
Method Article
エレクトロポ生きた微生物で内在化および蛍光生体分子の観測
要約
Studies of biomolecules in vivo are crucial for understanding molecular function in a biological context. Here we describe a novel method allowing the internalization of fluorescent biomolecules, such as DNA or proteins, into living microorganisms. Analysis of in vivo data recorded by fluorescence microscopy is also presented and discussed.
要約
The ability to study biomolecules in vivo is crucial for understanding their function in a biological context. One powerful approach involves fusing molecules of interest to fluorescent proteins such as GFP to study their expression, localization and function. However, GFP and its derivatives are significantly larger and less photostable than organic fluorophores generally used for in vitro experiments, and this can limit the scope of investigation.
We recently introduced a straightforward, versatile and high-throughput method based on electroporation, allowing the internalization of biomolecules labeled with organic fluorophores into living microorganisms. Here we describe how to use electroporation to internalize labeled DNA fragments or proteins into Escherichia coli and Saccharomyces cerevisiæ, how to quantify the number of internalized molecules using fluorescence microscopy, and how to quantify the viability of electroporated cells. Data can be acquired at the single-cell or single-molecule level using fluorescence or FRET. The possibility of internalizing non-labeled molecules that trigger a physiological observable response in vivo is also presented. Finally, strategies of optimization of the protocol for specific biological systems are discussed.
概要
生きた細胞内部の大部分の蛍光の研究では、GFP 1などの蛍光タンパク質(FPS)、とのタンパク質融合に依存します。これらの蛍光タグは、遺伝子発現または膜輸送2-7のようなプロセスに関与するタンパク質のコピー数、拡散パターンまたは局在化の研究を可能にする。 FPSが高いラベリング特異性、容易な実装を提供し、様々な光物理的および化学的性質1を有する変異体の大規模な在庫でご利用いただけます。しかし、有機蛍光色素分子は、in vitroで彼らの大きな光安定性のための実験(最大のFPよりも安定100倍)8,9、小さいサイズ(最大のFPよりも小さなボリュームを100倍)と分子内ラベリングを容易にするために主要な選択肢のまま(主にシステイン残基の使用による)。これらの要因はすべて、単一分子蛍光のために特に重要であり、FRETが10を検討する 。
いくつかの内部移行方法のCOMBIN有機標識およびin vivo検出に有利で るが過去10年間に導入されている。しかしながら、このような方法は、非天然アミノ酸15の使用を必要とするか、 例えば、(大きな単一膜の真核細胞に限定され、比較的大きなポリペプチドタグ( 例えば 、TMP、HALO、または20kDaのSNAPタグ)11-14を使用するいずれか。 、ロード、注射器のロード、マイクロインジェクション)16-19をこすり。
このプロトコルは、新規な、 インビボでの観察と結合する有機蛍光体の長所を簡単かつ高スループットの内在化方法を記載している。この技術を開発するために、我々は、Eのように、一般に微生物をロードするために、プラスミドDNA 20,21を用いて細胞を形質転換するために使用されるエレクトロポレーションの手順を適合さcoliまたはS.有機的にラベル生体分子と出芽酵母 。プロトコルは4簡単な手順で構成されています。ラベルの生体分子と細胞のインキュベーション、エレクトロポレーション、細胞の回収、および細胞洗浄は、非内在化生体分子を除去する。ここで、我々は、細胞ベースの単一分子蛍光を研究するために、このエレクトロポレーションプロトコル、ならびに細胞イメージングおよびデータ分析プロセスを提示し、信号をFRET。
エレクトロポレーションは、生体分子( 図1)20,21細胞に進入できるような一過性の膜孔を形成するために低イオン強度細胞懸濁液を横切って高電圧電場を放電に依存している。ただ、プラスミドDNAと細菌または酵母の形質転換と同様に、細胞は、それらのelectrocompetencyを確実にするために電気穿孔する前に用意しなければならない。この手順は、水で数回の洗浄工程からなる、膜透過性を増加させ、エレクトロポレーションキュベット中でのアーク放電を回避するために、細胞溶液のイオン強度を低下させる。このプロトコルでは、細胞は、(PROTOCOL参照:1.1)を下記のように調製することができるか、商業プロバイダから購入S。

図1:内在化プロトコルの概略図左から右へ:エレクトロコンピテント細胞(二重に標識されたDNA断片と、この例では、細菌)のアリコートに標識された生体分子の数マイクロリットルを加える。氷上で1から10分間インキュベートし、予め冷却したエレクトロポレーションキュベットに移す。エレクトロポレーションし、すぐ後に、細胞に0.5〜1ミリリットル豊富な培地を追加します。細胞が回復させるために37℃(または生物によって必要な温度、 例えば、酵母のための29℃)でインキュベート。余分な非内部化標識分子を除去するための5回の洗浄工程を行う。 PBS緩衝液およびピペットアガロースパッド上に10μlの100〜200μlの最終ペレットを再懸濁。 (広視野モードまたはHILOモード)蛍光顕微鏡での清掃カバーガラスと画像とのパッドをカバーしています。
エレクトロ細胞はほとんどの生化学研究室で見られる標準エレクトロポを使用して行うことができるだけでエレクトロポレーションの前にラベル生体分子、とインキュベートする。直ちにエレクトロポレーション後、細胞を、( 図1)を洗浄する前に、リカバリを可能にする富栄養培地中でインキュベートする。非内在化標識生体分子の過剰な第1の塩のかなり高い濃度といくつかの界面活性剤を含有する緩衝液中で洗浄することにより除去される(3.3 PROTOCOL参照 )。塩の存在は、そうでなければ、外膜に付着も非内部化ラベル生体分子によって形成され、非特異的な静電相互作用を破壊。同様に、洗浄緩衝液中の界面活性剤の存在は、非特異的疎水性相互作用を破壊する。
DNAの内在化は、( 図2)簡単ですがエレクトロポレーションを用いて標識されたタンパク質を内部移行する際、予防措置が取られる必要がある。まず、有機的に標識されたタンパク質のストック·サンプルは、まだ遊離色素の小さな割合が含まれる場合があります。遊離色素分子は、タンパク質よりもはるかに小さいので、優先的に内在化されるかもしれない。観察に内在蛍光分子の大部分は、目的のタンパク質に対応するように、初期のタンパク質試料は、〜2%遊離色素( 図5)22未満含まれている必要があります。非内在化さ標識タンパク質の過剰もエレクトロポレーション後、外側細胞膜に固執することができます。この現象は、タンパク質特異的であり、それぞれの新しいタンパク質をチェックする必要がある。我々は(:3.3.3プロトコルを参照してください)ロードされた細胞試料からの非内部化タンパク質の除去を可能にするいくつかのオプションを提案する。
最後に、細胞をリン酸緩衝液を少量に再懸濁し、蛍光顕微鏡でその画像化を可能にする、アガロースのパッド上にピペット。アガロースパッド上固定化は、SIMPLですEとそれらの完全性を損なうことなく、カバースリップ上の細胞を画像化する効率的な方法。パッドは、低蛍光培地が含まれている必要があります。
細胞イメージングを行うことができるいずれかの広視野、全内部反射蛍光(TIRF)またはHILO(高傾斜及び積層光学シート)を使用して顕微鏡検査。 HILO構成では、レーザビームは、TIRFよりも試料に深く浸透し、まだ大きな信号対雑音比23を可能にする、広視野用として試料全体を点灯しない。使用されるレーザパワーと時間分解能に応じて、内在化生体分子は、(段階的に、光退色分析を用いて、 図3)をカウント局在化、または24-28を追跡することができる。フルオロフォアのFRET対で二重に標識された構築物の内在化は、単一細胞または単一分子レベル( 図6)でのFRETの両方の定量化を可能にする。
異なるパラメータを変化させることができる所望の出力に応じて、及び生物学的システムを研究。まず、細胞当たりの内在化材料の量は、標識された生体分子の濃度を変えることによって調整することができるエレクトロポレーションの前( 図2)を細胞に添加した。エレクトロポレーションの電界強度も荷重効率および細胞生存率の両方に影響する。予想されるように、増加する電界強度と積載効率が増加しながら、エレクトロポレーションした細胞の生存率は、( 図4A)を減少させる。両方のパラメータは、エレクトロポレーションの後にロードされ、分裂細胞の割合を記録することによって定量することができる。蛍光イメージングと相まって、この生存率アッセイはまた、 生きた細胞内に内在化生体分子の観察を検証し、数世代( 図4B)にわたって連続観測を可能にします。
要約すると、このプロトコルは、に蛍光標識DNAおよびタンパク質分子の内在化を可能にする大腸菌またはS. cerevisiaeの 26。有機フルオロフォアで標識された個々の分子は、一桁長い時間スケールのためのFPよりも高い時空間解像度で追跡することができる。最後に、この方法は、広視野、TIRFおよび共焦点検出、ならびにALEX(レーザー励起28,29を交互に)としてパルス励起スキームと互換性がある。
プロトコル
1.細胞の準備
- ラボ製エレクトロ細菌の調製
- そのようなM9またはEZリッチな培地などの低蛍光媒体への関心の大腸菌株の単一コロニーから5〜10ミリリットル一晩前培養を準備します。
- 午前中は、OD 600が 0.02で始まりnmのように一晩前培養を持つ新しい400ミリリットルの培養に接種。 1 MのMgCl 2の1 M MgSO 4をと2.5ミリリットルの2.5ミリリットル低蛍光培地400ミリリットルに追加。
- 37℃で成長し、0.4〜0.6に達する程度のOD 600まで、250 rpmで。
- 10〜15分間氷水浴中での培養を冷却することにより成長を停止させる。
注意:今すぐ上から、4℃(氷上で)でのすべてのステップを実行する。 - 遠心1000×gで15分間培養する。上清を捨て、250ミリリットルに細胞ペレットを再懸濁冷却し、滅菌蒸留H 2 O
- 遠心分離および再懸濁がTステップ繰り返し100ミリリットル、次いで50 mlの水の量を減少させるwice。
- 遠心1000×gで10分間培養する。上清を捨て、25ミリリットル冷却し、滅菌蒸留H 2 O + 10%グリセロール中で細胞ペレットを再懸濁します。
- 遠心分離を繰り返して再懸濁し、10mlの5 mlであり、最終的に500μlの10%グリセロール溶液の体積を減少させる、三回繰り返す。
- -80℃で液体窒素とストア内の20μlの各、フラッシュ凍結のアリコート中の細胞を分注し。
- 市販のエレクトロ細菌細胞
- 無菌冷水蒸留水で1:1に市販の細胞アリコートを希釈する。 -80℃で20μlのアリコートとストアを行います。
- エレクトロ酵母の準備
注:エレクトロS.セレビシエ細胞を各エレクトロポレーション実験の前に調製され、E.用として-80℃で保存することができない大腸菌 。- 開始するには、接種5所望の関心株の単一コロニーを0ミリリットルYPD培地。
- 30℃、0.6〜0.8に達した程度 OD 600まで、250 rpmでインキュベートする。
- 4℃で5分間1000×gで遠心分離細胞。
- 25ミリリットルでペレットを再懸濁冷却し、滅菌蒸留H 2 O
- 洗浄工程は、水25mlで二回再懸濁し、1 M.におけるソルビトールの冷却溶液2mlに再懸濁回繰り返す
- 1Mソルビトール250μlの中で細胞を再懸濁し、50μlのアリコートにセルを分割します。
2.アガロースパッド調製
- バックグラウンドの蛍光粒子を除去するため、500℃で1時間炉内でカバースリップを燃やす。カバースリップは、アルミホイルで覆われ、室温で数週間保存することができる「クリーン焼け」。
注:このようなプラズマ洗浄またはピラニア溶液などの他の一般的な洗浄方法は、背景である限り使用され得る清掃スライドの蛍光は、準ヌルのまま。 - 蒸留水の溶液(70℃) - 電子レンジで2%アガロースを溶融して低蛍光アガロース溶液を調製する。すぐに2X低蛍光培地500μlに明確な2%アガロース溶液500μlを加え、穏やかに混合。
- それがダウンし、硬化冷却する前に、速やかにこのアガロースをピペット - 顕微鏡カバースリップ(ノー1.5厚さ)に培地溶液をおよそ2cmの直径及び数ミリメートルの高さのパッドを形成するために。泡を避け、必要に応じてピペットチップでそれらをポップ。
- 第二の「焼け」カバースリップ(ノー1.5厚さ、図1を参照)を有するパッドを平坦。
注:このアッパーカバースリップは、平らな均質パッドを形成し、細胞が準備されている間にほこりや乾燥から保護します。などEZリッチな培地としてM9またはリッチメディアなどの最少培地は、彼らの低い蛍光について試験されている。
3. Electroporatイオン
- インキュベーション
- テント細胞(20μlの細菌または50μlの酵母)の単アリコートに、低塩緩衝液(<50 mMの塩)に格納されている標識分子の5μLまで追加し、氷上で10分間インキュベートする。
注:ストック溶液における蛍光標識された分子の濃度、したがって、エレクトロポレーションの前に細胞に添加し、標識分子の量を直接充填効率(図3、及び考察)と相関している。いくつかのタンパク質は、低塩条件との相溶性であるように、保存緩衝液の塩濃度を増加させることかもしれないが、細胞のエレクトロポレーションの前に追加標識分子の量は、その後減少させる必要がある。 - (それぞれ、細菌や酵母のための0.1と0.2cmの間隔)を予め冷却したエレクトロポレーションキュベットへの細胞および標識された生体分子の混合物を転送します。ゆっくりと溶液からの任意の潜在的な気泡を除去するためにベンチにキュベットをタップします。
- 立方を配置エレクトロにVetteのとの溶液に高電圧の電気パルスを適用する(0.9から1.8 kVの/ cmの、電圧を選択することの詳細については説明を参照)。このようなパルスは、標識された生体分子が細胞内に拡散させ、細胞膜における一過性の孔を形成する。
- エレクトロに表示されて時定数は4から6ミリ秒の間にあることを確認してください。低い時定数は、多くの場合、高すぎる塩濃度および/またはキュベット内の気泡の存在に起因して、非常に低いまたは細胞の無負荷につながる。
- テント細胞(20μlの細菌または50μlの酵母)の単アリコートに、低塩緩衝液(<50 mMの塩)に格納されている標識分子の5μLまで追加し、氷上で10分間インキュベートする。
- 回復
- すぐにエレクトロポレーションした後、そのようなSOC、EZリッチな培地、YPDまたは細胞への豊富なメディアとしての豊富な培地500μlを追加します。
- 2から10分間、酵母に細菌や29℃、37℃でサンプルをインキュベートする。ユーザは、成長およびエレクトロポレーション後の分裂細胞の割合を評価したい生存率測定のために、ワットなどの長い回復時間(1時間まで)を使用しE最初の細胞分裂前にこのような遅延時間を観察します。
- 洗浄工程
- 3300×gで、4℃で1分間細胞をスピンダウンして、任意の非内部化生体分子を除去するために細胞を洗浄。上清を捨て、500μlのPBSで細胞を懸濁します。
注:各サンプルについて、標識された生体分子の同じ量と共にインキュベートが、エレクトロポレーション、メインサンプルとまったく同じように洗浄していない細胞のネガティブコントロールを調製。 - 前の手順を3回繰り返します。
- タンパク質の内在化の場合には、関心対象の標識されたタンパク質の特性及び挙動に応じて、洗浄手順を最適化する。次の手順は、可能な最適化の一例である:
- 細胞外膜22に付着する可能性があり、非内在化タンパク質を除去し、100mMのNaClおよび0.005%トリトンX100を含むPBS、<を使用して最初の3回の洗浄サイクルを実行するSUP> 26。
- フィルタにエレクトロポレーションした細胞をピペットを1.5 mlのマイクロ遠心チューブの内側に装着し、0.22μmの細孔径フィルターでエレクトロポレーションした細胞をフィルタリングします。 800×gで、4℃で3分間スピンダウン。フロースルーを捨てます。細胞の上に500μlの新たなPBSを追加し、以前のように再びそれらにスピンし、22一度これらの手順を繰り返します。
- 非内在化タンパク質の消化を可能にする第1洗浄サイクル中にプロテアーゼK(500μlのPBS中に10ng)を少量加える。
- 細胞外膜22に付着する可能性があり、非内在化タンパク質を除去し、100mMのNaClおよび0.005%トリトンX100を含むPBS、<を使用して最初の3回の洗浄サイクルを実行するSUP> 26。
- 3300×gで、4℃で1分間細胞をスピンダウン。上清を捨て、PBS150μlの中に細胞を懸濁します。
- 上部のカバースリップを除去し、液滴によって細胞懸濁液滴の10μlのを広げることによって、アガロースパッド上にロード細胞溶液を広げた。 T ON(顕微鏡対物仕様に一致する1.5厚さ、)未使用のきれいな焼かれたカバーガラスを交換してください非常に軽くスライド上のパッドを押すの彼がトップ。
- 異なるサンプルを画像化しながら、不透明なボックス内のパッドを格納することで、光から電気穿孔された細胞を保護します。
- 3300×gで、4℃で1分間細胞をスピンダウンして、任意の非内部化生体分子を除去するために細胞を洗浄。上清を捨て、500μlのPBSで細胞を懸濁します。
4.顕微鏡データの取得
注:生きた微生物中の単細胞および単一分子蛍光顕微鏡は、任意の適切な蛍光顕微鏡(特注のまたは商業)で行うことができる。
- [設定]
- 広またはHILO照明
- すべてのTIRF /単一分子顕微鏡による画像サンプル。
注:例として、我々は実験室でTIRFのセットアップとカスタマイズされた倒立顕微鏡を使用しています。 532 nmおよび637 nmのダイオードレーザーからのビームを合わせ、対物レンズの後焦点面に集束する前にコリメートされる。試料からの蛍光は、ロングパスとノッチフィルタを使用して励起光から分離され、同一の対物レンズを介して収集し、再分割されダイクロイックミラーを用いて、dおよび緑チャネル。 2つのチャネルが、電子増倍電荷結合素子(EM-CCD)カメラのチップの別個の半体上に結像される。ビデオ運動モードを使用して記録されている。白色光画像は、白色光ランプ、照明源として顕微鏡に取り付けられた凝縮器を使用して得られる。 - 一般的な単一分子観察のため、(ヒロイメージングに対する TIRFの詳細については説明を参照)TIRFまたはHILO 23に顕微鏡の照明モードを設定します。むしろ、カバースリップと接触し、その下部膜よりも内側の細胞、4.5.4を参照TIRF顕微鏡上HILOモードを設定するには、カバーガラス表面よりもわずかに高い焦点(像をシフトするためにわずかに励起光の入射角を減少させる)。
- 細胞レベルの分析、長い単一分子追跡実験または段階的退色分析のために、広視野モードensuriに顕微鏡の照明モードを設定する全体の細胞容積の連続観測をngのため、すべての内部化標識分子の。
- すべてのTIRF /単一分子顕微鏡による画像サンプル。
- 典型的には、0.5〜3 mWの周り利用励起パワー(〜50〜400 W / cm 2)であった。
注:より高いレーザパワーが高い時空間分解能または段階的に光退色解析に必要となる場合がありながら、低レーザパワーは、長寿命の蛍光観察を達成するために有用であると(1分にわたって)を追跡する。 - より一般的な観察と強度の定量化のために100 msに実験を追跡するための15ミリ秒に至るまでの露光時間を使用してください。注:他のフレームレートとモードは、ストロボ照明として使用することができ、特に高速の拡散種30を研究するために。
- TIRF顕微鏡では、約100ナノメートル/ピクセルのピクセル長、結果の倍率で電子増倍CCD(EMCCD)カメラの蛍光チャネルを記録。 TIRFのセットアップは、参照26でより詳細に記述している。
- 広またはHILO照明
- データ収集
- スイッチを切るか、実験の開始まで、レーザー照射を遮断する。原因露出オーバーにカメラへの損傷を防ぐためにオフEMCCDカメラゲインを切り替えます。
- 客観的に近いセルをもたらすために、下向きに細胞に覆われた側で、逆さまに顕微鏡ステージ上のアガロースパッドサンドイッチを置きます。透過光顕微鏡モード28のセルにフォーカスを設定。セルの位置を特定するために、白色光画像化の下で見ると、各セルの画像を記録するには、白色光をオフにする前に概説する。
- 実験室周囲光からサンプルを保護します。
- HILO励磁モードの場合は、カバースリップの表面に近接した試料の部分のみを照明することにより、各最大信号対雑音比の励起ビームの角度を調整する。
- HILO照明を達成するために、100×NA 1.4の対物28(より高いヌメの後焦点面にレーザビームを集束そのような1.45または1.49などrical開口部)も好適である。ビームが角度で目標を終了しますように、ビームに対して垂直に焦点レンズをシフトすることで、焦点は客観的な中心部から離れて移動します。
- 細胞外の背景信号対信号対雑音比、細胞内蛍光強度を最大にするためにレンズ位置を調整する。
- カメラゲインを切り替え、レーザーのスイッチを入れる前に、データ収集を開始します。
- データを記録しながら、前または蛍光データを記録した後、各FOVの白色光画像を取得する。これは、蛍光チャネルでセル境界を識別するのに役立ちます。
- 生存率推定のために
- 細胞はエレクトロポレーション後に成長できるように、アガロースパッドに低蛍光豊富な培地を使用してください。
- 、 大腸菌用研究微生物(37℃に最適な温度に顕微鏡を平衡化するS.セレビシエのための29℃)目的のヒータシステム。
- レコードの両方の白色光および蛍光画像を30分毎に、全体のデータ記録時ビューの全く同じフィールド上に残ることを確認すること。細胞が分裂を開始する前に≈1時間の遅れは、一般的に観察される。
- セル当たりの内部化生体分子の数を数える
- 高い値(2~3ミリワット)と長い露出時間(100ミリ秒)のレーザーパワーを設定する。
- セル全体を照明するためにモードを広視野への照明を設定します。
- ステップ4.2で説明したようにレコード·ムービーは蛍光退色が完了した後に追加のフレーム(50〜100フレーム)を記録して確認すること。
5.データ分析
- 一般的解析
- そのようなフリーソフトImageJのように、イメージングソフトウェアを使用して、白色光と蛍光励起の両方で、記録した画像や動画を分析します。
- ImageJのでは、ファイル内の顕微鏡(TIF形式)に記録された画像や動画>開く>あなたのファイルの場所を開く。
- 質的にすべての蛍光画像はイメージで同じ明るさとコントラストの設定で表示されていることを確認し、コンピュータ画面上の蛍光強度を比較するには>>明るさ/コントラストを調整します。 、選択した画像のために手動または自動で設定を調整してボタンを押し、「設定」と「他のすべての画像に伝播」オプションを選択します。
- 抽出するために、情報の種類を設定します。> SET計測を分析し、選択します(少なくとも)「エリア」、「標準偏差」、「ミン·マックスグレー値」と「灰色の平均値」。
- 、細胞蛍光強度を比較するのImageJのフリーハンド選択ボタンを使用して関心の面積Sを選択して、分析>測定におけるセル強度を抽出します。結果のテーブルは、測定値を含んで保存された、および/または他のソフトウェアをコピーすることができる。 「平均」値は、選択された領域内の画素当たりの平均強度に対応しており、直接の細胞間、または細胞と背景の間で比較することができる。
- エレクトロポレーション細胞試料では、細胞は、ピクセル当たりの平均強度は、ネガティブコントロールのピクセルプラス3倍の標準偏差(AVが(私はセルをロードした )> AV(I -EP)+あたりの平均強度よりも大きい場合にロードされたとみなされる3 * STDDEV(I -EP))。
- サンプルの品質および負荷を評価するために、偽色の蛍光オーバーレイ画像や動画を作成します。
- ImageJのでは、そのような白色光画像と画像>色で同じFOVに対応する蛍光画像などのオーバーレイ画像>チャンネルをマージします。赤チャンネル、C2(グリーンチャンネル用グリーン...等のためにC1(赤)、白色光のために、各画像(C4(グレー)の色を選択します。)。
- チェックオーバーレイ蛍光はセル境界内に配置されている画像(白色光画像)と、そのバックグラウンド蛍光が低く、均一である(セル境界の外側ない輝点)に関する。
- 多数の細胞を分析する前に、負のサンプルに対応する質的に画像が空のセルの画像に類似しており、エレクトロポレーションした細胞よりもはるかに低い強度を表示するチェック。
- 生存実験のために、手動で(4.2.6を参照)は、時間の経過成長同一視野から、非分裂が、(同一の)目に見えて、無傷で損傷を受けた(死んだ)分裂細胞の割合を数える。
- 十分な統計情報を収集するために、サンプル(エレクトロポレーションし、ネガティブコントロールと空のセル)あたり少なくとも200細胞の生存率を評価します。
- そのようなフリーソフトImageJのように、イメージングソフトウェアを使用して、白色光と蛍光励起の両方で、記録した画像や動画を分析します。
- 細胞ベースの分析
- 段階的な光退色分析により、細胞当たり内在化生体分子の数を数える
- のSelectI別セグメント細胞ImageJののフリーハンド選択ボタンを使用して、関心領域をngのと(細胞膜に相当)を正確にセルを囲む形状を描く。
- 画像>スタック>プロットZ軸プロファイルに時間をかけて細胞強度を抽出します。結果のグラフは、その特定のセルのための光退色曲線結果として各ムービーフレームに対するセル境界内の面積のピクセルあたりの平均強度を表す。それは、より低い漸近線(バックグラウンド蛍光)に達するセル強度の初期の指数関数的減少が含まれています。測定値と「保存」または「コピー」をクリックすることにより保存され、および/または他のソフトウェアをコピーすることができる。
- コピーしてスプレッドシートの列(I 生 )に漂白値を貼り付けます。
- 最後の50〜100フレーム(下の漸近線)のために得られたI 生の値を平均化することによって(I 自動 ) を光退色後に残っているピクセルあたりの平均自家蛍光を計算します。
- S初期の光退色曲線からそのセルの光退色後に残っているピクセルあたりの平均自家蛍光をubtract:私は漂白 =私は生の-私のオート。
- 原因シングルフォア26の漂白に平均ステップサイズ(単一の蛍光団の強度)を評価するために、10未満の量子化された工程を示す(私はフレーム対 漂白 )は、ベースラインを差し引いた退色timetracesを使用してください。
- 単一のフルオロフォアの強度で(Iは、t = 0での漂白 )最初のベースラインを差し引いたセルの強度で割ることにより、細胞あたり内部移行分子の数を評価する。
- 単細胞FRET効率
- (ドナー励起時)の両方が、ドナーとアクセプターの発光チャネルにおけるピクセル当たりの平均細胞強度を測定し、各チャネルのセル境界内の5.1.1.4に説明するように。
- 各chのバックグラウンドの平均ピクセル強度を測定するスライドの空白領域からannel。
- ピクセルあたりの平均強度からこのバックグラウンド強度を差し引く。ドナー励起時合計(アクセプタ+ドナー)で割ったバックグラウンドを差し引いたアクセプター強度バックグラウンドを差し引いた強度を計算することにより、各セルのためのFRETを計算するためにこれらのバックグラウンドを差し引いた蛍光強度を使用してください:私は(私は+ I ドナーアクセプター )/ アクセプタ
- 段階的な光退色分析により、細胞当たり内在化生体分子の数を数える
- 単一分子分析
- 単一分子追跡と拡散分析
注:生細胞における蛍光分子の拡散とその見かけの拡散係数を評価するために追跡するためのプロトコルは、28 26に記載されている。- 簡単に言えば、2D楕円形のガウスによって、各フレーム内の単一の蛍光団の画像をフィット。彼らは5-7ピクセル(0.48から0.67ミクロン)のウィンドウ内の連続したフレームに現れた場合、リンクがトラックに分子を局在する。 memorを使用してください1フレームのYパラメータが原因点滅または逃したローカライゼーションにフォアの過渡消失を考慮するため。
- インビボsmFRET分析において
- 手動ImageJの中で映画を経て映画から細胞内部の局所的な分子を同定し、FRET(アクセプター)チャンネルで不動(またはかなり不動)分子を同定する。
- 不動の分子に対応するアクセプターおよびドナーチャネル中の強度を抽出するために、(〜3ピクセルの半径を使用して、各単一フォアの周りの円)のImageJの「オーバル」選択ボタンを使用して、各チャンネル内の分子の周りの領域を選択し、分析>測定中の分子の強度を抽出します。結果のテーブルは、測定値が含まれ、保存され、および/または他のソフトウェアをコピーすることができる。
- 分析したすべてのフレーム上のスライドの空白領域内の同じ大きさの円からの平均ピクセル強度からチャネルあたりのバックグラウンド値を計算します。
- (5.2.1.7を参照)単一セルのFRET場合のように、蛍光およびFRET時間トレース用(ドナー励起時)にドナーとアクセプターのチャネルでバックグラウンドを差し引いた蛍光値を使用してください。
注:自動化された強固な分析およびアルゴリズムが文献26-28、31に記載されている。
- 単一分子追跡と拡散分析
結果
試料調製
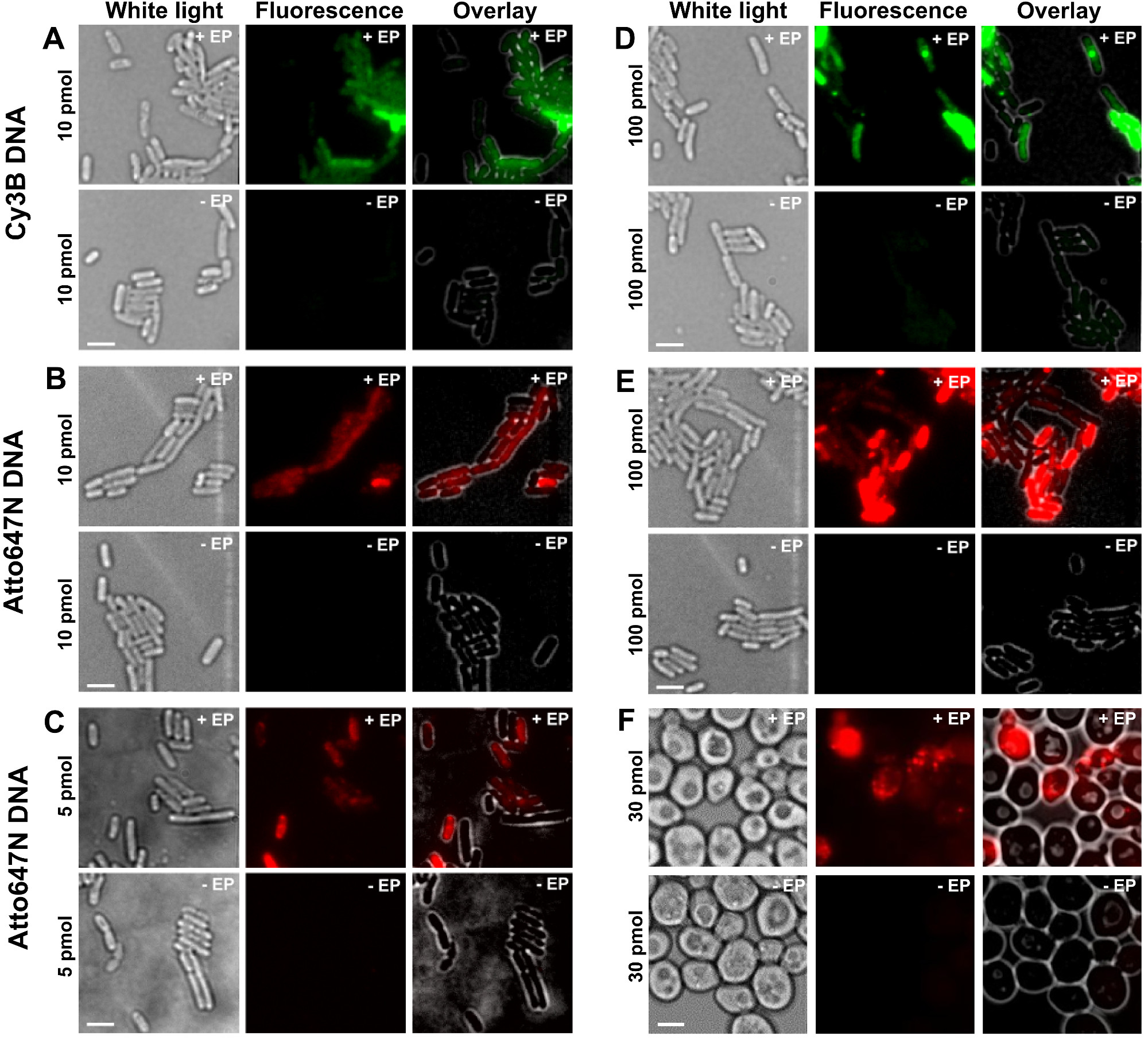
プロトコルの異なるステップは、 図1の回路図として示されている。一例として、我々は二重標識(ドナー色素およびアクセプター色素)でDNA断片を細菌の負荷を示した。代表的なDNAの内在化の結果を図2に示す。各エレクトロポレーションサンプルは、空のセルのためのデータと非エレクトロポレーションした細胞はまた、( 図2)を記録した。 「空のセルは、「蛍光生体分子とインキュベートすることも、エレクトロポレーションどちらの細胞をエレクトロに対応。蛍光チャネルにおけるそれらの強度は、同一の実験条件(レーザパワー、時間分解能、温度など)下での自己蛍光のレベルを反映する。 「非エレクトロポレーションした細胞」(別名-EP、 すなわち 、マイナスEP)は、細胞を蛍光biomolecとインキュベートされたエレクトロいるネガティブコントロールに対応ulesではなく、電気穿孔した。これらの非エレクトロポレーションした細胞は、空の細胞の自家蛍光と同様、ロードされ、エレクトロポレーションした細胞で表示される蛍光強度よりも有意に低い蛍光レベルを示すべきである。これは、外側の細胞膜に付着している可能性のある非内在化標識生体分子の除去を確認する。

図2:細菌において異なる濃度で異なる蛍光体で標識された二本鎖DNAの内部化(AE)及び酵母(F)のための代表的な結果を左から右へ:白色光、蛍光及びオーバーレイ画像。 - / + EPが/エレクトロポレーションとせずにインキュベーションを表す。スケールバー:3程度である。 A. CY3B二本鎖DNA、10pmolの、E.大腸菌 。 B. ATTO647N二本鎖DNA、10pmolの、E.大腸菌 。 C. Alexa647の二本鎖DNA、5 pmolの、大腸菌。 D. CY3B dsDNAを、100ピコモル、大腸菌。 E. ATTO647N DSDNA、100ピコモル、E.大腸菌 。 F. ATTO647N二本鎖DNA、30ピコモル、酵母。この図は、参照26から変更されている。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}
セル当たりの内部化生体分子の数を数える
光退色分析を使用して細胞あたり内部移行標識生体分子の数を推定するための手順は、一緒に標識されたDNAの異なる濃度で得られた代表的な結果を、図3及び補足動画2に示されている。インキュベート標識されたDNAの初期量を持つ細胞積載効率が増加し、「単一分子」レベル(10 <10、補足ムービー2B)「アンサンブル」レベル(へ>チューニングするために、細胞当たり標識分子の数をユーザーに許可する、補足ムービー2A)。ロードされた細胞の割合を推定する確実な方法をtO、非エレクトロポレーションした細胞の平均細胞強度プラス3倍の標準偏差( すなわち 、のAv(I-EP)+ 3 * STDDEV(I-EP)のAv =平均より上の細胞強度を表示するエレクトロポレーションした細胞の数を数えるI =ピクセルあたりの強度、基準色。開発=標準偏差-EP =非エレクトロポレーション) を図3に示すように。

図3:退色分析を使用して内在化分子の数を数える(A)単セル退色分析。蛍光強度timetracesの例(青:生データ;赤色:適合;挿入図:WL前および漂白後ATTO647N標識二本鎖DNAを装填した大腸菌の蛍光画像)。上:シングルステップ漂白イベント。ミドル:漂白を示す±3分子を含む細胞点滅。底部:6未満の区別可能なステップを含む57細胞からの自動化された段階フィッティングアルゴリズムからのシングルステップ高強度の少なくとも10分子に対応する> 10のステップを含む細胞(B)のヒストグラム。単一ガウスフィットは、毎秒8100光子の単一のフルオロフォアの強度に対応し、11±3 auの中心とする。アスタリスクは、単一の蛍光団強度で初期の蛍光強度で除して計算しATTO647N dsDNAの異なる量、でエレクトロセル当たりに内在分子の50 AU(C)ヒストグラム上または同等のすべてのステップの高さを収集ビンをマーク。上から下へ:空のセル( すなわち 、蛍光分子とインキュベートしていないし、エレクトロポレーションではない)、非電気穿孔(ただし、蛍光分子、名前-EPとインキュベート)、およびエレクトロポレーションした細胞は、10〜100ピコモル(EP +という名前の)二本鎖DNAとインキュベートした。空の非エレクトロポレーションした細胞を対応自家蛍光に、エレクトロポレーションした細胞は、100ピコモル(≥4分子、アスタリスクでマークされたビンを参照)での高負荷細胞のより高い割合で、内部移行した分子の広い分布を示したのに対し。内在化効率(int型を有する細胞の割合> + 3×基準色。開発を意味する。非-EP試料)10及び100pmolのサンプルについて、それぞれ、94%および90%であった。 10 pmolのdsDNAのための121±106分子、および100 pmolのdsDNAのための176±187分子:細胞あたりの内部化分子の数を意味する。セッティング:100ミリ秒露出、広視野照明。スケールバー:1μmである。この図は、参照26から変更されている。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}
セルの負荷および生存
標識された生体分子の量を変えることに加えて、エレクトロポレーションの前に細胞に添加ユーザーは、エレクトロポレーション中に異なる磁場強度を選択することによって調整するに内在する分子の量を( 図4、補足ムービー1)することができます。より高い電界強度が大きく、内在化効率をもたらすが、細胞生存率のわずかな減少をもたらす。タンパク質の内在化のために、ろ過工程の使用は非内部化標識タンパク質を(3.3.3.1.1参照)を排除できる場合があります。そのような場合、細胞ろ過は、観察された蛍光タンパク質が実際に細菌の細胞質の内部に内在化されることを保証する。我々は(詳細については、REF 22を参照)、濾過も細胞生存率に悪影響を与えることは、注意してください。

図4:セル·ローディングおよび生存時のエレクトロポレーション電圧の影響(A)のロードeffic上のエレクトロポレーションの電界強度の影響を表すバーグラフ。iency(赤いバー)と細胞(緑色のバー)の生存。 37℃でアガロースパッドの上に、1時間後の非電気穿孔細胞(0 kVの/ cm)の除算の84±8%。同じ条件の下では、78±3%、0.9 kVの/ cmであり、1.8 kVの/ cm 2でエレクトロポレーションした細胞の49±3%、それぞれ1時間後に分割します。細胞の92±6%が1.8 kVの/ cmの時にロードされている間、積載効率のために、細胞の73±8%は、0.9 kVの/ cm 2でロードされます。エラーバーは、3つの独立した測定値から算出した標準偏差を表す。 200以上の細胞を各サンプルと各繰り返しについて分析した。数世代にわたって細胞生存率のわずかな損害にエレクトロポレーション電圧の全体の積載効率が増加する。(B)細胞ベースの蛍光測定は、全体的な蛍光強度の両方の娘細胞間で均等に共有されていることを示している。セル1及び2は、t = 0でのスケールでの白色光画像における細胞数(左)および蛍光画像を指しバー:1μm)を。この図は、参照26から変更されている。
タンパク質の内在化
タンパク質の内在化のための代表的な結果は、 図5A&Bである。これは、エレクトロポレーションの前にできるだけタンパク質試料からの残りの空き(未反応)色素をできるだけ多く除去するために特に重要である。 図5A&B、CY3B標識ノウ断片試料(KF、 大腸菌 DNAポリメラーゼIのクレノウ断片であるCY3B-KF、66 kDa)のわずか1%の遊離染料を含むの例では、全体的なセル·ローディングにそのような色素の寄与は無視できる。遊離染料の当量でエレクトロ(標識タンパク質の同量とインキュベート)非エレクトロポレーションした細胞の両方で関心のエレクトロポレーションサンプルの比較だけでなく、細胞が観察された蛍光分子ことを保証するために2必要なコントロールを構成する確かに標識タンパク質を内在化される。

図5:生きた細菌中のタンパク質ナリ(A)ビューの代表的な蛍光オーバーレイフィールド。。細胞は、わずか1%の遊離CY3B染料を含まれるタンパク質ストック溶液から50ピコモルRNAPωサブユニットと1.4 kVの電圧でエレクトロ。非電気穿孔(非-EP)と空のセルは、以前のように定義されている。無料色素はRNAPωエレクトロポサンプル中と同じ濃度で内部移行されました。 1mWの、(A)におけるサンプルについて補正されていないセル平均強度の50ミリ秒の露出(B)分布における広視野モード、532 nmの励起での撮像、総細胞数の割合で与えられる。サンプルあたり400以上の細胞がセグメント化された。この図は、参照22から変更されている。unlaの(C)内在化T7プロモーターの制御下にあるpRSET-EmGFPコードするプラスミドエメラルドGFP(EmGFP)を有するエレクトロDH5αにbeled T7 RNAポリメラーゼ(T7 RNAP、98 kDaで)。左:アッセイの概略図。ミドル:蛍光オーバーレイ。右:非電気穿孔サンプル(上)のための細胞ベースの蛍光強度のヒストグラムは、細胞T7 RNAP(下部)とインキュベートし、エレクトロポレーション;エレクトロポレーションした細胞の約11%が高い蛍光強度を示した(INTを持つ細胞の割合を。> + 3X基準色。開発。非-EPサンプルの平均)EmGFPの発現を示す。 3〜:アスタリスクは、上記のすべての強度以上1100 AUのスケールバーを収集ビンを示している。この図は、参照26から変更されている。
図5Cは、タンパク質エレクトロポレーションの別のアプリケーションを提示します。ここでは、エレクトロポレーションしたタンパク質は、非標識であるが、その内在化が観察可能な蛍光応答をトリガします。この実験は、前の検証SENCEと細胞質内エレクトロポレーション、タンパク質の機能性。ラベルなしT7 RNAポリメラーゼ(98 kDa)が、Eに内部移行したT7プロモーター26の制御下で蛍光タンパク質EmGFPコードするプラスミドを含む大腸菌株 DH5α。 T7 RNAポリメラーゼの遺伝子はDH5αに存在しないように、我々の実験でEmGFP発現は、機能的なT7 RNAポリメラーゼは、エレクトロポレーション( 図5C)を介して細胞内に導入することを必要とする。 1ピコモルのT7 RNAPとエレクトロポレーションに続いて、>細胞(青色のバー、 図5C)の11%は、陰性対照(T7 RNAP同量のインキュベーションではなく、エレクトロポレーション)よりも高い蛍光を示した。この結果は、エレクトロポレーションによって内在T7 RNAP分子の割合は、in vivoでその完全性を保持し、細胞質中でそれらの意図された機能を実行することができることを立証する。
生体内では単一分子でFRET単一細胞レベル
最後に、生きた細菌中での二重標識種の内在化および分析は、蛍光タンパク質の融合は、生体内 smFRET研究のための理想的ではないとして、エレクトロポレーションを用いて生きた細胞内に二重標識生体分子を提供する能力が1つである図6に提示され、補足ムービー3です。この方法の大きな資産を。 図6(a)は、単一セルは、異なるFRET DNA標準(ドナー-アクセプターFRETペアとしてCY3BとAtto647N蛍光プローブを使用)を搭載した細菌の分析をFRET提示。細胞は、0.17、0.48、および0.86 インビトロ (以前は26を決定した)の見かけのFRET効率を有する3つの短い二重標識二本鎖DNA FRET基準の20ピコモル(E *)でエレクトロされている。 図6A(全てのDNAは、(左図6A、)を効率よく細胞に入り、それぞれの単セルEの主ピークは、*分布はインビトロの結果とよく一致する、右)。中悪性および高FRETサンプルにおける、低いおそらく受容漂白及び光物理的不活動、可変セル負荷の組み合わせに、観察される予想よりE *を(したがって、可変信号対雑音比)およびDNA分解による細胞集団。

図6:単一セルと単一分子のための代表的な結果は、生きた細菌で観察をFRETシングル細菌中のアンサンブルとsmFRET研究20pmolの低いを示す3つのDNA FRET基準の各(〜0.17)を搭載した細胞の(A)の分析、。。中間体(〜0.48)、および高い(〜0.86)FRET( インビトロでの単一分子測定を用いて測定した。REF 26を参照)。左:白色光と赤/緑(FRET)蛍光オーバーレイ画像(スケールバー:3μm)を。異なる細胞からのFRET値の例としては、(白)が示されている。リグ (上から下)htは:補正前の細胞ベースのFRET(E *)は、DNA標準をFRETドナーのみ (暗緑色)、低(ライトグリーン)、中間体(黄色)、および高(赤)のためのヒストグラム(B-D) 生体 smFRET で 。細胞は、0.25 pmolの中間-FRET DNA(パネルB)、0.25 pmolの高いFRET DNA(パネルC)、および5ピコモルの二重標識KF(パネルD)にロードされる。左の列:アクセプター光退色前後のシングルフレームの緑/赤の蛍光のオーバーレイ。中央の列:黄色い丸中の分子に対応する時間の痕跡。 FRETの効率は、ドナー発光強度、およびアクセプターの発光強度は、それぞれ、青、緑、赤で表示されている。右欄:ドナーのFRETヒストグラムのみ分子(緑色)およびドナー - アクセプター分子(黄色、赤、灰色)各試料について20時間トレースから。スケールバー:Aの3程度、B-Dのための1程度である。この図は、参照26から変更されている。208fig6large.jpg「ターゲット= "_空白">この図の拡大版をご覧になるにはこちらをクリックしてください。
FRETペア、図のように中距離および高FRET DNA標準( 図6B、C)または二重標識KF(Alexa647の/ CY3Bの蛍光団の5ピコモルのDNAまたはタンパク質のサンプル、低量(0.25ピコモル)のために、生体内で smFRETを観察する6D)は、Eに電気穿孔されている大腸菌 。このような濃度は、追跡、および単一分子の監視FRET、直接のローカライズを可能にし、いくつかの(N = 1-10)標識分子でロード多くの細胞につながった。他の人が不動現れたり(補足ムービー3)ゆっくりと拡散のに対し、いくつかの分子は、自由に拡散。 1から30秒のための最後の固定化された二重標識生体分子のTimetraces( 図6、中央)とsmFRETの顕著な特徴を示していますアクセプター漂白(例えば、t-〜16秒の際に、ドナーとアクセプター蛍光の反相関の変化を、 図6B、ミドル)、単一続く(例えば、T〜; 19秒; 図6B) -ステップドナー漂白。そのようなtimetraces( 図6、右)in vitro試験 26,31,32 に掲載さとよく一致している平均値の結果から生成されたFRETのディストリビューション。これらの結果は、内部移行したDNAやタンパク質に関する定量的smFRET研究のための能力を確立し、(T7 RNAP内在化実験によって支持される)タンパク質は、エレクトロポレーションおよび内在の際、整合性と構造を維持することを示唆している。
補足ムービー1:細胞生存率左:白色光画像。右:蛍光画像。アニメーションGIFは、10ピコモルのAtto647標識DNAを搭載した細菌のエレクトロポレーション(1.8 kVの/ cm)の後に分裂を示すのアニメーション。蛍光の全体の見かけの減少は、細胞分裂の際に標識されたDNAの希釈にも部分的に中に発生する光退色に起因する測定。
補足ムービー2:セルベースの光退色研究A.。高負荷のセルの代表例(> 100 Atto647N標識DNA分子を含有する)。左上は、目的の細胞の白色光画像(赤い四角形)。右上、数分間にわたって彼らの蛍光減衰を示す負荷細胞の映画。ボトムは、目的の細胞の全体的な蛍光強度の減衰の時トレース。有機フルオロフォアは、寿命のFPをより2桁(ここで、〜Atto647N 41秒)光退色示すことができる。 B. 10未満の標識分子(この場合は3)を装填し、細胞の代表的な例。パネルA.ボトム、関心単一工程の漂白を示す、および/または単一の有機フルオロフォアに対応する点滅の細胞の全体的な蛍光強度の時間トレースと同じトップ。これらのステップの平均高さは〜ここに(単一分子の単一強度に対応する12オー)細胞当たりの内在化分子の初期数を推定するために使用される。 300μW電力とフレームあたり100ミリ秒で連続赤色レーザー励起下作品。
補足ムービー3:I n は 単一分子FRETの体内トップ:0.25ピコモル( 図6Cのように)高FRET DNAを搭載した細胞は、継続的に1 MWの緑色(532nm)をレーザーを用いnTIRF照明下フレームあたり50ミリ秒で監視。各フレームは、各チャンネルの緑/赤(FRET)、蛍光のオーバーレイである。拡散不動赤(無傷)および緑色(単一活性標識)DNA分子を観察することができる。下:黄色い丸中の分子に対応する時間トレース。 FRETの効率は、ドナー発光強度、およびアクセプターの発光強度は、それぞれ、青、緑、赤で表示されている。反相関アクセプター漂白イベント(赤から緑色の遷移は)単一分子FRETの署名に対応している。
ディスカッション
多くのパラメータは、細胞のエレクトロポレーションおよび対象の生物学的システムと実験(細胞レベルまたは単一分子分析)の正確な性質に応じて、データ取得中に変化させることができる。細菌にDNAをエレクトロポレーションする際に、例えば、標識されたdsDNAフラグメントの0.25〜5ピコモル(事前に光退色を必要とすることなく、 すなわち 、)直接の単一分子の検出を可能にする、低い内在化効率をもたらす。 5 pmolのdsDNAの上方には、細胞は、単一細胞分析のための体制が適し、高負荷になる傾向がある。すべての標識されたDNAはまた、DNAのストック溶液から遊離染料(未反応のフルオロフォア)の痕跡を除去するために、先にゲル精製しなければならない。また、特にsmFRET実験のためのDNAの分解、潜在的な問題は、非天然の核酸、またはそのようなヘアピンループとしてエキソヌクレアーゼアクセス可能な末端を保護するモチーフがDNAを使用することによって対処することができる。
もうadjustablエレクトロポレーションの電子パラメータは、エレクトロポレーションの際に印加される電界強度である。低い電界強度(〜1kVの/ cm)は、単分子研究のための適切な低充填効率につながる。 (1.8 kVの/センチまで)より高い電界強度が積載効率を増加させる。しかしながら、エレクトロポレーション後の電界強度と細胞生存率との間に逆相関がある( 図4参照)。参考のために、細菌および酵母エレクトロポレーションのために使用される通常の電界強度は〜1.5 kVの/ cmである。時定数は、すぐに任意のアーキング現象がキュベットで起こるように低下するので、この減衰の長さを表す時定数は、追従するための便利なパラメータである。通常の設定の下では、時定数は4ミリ秒以上でなければなりません。低い値は、低い積載効率あるいは非ロードされた損傷を受けた細胞につながる。ほとんどのエレクトロポレーターは、両方の曲に変更することができる(例えば、「パルス切り捨て」または「パルス状」のような)他の自由度を提供セル·ローディングおよび生存。私たちは、その膜は(単一脂質二重層)は実際それほど複雑ではないので同じような手順では、また、適切なエレクトロの設定を用いて哺乳動物細胞への標識生体分子の内在化を可能にするはずである、細菌や酵母の両方にこの方法を適用し、エレクトロポレーションは、すでにこのような細胞で使用されているため21。
標識されたタンパク質を内在化すると、すべての遊離の色素は、標識されたタンパク質のストック溶液エレクトロポレーションの前に除去する必要がある。遊離色素分子は、それらの小さいサイズに、目的のタンパク質よりも優先的に内在化し、(それらの予想より速い拡散にもかかわらず)は、データ分析の間を区別するのが困難であることができる。有機的に標識されたタンパク質のサンプルのためのガイドは、エレクトロポレーションのために適切であるとして、遊離色素の残存量が2%未満であるべきである22(SDS-PAGEの蛍光スキャニングを用いて検出)。このプロセスは、特に重要であるいくつかの分子は、エレクトロポレーション、細菌や酵母の外膜に付着することがありますように。この点において、陰性対照試料は、(任意の蛍光標識された生体分子と共にインキュベートもエレクトロポレーションされていない細胞、 図2)は、空のセルの自己蛍光のレベルとして、理想的な低エレクトロポレーションした細胞よりも明らかに低い細胞当たりの蛍光強度を表示すべきである。
二本鎖DNAの場合と同様に、標識されたタンパク質の内在化効率は、エレクトロポレーションの前に細胞に加え、生体分子の量にリンクされる。しかしながら、そのような大きさおよび電荷などの他のパラメータは、内在化において役割を果たす。小さ なタンパク質は、(kDaの98まで)より大きなタンパク質に対して、高い内部移行効率を示す成功し、より低い効率( 図5)26と内部移行することができる。タンパク質の等電点、細胞膜との相互作用の可能性、また、他の物理化学的パラメータエレクトロポレーション時の影響セル·ローディング。その結果、ユーザーは標識タンパク質(>50μM)の高い初期濃度が成功ロードするための最善の機会を与えることを知って、自分のシステムのための実験を最適化する必要があります。エレクトロポレーションも細胞(ラベルまたは非標識のいずれか)に、タンパク質および他の生体分子を導入することにより、細胞機能を撹乱し、分析するための新しいツールを提供しています。存在するT7 RNAポリメラーゼの実験( 図5C)、我々は電気穿孔法を用いてインビボで遺伝子発現を変更することができる生体分子を導入することができ、実験のような例。
単一分子蛍光実験を行う際には、カバースリップの表面(〜100nm)を上記の薄切片内の励磁のみフルオロフォアで最高の信号対雑音比を提供するように、TIR照明は、通常、他の照明モードよりも好まれる。しかし生きた微生物の中に拡散イメージングラベル生体分子が再かもしれない( 大腸菌用0.8ミクロンまで)帖より深い照明。高い信号対雑音比を維持しながら、より深い照明は、HILOモードで達成される。一方、広視野画像は、ユーザが高いレーザパワー全体ロードされたセルを光退色し、生産ユニタリ強度によって初期細胞の蛍光強度を割ることによって内在化される分子の数を推定された段階的な光退色の分析のために特に重要である単一分子(単一光漂白工程と、 図3)による。それらの軌道がセル全体の体積をカバーする場合でも、広視野イメージングはまた、関心のある拡散分子を局在化するために、長期的な分子の追跡に必要とされる。
このプロトコルでは、エレクトロポレーション、細胞内で核酸を送達するための生物学者や生化学者のための標準的な技術は、種々の細胞型蛍光生体分子を送達するための簡単な方法を構成する方法を提示する。目小説、ハイスループット技術は、それらのネイティブ環境で標識された分子を観察するユニークなツールを提供しています。広い波長範囲をカバーするフルオロフォアで標識された生体分子の付加では、エレクトロポレーションは、非天然のヌクレオチドおよびアミノ酸、金属キレート剤、架橋剤、及びケージング基のような多くの化学基で修飾された分子を送達することができる。対象の生物学的システムは、細胞の発達に必須ではない場合には内在化後に観察されたタンパク質は、細胞内タンパク質プールのすべて(またはほとんど)を表すことを保証する、標的タンパク質をコードする遺伝子はまた、削除することができる(またはノックダウン) 。本質的には、エレクトロポレーションは、「移植」生きている細胞へのin vitroでのバイオコンジュゲートの柔軟性、したがって、合成生物学、システム生物学の取り組み、および生体内の単一分子検出に利益をもたらすことができます。
開示事項
The authors have nothing to disclose.
謝辞
We thank Stephan Uphoff for discussions.
R.C. was supported by Linacre College, Oxford University. A.P. was supported by the German Academic Exchange Service (DAAD), the German National Academic Foundation and EPSRC. M.S. was supported by the Wellcome Trust. A.N.K. was supported by a UK BBSRC grant (BB/H01795X/1), and a European Research Council Starter grant (261227).
資料
| Name | Company | Catalog Number | Comments |
| ElectroMax DH5-alpha Comptent cells | Invitrogen | 11319-019 | or any other commercial or lab-mage electrocompetant bacteria or yeast. |
| EZ Rich Defined Madia | Teknova | M2105 | low fluorescence rich media |
| MicroPulser Electroporation Apparatus | Biorad | 165-2100 | or any classical electroporator for microorganism transformation |
| Certified Molecular Biology agarose | Biorad | 161-3100 | low fluorescence agarose for agarose pad |
| Microscope coverslips No 1.5 thickness | Menzel | BB024060SC | remove background particles by heating slides in furnace at 500 °C for 1h |
| Single-molecule fluorescence microscope | Home-built | described in REFs | |
| Localization software | Custom-written, available online | MATLAB and C++ software package that can be adapted for localization analysis. | |
| Tracking software | Available online | MATLAB implementation by Blair and Dufresne. |
参考文献
- Tsien, R. Y. The green fluorescent protein. Annu Rev Biochem. 67, 509-544 (1998).
- Leake, M. C., et al. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature. 443, 355-358 (2006).
- Taniguchi, Y., Kawakami, M. Application of HaloTag protein to covalent immobilization of recombinant proteins for single molecule force spectroscopy. Langmuir. 26, 10433-10436 (2010).
- Xie, X. S., Choi, P. J., Li, G. W., Lee, N. K., Lia, G. Single-molecule approach to molecular biology in living bacterial cells. Annual review of biophysics. 37, 417-444 (2008).
- Lee, J. H., et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 343, 1360-1363 (2014).
- Miesenbock, G., De Angelis, D. A., Rothman, J. E. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 394, 192-195 (1998).
- Sauer, M. Localization microscopy coming of age: from concepts to biological impact. J Cell Sci. 126, 3505-3513 (2013).
- Dempsey, G. T., Vaughan, J. C., Hao Chen, K., Zhuang, X. Evaluation of fluorophores for optimal performance in localizationbased super-resolution imaging. Nat Meth. 8, 1027-1041 (2011).
- Shaner, N. C., Steinbach, P. A., Tsien, R. Y. A guide to choosing fluorescent proteins. Nat Meth. 2, 905-909 (2005).
- Landgraf, D., Okumus, B., Chien, P., Baker, T. A., Paulsson, J. Segregation of molecules at cell division reveals native protein localization. Nat. Methods. 9, 480-482 (2012).
- Jaitin, D. A., et al. Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science. 343, 776-779 (2014).
- Aldridge, S., et al. AHT-ChIP-seq: a completely automated robotic protocol for high-throughput chromatin immunoprecipitation. Genome Biol. 14, R124(2013).
- Keppler, A., et al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 21, 86-89 (2003).
- Wombacher, R., et al. Live-cell super-resolution imaging with trimethoprim conjugates. Nat. Methods. 7, 717-719 (2010).
- Zhang, Z., et al. A new strategy for the site-specific modification of proteins in vivo. Biochemistry. 42, 6735-6746 (2003).
- McNeil, P. L., Murphy, R. F., Lanni, F., Taylor, D. L. A method for incorporating macromolecules into adherent cells. J Cell Biol. 98, 1556-1564 (1984).
- Clarke, M. S., McNeil, P. L. Syringe loading introduces macromolecules into living mammalian cell cytosol. J Cell Sci. 102, 533-541 (1992).
- Sakon, J. J., Weninger, K. R. Detecting the conformation of individual proteins in live cells. Nat. Methods. 7, 203-205 (2010).
- Taylor, L. S. Electromagnetic syringe. IEEE Trans. Biomed. Eng. 25, 303-304 (1978).
- Dower, W. J., Miller, J. F., Ragsdale, C. W. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 16, 6127-6145 (1988).
- Neumann, E., Schaefer-Ridder, M., Wang, Y., Hofschneider, P. H. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J. 1, 841-845 (1982).
- Sustarsic, M., et al. Optimized delivery of fluorescently labeled proteins in live bacteria using electroporation. Histochem Cell Biol. , (2014).
- Tokunaga, M., Imamoto, N., Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods. 5, 159-161 (2008).
- Sinha, A., et al. A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature. 000, 1-5 (2014).
- English, B. P., et al. Single-molecule investigations of the stringent response machinery in living bacterial cells. Proc Natl Acad Sci U S A. 108, E365-E373 (2011).
- Crawford, R., et al. Long-lived intracellular single-molecule fluorescence using electroporated molecules. Biophys J. 105, 2439-2450 (2013).
- Uphoff, S., Reyes-Lamothe, R., Garza de Leon, F., Sherratt, D. J., Kapanidis, A. N. Single-molecule DNA repair in live bacteria. Proc Natl Acad Sci U S A. 110, 8063-8068 (2013).
- Uphoff, S., Sherratt, D. J., Kapanidis, A. N. Visualizing Protein-DNA Interactions in Live Bacterial Cells Using Photoactivated Single-molecule Tracking. J Vis Exp. , (2014).
- Hohlbein, J., Gryte, K., Heilemann, M., Kapanidis, A. N. Surfing on a new wave of single-molecule fluorescence methods. Phys Biol. 7, 031001(2010).
- Xie, X. S., Yu, J., Yang, W. Y. Perspective - Living cells as test tubes. Science. 312, 228-230 (2006).
- Santoso, Y., et al. Conformational transitions in DNA polymerase I revealed by single-molecule FRET. Proc Natl Acad Sci U S A. 107, 715-720 (2010).
- Hohlbein, J., et al. Conformational landscapes of DNA polymerase I and mutator derivatives establish fidelity checkpoints for nucleotide insertion. Nature communications. 4, 2131(2013).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved