
- 아침에, OD 600이 0.02에서 시작 내지 않도록 하룻밤 예비 배양과 새로운 400 ml의 문화를 접종. 1 M 황산 2.5 ㎖의 1 M의 MgCl 2의 2.5 ml의 매체 낮은 형광 400 ml의 추가합니다.
- 37 ° C에서 성장과 0.6, 0.4에 도달 nm의 OD 600까지 250 RPM.
- 10-15 분 동안 빙수 조에서 배양 차게하여 성장을 중지.
지금부터, (얼음) 4 ° C에서 모든 단계를 수행 :합니다. - g X 1000 문화 15 분 원심 분리기. 뜨는을 취소하고 H 2 O 증류 냉장 및 멸균 250 ML에있는 세포 펠렛을 재현 탁
- 원심 분리를 반복하고 재 부상은 t 단계wice 후 50 ㎖를 100 ㎖의 물 부피를 감소시키고.
- g X 1000 문화 10 분 원심 분리기. 뜨는을 취소하고 25 ml의 냉장 및 멸균 증류수 H 2 O + 10 % 글리세롤에있는 세포 펠렛을 resuspend.
- 원심 분리를 반복하고 재현 탁 10 ㎖, 5 ㎖, 최종적으로 500 ㎕의 10 % 글리세롤 용액의 부피를 감소시키는 세 번 단계.
- -80 ° C에서 액체 질소 저장 20 μL 각, 플래시 동결 분량의 셀 나누어지는.
- 상업 electrocompetent 박테리아 세포
- 상업용 세포 분취 한 희석 : 1 냉장 멸균 증류수. -80 ° C에서 20 μL 씩 저장을합니다.
- electrocompetent 효모 준비
참고 : Electrocompetent S.을 cerevisiae의 세포는 각각의 전기 실험을하기 전에 준비와 E에 관해서는 -80 ° C에 저장 될 수 없다 대장균.- 시작하려면 5 접종관심 원하는 균주의 단일 콜로니로 0 ml의 YPD 배지.
- 30 ° C 0.8 0.6에 도달 nm의 OD 600까지 250 rpm으로 품어.
- 4 ℃에서 5 분 동안 1,000 XG에 원심 분리기 세포.
- H 2 O 증류 냉장 및 멸균 (25) 용액에 펠렛을 재현 탁
- 두 번에 25 ml의 물을 재 부유 1 M.에서 소르비톨의 냉각 된 용액 2 ㎖에 두 번 재현 탁 세척 단계를 반복
- 1 M 소르비톨 250 μL에 재현 탁하고 세포를 50 ㎕의 분취 액으로 세포를 분리.
2. 아가로 오스 패드 준비
- 배경 형광 입자를 제거하기 위해 1 시간 동안 500 ° C에서 노 커버 슬립을 구울 수 있습니다. 된 커버는 알루미늄 호일에 덮여 실온에서 주 동안 저장 될 수있다 "청소 불".
주 : 이러한 플라즈마 세정 또는 피라냐 용액과 같은 다른 일반적인 세정 방법은 배경으로 한 사용될 수도청소 슬라이드의 형광을 준 널 남아있다. - 마이크로파 오븐에서 2 % 아가 로스를 용융하여 낮은 형광 아가 용액을 준비 - 증류수 수용액 (70 ° C)를. 즉시 2X 낮은 형광 배지 500 μL를 2 % 아가 로스 맑은 용액 500 μL를 추가하고 부드럽게 혼합한다.
- 무너 냉각 및 경화 전에 즉시이 아가 피펫 - 현미경 커버 슬립 (두께 1.5 없음) 매체에 액을 약 2cm 직경 및 밀리미터 높이의 패드를 형성하기 위해. 거품을 피하고 필요한 경우 피펫 팁을 팝업.
- 두 번째 "불"커버 슬립과 패드를 평평하게 (없음 1.5 두께, 그림 1 참조).
참고 :이 상단 커버 슬립은 평면 균일 패드를 형성 세포가 준비되는 동안 먼지와 건조로부터 보호하는 데 도움이됩니다. 같은 M9 또는 EZ 리치 정의 중간 풍부한 매체로 최소 매체는 낮은 형광 테스트되었습니다.
3. Electroporat이온
- 잠복
- 유능한 세포 (20 ㎕를 박테리아 또는 50 μL 효모)의 단일 나누어지는에 저염 버퍼 (<50 mM의 소금)에 저장된 표지 분자의 5 μL까지 추가하고 얼음에 10 분을 배양한다.
주 : 원액에서 형광 표지 된 분자의 농도에 따라서 전기 천공 전에 직접 적재 효율과 상관 셀에 추가 표지 분자의 양 (도 3 및 토론). 일부 단백질은 저염 조건 이하 호환되는 바와 같이, 저장 버퍼에서 염 농도가 증가 될 수 있지만, 종래 전기 세포에 첨가 표지 분자의 체적은 감소 될 필요가있다. - (각각, 박테리아와 효모 0.1 및 0.2 cm 간격) 사전 냉장 전기의 큐벳에 세포 표지 생체 분자의 혼합물을 전송합니다. 조심스럽게 용액으로부터 잠재적 인 거품을 제거하기 위해 벤치에 큐벳을 누릅니다.
- CU를 배치electroporator에의 Vette 및 솔루션에 고전압 펄스를 적용 (0.9-1.8 kV의 / cm로는 전압을 선택하는 방법에 대한 자세한 내용은 토론 참조). 이러한 펄스는 표지 된 생체 분자가 세포 내로 확산 할 수 있도록 세포막 과도 기공을 형성한다.
- electroporator에 표시되는 시간 상수는 4 ~ 6 밀리 초 사이에 있는지 확인하십시오. 낮은 시상수는 종종 너무 높은 염 농도 및 / 또는 큐벳 중의 기포의 존재에 기인 한 매우 낮은 또는 세포의 로딩없이 발생할 것이다.
- 유능한 세포 (20 ㎕를 박테리아 또는 50 μL 효모)의 단일 나누어지는에 저염 버퍼 (<50 mM의 소금)에 저장된 표지 분자의 5 μL까지 추가하고 얼음에 10 분을 배양한다.
- 회복
- 즉시 전기 후, 같은 SOC, EZ 리치 정의 중간 YPD 또는 세포에 대한 풍부한 매체 등 다양한 매체의 500 μl를 추가합니다.
- 박테리아 37 ° C에서 샘플 2 ~ 10 분 동안 효모 29 ° C를 품어. 사용자가 증가하고 전기 천공 후 세포의 분할 비율을 평가하고자 생존력 측정의 경우, w 같은 더 긴 복구 시간 (최대 1 시간)을 사용하여즉 제 1 셀 분할 전에 해당 지연 시간을 관찰한다.
- 세척 단계
- 3300 XG, 4 ℃에서 1 분 동안 세포를 회전시켜 아닌 내면화 된 생체 분자를 제거하는 세포를 씻으십시오. 뜨는을 취소하고 500 μl의 PBS에 세포를 재현 탁.
주 : 각 샘플에 대해, 생체 분자 표지 된 동일한 양의 배양하지만 일렉트로 메인 샘플과 정확하게 동일한 방식으로 세포를 세척하지 음성 대조군을 준비한다. - 이전 단계를 3 번 반복합니다.
- 내재화 된 단백질의 경우, 관심의 표지 단백질의 특성 및 행동에 따라 세척 과정을 최적화한다. 다음 단계는 가능한 최적화의 예이다 :
- <, PBS는 세포 외막 (22)에 부착 할 수있는 비 내재화 단백질을 제거하기 위해 100 mM의 NaCl을 0.005 % 트리톤 (Triton) X100을 포함하여 제 3 세척 사이클을 수행SUP> 26.
- 필터로 전기 천공 피펫 팅하여 세포를 1.5 ml의 마이크로 원심 튜브 내부에 끼워 0.22 μm의 공경의 필터로 전기 천공 세포 필터. 800 XG에 3 분, 4 ° C에 대한 스핀 다운. 흐름을 통해 폐기하십시오. 세포를 통해 500 μl의 새로운 PBS를 추가하고 이전과 같이 다시 한 번 그들을 회전, 22 번이 단계를 반복합니다.
- 비 내면화 단백질의 소화를 허용하도록 1 차 세척 사이클 중에 프로테아제 K의 소량 (500 μL의 PBS 10 NG)를 추가한다.
- <, PBS는 세포 외막 (22)에 부착 할 수있는 비 내재화 단백질을 제거하기 위해 100 mM의 NaCl을 0.005 % 트리톤 (Triton) X100을 포함하여 제 3 세척 사이클을 수행SUP> 26.
- 3300 XG에 1 분 4 ° C의 세포를 스핀 다운. 뜨는을 취소하고 PBS 150 μL의 세포를 재현 탁.
- 상부 커버 슬립을 제거하고 액적하여 세포 현탁액 방울의 10 μl를 확산하여 아가 패드로드 셀 솔루션을 확산. 사용하지 않은 깨끗한 구운 커버 슬립 (없음 1.5 두께, 현미경 목표 명세서와 일치) t에 교체아주 부드럽게 슬라이드 패드와 언론의 그는 최고.
- 다른 샘플을 이미징 동안 불투명 한 상자에 패드를 저장하여 빛 일렉트릭 세포를 보호합니다.
- 3300 XG, 4 ℃에서 1 분 동안 세포를 회전시켜 아닌 내면화 된 생체 분자를 제거하는 세포를 씻으십시오. 뜨는을 취소하고 500 μl의 PBS에 세포를 재현 탁.
4. 현미경 데이터 수집
참고 : 살아있는 미생물의 단일 셀 및 단일 분자 형광 현미경은 적절한 형광 현미경에서 수행 할 수 있습니다 (주문 제작 또는 상업적).
- 설정
- 위드 또는 HILO 조명
- 어떤 TIRF / 단일 분자 현미경 이미지 샘플.
참고 : 예를 들어, 우리는 TIRF 셋업과 실험실에서 사용자 정의 거꾸로 현미경을 사용합니다. 532 nm 내지 637 nm의 다이오드 레이저로부터의 빔은 대물 합하고의 초점면에 포커싱 전에 시준된다. 샘플이 형광 롱 패스 및 노치 필터를 이용하여 여기 광으로부터 분리, 동일한 목적을 통해 수집하고, 재로 분할이색 거울을 사용하여 D와 녹색 채널. 두 채널들은 전자 승산 전하 결합 소자 (EM-CCD) 카메라를 별도의 칩 상에 묘화 반쪽된다. 동영상 운동 모드를 사용하여 기록됩니다. 백색광 이미지 백색광 램프 및 조명 원으로서 현미경에 부착 된 콘덴서를 사용하여 얻어진다. - 일반적으로 단일 분자 관찰, TIRF 또는 HILO 23 현미경의 조명 모드 (힐로 영상 대 TIRF에 대한 자세한 내용은 토론 참조)을 설정합니다. 오히려 커버 슬립에 접하는 하부 막보다 내부 셀, 4.5.4 참조 TIRF 현미경에 HILO 모드를 설정하기 위해, 커버 슬립의 표면보다 약간 높은 초점 (화상 시프트 약간 여기 광의 입사각을 감소 ).
- 세포 수준의 분석을 위해, 긴 단일 분자 실험 또는 단계적 광표백 분석 트래킹 위드 모드로 ensuri 현미경의 조명 모드를 설정할전체 세포 부피의 연속 관찰 겨 따라서 모든 내면화 표지 분자.
- 어떤 TIRF / 단일 분자 현미경 이미지 샘플.
- 일반적으로 0.5 ~ mW의 주위에 사용 여기에 전원 (~ 50-400 W / cm 2).
참고 : 낮은 레이저 파워가 수명이 긴 형광 관찰과 높은 레이저 파워가 높은 시공간 해상도 또는 단계적 광표백 분석을 위해 필요할 수 있습니다 동안 (일분 이상)를 추적을 달성하는데 유용하다. - 15 MS에서 더 일반적인 관측 및 강도 정량화 100 밀리로 실험을 추적까지 노출 시간을 사용합니다. 참고 : 다른 프레임 속도와 모드는 특히 빠르게 확산 종 (30)을 공부, 스트로보 조명 등을 사용할 수 있습니다.
- TIRF 현미경에서, ~ 100 nm 인 / 화소의 화소 길이 초래 배율 전자 승산 CCD (EMCCD) 카메라 형광 채널을 기록한다. TIRF 설정 참조 (26)에서 더 자세한 내용을 기술 할 것입니다.
- 위드 또는 HILO 조명
- 데이터 수집
- 끄거나 실험 개시까지 레이저 조명 블록. 때문에 과다 노출로 카메라에 손상을 방지하기 위해 오프 EMCCD 카메라 이득을 전환합니다.
- 목표에 가까운 셀을 가져 오기 위하여, 거꾸로 현미경 무대에, 아래로 향 셀 덮인면 아가로 오스 패드 샌드위치를 놓습니다. 투과광 현미경 모드 (28)의 셀에 포커스를 설정합니다. 셀을 찾기 위해 백색광 영상보기에서의 각 셀의 이미지를 기록는 흰색 빛을 전환하기 전에 간략하게 설명합니다.
- 실험실 주변 광에서 샘플을 보호합니다.
- 힐로 여진 모드의 경우, 커버 슬립의 표면에 가까운 샘플의 부분 만 광을 조사하여, 각각의 최대 신호 대 잡음 비에 여기 빔의 각도를 조정한다.
- HILO 조명을 달성하기 위해, 100 × 1.4 NA 대물 28의 (높은 누메의 초점면에 레이저 광을 집중이러한 1.45 또는 1.49 같은 RICAL 구멍)도 적합하다. 빔 각도 대물 종료되도록 빔에 수직 인 포커싱 렌즈를 이동시킴으로써, 초점은 대물 중심으로부터 멀어지는.
- 세포 외 백그라운드 신호 대 신호대 잡음비, 세포 내 형광 강도를 최대화하기 위해 렌즈 위치를 조정한다.
- 카메라 게인을 전환하고 레이저를 전환하기 전에 데이터 수집을 시작합니다.
- 데이터를 기록하는 동안, 전 또는 형광 데이터를 기록한 후, 각 FOV의 백색광 화상을 취득; 이것은 형광 채널에서 셀 경계를 식별하는 것을 돕는다.
- 생존력 추정
- 세포가 전기 후 성장 할 수 있도록 아가로 오스 패드에 저 형광 다양한 매체를 사용합니다.
- 연구 미생물 (37 ° C 대장균, 29 ° C S. cerevisiae가 용을위한 최적의 온도에 현미경을 평형) 대물 히터 시스템.
- 기록 모두 흰색 빛과 형광 이미지를 매 30 분, 전체 데이터 기록시보기의 동일한 필드에 남아 확인하고. 세포 분열을 시작하기 전에 ≈1 시간의 지연은 일반적으로 관찰된다.
- 셀당 내면화 생체 분자의 수를 카운팅
- 높은 값 (2-3 MW)와 긴 노출 시간 (100 밀리)에 레이저 파워를 설정합니다.
- 셀 전체를 조명하기 위해 모드 위드 할 수있는 조명을 설정합니다.
- 같은 기록 영화는 형광 광표백의 완료 후 추가 프레임 (50 ~ 100 프레임)을 기록 할 확인하는 단계 4.2에 설명.
5. 데이터 분석
- 일반 분석
- 이러한 자유 소프트웨어 ImageJ에 같은 이미징 소프트웨어를 사용하여 흰색 빛과 형광 가진에 모두 녹화 된 영상과 영화를, 분석한다.
- ImageJ에에서 파일의 현미경 (TIF 형식)에 기록 된 이미지 나 동영상> 열기> 귀하의 파일 위치를 엽니 다.
- 질적으로 컴퓨터 화면에 형광 강도를 비교하기> 모든 형광 이미지는 이미지에서 동일한 밝기 및 대비 설정으로 표시되어 있는지 확인합니다> 밝기 / 대비를 조정합니다. 선택된 이미지를 수동 또는 자동으로 설정을 조정 버튼을 누릅니다 "설정"과 "다른 모든 이미지에 전파"옵션을 선택합니다.
- "최소 및 최대 회색 값"및 "회색 값을 평균"분석> 설정 측정하고, (적어도) "영역", "표준 편차"를 선택 : 추출하는 정보의 유형을 설정합니다.
- > 측정을, 세포 형광 강도를 비교 지역의 ImageJ에의 자유형 선택 버튼을 사용하여 관심을 선택하고 셀 강도가의 분석 추출합니다. 결과 테이블은 상기 측정 값을 포함하고저장 및 / 또는 다른 소프트웨어에 복사 할 수 있습니다. "평균"값은 선택 영역에서 화소 당 평균 휘도에 대응하고, 직접적으로 셀 사이 또는 셀과 배경 사이의 비교 될 수있다.
- 일렉트로 세포 샘플에서, 셀은 화소 당 평균 강도는 대조군의 픽셀 및 3 배의 표준 편차 (AV은 (I 셀 로딩)> 아베 (I -EP) + 당 평균 강도보다 큰 경우 로딩 된 것으로 간주 3 * STDDEV (I -EP)).
- 샘플의 품질과 로딩을 평가하기 위해 거짓 색 형광 오버레이 이미지 및 동영상을 구축 할 수 있습니다.
- ImageJ에, 그러한 백색광 화상과 이미지> 컬러 동일한 FOV에 대응하는 형광 화상으로서 오버레이 이미지> 채널을 병합. 백색광에 대한 각각의 이미지 (C4 (회색의 색상)을 선택, C1 붉은 채널 (빨간색), C2 (녹색 채널 ... 등 녹색.).
- 확인오버레이 형광 셀 경계 내에있는 이미지 (흰색 빛 이미지)와 그 배경 형광이 낮고 균일 (셀 경계 밖에있는 밝은 반점)에.
- 다수의 셀을 분석하기 전에 빈 셀 화상과 유사한 음성 시료 성적에 대응되는 이미지를 확인하고 세포를 전기 천공보다 훨씬 낮은 농도를 표시.
- 생존 실험을 위해, 수동 (4.2.6 참조) 시간이 지남에 따라 성장 같은 시야에서, 비 분할하지만 (동일) 눈에 띄게 손상 및 손상 (죽은) 셀 분할의 비율을 계산합니다.
- 충분한 통계를 수집하기 위해 샘플 (electroporation하여, 음성 대조군과 빈 셀) 당 최소 200 세포의 생존 능력을 평가합니다.
- 이러한 자유 소프트웨어 ImageJ에 같은 이미징 소프트웨어를 사용하여 흰색 빛과 형광 가진에 모두 녹화 된 영상과 영화를, 분석한다.
- 세포 기반 분석
- 단계적 광표백 분석으로 셀당 내재화 생체 분자의 수를 카운팅
- selecti 세그먼트 - 세포ImageJ에의 자유형 선택 버튼을 사용하여 관심 영역을 ng를 정확하게 셀 (세포막 할 수있는 동등한)를 둘러싸는 모양을 그립니다.
- 이미지> 스택> 플롯 Z 축 프로파일에 시간이 지남에 따라 세포 강도의 압축을 풉니 다. 얻어진 그래프는 그 특정 셀에 대한 광표백 곡선에서 얻어진 각각의 동영상 프레임에 비해 셀 경계 영역 내의 화소 당 평균 강도를 나타낸다. 그것은 낮은 점근선 (형광 배경)에 도달하는 세포 강도의 초기 지수 감소가 포함되어 있습니다. 측정 값과는 "저장"또는 "복사"를 클릭하여 저장 및 / 또는 다른 소프트웨어에 복사 할 수 있습니다.
- 스프레드 시트 열에 복사 및 표백 값을 붙여 넣기 (I 원시).
- 지난 50 ~ 100 프레임 (낮은 점근선)에서 얻은 I 원료 값을 평균하여 (I 자동)을 photobleaching에 후 나머지 픽셀 당 평균 형광도를 계산합니다.
- 에스초기 광표백 곡선에서 해당 셀에 대한 photobleaching에 후에 남아있는 픽셀 당 평균 형광도를 ubtract : 나는 표백 = 나는 원시 - 나는 자동차.
- 사용 기준 감산 (I 프레임 대 표백) timetraces을 photobleaching에 의한 단일 형광 (26)의 표백에 평균 스텝 크기 (단위 형광 강도)를 평가하기보다 10 양자화 단계를 표시합니다.
- 단일 형광 강도에 의해 초기 기준 셀 감산 강도 (I는 t = 0에서 표백) 나누어서 셀당 내재화 분자의 수를 평가한다.
- 단일 셀 FRET 효율성
- (도너 여진시) 모두 도너와 억 셉터 발광 채널 내의 화소 당 평균 셀 강도를 측정하고, 각 채널에 대한 셀 경계 내에서 5.1.1.4로 설명.
- 각 채널에 대한 배경 평균 픽셀 강도를 측정슬라이드의 빈 영역에서 annel.
- 화소 당 평균 강도에서이 배경 강도를 뺀다. 기증자 여기에 따라 총 (셉터 + 공여)로 나눈 배경 감산 수용체 강도 배경 감산 강도를 계산하여 각 셀에 대한 FRET을 계산하기 위해 이러한 배경 감산 형광 강도를 사용하여 I (나는 + I 기증자 수용체) / 수용체
- 단계적 광표백 분석으로 셀당 내재화 생체 분자의 수를 카운팅
- 단일 분자 분석
- 단일 분자 추적 및 확산 분석
주 : 살아있는 세포에 형광 분자를 확산 및 명백한 확산 계수를 평가하는 추적 프로토콜 28 26 설명되었다.- 간단히 말해서, 2D 타원 가우시안 각 프레임에 하나의 형광의 이미지에 맞게. 그들은 5-7 픽셀 (0.48-0.67 μm의)의 창 내에서 연속적인 프레임에 나타나는 경우 링크는 트랙에 분자를 지역화. 로는 메모리를 사용하여1 프레임의 Y 매개 변수로 인해 점멸 또는 누락 현지화에 형광의 과도 실종을 설명합니다.
- 생체 smFRET 분석
- 수동 ImageJ에있는 영화를 통해 이동하여 영화에서 현지화 된 분자를 내부 세포를 식별하고 FRET (수용체) 채널에 고정되어 (또는 매우 부동의) 분자를 식별합니다.
- 부동 분자에 대응 셉터와 도너 채널의 강도를 추출하기 위해, (~3 픽셀 반경을 이용하여, 각각의 단일 형광 물질 주위에 원) ImageJ에의 "타원형"선택 버튼을 이용하여 각 채널에서의 분자의 주위 영역을 선택하고 > 분자 강도가의 분석 측정의 압축을 풉니 다. 결과 테이블 측정 값을 포함하고, 저장 및 / 또는 다른 소프트웨어에 복사 할 수있다.
- 분석 된 모든 프레임 위에 슬라이드의 빈 영역의 크기가 같은 원으로부터 평균 픽셀 강도로부터 채널당 배경 값을 계산.
- 단일 셀에서와 같이, 형광 (도너 여기시) 도너와 억 셉터 채널의 배경 형광 감산 된 값을 사용하여 시간 추적을 FRET은 (5.2.1.7 참조) 경우를 FRET.
참고 : 자동화 및 강력한 분석 및 알고리즘은 참고 문헌 26 ~ 28, 31에 기재되어있다.
- 단일 분자 추적 및 확산 분석
결과
샘플 준비
프로토콜의 다른 단계는도 1에 도식으로 제시한다. 예를 들어, 우리는 이중 표지 (도너와 억 셉터 염료) DNA 단편의 박테리아 하중을 나타낸다. DNA 내재화를위한 대표 결과는 그림 2에 나타내었다. 각 electroporation하여 샘플의 경우, 빈 셀 및 비 electroporation하여 세포에 대한 데이터는 (그림 2)을 기록 하였다. "빈 셀은"생체 분자 형광하지도 않으며 전기 천공으로 세포를 배양하지 electrocompetent 대응함 채널에서의 형광 강도는 동일 실험 조건 (레이저 파워, 시간 해상도, 온도 등)에서자가 형광 수준을 반영한다. "비 일렉트로 세포"(또한 -EP, 즉 마이너스 EP)는 세포 형광 biomolec 함께 인큐베이션 된 electrocompetent되는 음성 대조군에 대응개 모듈 있지만, 일렉트로 없습니다. 이러한 비 - 세포를 전기 천공 빈 셀의 자기 형광로드와 유사한, 세포를 전기 천공에 의해 표시되는 형광 강도보다 상당히 낮은 형광 수준을 나타낼 것이다. 이것은 세포 외막에 접착 있었을 비 표지 된 생체 분자의 내재화 제거를 확인한다.

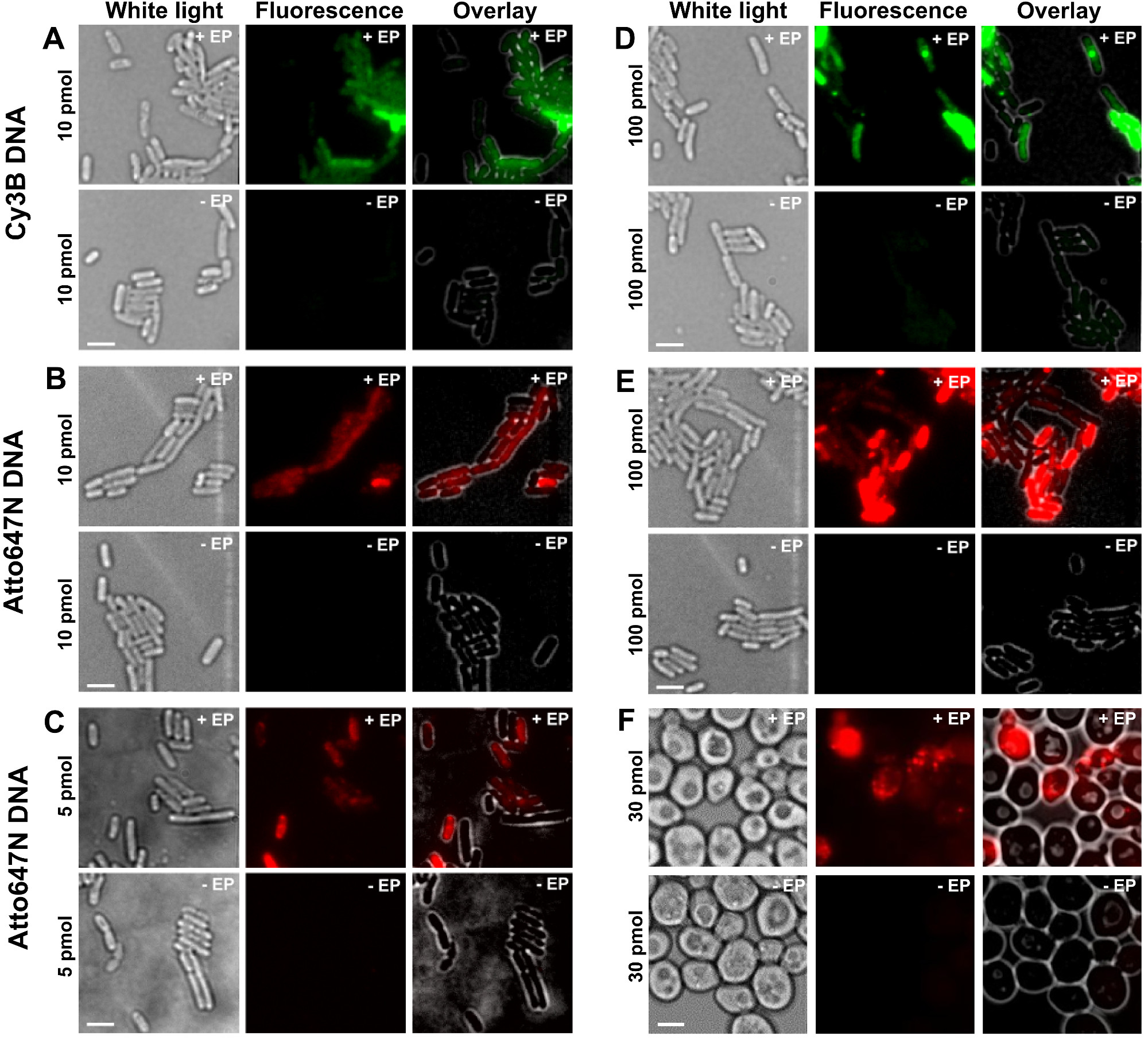
그림 2 : 박테리아에서 서로 다른 농도에서 다른 형광 물질로 표지 dsDNA의 내재화 (AE) 및 효모 (F)를위한 대표 결과 왼쪽에서 오른쪽으로. 백색광, 형광 및 오버레이 이미지. - / + EP는 전기와 함께 / 않고 배양을 의미한다. 스케일 바 : 3 μm의. A. CY3B dsDNA, 10 pmol의, E. 대장균. B. ATTO647N dsDNA, 10 pmol의, E. 대장균. C. Alexa647 dsDNA 5 pmol의, E. 콜라이. D. CY3B dsDNA, 100 pmol의, E. 콜라이. E.의 ATTO647N ds는DNA, 100 pmol의, E. 대장균. F. ATTO647N dsDNA, 30 pmol의, 효모. 이 그림은 참조 (26)에서 수정되었습니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
셀당 내면화 생체 분자의 수를 카운팅
절차는 함께 표지 된 DNA와 상이한 농도로 얻어진 대표적인 결과,도 3 및 보충이 영화에 제시되어 photobleaching에 분석을 이용하여 셀당 내면화 표지 된 생체 분자의 수를 추정한다. 셀 로딩 효율 인큐베이션 표지 된 DNA의 초기 량에 따라 증가, "단일 분자"수준 (<10, 보충 영화 2B) "앙상블"수준 (> 10로 조정하는 사용자에게 셀당 표지 된 분자의 수를 허용 , 보충 영화 2A). 로드 된 세포의 비율을 추정하는 방법은 강력한 t이고O, 비 - 일렉트로 세포의 평균 기포 강도 및 3 배에 달하는 표준 편차 (즉, 아베 (I-EP) + 3 * STDDEV (I-EP) 아베 = 평균 이상의 셀 강도를 표시하는 일렉트로 세포의 수를 세 I = 픽셀 당 강도, Dev.에. = 표준 편차 -EP = 비 일렉트로) (그림 3).

도 3 :. photobleaching에 내재화 분석을 이용하여 분자의 수를 계수 (A)는 단일 셀 photobleaching에 분석. 형광 강도 timetraces의 예 (파란색 : 원시 데이터, 빨간색 : 적합; 세트 : WL과 전 표백 후 ATTO647N 표지 dsDNA로드 대장균의 형광 이미지). 탑 : 한 단계 백화 현상. 중동 : 표백을 보여주는 ± 3 분자를 포함하는 세포점멸. 바닥 : 적어도 10 분자에 대응> 10 단계를 포함하는 셀 6 미만 구별 단계를 포함하는 셀 (57)로부터 자동 - 피팅 알고리즘 단계에서 한 단계 높이의 강도 (B)의 히스토그램.. 단일 가우스 착용감 초당 8100의 단일 광자 형광 강도에 대응하는, 11 ± 3 AU 중심된다. 별표는 위 또는 단일 형광 강도에 의해 초기 형광 강도를 나누어 계산 ATTO647N dsDNA 상이한 양으로 일렉트로 셀당 내면화 분자의 동일한 50 AU (C) 히스토그램 모든 단차를 수집 함을 표시한다. 위에서 아래로 : 빈 셀 (즉, 형광 분자로 배양되지 않고 일렉트로되지 않음), 비 일렉트로 (그러나 형광 분자라는 -EP 배양) 및 electroporation하여 세포가 10 100 pmol의 (EP + 이름) dsDNA와 함께 배양 하였다. 빈 비 일렉트로 세포 대응자가 형광에, 일렉트로 반면 세포 (≥ 4 분자 별표 마크 빈 참조) 100 pmol의에서 높은로드 셀의 높은 비율이 내면화 된 분자의 넓은 분포를 보여줍니다. 국제화 효율 10, 100 pmol의 샘플 (INT와 세포의 비율은. + 3 배 Dev.에. 비 -EP 샘플의 의미>)는 각각 94 %와 90 %였다. 10 pmol의 dsDNA 121 ± 106 분자, 100 pmol의 dsDNA 176 ± 187 분자 : 셀 당 내면화 분자의 수를 의미한다. 설정 : 100 MS 노출, 위드 조명. 스케일 바 : 1 μm의. 이 그림은 참조 (26)에서 수정되었습니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
셀 로딩과 생존
표지 된 생체 분자의 변화량 이외에 종래 전기 천공 세포에 첨가,사용자는 전기 중 (그림 4, 보충 동영상 1) 다른 필드 강점을 선택하여 조정 내면화 분자의 양을 수 있습니다. 높은 필드 강도는 더 국제화 효율성으로 이어질하지만 세포 생존 능력의 약간의 감소로 이어집니다. 단백질의 내재화를 들어, 여과 공정의 사용은 비 표지 된 단백질을 내면화 (3.3.3.1.1 참조)를 제거하는 데 도움이된다. 이러한 경우, 셀 여과 관측 형광 단백질이 실제로 박테리아 세포질 내부에서 내재화되는 것을 보장; 우리는 (자세한 내용은, REF 22 참조) 그러나, 여과는 세포 생존 능력에 부정적인 영향을 가지고 있습니다.

그림 4 : 셀 로딩과 생존시 전기 전압의 영향 (A)로드 effic에 전기 자기장 강도의 효과를 나타내는 막대 차트.iency (빨간색 막대)와 세포 (녹색 막대)의 생존. 37 ° C에서 아가 패드 1 시간 후 비 - 일렉트로 셀 (0 kV로 / cm)에 나누어 84 ± 8 %. 동일한 조건 하에서 78 ± 3 % 및 0.9 kV로 / cm 내지 1.8 kV로 / cm에서 전기 천공 셀 49 ± 3 %를 각각 1 시간 후 나눈다. 세포를 92 ± 6 %가 1.8 kV로 / cm에로드하는 동안 적재 효율, 셀 (73)은 ± 8 %, 0.9 kV로 / cm로 로딩된다. 오차 막대는 세 독립적 인 측정으로부터 계산 된 표준 편차를 나타내고; 200 개 이상의 세포는 각각의 샘플과 각각의 반복에 대해 분석 하였다. 여러 세대에 걸쳐 세포 생존을 약간 손해에 전기 전압 전반적인 적재 효율이 증가한다. (B) 형광 세포 기반 측정은 전반적인 형광 강도가 두 개의 딸세포간에 똑같이 공유되는 것을 보여준다. 셀 1과 2는 t = 0에서의 스케일 (왼쪽) 백색광 이미지에서 세포 수 및 형광 영상을 의미바 : 1 ㎛). 이 수치는 참조 (26)에서 수정되었습니다.
단백질 국제화
단백질 내재화를위한 대표 결과는도 5A & B에 있습니다. 이것은 이전에 가능 일렉트로 같은 단백질 샘플로부터 가능한 나머지 (미) 염료를 가능한 많이 제거하는 것이 특히 중요하다. 도 5a & B, CY3B 표지 클레 나우 단편 샘플 (KF는 대장균 DNA 중합 효소 I의 클레 나우 단편 CY3B-KF, 66 kDa의) 1 % 무료 염료를 포함의 예에서; 전체 셀 로딩에 염료의 기여는 무시할 수있다. 자유 염료의 등량 전기 천공 (표지 된 동일한 양의 단백질과 함께 배양) 비 일렉트로 세포 모두 관심 electroporation하여 샘플의 비교뿐만 아니라 세포 관찰 형광 분자되도록하여 두 개의 필수 구성 컨트롤참으로 표지 단백질을 내장되어 있습니다.

그림 5 : 라이브 박테리아의 단백질 국제화 (A) 뷰의 대표적인 형광 오버레이 필드.. 세포는 1 % 무료 CY3B 염료가 포함 된 단백질 원액에서 50 pmol의 RNAP ω 서브 유닛으로 1.4 kV의 전압에서 전기 천공. 비 전기 천공 (비 -EP)과 빈 셀은 이전과 같이 정의된다. 자유 염료 RNAP ω 일렉트로 샘플과 동일한 농도로 내재화시켰다. 1 mW의 (A)의 샘플에 대한 보정 세포 평균 강도의 50 밀리 노출. (B) 분포 넓은 필드 모드, 532 nm의 여기에서 이미징, 총 세포 수의 비율에 따른다. 샘플 당 400 개 이상의 세포를 분할했다. 이 수치는 참조 (22)에서 수정되었습니다. unla의 (C) 국제화T7 프로모터의 통제하에 pRSET-EmGFP 플라스미드 인코딩 에메랄드 GFP (EmGFP)를 들고 electrocompetent DH5α에이라고 표기 T7 RNA 중합 효소 (T7 RNAP, 98 kDa의). 왼쪽 : 분석의 개략도. 중동 : 형광 오버레이. 오른쪽 : 배양 및 T7 RNAP (아래)와 일렉트로 비 electroporation하여 샘플 (위)과 전지 셀 기반 형광 강도의 히스토그램; 일렉트릭 세포의 약 11 %가 높은 형광 강도 표시 (INT와 세포의 일부를.> + 의미 배 Dev.에. 비 -EP 샘플의) EmGFP의 발현을 나타내는. 3 μm의 : 별표 위 또는 동일한 1,100 AU 스케일 바의 모든 강도를 수집 쓰레기통을 나타냅니다. 이 수치는 참조 (26)에서 수정되었습니다.
그림 5c는 단백질 전기의 또 다른 응용 프로그램을 제공합니다. 여기에, 일렉트릭 단백질은 레이블이없는, 그러나 그것의 내면화는 관찰 형광 반응을 트리거합니다. 이 실험은 사전 검증가 나타나지 않는 및 세포질에 electroporation하여 단백질의 기능을 제공합니다. 레이블이없는 T7의 RNA 중합 효소 (98 kDa의)는 E.으로 내면화되었다 T7 프로모터 (26)의 제어하에 형광 단백질 EmGFP위한 코드하는 플라스미드를 함유하는 대장균 균주 DH5α. T7 RNA 중합 효소의 유전자가 DH5α에 존재하기 때문에, 우리의 실험에서 EmGFP 식 기능 T7의 RNA 중합 효소는 전기 (그림 5C)를 통해 세포 내로 도입되어 있어야합니다. 1 pmol의 T7 RNAP, 세포의> 11 %로 전기를 이하의 (청색 막대도 5C)은 음성 대조군보다 높은 형광 나타냈다 (T7 RNAP 동일한 양 배양을하지만 일렉트로 생략). 이 결과는 일렉트로 내재화 T7 RNAP 분자의 비율이 생체 내에서 자신의 무결성을 유지하고 세포질에서 의도 기능을 수행 할 수 있다는 것을 정한다.
생체 내에서 단일 분자 FRET및 단일 세포 수준
마지막으로, 살아있는 박테리아에 이중 라벨 종의 내재화 및 분석 3. 형광 단백질 융합 생체 내 smFRET 연구에 적합하지 않은 그림 6과 보충 영화에 제시되어, 전기를 사용하여 살아있는 세포에 이중 라벨 생체 분자를 제공 할 수있는 능력은 하나입니다 이 방법의 가장 큰 자산. 그림 6a는 단일 셀 (기증자 - 수용체 FRET 쌍으로 CY3B 및 Atto647N의 형광체를 사용하는) 다른 FRET DNA 표준로드 박테리아의 분석을 FRET 제공합니다. 세포는 0.17, 0.48의 명백한 FRET 효율 세 짧은 이중 표지 dsDNA FRET 기준의 20 pmol의 (E *)로 전기 천공하고, 0.86는 시험 관내 (이전 26 결정). 모든 DNA를 (그림의 6A, 왼쪽)와 각각의 단일 셀 E의 * 분포의 주 피크 효율적으로 세포를 입력합니다 (체외 결과와 잘 그림 6A를 동의, 오른쪽). 중급 및 고급 FRET 샘플에서는, 하부 아마도 인해 셉터 표백 광 물리 비활성, 가변 셀 로딩의 조합, 관찰 예상보다 E의 * (따라서, 가변 신호 대 잡음 비) 및 DNA 분해와 세포 집단 .

그림 6 :.. 단일 셀 및 단일 분자에 대한 대표 결과가 앙상블과 smFRET 연구 한 박테리아에 살고있는 박테리아에서 관찰 FRET 저를 보이는 각각의 세 가지 DNA FRET 기준의 20 pmol의로드 셀 (A) 분석 (~ 0.17) 중간체 (~ 0.48), 높은 (~ 0.86) FRET (체외 단일 분자 측정으로 측정 한, REF 26 참조). 왼쪽 : 흰색 빛과 그린 / 레드 (FRET) 형광 오버레이 이미지 (스케일 바 : 3 μm의). 다른 세포로부터의 FRET 값의 예는 (흰색) 표시된다. 조작 (위에서 아래로) HT :. 수정되지 않은 셀 기반 FRET (E *)는 DNA 표준을 FRET 기증자 만 (진한 녹색), 저 (밝은 녹색), 중간 (노란색), 고 (빨간색)에 대한 히스토그램 (B-D) 생체 smFRET합니다. 세포는 0.25 pmol의 중간 FRET DNA (패널 B), 0.25 pmol의 높은 FRET DNA (패널 C)와로드, 5 pmol의는 KF (패널 D)를 이중 표지. 왼쪽 열 : 전 수용체 광표백 후 단일 프레임의 그린 / 레드 형광 오버레이. 가운데 열 : 노란색 원 분자에 해당하는 시간을 추적합니다. FRET 효율, 기증자 방출 강도 및 수용체 방출 강도는 각각, 녹색, 파란색과 빨간색으로 표시됩니다. 오른쪽 열 : 기증자 만 분자 (녹색) 및 각 샘플에 대한 20 시간 추적에서 (노란색, 빨간색, 회색) 기증자 - 수용체 분자의 FRET 히스토그램. 스케일 바 : 3 μm의, B-D 1 μm의. 이 수치는 참조 (26)에서 수정되었습니다.208fig6large.jpg "대상 ="_ 빈 ">이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
DNA 또는 단백질 중급 및 하이 FRET DNA 표준 샘플, 소량 (0.25 pmol의) (그림 6B, C) 또는 FRET 쌍으로 이중 표지 KF (Alexa647 / CY3B의 형광의 5 pmol의 그림에 대한 생체 내에서 smFRET을 준수합니다 6D는) E.에 electroporation된다 대장균. 이러한 농도는 추적 및 단일 분자 모니터링 퍼즐, 직접 현지화를 허용, 몇 (N = 1-10) 표지 분자와로드 많은 세포로 이어졌다. 다른 사람이 움직이지 나타나거나 천천히 (보충 영화 3) 확산 반면 일부 분자는 자유롭게 확산. 1 ~ 30 초 동안 마지막과 smFRET의 특징을 보여 고정 이중 표지 생체 분자 (그림 6, 중)의 Timetraces : 예를 들어 수용체 표백시 기증자 및 수용체 형광 anticorrelated 변경 (, t는 ~ 16 초, 그림 6B, 중간 ), 단일 다음(예를 들어, t ~ 19 초, 그림 6B) - 스텝 기증자 표백. 이러한 timetraces (그림 6, 오른쪽)의 시험 관내 (in vitro) 연구 26,31,32 년에 출판과 매우 잘 일치에 평균 값의 결과에서 생성 FRET 분포. 이러한 결과는 내면화 된 DNA와 단백질의 정량 smFRET 연구에 대한 기능을 설정하고 (T7의 RNAP 국제화 실험에서 지원) 단백질이 전기 및 국제화에 자신의 무결성과 구조를 유지하는 것이 좋습니다.
보충 영화 1. 세포 생존 왼쪽 : 백색광 이미지. 오른쪽 : 형광 이미지. 애니메이션 GIF 10 pmol의의 Atto647 표지 DNA로드 박테리아의 전기 (1.8 kV의 / cm) 후 분열을 보여주는 애니메이션. 형광의 감소는 전반적인 명백 서로 부분적 중에 발생 광표백에 또한 세포 분열시 표지 된 DNA의 희석으로 인해이고측정.
보충 영화 2 : 세포 기반 광표백 연구 A.. (> 100 Atto647N 표지 DNA 분자를 포함하는) 과도하게로드 셀의 대표적인 예. 왼쪽 위, 관심 (빨간색 사각형)의 셀의 백색광 이미지. 오른쪽 상단, 몇 분에 걸쳐 형광 붕괴를 보여주는로드 세포의 영화. 관심 셀의 전체 형광 강도 감쇠의 아래에, 시간 추적. 유기 형광 물질은 수명을 FPS보다 높은 크기의이 주문 (여기에, ~ Atto647N 41 초) photobleaching에 나타낼 수있다. 10 미만 표지 분자 (이 경우에는 3)로드 셀 B. 대표적인 예. 패널 A. 바닥, 단일 단계 표백을 보여주는 및 / 또는 단일 유기 형광에 해당 깜박 관심있는 셀의 전체 형광 강도의 시간 추적과 동일한 최고. 이 단계의 평균 높이가 ~ 여기 (단일 분자 단위 강도에 해당AU (12))은 셀 당 분자의 내재화 초기 수를 추정하기 위해 사용된다. 300 μW 전력에서 연속 적색 레이저 여기에서 영화와 프레임 당 100 밀리 초.
보충 영화 3 :. 나는 N 단일 분자 FRET 최고 생체 내 : 세포는 지속적으로 nTIRF 조명 아래 프레임 당 50 밀리에서 모니터 0.25 pmol의 (그림 6C에서와 같이) 고 FRET DNA는 1 mW의 녹색 (532 ㎚) 레이저를 사용하여로드. 각 프레임은 각 채널의 그린 / 레드 (FRET) 형광 오버레이입니다. 확산 및 움직이지 빨간색 (그대로)과 녹색 (단일 활성 라벨) DNA 분자는 관찰 할 수있다. 아래 : 노란색 원의 분자에 해당하는 시간 추적. FRET 효율, 기증자 방출 강도 및 수용체 방출 강도는 각각, 녹색, 파란색과 빨간색으로 표시됩니다. 안티 상관 수용체 백화 현상 (빨간색 - 투 - 녹색 전환) 단일 분자 FRET의 서명에 해당합니다.
토론
많은 매개 변수는 셀 전기 및 데이터 수집 관심 생물학적 시스템 및 실험 (셀 레벨 또는 단일 분자 분석)의 정확한 성질에 따라 동안 변화 될 수있다. 세균 내로 DNA를 electroporating 때 예를 들어, 표지 된 dsDNA 단편의 0.25-5 pmol의이 (미리 photobleaching에 대한 필요없이, 즉.) 직접 단일 분자 검출을 허용하는 내재화 낮은 효율에 이르게. 5 pmol의 dsDNA보다도 무겁게 단일 세포 - 세포 분석을위한 체계가 더 적합로드되는 경향이있다. 표지 된 DNA는 모든 이전에 겔 - 정제 된 자유 염료의 흔적을 제거하기 위해 있어야 DNA 스톡 용액으로부터 (형광 비 반응). 게다가, 특히 smFRET 실험에 대한 DNA 저하, 잠재적 인 문제, 부 자연스러운 핵산, 또는 머리 핀의 자형 루프로 엑소 뉴 클레아 접근 말단을 보호 모티브의 DNA를 사용하여 해결할 수 있습니다.
또 다른 adjustabl일렉트로 목록 매개 변수는 E 일렉트로 동안인가 전계 강도이다. 낮은 전계 강도 (~ 1 kV의 / cm)는 단일 분자 연구에 적합한 낮은 부하 효율로 이어질 것입니다. (1.8 kV로 / cm까지) 더 높은 전기장 세기가 적재 효율을 증가시킬 것이다; 그러나, 전기 천공 후 전기장 세기 및 세포 생존율 사이의 역 상관 관계가있다 (도 4 참조). 참고로, 박테리아, 효모 및 전기 천공에 사용되는 통상의 전계 강도는 ~ 1.5 kV로 / cm이다. 시정 방울 즉시 임의의 아킹 현상이 발생 큐벳으로 보낸이 붕괴의 길이를 나타내는 시간 상수는, 따를 편리한 파라미터이다. 정상 설정에서 시간 상수는 4 밀리 초보다 커야한다; 낮은 값은 낮은 부하 효율 또는 비로드 손상된 세포로 이어질 것입니다. 대부분의 electroporators 모두 조정 수정할 수 있습니다 (예 : "펄스 절단"또는 "펄스 모양"등의) 자유의 다른 학위를 제공셀 로딩과 생존. 그러나 우리는 유사한 절차 또한 멤브레인 (단일 지질 이중층) 실제로는 덜 복잡하고 전기부터 이미 세포와 함께 사용되어 있기 때문에 적절한 electroporator 설정을 사용하여 포유 동물 세포에 표지 된 생체 분자의 내면화를 허용해야합니다, 모두 박테리아와 효모에이 방법을 적용 (21).
표지 단백질을 내면화 할 때, 모두 무료 염료 표지 단백질 원액 이전 전기에서 제거해야합니다. 자유 염료 분자가, 그들의 더 작은 크기에, 관심있는 단백질의 내재화를 통해 우선적으로, 그리고 (자신의 확산에도 불구하고 예상 빠른) 데이타 분석에서 구별하는 것이 어려울 수있다. 유기적 표지 단백질의 샘플에 대한 설명서는, 전기 천공에 적합하도록, 자유 염료 잔량이 2 % 미만이어야 22 (SDS-PAGE의 형광 주사를 사용하여 검출). 이 프로세스는 특히 중요일부 분자는 일렉트로 세균이나 효모의 외막에 충실 할 수있다. 이러한 관점에서, 음성 대조군 샘플 (임의 형광 표지 된 생체 분자와 함께 배양이나 전기 천공되지 않은 세포도 2) 빈 셀의 자기 형광도 수준 이상적으로 낮은 전기 천공 세포보다 분명히 낮은 셀당 형광 강도를 표시한다.
dsDNA와 마찬가지로, 표지 단백질의 내재화 효율 일렉트로 이전 세포에 첨가 된 생체 분자의 양에 연결된다. 그러나, 이러한 크기 및 전하와 같은 다른 파라미터들은, 내재화의 역할을한다. 작은 단백질은 더 큰 단백질 반면 (그림 5) (26) (98 kDa의까지) 성공적으로 내면화 될 수 있지만, 낮은 효율, 높은 국제화 효율을 나타낸다. 단백질 isolelectric 점, 세포막 전위와의 상호 작용 또는 다른 물리 화학적 및 파라미터전기 동안 영향 셀 로딩. 그 결과, 사용자는 표지 단백질 (> 50 μM)의 높은 초기 농도가 성공적으로 로딩을위한 최고의 기회를 줄 것이다 것을 알고, 자신의 시스템에 대한 실험을 최적화해야합니다. 전기도 교란 및 세포 (중 레이블 또는 레이블)에 단백질 등의 생체 분자를 도입하여 세포의 기능을 분석 할 수있는 새로운 도구를 제공합니다. 본 T7 RNA 폴리머 라제의 실험 (도 5C) 우리는 전기를 이용하여 생체 내에서 유전자 발현을 변경 생체 분자를 도입 할 수있다 등의 실험 예.
단일 분자 형광 실험 수행 때 커버 슬립면 (~ 100 ㎚) 상기 얇은 섹션 내 흥분 만 형광 의해 최상의 신호 대 잡음비를 제공하는 것처럼, TIR 조명은 일반적으로 다른 조명 모드를 통해 선호된다. 그러나 살아있는 미생물의 내부 확산 영상 표시 생체 분자는 다시 수첩 깊은 조도 (최대 E. 콜라이 0.8 ㎛이다). 높은 신호 대 잡음비를 유지하면서 깊은 조명은, 힐로 모드에서 달성된다. 한편, 넓은 시야의 촬상은 사용자가 높은 레이저 파워와 전체로드 셀 photobleaching에 및 생산 단위의 강도에 의해 초기 셀 형광 강도로 나누어 내재화 분자의 수를 예상된다 단계적 광표백 분석에 특히 중요 단일 분자 (단일 광표백 단계, 그림 3)에 의해. 그 궤적이 셀 전체 볼륨을 커버해도 위드 촬상 또한 관심 분자 확산 지역화하기 위하여 장기간 추적 분자 요구된다.
이 프로토콜에서, 우리는 전기, 세포에서 핵산을 전달하기위한 생물학 및 생화학의 표준 기법은 다양한 세포 유형에서 형광 생체 분자를 전달하기위한 간단한 방법을 구성하는 방법을 제시한다. 목소설, 높은 처리량 기술은 그 나라의 환경 표지 분자를 관찰 할 수있는 독특한 도구를 제공합니다. 형광 물질은 넓은 파장 범위를 포함 표지 또한 생체 분자에있어서, 이러한 비 천연 뉴클레오타이드 및 아미노산, 금속 킬레이트 제, 가교제, 및 caging 기 등 많은 화학 기, 변성 분자를 제공 할 수있는 전기. 관심있는 생물학적 시스템 세포 발달에 필수적이지 않으면 내재화 후 관찰 된 단백질은 세포 내 단백질 풀의 모든 (또는 대부분의) 표현을 보장, 표적 단백질을 코딩하는 유전자는 또한 삭제 될 수있다 (또는 노크 다운) . 본질적으로, 전기는 "이식"살아있는 세포로 체외 bioconjugates의 유연성 때문에 합성 생물학, 시스템 생물학에 노력하고, 생체 내 단일 분자 검출을 혜택을 누릴 수 있습니다.
공개
The authors have nothing to disclose.
감사의 말
We thank Stephan Uphoff for discussions.
R.C. was supported by Linacre College, Oxford University. A.P. was supported by the German Academic Exchange Service (DAAD), the German National Academic Foundation and EPSRC. M.S. was supported by the Wellcome Trust. A.N.K. was supported by a UK BBSRC grant (BB/H01795X/1), and a European Research Council Starter grant (261227).
자료
| Name | Company | Catalog Number | Comments |
| ElectroMax DH5-alpha Comptent cells | Invitrogen | 11319-019 | or any other commercial or lab-mage electrocompetant bacteria or yeast. |
| EZ Rich Defined Madia | Teknova | M2105 | low fluorescence rich media |
| MicroPulser Electroporation Apparatus | Biorad | 165-2100 | or any classical electroporator for microorganism transformation |
| Certified Molecular Biology agarose | Biorad | 161-3100 | low fluorescence agarose for agarose pad |
| Microscope coverslips No 1.5 thickness | Menzel | BB024060SC | remove background particles by heating slides in furnace at 500 °C for 1h |
| Single-molecule fluorescence microscope | Home-built | described in REFs | |
| Localization software | Custom-written, available online | MATLAB and C++ software package that can be adapted for localization analysis. | |
| Tracking software | Available online | MATLAB implementation by Blair and Dufresne. |
참고문헌
- Tsien, R. Y. The green fluorescent protein. Annu Rev Biochem. 67, 509-544 (1998).
- Leake, M. C., et al. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature. 443, 355-358 (2006).
- Taniguchi, Y., Kawakami, M. Application of HaloTag protein to covalent immobilization of recombinant proteins for single molecule force spectroscopy. Langmuir. 26, 10433-10436 (2010).
- Xie, X. S., Choi, P. J., Li, G. W., Lee, N. K., Lia, G. Single-molecule approach to molecular biology in living bacterial cells. Annual review of biophysics. 37, 417-444 (2008).
- Lee, J. H., et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 343, 1360-1363 (2014).
- Miesenbock, G., De Angelis, D. A., Rothman, J. E. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 394, 192-195 (1998).
- Sauer, M. Localization microscopy coming of age: from concepts to biological impact. J Cell Sci. 126, 3505-3513 (2013).
- Dempsey, G. T., Vaughan, J. C., Hao Chen, K., Zhuang, X. Evaluation of fluorophores for optimal performance in localizationbased super-resolution imaging. Nat Meth. 8, 1027-1041 (2011).
- Shaner, N. C., Steinbach, P. A., Tsien, R. Y. A guide to choosing fluorescent proteins. Nat Meth. 2, 905-909 (2005).
- Landgraf, D., Okumus, B., Chien, P., Baker, T. A., Paulsson, J. Segregation of molecules at cell division reveals native protein localization. Nat. Methods. 9, 480-482 (2012).
- Jaitin, D. A., et al. Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science. 343, 776-779 (2014).
- Aldridge, S., et al. AHT-ChIP-seq: a completely automated robotic protocol for high-throughput chromatin immunoprecipitation. Genome Biol. 14, R124 (2013).
- Keppler, A., et al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 21, 86-89 (2003).
- Wombacher, R., et al. Live-cell super-resolution imaging with trimethoprim conjugates. Nat. Methods. 7, 717-719 (2010).
- Zhang, Z., et al. A new strategy for the site-specific modification of proteins in vivo. Biochemistry. 42, 6735-6746 (2003).
- McNeil, P. L., Murphy, R. F., Lanni, F., Taylor, D. L. A method for incorporating macromolecules into adherent cells. J Cell Biol. 98, 1556-1564 (1984).
- Clarke, M. S., McNeil, P. L. Syringe loading introduces macromolecules into living mammalian cell cytosol. J Cell Sci. 102, 533-541 (1992).
- Sakon, J. J., Weninger, K. R. Detecting the conformation of individual proteins in live cells. Nat. Methods. 7, 203-205 (2010).
- Taylor, L. S. Electromagnetic syringe. IEEE Trans. Biomed. Eng. 25, 303-304 (1978).
- Dower, W. J., Miller, J. F., Ragsdale, C. W. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 16, 6127-6145 (1988).
- Neumann, E., Schaefer-Ridder, M., Wang, Y., Hofschneider, P. H. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J. 1, 841-845 (1982).
- Sustarsic, M., et al. Optimized delivery of fluorescently labeled proteins in live bacteria using electroporation. Histochem Cell Biol. , (2014).
- Tokunaga, M., Imamoto, N., Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods. 5, 159-161 (2008).
- Sinha, A., et al. A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature. 000, 1-5 (2014).
- English, B. P., et al. Single-molecule investigations of the stringent response machinery in living bacterial cells. Proc Natl Acad Sci U S A. 108, E365-E373 (2011).
- Crawford, R., et al. Long-lived intracellular single-molecule fluorescence using electroporated molecules. Biophys J. 105, 2439-2450 (2013).
- Uphoff, S., Reyes-Lamothe, R., Garza de Leon, F., Sherratt, D. J., Kapanidis, A. N. Single-molecule DNA repair in live bacteria. Proc Natl Acad Sci U S A. 110, 8063-8068 (2013).
- Uphoff, S., Sherratt, D. J., Kapanidis, A. N. Visualizing Protein-DNA Interactions in Live Bacterial Cells Using Photoactivated Single-molecule Tracking. J Vis Exp. , (2014).
- Hohlbein, J., Gryte, K., Heilemann, M., Kapanidis, A. N. Surfing on a new wave of single-molecule fluorescence methods. Phys Biol. 7, 031001 (2010).
- Xie, X. S., Yu, J., Yang, W. Y. Perspective - Living cells as test tubes. Science. 312, 228-230 (2006).
- Santoso, Y., et al. Conformational transitions in DNA polymerase I revealed by single-molecule FRET. Proc Natl Acad Sci U S A. 107, 715-720 (2010).
- Hohlbein, J., et al. Conformational landscapes of DNA polymerase I and mutator derivatives establish fidelity checkpoints for nucleotide insertion. Nature communications. 4, 2131 (2013).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유