
Method Article
Internalisierung und Beobachtung von Fluoreszenz Biomoleküle in lebenden Mikroorganismen durch Elektroporation
In diesem Artikel
Zusammenfassung
Studies of biomolecules in vivo are crucial for understanding molecular function in a biological context. Here we describe a novel method allowing the internalization of fluorescent biomolecules, such as DNA or proteins, into living microorganisms. Analysis of in vivo data recorded by fluorescence microscopy is also presented and discussed.
Zusammenfassung
The ability to study biomolecules in vivo is crucial for understanding their function in a biological context. One powerful approach involves fusing molecules of interest to fluorescent proteins such as GFP to study their expression, localization and function. However, GFP and its derivatives are significantly larger and less photostable than organic fluorophores generally used for in vitro experiments, and this can limit the scope of investigation.
We recently introduced a straightforward, versatile and high-throughput method based on electroporation, allowing the internalization of biomolecules labeled with organic fluorophores into living microorganisms. Here we describe how to use electroporation to internalize labeled DNA fragments or proteins into Escherichia coli and Saccharomyces cerevisiæ, how to quantify the number of internalized molecules using fluorescence microscopy, and how to quantify the viability of electroporated cells. Data can be acquired at the single-cell or single-molecule level using fluorescence or FRET. The possibility of internalizing non-labeled molecules that trigger a physiological observable response in vivo is also presented. Finally, strategies of optimization of the protocol for specific biological systems are discussed.
Einleitung
Die meisten Fluoreszenzuntersuchungen in lebenden Zellen hängen von Proteinfusionen mit fluoreszierenden Proteinen (FRP), wie GFP 1. Diese fluoreszierenden Tags erlauben Untersuchungen der Kopienzahl, Diffusionsmuster oder Lokalisierung von Proteinen in Prozesse wie die Genexpression oder Membrantransport 7.2 beteiligt. Rahmenprogramme bieten eine hohe Kennzeichnungs Spezifität, einfach implementiert werden und sind in einem großen Bestand an Varianten mit unterschiedlichen photophysikalischen und chemischen Eigenschaften 1 zur Verfügung. Allerdings bleiben organische Fluorophore die erste Wahl für die in vitro durch ihre größere Fotoexperimente (bis zu 100-mal stabiler als RP) 8,9, geringe Größe (bis zu 100-fach geringeres Volumen als RP) und die einfache intramolekulare Kennzeichnung (vor allem durch die Verwendung von Cystein-Resten). Alle diese Faktoren sind besonders wichtig für die Einzelmolekül-Fluoreszenz und FRET untersucht 10.
Mehrere Internalisierung Methoden combining die Vorteile von Bio-Kennzeichnung und in-vivo-Nachweis haben sich im letzten Jahrzehnt eingeführt wurden; aber solche Methoden entweder beschäftigen relativ große Polypeptide Tags (zB TMP, Halogen oder 20 kDa SNAP Tags) von 11 bis 14, erfordern die Verwendung von unnatürlichen Aminosäuren 15 oder an großen, Single-Membran eukaryotischen Zellen beschränkt (z. , Scrape Loading, Spritze Belastung, Mikroinjektion) 16-19.
Dieses Protokoll beschreibt eine neue, einfache und Hochdurchsatz-Verfahren Verinnerlichung, dass Paare die Vorteile organischer Fluorophore mit in vivo Beobachtung. Um diese Technik zu entwickeln, passten wir die Elektroporation Verfahren häufig verwendet, um Zellen mit Plasmid DNA 20,21 um Mikroorganismen, wie E. laden Transformations coli oder S. cerevisiae mit organisch markierten Biomolekülen. Das Protokoll besteht aus 4 Schritten: Inkubation von Zellen mit markierten BiomoleküleElektroporation, Zellgewinnung und Zellwasch auf nicht verinnerlicht Biomolekülen zu entfernen. Hier präsentieren wir dieses Elektroporation Protokoll, sowie die Cell Imaging und Datenanalyseverfahren, um zellbasierte und Einzelmolekül-Fluoreszenz zu studieren und FRET-Signale.
Elektroporation beruht auf Entladen eines elektrischen Hochspannungsfeld in einer niedrigen Ionenstärke Zellsuspension transienter Membranporen durch die Biomoleküle Zellen eindringen (Figur 1) 20,21 bilden. Genau wie bei Transformation von Bakterien oder Hefe, die mit Plasmid-DNA, haben Zellen vor der Elektroporation ihre electrocompetency zu gewährleisten, hergestellt werden. Dieses Verfahren, das aus mehreren Waschschritten mit Wasser, erhöht die Durchlässigkeit der Membran und verringert die Ionenstärke der Zelllösung, um Überschlagen im Elektroporationsküvette vermeiden. In diesem Protokoll können Zellen, wie unten beschrieben (siehe PROTOCOL: 1,1), hergestellt werden oder von kommerziellen Anbieter gekaufts.

Abbildung 1: Schematische Darstellung der Internalisierung Protokoll Von links nach rechts:. Es werden einige Mikroliter der markierten Biomolekülen auf das Aliquot elektrokompetenter Zellen (doppelt markierte DNA-Fragmente und Bakterien in diesem Beispiel); Inkubation 1 bis 10 Minuten auf Eis und Transfer in ein vorgekühltes Elektroporationsküvette; elektroporieren und fügen 0,5-1 ml Vollmedium zu den Zellen unmittelbar nach; Inkubieren bei 37 ° C (oder der durch den Organismus erforderliche Temperatur, beispielsweise 29 ° C für Hefe) die Zellen zu gewinnen lassen; 5 Waschschritte durchführen, um überschüssige nicht internalisierte markierten Moleküle zu entfernen; resuspendieren endgültige Pellet in 100-200 ul PBS-Puffer und Pipette 10 ul auf einem Agarose-Pad; decken Sie das Pad mit einem Deckglas reinigen und Bild auf einem Fluoreszenzmikroskop (in Weitfeld-Modus oder HILO-Modus).
Elektrokompetente Zellen mit den markierten Biomoleküle nur vor der Elektroporation, die Verwendung von Standard electroporators in den meisten biochemischen Laboratorien durchgeführt werden kann, inkubiert. Unmittelbar nach der Elektroporation werden die Zellen in einem reichen Medium, das die Wiederherstellung vor der Wäsche (1) inkubiert. Der Überschuß an nicht internalisierte markierten Biomolekülen wird zuerst durch Waschen in einem Puffer, der eine ziemlich hohe Salzkonzentration und eine Reinigungsmittel entfernen (siehe PROTOKOLL: 3,3). Die Anwesenheit von Salz stört unspezifische elektrostatische Wechselwirkungen von nicht internalisierte markierten Biomolekülen, die sonst auf der äußeren Membran haften gebildet. Auch die Gegenwart von Detergens in der Waschpuffer stört unspezifische hydrophobe Wechselwirkungen.
Während DNA-Internalisierung ist unkompliziert (Abbildung 2), als Internalisierungsfaktor markierten Proteine unter Verwendung von Elektroporation die ergriffen werden müssen Vorsichtsmaßnahmen. Erstens könnte die Aktie Probe organisch markierten Proteins noch eine geringe Prozentsatz an freiem Farbstoff. Freien Farbstoffmoleküle sind viel kleiner als Proteine und könnte daher bevorzugt internalisiert werden. Um sicherzustellen, dass die überwiegende Mehrheit der beobachteten internalisiert fluoreszierenden Molekülen zu dem Protein von Interesse entspricht, sollte die anfängliche Proteinprobe weniger als ~ 2% enthalten freie Farbstoff (5) 22. Der Überschuß an nicht internalisierte markierten Proteine können auch an der äußeren Zellmembran nach der Elektroporation kleben; Dieses Phänomen ist proteinspezifische und muss für jede neue Protein kontrolliert werden. Wir schlagen vor, mehrere Optionen, die die Entfernung von nicht internalisierte Proteine aus der geladenen Zellprobe zu ermöglichen (siehe PROTOKOLL: 3.3.3).
Schließlich werden die Zellen in einem kleinen Volumen von Phosphatpuffer pipettiert und auf einem Agarose-Pad, wodurch die Abbildung auf einem Fluoreszenzmikroskop. Immobilisierung auf Agarose-Pads ist ein simple und effiziente Art der Bildgebung von Zellen auf einem Deckglas ohne Schädigung ihrer Integrität. Die Unterlage sollte eine niedrige Fluoreszenzkulturmedium enthalten.
Cell Imaging kann entweder im Weitfeld, Totalreflexions-Fluoreszenz (TIRF) oder über HILO (stark geneigt und laminierte optische Sheet) Mikroskopie durchgeführt werden. Im HILO Konfiguration dringt der Laserstrahl tiefer in die Probe als im TIRF, noch nicht die gesamte Probe wie für Weitfeld zu beleuchten, so dass ein größeres Signal-zu-Rausch-Verhältnisses 23. In Abhängigkeit von der Laserleistung und Zeitauflösung verwendet wird, kann verinnerlicht Biomoleküle angerechnet werden (mit Hilfe schrittweise Photobleaching-Analyse, Abbildung 3), lokalisiert oder verfolgt 24-28. Internalisierung von doppelt markierten Konstrukte mit einem FRET-Paar von Fluorophoren erlaubt die Quantifizierung von FRET sowohl auf Einzelzelle oder der Einzelmolekülebenen (Abbildung 6).
Verschiedene Parameter können variiert werden,abhängig von der gewünschten Leistung und dem biologischen System untersucht. Erstens kann die Menge des Materials pro Zelle internalisiert durch Verändern der Konzentration von markierten Biomolekülen zugegeben, um die Zellen vor der Elektroporation (2) eingestellt werden. Elektroporation Feldstärke wird auch beeinflussen sowohl die Ladeeffizienz und die Lebensfähigkeit der Zellen; wie erwartet, während die Ladeeffizienz erhöht sich mit zunehmender Feldstärke, die Lebensfähigkeit der elektroporierten Zellen ab (4A). Beide Parameter können durch Aufzeichnung der Prozentsatz der belasteten und sich teilenden Zellen nach der Elektroporation zu quantifizieren. Diese Lebensfähigkeitstest in Verbindung mit Fluoreszenz-Bildgebung überprüft auch die Beobachtung von verinnerlicht Biomoleküle in lebenden Zellen und ermöglichen eine kontinuierliche Beobachtung über mehrere Generationen (4B).
Zusammenfassend lässt dieses Protokoll die Internalisierung von fluoreszenzmarkierten DNA-und Proteinmoleküle inE. coli oder S. cerevisiae 26. Einzelmoleküle mit organischen Fluorophoren markiert mit hoher Raum-Zeit-Auflösung für Zeitskalen eine Größenordnung länger als RP verfolgt werden. Schließlich ist dieses Verfahren mit Weitfeld, TIRF und konfokale Detektion sowie gepulste Anregung Systeme wie ALEX (Wechsellaseranregung 28,29) kompatibel.
Protokoll
1. Zellvorbereitung
- Vorbereitung der im Labor hergestellten elektro Bakterien
- Vorbereiten einer 5-10 ml über Nacht-Vorkultur von einer Einzelkolonie des E. coli-Stamm von Interesse in einer Niederfluoreszenzmedium wie M9 oder EZ reich definiertem Medium.
- Am Morgen impfen einen neuen 400 ml Kultur mit der Vorkultur über Nacht, so dass OD 600 nm bei 0,02 beginnt. Zu den 400 ml Niederfluoreszenzmedium 2,5 ml 1 M MgSO 4 und 2,5 ml von 1 M MgCl 2.
- Wachsen bei 37 ° C und 250 Umdrehungen pro Minute, bis die OD 600 nm 0,4 bis 0,6 erreicht.
- Stoppen das Wachstum durch Abkühlen der Kultur in einem Eis-Wasserbad für 10-15 Minuten.
Hinweis: Von nun an führen alle Schritte bei 4 ° C (auf Eis). - Zentrifugieren Sie die Kultur 15 min bei 1000 x g. Überstand verwerfen und Zellpellet in 250 ml gekühlt und steril destilliertem H 2 O
- Wiederholen Sie die Zentrifugation und Resuspension Schritte twice, Verringern des Volumens von Wasser auf 100 ml und dann 50 ml.
- Zentrifugieren Sie die Kultur 10 min bei 1000 x g. Überstand verwerfen und Zellpellet in 25 ml gekühlte und sterilem destilliertem H 2 O + 10% Glycerin.
- Wiederholen der Zentrifugation und Resuspension Schritte dreimal Verringern des Volumens von 10% Glycerin-Lösung auf 10 ml, 5 ml, und schließlich auf 500 & mgr; l.
- Aliquot der Zellen in Aliquots von 20 ul jeder, flash-freeze in flüssigem Stickstoff und bei -80 ° C.
- Kommerzielle elektroBakterienZellen
- Verdünnen einer kommerziellen Zelle aliquoten 1: 1 mit sterilem destilliertem Wasser gekühlt. Stellen Sie 20 ul Aliquots und bei -80 ° C.
- Vorbereitung elektro Hefe
Anmerkung: Elektrokompetente S. cerevisiae-Zellen werden vor jedem Experiment Elektroporation vorbereitet und kann bei -80 ° C für E. gespeichert werden coli.- Um zu beginnen, zu impfen 50 ml YPD-Medium mit einer Einzelkolonie des gewünschten Stamm von Interesse.
- Inkubiere bei 30 ° C und 250 rpm bis zu einer OD 600 nm 0,6 bis 0,8 erreicht.
- Zentrifuge Zellen bei 1000 × g für 5 min bei 4 ° C.
- Das Pellet in 25 ml gekühlte und sterilem destilliertem H 2 O.
- Wiederholen der Waschschritte resuspendiert zweimal in 25 ml Wasser und Resuspendieren zweimal in 2 ml einer gekühlten Lösung von Sorbit in 1 M.
- Resuspendieren der Zellen in 250 ul 1 M Sorbit und spalten die Zellen in 50 ul Aliquots.
2. Agarose-Pad Vorbereitung
- Um die Hintergrundleuchtstoffpartikel zu entfernen, brennen ein Deckglas in einem Ofen bei 500 ° C für 1 Stunde. "Clean verbrannt" Deck können über Wochen bei Raumtemperatur in Aluminiumfolie abgedeckt gelagert werden.
Hinweis: Andere übliche Reinigungsverfahren wie Plasmareinigung oder Piranha-Lösung verwendet werden können, solange die HinterFluoreszenz der gereinigten Objektträger bleibt quasi null. - Vorbereiten eines Niederfluoreszenz Agaroselösung durch Schmelzen in einem Mikrowellenofen 2% Agarose - destilliertes Wasser-Lösung (70 ° C). Sofort im 500 ul des klaren 2% igen Lösung zu 500 ul 2X geringe Fluoreszenz Kulturmedium und vorsichtig mischen.
- Bevor es auf und härten kühlt, zeitnah zu pipettieren dieses Agarose - Mediumlösung auf einen Mikroskopdeckglas (No 1,5 Dicke), um ein Polster von ca. 2 cm Durchmesser und einigen Millimetern Höhe zu bilden. Vermeiden Sie Luftblasen und Pop sie mit Pipettenspitzen, wenn nötig.
- Glätten Sie das Pad mit einem zweiten "verbrannt" Deckglas (No 1,5 Dicke, siehe Abbildung 1).
Hinweis: Diese obere Deckglas hilft eine ebene homogene Pad und zum Schutz vor Staub und Trocknen während Zellen sind in Vorbereitung. Minimalmedium, wie M9 oder Vollmedium wie EZ Reichhaltiges definiertem Medium wurden für ihre geringe Fluoreszenz getestet.
3. ElectroporatIon
- Incubation
- Fügen Sie bis zu 5 ul der markierten Moleküle in einem Niedrigsalzpuffers (<50 mM Salz) zu einem einzelnen Aliquot kompetenter Zellen (20 ul Bakterien oder 50 ul Hefe) gespeichert und Inkubation 10 min auf Eis.
Anmerkung: Die Konzentration der fluoreszenzmarkierten Moleküle in den Stammlösungen und damit die Menge des markierten Moleküle an die Zelle vor der Elektroporation direkt mit Beladungseffizienz korreliert hinzugefügt (Abbildung 3, und Diskussion). Da einige Proteine mit niedrigem Salz Zustand weniger verträglich, kann die Salzkonzentration in der Pufferspeicher erhöht werden, aber das Volumen der markierten Moleküle an die Zellen vor der Elektroporation hinzugefügt muß dann abgesenkt werden. - Überführt die Mischung von Zellen und markierten Biomolekülen in eine vorgekühlte Elektroporationsküvette (0,1 und 0,2 cm Abstand für Bakterien und Hefe, jeweils). Klopfen Sie leicht die Küvette auf der Bank, um mögliche Luftblasen aus der Lösung zu entfernen.
- Legen Sie die cuvette in die Elektroporator und Anwendung einer elektrischen Hochspannungsimpuls an die Lösung (0,9 bis 1,8 kV / cm, siehe Diskussion für mehr Details über die Wahl Spannung). Ein solcher Impuls bildet transienten Poren in Zellmembranen kann markierten Biomolekülen in die Zellen zu diffundieren.
- Überprüfen Sie, dass die Zeitkonstante auf der Elektroporator angezeigt ist zwischen 4-6 ms. Niedrigere Zeitkonstanten sind häufig durch zu hohe Salzkonzentration und / oder der Anwesenheit von Blasen in der Küvette und wird mit sehr geringen oder keiner Belastung der Zellen führen.
- Fügen Sie bis zu 5 ul der markierten Moleküle in einem Niedrigsalzpuffers (<50 mM Salz) zu einem einzelnen Aliquot kompetenter Zellen (20 ul Bakterien oder 50 ul Hefe) gespeichert und Inkubation 10 min auf Eis.
- Erholung
- Unmittelbar nach der Elektroporation, fügen 500 ul Vollmedium wie SOC, EZ Reichhaltiges definiertem Medium, YPD oder einem reichen Medium zu den Zellen.
- Inkubieren der Probe bei 37 ° C für Bakterien und 29 ° C für Hefe für 2 bis 10 min. Für Viabilitätsmesswerte, wo der Benutzer den Prozentsatz der Zellen, die nach der Elektroporation und Dividieren auswerten will, verwenden eine längere Erholungszeit (bis zu 1 Stunde) als we beobachten wie Verzögerungszeiten, bevor die erste Zellteilung.
- Waschschritte
- Waschen Sie die Zellen, um alle nicht verinnerlicht Biomolekülen durch Abzentrifugieren der Zellen für 1 min bei 3300 xg und 4 ° C zu entfernen. Überstand verwerfen und die Zellen resuspendieren in 500 ul PBS.
Anmerkung: Für jede Probe vorzubereiten eine Negativkontrolle von Zellen mit der gleichen Menge an markierten Biomolekülen inkubiert aber nicht elektroporiert und gewaschen genau die gleiche Weise wie der Hauptprobe. - Wiederholen Sie die vorherigen Schritte, 3-mal.
- Im Falle von Protein Internalisierung Optimierung der Waschvorgang in Abhängigkeit von den Eigenschaften und das Verhalten des markierten Proteins von Interesse. Die folgenden Schritte sind Beispiel für mögliche Optimierungen:
- Führen die ersten 3 Waschzyklen unter Verwendung von PBS, enthaltend 100 mM NaCl und 0,005% Triton X100 auf nicht internalisierten Proteine, die an der äußeren Zellmembran 22 haften, zu entfernen, 26.
- Filtern Sie die elektroporierten Zellen mit einer 0,22 um Porendurchmesser Filter in ein 1,5 ml Mikrozentrifugenröhrchen pipettiert, die elektroporierten Zellen in den Filter eingebaut. Spin down 3 min bei 800 xg und 4 ° C. Verwerfen Sie den Durchfluss durch. Fügen Sie 500 ul neue PBS über die Zellen und drehen sie wieder wie zuvor, und wiederholen Sie diese Schritte einmal 22.
- Hinzufügen einer kleinen Menge an Protease K (10 ng in 500 ul PBS) während des ersten Waschzyklus, um die Verdauung von jedem nicht internalisierte Proteins zu ermöglichen.
- Führen die ersten 3 Waschzyklen unter Verwendung von PBS, enthaltend 100 mM NaCl und 0,005% Triton X100 auf nicht internalisierten Proteine, die an der äußeren Zellmembran 22 haften, zu entfernen, 26.
- Spin down die Zellen für 1 Minute bei 3300 xg und 4 ° C. Überstand verwerfen und die Zellen resuspendieren in 150 ul PBS.
- Verbreiten Sie die geladenen Zell Lösung auf dem Agarose-Pad, indem Sie die obere Deckglas und Verbreitung 10 ul der Zellsuspension Tröpfchen durch Tröpfchen. Ersetzen Sie eine unbenutzte sauber verbrannt Deckglas (nicht vorhanden 1,5 Dicke, passend zum Mikroskopobjektiv-Spezifikation) auf T-er Oberseite des Pads und drücken Sie ganz sanft auf der Folie.
- Schützen Sie die elektroporierten Zellen vor Licht, indem die Pads in einem undurchsichtigen Kasten, während die Abbildung verschiedener Proben.
- Waschen Sie die Zellen, um alle nicht verinnerlicht Biomolekülen durch Abzentrifugieren der Zellen für 1 min bei 3300 xg und 4 ° C zu entfernen. Überstand verwerfen und die Zellen resuspendieren in 500 ul PBS.
4. Mikroskop Datenerfassung
Hinweis: Einzellige und Einzelmolekül-Fluoreszenzmikroskopie in lebenden Mikroorganismen können auf jedem geeigneten Fluoreszenzmikroskop durchgeführt werden (custom-built oder kommerziell).
- Einstellungen
- Widefield oder HILO Beleuchtung
- Bildproben mit jedem TIRF / Einzelmolekül-Mikroskop.
Hinweis: Als Beispiel verwenden wir im Labor eine angepasste inverse Mikroskop mit TIRF-Set-up. Strahlen von einer 532-nm und einem 637-nm-Diodenlaser werden vereinigt und vor der Fokussierung auf der hinteren Brennebene der Objektiv kollimiert. Die Fluoreszenz der Probe wird durch das gleiche Objektiv gesammelt, aus dem Anregungslicht unter Verwendung eines Langpass und ein Kerbfilter getrennt und in wieder gespaltend und grünen Kanäle mit einem dichroitischen Spiegel. Die beiden Kanäle sind auf getrennte Hälften des Chips einer Elektronenvervielfachungsladungsgekoppelte Einrichtung (EM-CCD-Kamera) abgebildet. Videos werden mit der kinetischen Modus aufgenommen. Weißlichtbilder werden mit einer Weißlichtlampe und einen Kondensator mit dem Mikroskop als Beleuchtungsquelle angeschlossen erhalten. - Für allgemeine Einzelmolekülbeobachtung, stellen Sie den Beleuchtungsmodus des Mikroskops zu TIRF oder HILO 23 (siehe Diskussion für mehr Details über TIRF gegen HILO Imaging). Um eine HILO Modus auf einem TIRF Mikroskop gesetzt, leicht den Einfallswinkel des Anregungslichts zu verringern, um den Fokus etwas höher als die Deckfläche (Bild nach dem Zellinneren statt seiner unteren Membran in Kontakt mit dem Deckglas, siehe 4.5.4 ).
- Für Zell-Level-Analyse, lange Einzelmolekül-Tracking Experimente oder schrittweise Bleichen Analyse, stellen Sie den Beleuchtungsmodus des Mikroskops auf eine Weitfeld-Modus ensuring die kontinuierliche Beobachtung des gesamten Zellvolumens und damit aller internalisiert markierten Moleküle.
- Bildproben mit jedem TIRF / Einzelmolekül-Mikroskop.
- Typischerweise Verwendung Anregungsleistungen rund 0,5-3 mW (~ 50-400 W / cm 2).
Hinweis: Niederlaserleistungen sinnvoll, langlebige Fluoreszenz Beobachtung und Verfolgung (mehr als 1 Minute), während höhere Laserleistungen kann für höhere Raum-Zeit-Auflösung oder schrittweise Bleichen Analyse erforderlich zu erreichen. - Verwenden Sie Belichtungszeiten von 15 ms für die Verfolgung von Experimenten auf 100 ms für weitere allgemeine Beobachtung und Intensität Quantifizierung. Hinweis: Weitere Bildraten und Betriebsarten verwendet werden, wie stroboskopische Beleuchtung solche, insbesondere für die Untersuchung schnell diffundierenden Spezies 30.
- In der TIRF-Mikroskop, notieren Sie die Fluoreszenzkanal auf einer elektronen Multiplikation CCD (EMCCD) Kamera bei einer Vergrößerung was zu einer Pixellänge von ~ 100 nm / Pixel. Die TIRF-Setup ist zu beschreiben in mehr Details in Bezug 26.
- Widefield oder HILO Beleuchtung
- Datenerfassung
- Abschalten oder Sperrung der Laserbeleuchtung bis zum Beginn des Experiments. Schalten Sie das EMCCD Kameraverstärkung aus, um Schäden an der Kamera durch Überbelichtung zu verhindern.
- Legen Sie die Agarose-Pad-Sandwich auf den Kopf auf dem Mikroskoptisch, mit nach unten Zelle bedeckten Seite, um die Zelle in der Nähe des Ziels zu bringen. Stellen Sie den Fokus auf die Zellen im Durchlichtmikroskopie-Modus 28. Nehmen Sie ein Bild von jeder Zelle aus gesehen unter Weißlicht-Bildgebung, um die Zelle zu lokalisieren skizziert vor dem Ausschalten des weißen Lichts.
- Schützen Sie die Probe vom Labor Umgebungslicht.
- Für HILO Erregungsmodus, den Winkel des Anregungsstrahls zu jedem Maximum-Signal-zu-Rausch-Verhältnis einzustellen durch Beleuchten nur den Abschnitt der Probe in der Nähe der Deckfläche.
- Nach Hilo Ausleuchtung zu erreichen, konzentrieren den Laserstrahl in der hinteren Brennebene eines 100x NA 1.4 Ziel 28 (höhere numerische Aperturen wie 1,45 oder 1,49 sind ebenfalls geeignet). Durch Verschieben der Fokussierungslinse senkrecht zum Strahl bewegt sich der Schwerpunkt neben der Objektivmitte, so dass der Strahl aus dem Ziel mit einem Winkel.
- Stellt die Linsenposition, um das Signal-zu-Rausch-Verhältnis, intrazellulären Fluoreszenzintensität gegenüber der extrazellulären Grundsignal zu maximieren.
- Schalten Sie die Kamera Gain und starten die Datenerfassung vor dem Einschalten des Lasers.
- Während der Aufnahme Daten, erwerben ein Weißlichtbild jeder FOV vor oder nach der Aufzeichnung der Fluoreszenzdaten; dies hilft, die Zellgrenzen in den Fluoreszenzkanäle zu identifizieren.
- Für Lebensfähigkeit Schätzung
- Verwenden Sie einen Low-Fluoreszenz reichen Medium in der Agarose-Pad, damit Zellen nach der Elektroporation wachsen.
- Äquilibrieren des Mikroskops auf die optimale Temperatur für den untersuchten Mikroorganismus (37 ° C für E. coli, 29 ° C für S. cerevisiae) Mit dem Ziel Heizsystem.
- Nehmen sowohl weißes Licht und Fluoreszenzbilder alle 30 Minuten, achten Sie darauf, auf genau die gleiche Sichtfeld während der gesamten Datenaufzeichnung bleiben. Eine Verzögerung von ≈1 Stunde wird in der Regel eingehalten, bevor Zellen beginnen sich zu teilen.
- Zählen der Anzahl von verinnerlicht Biomolekülen pro Zelle
- Stellen Sie die Laserleistung zu hohen Werten (2-3 mW) und lange Belichtungszeit (100 ms).
- Stellen Sie die Beleuchtung, um die gesamte Zelle zu beleuchten Weitfeld-Modus.
- Nehmen Sie die Videos, wie in Schritt 4.2 beschrieben, und achten Sie auf weitere Bilder (50 bis 100 Frames) nach Abschluss des Fluoreszenzphotobleichen aufzuzeichnen.
5. Datenanalyse
- Allgemeine Analyse
- Analysieren Sie die aufgenommenen Bilder und Filme, die beide in weißem Licht und Fluoreszenzanregungen, mit einer Bildbearbeitungssoftware, wie beispielsweise die freie Software ImageJ.
- In ImageJ, öffnen Sie die Bilder oder Filme auf dem Mikroskop (TIF-Format) aufgezeichnet in Datei> Öffnen> Ihre Datei an.
- Um qualitativ vergleichen Fluoreszenzintensitäten auf einem Computerbildschirm, stellen Sie sicher, dass alle Fluoreszenzbilder mit den gleichen Helligkeits- und Kontrasteinstellungen in Bild angezeigt> Einstellen> Helligkeit / Kontrast. Passen Sie manuell oder automatisch die Einstellungen für ein ausgewähltes Bild, drücken Sie Taste "Set" und wählen Sie die Option "Propagieren auf alle anderen Bilder".
- Stellen Sie die Art der Informationen zu extrahieren: Analysieren> Set Messungen, und wählen Sie (mindestens) "Bereich", "Standardabweichung", "Min & Max Grauwert" und "Mittlerer Grauwert".
- Um Zellfluoreszenzintensitäten zu vergleichen, wählen Sie den Bereich s von Interesse mit dem Freihand-Wahltaste des ImageJ und extrahieren Sie die Zellintensitäten Analysieren> Messen. Die resultierende Tabelle enthält die Messwerte undkann gespeichert und / oder zu anderen Software kopiert. Die "mittlere" Wert entspricht der durchschnittlichen Intensität pro Pixel in dem ausgewählten Bereich und kann direkt zwischen den Zellen oder zwischen einer Zelle und dem Hintergrund verglichen werden.
- In einer elektroporiert Zellprobe wird ein betrachteten Zelle geladen, wenn seine mittlere Intensität pro Pixel größer als die Durchschnittsintensität pro Pixel der Negativkontrolle plus 3 Mal der Standardabweichung (Av (I geladen Zelle)> Av (I -EP) + 3 * StdDev (I -EP)).
- Erstellen Sie Falschfarbenfluoreszenz Overlay-Bilder und Filme, um die Qualität und Belastung der Proben zu bewerten.
- In ImageJ, Overlay-Bilder wie die Weißlichtbild und dem Fluoreszenzbild entsprechend demselben Sichtfeld in Bild> Farben> Merge Kanäle. Wählen Sie eine Farbe für jedes Bild (C4 (grau) für das weiße Licht, C1 (rot) für Rot-Kanal, C2 (grün für grünen Kanal ... etc.).
- Kreuze Anauf dem Überlagerungsbild, das die Fluoreszenz innerhalb der Zellgrenzen liegt (Weißlichtbild), und dass die Hintergrundfluoreszenz gering ist und homogen (keine hellen Punkte außerhalb der Zellgrenzen).
- Vor der Analyse einer großen Anzahl von Zellen, überprüfen Sie qualitativ die Bilder, die der negativen Probe werden ähnlich wie die leere Zelle Bildern und Anzeigen viel niedriger Intensitäten als die elektroporierten Zellen.
- Für Lebensfähigkeit Experimenten zählen manuell die Prozentsätze der Aufteilung, nicht teilende, aber sichtbar intakt (identischen) und beschädigt (toten) Zellen, die aus dem gleichen Blickfeld im Laufe der Zeit wächst (siehe 4.2.6).
- Bewerten Sie die Überlebensfähigkeit von mindestens 200 Zellen pro Probe (elektroporiert, Negativkontrolle und leere Zellen), um genügend Statistiken zu sammeln.
- Analysieren Sie die aufgenommenen Bilder und Filme, die beide in weißem Licht und Fluoreszenzanregungen, mit einer Bildbearbeitungssoftware, wie beispielsweise die freie Software ImageJ.
- Zellbasierte Analyse
- Zählen der Anzahl von verinnerlicht Biomolekülen pro Zelle durch schrittweises Bleichen Analyse
- Segment Zellen durch selecting den Bereich von Interesse mit dem Freihand-Wahltaste des ImageJ und zeichnen Sie eine Form der Zelle (entspricht Membranzelle) umgebenden genau.
- Extrahieren Sie die Zelle Intensitäten über die Zeit im Bild> Stapel> Plot Z-Achse Profil. Der resultierende Graph stellt die durchschnittliche Intensität pro Pixel für den Bereich innerhalb der Zellgrenze gegenüber jeder Filmrahmen, was zu einem Ausbleichen Kurve für diese bestimmte Zelle. Es enthält eine erste exponentielle Abnahme der Zellstärke Erreichen eines unteren Asymptote (Hintergrundfluoreszenz). Die Messwerte und können durch Klicken auf "Speichern" oder "Kopieren" gespeichert werden und / oder in andere Software kopiert.
- Kopieren Sie die Bleich Werte in eine Tabelle Spalte (I roh).
- Berechnen Sie die durchschnittliche Autofluoreszenz pro verbleibenden Pixel after photo (I auto) durch Mittelung der in den letzten 50 bis 100 Bildern (untere Asymptote) erhielt ich Rohwerte.
- Subtract den durchschnittlichen Autofluoreszenz pro Pixel after photo für diese Zelle von der ersten Photobleaching Kurve verbleibenden: Ich Bleichen = I raw - ich auto.
- Verwenden Basis subtrahiert Photobleichens timetraces (I Bleiche gegenRahmen) zeigt weniger als 10 Schritte quantisiert, um die mittlere Schrittgröße (unitären Fluorophor Intensität) beurteilen aufgrund der Bleiche von einem einzigen Fluorophor 26.
- Bewerten der Anzahl der Moleküle pro Zelle internalisiert durch Dividieren der ersten Basis subtrahiert Zellstärke (I Bleichen bei t = 0) durch die einheitliche Fluorophor Intensität.
- Einzellige FRET Effizienzen
- Sie messen die mittlere Zellen Intensität pro Pixel sowohl in der Donor- und Akzeptor-Emissionskanal (Anregung des Donors) und innerhalb der Zellgrenze für jeden Kanal, wie in 5.1.1.4 erklären.
- Messen Sie die durchschnittliche Pixelintensität für den Hintergrund in jeder channel von einem leeren Bereich des Objektträgers.
- Subtrahieren Sie dieses Hintergrundintensität von der durchschnittlichen Intensität pro Pixel. Mit diesen Hintergrund abgezogen Fluoreszenzintensitäten zu FRET für jede Zelle berechnet, indem der Hintergrund subtrahiert Akzeptor Intensität dividiert durch die Summe (Akzeptor + Spender) Hintergrund-subtrahierten Intensität auf Donoranregung: Ich Akzeptor / (I + I Akzeptor-Donor)
- Zählen der Anzahl von verinnerlicht Biomolekülen pro Zelle durch schrittweises Bleichen Analyse
- Einzelmolekülanalyse
- Einzelmolekül-Tracking und Diffusionsanalyse
Hinweis: Das Protokoll für die Verfolgung diffundierenden fluoreszierenden Molekülen in lebende Zellen und ihre scheinbare Diffusionskoeffizient auszuwerten beschrieben worden 26, 28.- Kurz gesagt, passen die Bilder der einzelnen Fluorophore in jedem Einzelbild von einem 2D-elliptischen Gauß. Link lokalisierten Moleküle zu einer Spur, wenn sie in aufeinanderfolgenden Rahmen in einem Fenster von 5-7 Pixel (0,48-0,67 um) erschienen. Verwenden Sie eine den Speichery-Parameter von 1 Rahmen für die vorübergehende Verschwinden eines Fluorophors durch blinkende oder verpasste Lokalisierung berücksichtigen.
- In vivo smFRET Analyse
- Manuell identifizieren lokalisierte Moleküle in Zellen von Filmen, indem Sie durch einen Film in ImageJ und unbeweglich (oder ziemlich unbeweglich) Moleküle zu identifizieren in der FRET (Akzeptor) Kanal.
- Um die Intensitäten in den Akzeptor und Geberkanal entsprechend einem unbeweglichen Molekül zu extrahieren, wählen Sie den Bereich um das Molekül in jedem Kanal unter Verwendung des "Oval" Wahltaste von ImageJ (Kreis um jeden einzelnen Fluorophor mit einem ~ 3-Pixel-Radius) und extrahieren die Molekülintensitäten Analysieren> Messen. Die resultierende Tabelle enthält die Messwerte und können gespeichert und / oder zu anderen Software kopiert.
- Berechnen der Hintergrundwerte pro Kanal von der mittleren Pixelintensität, die von einem Kreis mit der gleichen Größe in einem leeren Bereich des Schiebers über alle Frames analysiert.
- Verwenden Sie Hintergrund-subtrahierten Fluoreszenzwerte in den Donor- und Akzeptor-Kanäle (auf Anregung des Donors) für die Fluoreszenz und FRET-Zeitspuren, wie in der Einzelzell-FRET-Fall (siehe 5.2.1.7).
Hinweis: Automatisierte und robuste Analysen und Algorithmen in Referenzen 26-28, 31 beschrieben.
- Einzelmolekül-Tracking und Diffusionsanalyse
Ergebnisse
Probenvorbereitung
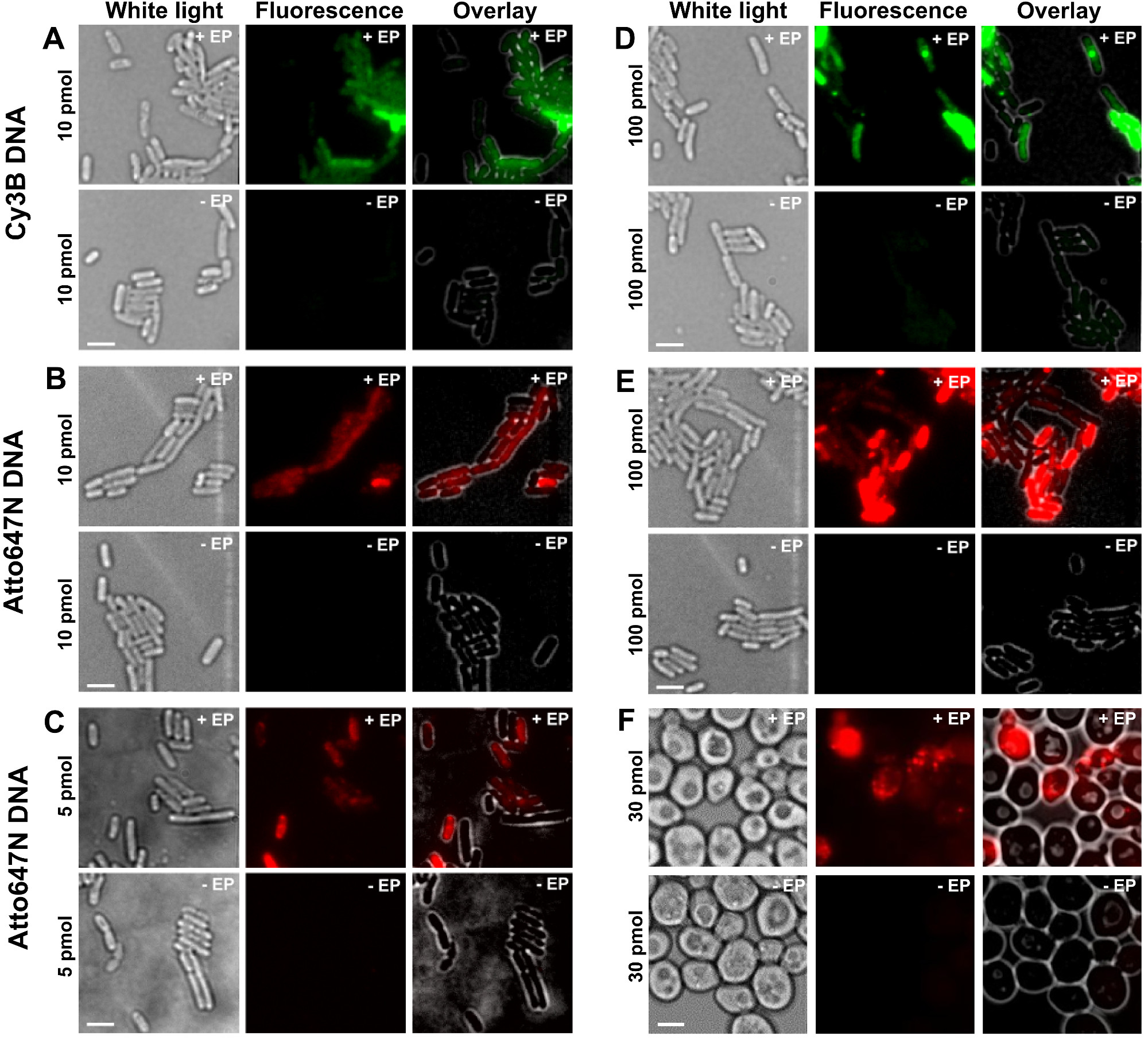
Die verschiedenen Stufen des Protokolls sind als Schaltbilder in Abbildung 1 dargestellt. Als ein Beispiel, das Laden von Bakterien, die mit doppelt markierte (Donor und Akzeptor-Farbstoffe) DNA-Fragmente dargestellt wir. Repräsentative Ergebnisse für DNA-Internalisierung sind in Abbildung 2 dargestellt. Für jedes elektroporiert Probe wurden Daten für Leerzellen und Nicht-elektroporierten Zellen ebenfalls aufgezeichnet (Figur 2). "Leere Zellen" entsprechen weder Zellen mit fluoreszierenden Biomolekülen noch elektroporiert inkubiert elektro; deren Intensität in dem Fluoreszenzkanal reflektiert das Autofluoreszenzpegel unter identischen experimentellen Bedingungen (Laserleistung, Zeitauflösung, Temperatur, etc.). "Non-elektroporierten Zellen" (auch -EP, dh minus EP) entsprechen einer negativen Kontrolle, bei der elektrokompetenten Zellen mit dem fluoreszierenden biomolec BebrütenModule aber nicht elektroporiert. Diese nicht-elektroporierten Zellen sollte einen Fluoreszenzniveau ähnlich der Autofluoreszenz der leeren Zellen und signifikant niedriger ist als die Fluoreszenzintensität, die durch geladene, elektroporierten Zellen dargestellt aufweisen. Dies bestätigt die Entfernung von irgendwelchen nicht internalisierte markierten Biomolekülen, die an die äußere Zellmembran angeklebt sein können.

Abbildung 2: Repräsentative Ergebnisse für die Internalisierung von dsDNA mit verschiedenen Fluorophoren in Bakterien bezeichnet in verschiedenen Konzentrationen (AE) und Hefe (F) Von links nach rechts:. Weißlicht, Fluoreszenz und Overlay-Bilder. - / + EP bezeichnet Inkubation ohne / mit Elektroporation. Maßstabsbalken: 3 um. A. Cy3B dsDNA, 10 pmol, E. coli. B. ATTO647N dsDNA, 10 pmol, E. coli. C. Alexa647 dsDNA, 5 pmol, E. coli. D. Cy3B dsDNA, 100 pmol, E. coli. E. ATTO647N dsDNA, 100 pmol, E. coli. F. ATTO647N dsDNA, 30 pmol, Hefe. Diese Zahl hat sich von der Referenz 26 modifiziert. Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.
{kind=link}
Zählen der Anzahl von verinnerlicht Biomolekülen pro Zelle
Das Verfahren, um die Anzahl von internalisierten markierten Biomolekülen pro Zelle unter Verwendung von Photobleich Analyse ist in Abbildung 3 und Ergänzungs Movie 2 dargestellt abzuschätzen, zusammen mit verschiedenen Konzentrationen von markierten DNA erhaltenen Ergebnisse repräsentativ. Zelle Ladewirkungsgrad steigt mit der Ausgangsmenge an inkubiert markierter DNA, so dass der Anwender die Feineinstellung der Anzahl der markierten Moleküle pro Zelle von einer "Einzelmolekül" Niveau (<10, Ergänzender Film 2B) zu einem "Ensemble" Niveau (> 10 , Ergänzender Film 2A). Eine robuste Möglichkeit zur Schätzung der Prozentsatz der geladenen Zellen to zählen die Anzahl der elektroporierten Zellen, die eine Zellstärke über dem mittleren Zellenintensität nicht elektroporierten Zellen plus 3-fache ihrer Standardabweichung (dh, Av (I-EP) + 3 * StdDev (I-EP), wobei Av = mittel, I = Intensität pro Pixel, Std. Abw. = Standardabweichung und -EP = nicht elektroporiert), wie in Abbildung 3 dargestellt.

Abb. 3: Zählen der internalisierte Moleküle mit Photobleaching-Analyse (A) Einzellige Photobleaching-Analyse. Beispiele für Fluoreszenzintensität timetraces (blau: Rohdaten; rot: fit, Einlässe: WL und Fluoreszenzbilder von E. coli mit ATTO647N markierten dsDNA geladen vor und nach dem Bleichen). Oben: Ein-Schritt-Bleich Veranstaltung. Mitte: Zelle mit ± 3 Moleküle, die Bleichund blinkt. Unten: cell> 10 Schritte, die mindestens 10-Moleküle (B) Histogramm der Einzelschritthöhe Intensitäten von einem automatisierten Schritt Anpassungsalgorithmus aus 57 Zellen, die weniger als 6 unterscheidbare Schritte.. Einzel-Gaußsche Anpassung wird bei 11 ± 3 au zentriert ist, entsprechend einem einheitlichen Fluorophor Intensität von 8100 Photonen pro Sekunde. Der Stern markiert die Behälter dem Sammeln aller Stufenhöhen größer oder gleich 50 au (C) Histogramm der internalisierten Moleküle pro Zelle, die mit unterschiedlichen Mengen an ATTO647N dsDNA, nach dem Aufteilen des Anfangsfluoreszenzintensität durch die einheitliche Fluorophor Intensität berechnet elektroporiert. Von oben nach unten: leere Zellen (dh nicht mit fluoreszierenden Molekülen inkubiert und nicht durch Elektroporation), nicht durch Elektroporation (aber mit fluoreszierenden Molekülen namens -EP inkubiert) und elektroporierten Zellen mit 10 und 100 pmol dsDNA (Namen + EP) inkubiert. Leere und nicht elektroporierten Zellen entsprechenum Autofluoreszenz, während elektroporierten Zellen zeigen eine breite Verteilung der verinnerlichten Moleküle, mit einem höheren Anteil an hoch belasteten Zellen bei 100 pmol (≥ 4 Moleküle finden Sternchen markierten bin). Internalisierung Effizienz (Fraktion von Zellen, die mit Int.> Mittelwert + 3x Std. Dev. Nicht -EP Probe) für die 10 und 100 pmol Proben betrug 94% bzw. 90% betrugen. Die mittlere Anzahl der verinnerlichten Molekülen pro Zelle: 121 ± 106 Moleküle für 10 pmol dsDNA und 176 ± 187 Moleküle für 100 pmol dsDNA. Einstellungen: 100 ms Belichtung, Weitfeldbeleuchtung. Maßstabsbalken: 1 um. Diese Zahl hat sich von der Referenz 26 modifiziert. Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.
{kind=link}
Zell Be- und Rentabilität
Neben der Änderung der Menge des markierten Biomoleküle vor der Elektroporation zu den Zellen gegeben, dieAnwender können noch die Menge der Moleküle internalisiert, indem Sie verschiedene Feldstärken bei der Elektroporation (Abbildung 4, Zusatz Film 1). Höhere Feldstärken führen zu mehr Effizienz Internalisierung, führen aber zu einem leichten Rückgang der Lebensfähigkeit der Zellen. Für die Protein Internalisierung kann die Verwendung eines Filtrationsschritt zur Beseitigung von nicht internalisierte markierten Proteinen (siehe 3.3.3.1.1). In solchen Fällen sorgt Zellfiltration daß beobachteten fluoreszierenden Proteinen sind in der Tat innerhalb des bakteriellen Zytoplasma internalisiert; wir weisen Sie jedoch darauf, dass die Filtration hat auch einen negativen Einfluss auf die Lebensfähigkeit der Zellen (für weitere Einzelheiten siehe REF 22).

Abbildung 4: Einfluss der Elektroporation Spannung beim Laden und Zelllebensfähigkeit (A) Balkendiagramm, das die Wirkung der Elektroporation Feldstärke zum Einlegen effic.iency (rote Balken) und Lebensfähigkeit der Zellen (grüne Balken). 84 ± 8% des nicht elektroporierten Zellen (0 kV / cm) Spaltung nach 1 h auf einem Agarose-Pad bei 37 ° C. Unter den gleichen Bedingungen, 78 ± 3% und 49 ± 3% der Zellen bei 0,9 kV / cm und 1,8 kV / cm elektroporiert dividieren nach 1 Stunde. Für Beladungseffizienz sind 73 ± 8% der Zellen bei 0,9 kV / cm geladen, 92 ± 6% der Zellen werden bei 1,8 kV / cm geladen. Die Fehlerbalken stellen die Standardabweichung aus drei unabhängigen Messungen berechnet; mehr als 200 Zellen wurden für jede Probe und jeder Wiederholung analysiert. Insgesamt Ladeeffizienzsteigerungen mit Elektroporation Spannung der geringen Kosten der Lebensfähigkeit der Zellen. (B) Zellbasierte Fluoreszenzmessungen über mehrere Generationen zu zeigen, dass die Gesamtfluoreszenzintensität wird gleichmäßig auf beide Tochterzellen weitergegeben. Zelle 1 und 2 bezieht sich auf die Anzahl von Zellen in der Weißlichtbild (links) und Fluoreszenzbild zum Zeitpunkt t = 0. MaßstabBar: 1 um). Diese Zahl hat sich von der Referenz 26 modifiziert.
Protein Internalisierung
Repräsentative Ergebnisse für Protein Internalisierung sind in den 5A & B. Es ist besonders wichtig, so viele der restlichen freien (nicht umgesetzten) Farbstoff aus der Proteinprobe wie möglich vor der Elektroporation zu entfernen. In den Beispielen in den Figuren 5A und B, die Cy3B markierten Klenow Fragment Probe (Cy3B-KF, wobei KF ist das Klenow-Fragment der E. coli DNA Polymerase I, 66 kDa) enthält nur 1% freien Farbstoffes; solche Farbstoff Beitrag zur Gesamtzellenbelastung ist vernachlässigbar. Vergleiche der elektroporierten interessierende Probe sowohl mit der nicht-elektroporierten Zellen (mit der gleichen Menge an markiertem Protein inkubiert werden), sowie Zellen, die mit der äquivalenten Menge des freien Farbstoffs elektroporiert bilden zwei erforderlichen Kontrollen, um sicherzustellen, dass die beobachteten Fluoreszenzmolekületatsächlich verinnerlicht markierten Proteine.

Abbildung 5: Protein Internalisierung in lebenden Bakterien (A) Vertreter Fluoreszenzüberlagerungsfeld von Ansichten.. Zellen bei 1,4 kV Spannung elektroporiert mit 50 pmol RNAP ω-Untereinheit aus einem Protein-Stammlösung, die nur 1% kostenlos CY3B Farbstoff enthalten. Nicht elektroporiert (nicht -EP) und leere Zellen wie zuvor definiert sind. Freien Farbstoffs wurde in der gleichen Konzentration wie in der RNAP ω elektroporiertem Probe internalisiert. Imaging in weiten Feldmodus mit 532 nm Anregung bei 1 mW, 50 ms Expositions. (B) Verteilung unkorrigierter Zelle gemittelte Intensitäten für Proben in (A), im Verhältnis des Gesamtzellzahl angegeben. Mehr als 400 Zellen pro Probe wurden segmentiert. Diese Zahl hat sich von der Referenz 22 geändert wurde. (C) Internalisierung von UNLABeled T7 RNA-Polymerase (T7 RNAP, 98 kDa) in elektro DH5a tragenden pRSET-EmGFP kodierendes Plasmid smaragd GFP (EmGFP) unter der Kontrolle eines T7-Promotors. Links: Schematische Darstellung des Assays. Mitte: Fluoreszenz-Overlay. Rechts: Histogramme der Zellbasis Fluoreszenzintensitäten für die nicht-elektroporiert Probe (oben) und die Zellen inkubiert und mit T7 RNAP (unten) elektroporiert; ca. 11% der elektroporierten Zellen zeigen hohe Fluoreszenzintensität (Anteil der Zellen mit Int.> Mittelwert + 3x Std. Abw. von nicht -EP Probe), die die Expression von EmGFP. Die Sternchen zeigen Körbe Erfassung aller Intensitäten größer oder gleich 1.100 au Maßstabsbalken: 3 um. Diese Zahl hat sich von der Referenz 26 modifiziert.
5C stellt eine weitere Anwendung der Protein Elektroporation. Hierbei ist die elektroporierten Protein unmarkierten, aber ihre Internalisierung triggert beobachtbares Fluoreszenzsignal. Es zeigt sich, das vorgeSinn und Funktionalität elektroporiert Proteine im Zytoplasma der Zelle. Unmarkiertem T7 RNA Polymerase (98 kDa) wurde in E. internalisiert coli Stamm DH5a enthält unter der Kontrolle eines T7-Promotors 26 ein Plasmid, das für eine fluoreszierende Protein EmGFP. Da das Gen für T7 RNA Polymerase in DH5a abwesend EmGFP Expression in unseren Experimenten erfordert, dass funktionelle T7 RNA Polymerase wird mittels Elektroporation (5C) in die Zellen eingeführt. Nach Elektroporation mit 1 pmol T7 RNAP,> 11% der Zellen (blau bar, 5C) zeigte Fluoreszenz höher als die Negativkontrolle (mit der gleichen Menge an T7 RNAP inkubiert, aber nicht durch Elektroporation). Dieses Ergebnis legt fest, dass ein Teil der T7 RNAP Molekülen durch Elektroporation internalisiert behalten ihre Integrität in vivo und können ihre beabsichtigten Funktionen in das Cytoplasma der Zelle durchzuführen.
In-vivo-FRET an der Einzelmolekül-und Single-Cell-Ebenen
Schließlich wird die Internalisierung und Analyse der doppelt markierten Spezies in lebenden Bakterien in 6 und Ergänzungs Film 3 dargestellt als Fluoreszenzproteinfusionen sind nicht ideal für in vivo Studien smFRET, ist die Möglichkeit, doppelt markierten Biomolekülen in lebende Zellen zu liefern unter Verwendung von Elektroporation einer der großen Vorteile dieses Verfahrens. 6A stellt Single-Cell-FRET-Analyse der Bakterien mit unterschiedlichen FRET-DNA-Standards (mit Cy3B und ATTO647N Fluorophore als Donor-Akzeptor-FRET-Paar) geladen. Die Zellen werden mit 20 pmol von drei kurzen doppelt markierte dsDNA FRET Standards FRET Effizienzen (E *) von 0,17, 0,48 elektroporiert, und 0,86 in vitro (vorher bestimmten 26). Alle DNA in die Zellen effizient (6A, links) und der Hauptpeak jeder einzellige E * Verteilung stimmt gut mit den in-vitro-Ergebnisse (6A, Rechts). In den mittel- und hoch FRET Proben, Zellpopulationen mit niedrigeren E * als erwartet beobachtet, vermutlich aufgrund einer Kombination von Akzeptorbleichen und photophysikalischen Inaktivität variable Zellenbelastung (also variable Signal-zu-Rausch-Verhältnis) und den DNA-Abbau .

Abb. 6:. Repräsentative Ergebnisse für Einzelzellen-und Einzelmolekül-FRET-Beobachtung in lebenden Bakterien Ensemble und smFRET Studien in einzelnen Bakterien (A) Analyse der Zellen mit 20 pmol jedes der drei DNA-FRET-Standards beladen eine niedrige (~ 0,17) Zwischen (~ 0,48) und hohen (~ 0,86) FRET (gemessen unter Verwendung von in vitro Einzelmolekülmessungen, siehe REF 26). Links: weißes Licht und grün / rot (FRET) Fluoreszenz-Overlay-Bilder (Maßstabsbalken: 3 um). Beispiele für FRET Werte aus verschiedenen Zellen angegeben (weiß). Ausrüstung ht (oben nach unten):. unkorrigierte zellbasierten FRET (E *) Histogramme für Spender nur (dunkelgrün), niedrig (hellgrün), Mittelstufe (gelb) und hoch (rot) FRET-DNA-Standards (B-D) In vivo smFRET. Die Zellen werden mit 0,25 pmol Zwischen-FRET-DNA (Feld B), 0,25 pmol High-FRET-DNA (Panel C) geladen und 5 pmol doppelt markierte KF (Panel D). Linke Spalte: grün / rote Fluoreszenz Überlagerung der Einzelbild vor und nach der Akzeptor-Photobleichen. Mittlere Spalte: Zeit Spuren entsprechend dem Molekül in der gelben Kreis. FRET Effizienz, Geberemissionsintensitäten und Akzeptor-Emissionsintensitäten sind in blau, grün und rot dargestellt sind. Rechte Spalte: FRET-Histogramme der Spender nur Moleküle (grün) und Donor-Akzeptor-Moleküle (gelb, rot und grau) von 20 Zeitspuren für jede Probe. Maßstabsbalken: 3 & mgr; m für die A, 1 & mgr; m für die B-D. Diese Zahl hat sich von der Referenz 26 modifiziert.208fig6large.jpg "target =" _ blank "> Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.
Um smFRET in vivo für DNA- oder Proteinproben, geringe Mengen (0,25 pmol) von Mittel- und Hoch FRET DNA-Standards (6B, C) oder 5 pmol doppelsträngiger markierter KF (Alexa647 / Cy3B Fluorophore als FRET-Paar, Fig beachten 6D) in E. elektroporiert coli. Solche Konzentrationen führte zu vielen Zellen mit wenigen (n = 1-10) markierten Moleküle geladen werden, die eine direkte Lokalisierung, Verfolgung und Überwachung von FRET für einzelne Moleküle. Einige Moleküle diffundieren frei, während andere unbeweglich erscheinen oder langsam diffundieren (Ergänzende Movie 3). Timetraces immobilisierter doppelt markierte Biomoleküle (Abbildung 6, Mitte) dauern 1 bis 30 s und zeigen die Handschrift der smFRET: antikorreliert Veränderungen in der Donor und Akzeptor Fluoreszenz nach Akzeptorbleichen (zB t ~ 16 sec; 6B, Mitte ), gefolgt von Einzel-Schritt Donor Bleichmittel (beispielsweise t ~, 19 sec; 6B). FRET Ausschüttungen aus solchen timetraces (Abbildung 6, rechts) führen zu einem Mittelwert, der in sehr guter Übereinstimmung mit in-vitro-Studien 26,31,32 veröffentlicht erzeugt. Diese Ergebnisse stellen die Fähigkeit zur quantitativen smFRET Studien über internalisiert DNAs und Proteine, und lassen vermuten, dass Proteine behalten ihre Integrität und Struktur bei der Elektroporation und Internalisierung (wie durch die T7 RNAP Internalisierung Experimente unterstützt).
Ergänzende Film. 1: Zelllebensfähigkeit Links: Weißes Licht Bilder. Rechts: Fluoreszenzbilder. Animated GIF animiert, die die Teilung nach der Elektroporation (1,8 kV / cm) von Bakterien mit 10 pmol Atto647 markierte DNA geladen. Die scheinbare Gesamt Abnahme der Fluoreszenz aufgrund der Verdünnung des markierten DNA auf die Zellteilung und auch teilweise auf die Photozerstörung, die während jedes auftrittMessung.
Ergänzende Movie 2: Zellbasierte Ausbleichen Studien A.. Repräsentative Beispiel für eine stark belastete Zelle (mit> 100 ATTO647N-markierten DNA-Moleküle). Oben links, weißes Licht Bild der Zelle von Interesse (rotes Rechteck). Rechts oben, Film der beladenen Zellen, die ihre Fluoreszenzzerfall über mehrere Minuten. Unten, Zeitverlauf der Gesamtfluoreszenzintensität Zerfall der Zelle von Interesse. Organische Fluorophore weisen Photobleaching Lebensdauer 2 Größenordnungen höher als die FRP (hier ~ 41 sec für ATTO647N). B. Repräsentative Beispiel für eine Zelle, die mit weniger als 10 markierte Moleküle (3 in diesem Fall) geladen. Top, identisch mit der Abdeckung A. Unten, Zeitverlauf der Gesamtfluoreszenzintensität der Zelle von Interesse, die einzelnen Schritt Bleichen und / oder blinken entsprechend einzigen organischen Farbstoffen. Die durchschnittliche Höhe dieser Stufen entspricht der Einzelmolekül einheitlichen Intensität (hier ~12 au) verwendet, um die Anfangszahl der verinnerlichten Molekülen pro Zelle zu schätzen. Filme im Dauer roten Laseranregung bei 300 & mgr; W Leistung und 100 ms pro Frame.
Ergänzende Movie 3: I n vivo Einzelmolekül-FRET-Top:. Zellen mit 0,25 pmol High-FRET-DNA (wie in Abbildung 6C) kontinuierlich bei 50 ms pro Bild unter nTIRF Beleuchtung überwacht mit 1 mW Grün (532 nm) Laser geladen. Jeder Rahmen ist ein grün / rot (FRET) Fluoreszenzüberlagerung der einzelnen Kanäle. Diffundiert und unbeweglich rot (intakt) und Grün (einzelnes aktives Label) DNA-Moleküle zu beobachten. Unten: Zeit Spur entsprechend dem Molekül in der gelben Kreis. FRET Effizienz, Geberemissionsintensitäten und Akzeptor-Emissionsintensitäten sind in blau, grün und rot dargestellt sind. Anti-Korrelation Akzeptorbleichen Ereignis (rot nach grün Gänge) entspricht der Unterschrift der Einzelmolekül-FRET.
Diskussion
Viele Parameter können während der Zell Elektroporation und die Datenerfassung in Abhängigkeit von dem biologischen System von Interesse und der genauen Art des Experiments (Zellebene oder Einzelmolekülanalyse) variiert werden. Wenn zum Beispiel die Elektroporation von DNA in Bakterien, 0,25 bis 5 pmol der markierten dsDNA-Fragmenten führt zu einer geringen Effizienz der Internalisierung, die eine direkte Detektion von Einzelmolekülen (dh., Ohne die Notwendigkeit einer Photobleich vorher). Vor 5 pmol dsDNA, Zellen dazu neigen, stark belastet werden, eine Regelung für die Einzelzell-Analyse besser geeignet. Alle markierten DNAs sollten zuvor gelgereinigt, um jede Spur von freien Farbstoff zu entfernen (nicht-umgesetzte Fluorophor) von der DNA-Stammlösung. Außerdem potenzielle Probleme mit den DNA-Abbau, insbesondere für smFRET Experimente können mit DNAs mit unnatürliche Nukleinsäuren oder Motive, die Exonuclease-zugänglichen Enden zu schützen, wie Haarnadelschleifen richten.
Ein weiterer adjustablE-Parameter in der Elektroporation ist die Feldstärke während der Elektroporation angewendet. Niedriger Feldstärke (~ 1 kV / cm) wird auf einen niedrigen Ladeeffizienz für Einzelmoleküluntersuchungen angemessen zu führen. Höhere Feldstärken (bis zu 1,8 kV / cm) wird die Ladeeffizienz zu steigern; jedoch gibt es eine umgekehrte Korrelation zwischen Feldstärke und die Lebensfähigkeit der Zellen nach der Elektroporation (siehe Abbildung 4). Zur Bezugnahme ist eine normale Feldstärke für Bakterien und Hefe Elektroporation verwendet ~ 1,5 kV / cm. Die Zeitkonstante, die die Länge dieser Zerfall, ist ein bequemer Parameter zu folgen, da die Zeitkonstante sinkt, sobald eines Bogenphänomens in der Küvette erfolgt. Unter normalen Einstellungen sollte die Zeitkonstante größer als 4 ms ist; niedrigere Werte auf niedrige Ladeeffizienz oder sogar nicht-geladenen beschädigten Zellen führen. Meist electroporators bieten anderen Freiheitsgraden (wie "Impuls Abschneiden" oder "Impulsform"), der zur Abstimmung sowohl modifiziert werden könnenZelle Lade und Lebensfähigkeit. Wir wendeten diese Methode, um sowohl Bakterien und Hefen, jedoch ähnliche Verfahren soll auch die Internalisierung von markierten Biomolekülen in Säugerzellen unter Verwendung geeigneter Elektroporator Einstellungen seit der Membran ist eigentlich weniger komplex (Einzel Lipid-Doppelschicht), und da die Elektroporation bereits mit solchen Zellen verwendet 21.
Wenn Internalisierung markierten Proteinen, braucht alle freien Farbstoff aus dem markierten Protein-Stammlösung vor Elektroporation entfernt werden. Freien Farbstoffmoleküle, die aufgrund ihrer kleineren Größe können bevorzugt gegenüber der interessierenden Proteine internalisiert werden, und sind schwierig während der Datenanalyse (trotz ihrer voraussichtlich schneller Diffusion) zu unterscheiden. Als Anhaltspunkt für eine Probe von biologischem markierten Proteins geeignet zur Elektroporation zu sein, sollte die Menge des verbleibenden freien Farbstoffes unter 2% (detektiert unter Verwendung von Fluoreszenzabtastung einer SDS-PAGE) 22. Dieses Verfahren ist besonders wichtig,da einige Moleküle könnten an den äußeren Membranen von Bakterien oder Hefe elektroporiert haften. In dieser Hinsicht sollte die negative Kontrollprobe Fluoreszenzintensität pro Zelle deutlich niedriger als elektroporierten Zellen, idealerweise so niedrig wie der Autofluoreszenz Niveau der leeren Zellen (Zellen, die nicht mit fluoreszenzmarkierten Biomoleküle inkubiert wurden noch elektroporiert, Bild 2) an.
Wie bei dsDNA wird der Internalisierung Effizienz markierte Proteine zur Menge der Biomoleküle an die Zellen vor der Elektroporation hinzugefügt verbunden. Aber auch andere Parameter, wie Größe und Ladung, eine Rolle bei der Internalisierung. Kleine Proteine zeigen eine hohe Internalisierung Effizienzen, während größere Proteine (von bis zu 98 kDa) kann erfolgreich internalisiert werden, aber mit geringerer Effizienz (Figur 5) 26. Der isoelektrische Punkt des Proteins, mögliche Wechselwirkungen mit der Zellmembran und anderen physikalisch-chemischen Parametern auchEinfluss Zellbelastung während der Elektroporation. Infolgedessen müssen die Benutzer Experimente für ihre eigenen System zu optimieren, zu wissen, daß eine hohe Anfangskonzentration des markierten Proteins (> 50 uM) die besten Chancen für eine erfolgreiche Belastung ergeben. Elektroporation bietet auch ein neues Werkzeug, um zu stören und zu analysieren, die Zellfunktion durch die Einführung von Proteinen und anderer Biomoleküle in Zellen (entweder markiert oder unmarkiert). Die T7-RNA-Polymerase-Experimente (5C) vorhanden ist, so ein Beispiel für ein Experiment, wo wir ein Molekül, das die Genexpression in vivo unter Verwendung von Elektroporation ändern kann einzuführen.
Bei der Durchführung von Einzelmolekülfluoreszenzexperimenten wird TIR Beleuchtungs Regel über andere Beleuchtungsarten bevorzugt, da es die beste Signal-zu-Rausch-Verhältnis durch Erregen nur Fluorophore innerhalb einer dünnen Abschnitt oberhalb der Deckfläche (~ 100 nm). Allerdings Bildgebung markierten Biomolekülen diffundieren in lebenden Mikroorganismen könnte erneuterfordern tiefere Beleuchtung (bis zu 0,8 & mgr; m für E. coli). Tiefere Beleuchtung in HILO Modus erreicht, unter Beibehaltung eines hohen Signal-zu-Rausch-Verhältnis. Andererseits ist Weitfeldabbildungs besonders wichtig für schrittweises Bleichen Analyse, bei dem der Anwender die Schätzung der Anzahl der Moleküle internalisiert durch Photobleichen eines gesamten belasteten Zelle mit hoher Laserleistung und Dividieren des Anfangszellfluoreszenzintensität durch die einheitliche Intensität erzeugt von einem einzelnen Molekül (single Bleichen Schritt 3). Weitfeld-Bildgebung wird auch für die Langzeit Molekül Verfolgung, um die streuenden Moleküle von Interesse zu lokalisieren, auch erforderlich, wenn ihre Flugbahn decken das gesamte Zellvolumen.
In diesem Protokoll präsentieren wir, wie Elektroporation, eine Standardtechnik für Biologen und Biochemikern zur Abgabe von Nukleinsäuren in Zellen, stellt eine einfache Methode zur Bereitstellung von fluoreszierenden Biomolekülen in verschiedenen Zelltypen. Thist neu, bietet Hochdurchsatz-Technik ein einzigartiges Werkzeug, um markierte Moleküle in ihrer natürlichen Umgebung zu beobachten. Zusätzlich Biomolekülen mit Fluorophore mit einem breiten Spektrum von Wellenlängen gekennzeichnet, die Elektroporation können Moleküle mit vielen chemischen Gruppen, wie unnatürliche Nukleotide und Aminosäuren, Metallchelatoren, Vernetzer und Käfighaltung Gruppen modifiziert zu liefern. Wenn das biologische System in der Umgebung ist nicht wesentlich für die Zellentwicklung, kann das Gen-Kodierung für das Zielprotein ebenfalls gelöscht (oder zerlegt), so dass die nach Internalisierung beobachtet Proteine stellen alle (oder die meisten) der intrazellulären Proteinpool . Im Wesentlichen kann die Elektroporation "Transplantation" die Flexibilität des In-vitro-Biokonjugate in lebende Zellen und damit profitieren Bemühungen in der Synthetischen Biologie, Systembiologie und In-vivo-Detektion von Einzelmolekülen.
Offenlegungen
The authors have nothing to disclose.
Danksagungen
We thank Stephan Uphoff for discussions.
R.C. was supported by Linacre College, Oxford University. A.P. was supported by the German Academic Exchange Service (DAAD), the German National Academic Foundation and EPSRC. M.S. was supported by the Wellcome Trust. A.N.K. was supported by a UK BBSRC grant (BB/H01795X/1), and a European Research Council Starter grant (261227).
Materialien
| Name | Company | Catalog Number | Comments |
| ElectroMax DH5-alpha Comptent cells | Invitrogen | 11319-019 | or any other commercial or lab-mage electrocompetant bacteria or yeast. |
| EZ Rich Defined Madia | Teknova | M2105 | low fluorescence rich media |
| MicroPulser Electroporation Apparatus | Biorad | 165-2100 | or any classical electroporator for microorganism transformation |
| Certified Molecular Biology agarose | Biorad | 161-3100 | low fluorescence agarose for agarose pad |
| Microscope coverslips No 1.5 thickness | Menzel | BB024060SC | remove background particles by heating slides in furnace at 500 °C for 1h |
| Single-molecule fluorescence microscope | Home-built | described in REFs | |
| Localization software | Custom-written, available online | MATLAB and C++ software package that can be adapted for localization analysis. | |
| Tracking software | Available online | MATLAB implementation by Blair and Dufresne. |
Referenzen
- Tsien, R. Y. The green fluorescent protein. Annu Rev Biochem. 67, 509-544 (1998).
- Leake, M. C., et al. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature. 443, 355-358 (2006).
- Taniguchi, Y., Kawakami, M. Application of HaloTag protein to covalent immobilization of recombinant proteins for single molecule force spectroscopy. Langmuir. 26, 10433-10436 (2010).
- Xie, X. S., Choi, P. J., Li, G. W., Lee, N. K., Lia, G. Single-molecule approach to molecular biology in living bacterial cells. Annual review of biophysics. 37, 417-444 (2008).
- Lee, J. H., et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 343, 1360-1363 (2014).
- Miesenbock, G., De Angelis, D. A., Rothman, J. E. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 394, 192-195 (1998).
- Sauer, M. Localization microscopy coming of age: from concepts to biological impact. J Cell Sci. 126, 3505-3513 (2013).
- Dempsey, G. T., Vaughan, J. C., Hao Chen, K., Zhuang, X. Evaluation of fluorophores for optimal performance in localizationbased super-resolution imaging. Nat Meth. 8, 1027-1041 (2011).
- Shaner, N. C., Steinbach, P. A., Tsien, R. Y. A guide to choosing fluorescent proteins. Nat Meth. 2, 905-909 (2005).
- Landgraf, D., Okumus, B., Chien, P., Baker, T. A., Paulsson, J. Segregation of molecules at cell division reveals native protein localization. Nat. Methods. 9, 480-482 (2012).
- Jaitin, D. A., et al. Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science. 343, 776-779 (2014).
- Aldridge, S., et al. AHT-ChIP-seq: a completely automated robotic protocol for high-throughput chromatin immunoprecipitation. Genome Biol. 14, R124 (2013).
- Keppler, A., et al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 21, 86-89 (2003).
- Wombacher, R., et al. Live-cell super-resolution imaging with trimethoprim conjugates. Nat. Methods. 7, 717-719 (2010).
- Zhang, Z., et al. A new strategy for the site-specific modification of proteins in vivo. Biochemistry. 42, 6735-6746 (2003).
- McNeil, P. L., Murphy, R. F., Lanni, F., Taylor, D. L. A method for incorporating macromolecules into adherent cells. J Cell Biol. 98, 1556-1564 (1984).
- Clarke, M. S., McNeil, P. L. Syringe loading introduces macromolecules into living mammalian cell cytosol. J Cell Sci. 102, 533-541 (1992).
- Sakon, J. J., Weninger, K. R. Detecting the conformation of individual proteins in live cells. Nat. Methods. 7, 203-205 (2010).
- Taylor, L. S. Electromagnetic syringe. IEEE Trans. Biomed. Eng. 25, 303-304 (1978).
- Dower, W. J., Miller, J. F., Ragsdale, C. W. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 16, 6127-6145 (1988).
- Neumann, E., Schaefer-Ridder, M., Wang, Y., Hofschneider, P. H. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J. 1, 841-845 (1982).
- Sustarsic, M., et al. Optimized delivery of fluorescently labeled proteins in live bacteria using electroporation. Histochem Cell Biol. , (2014).
- Tokunaga, M., Imamoto, N., Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods. 5, 159-161 (2008).
- Sinha, A., et al. A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature. 000, 1-5 (2014).
- English, B. P., et al. Single-molecule investigations of the stringent response machinery in living bacterial cells. Proc Natl Acad Sci U S A. 108, E365-E373 (2011).
- Crawford, R., et al. Long-lived intracellular single-molecule fluorescence using electroporated molecules. Biophys J. 105, 2439-2450 (2013).
- Uphoff, S., Reyes-Lamothe, R., Garza de Leon, F., Sherratt, D. J., Kapanidis, A. N. Single-molecule DNA repair in live bacteria. Proc Natl Acad Sci U S A. 110, 8063-8068 (2013).
- Uphoff, S., Sherratt, D. J., Kapanidis, A. N. Visualizing Protein-DNA Interactions in Live Bacterial Cells Using Photoactivated Single-molecule Tracking. J Vis Exp. , (2014).
- Hohlbein, J., Gryte, K., Heilemann, M., Kapanidis, A. N. Surfing on a new wave of single-molecule fluorescence methods. Phys Biol. 7, 031001 (2010).
- Xie, X. S., Yu, J., Yang, W. Y. Perspective - Living cells as test tubes. Science. 312, 228-230 (2006).
- Santoso, Y., et al. Conformational transitions in DNA polymerase I revealed by single-molecule FRET. Proc Natl Acad Sci U S A. 107, 715-720 (2010).
- Hohlbein, J., et al. Conformational landscapes of DNA polymerase I and mutator derivatives establish fidelity checkpoints for nucleotide insertion. Nature communications. 4, 2131 (2013).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten