
Method Article
Internalisation et l'Observation des Biomolécules Fluorescent In Living micro-organismes par électroporation
Dans cet article
Résumé
Studies of biomolecules in vivo are crucial for understanding molecular function in a biological context. Here we describe a novel method allowing the internalization of fluorescent biomolecules, such as DNA or proteins, into living microorganisms. Analysis of in vivo data recorded by fluorescence microscopy is also presented and discussed.
Résumé
The ability to study biomolecules in vivo is crucial for understanding their function in a biological context. One powerful approach involves fusing molecules of interest to fluorescent proteins such as GFP to study their expression, localization and function. However, GFP and its derivatives are significantly larger and less photostable than organic fluorophores generally used for in vitro experiments, and this can limit the scope of investigation.
We recently introduced a straightforward, versatile and high-throughput method based on electroporation, allowing the internalization of biomolecules labeled with organic fluorophores into living microorganisms. Here we describe how to use electroporation to internalize labeled DNA fragments or proteins into Escherichia coli and Saccharomyces cerevisiæ, how to quantify the number of internalized molecules using fluorescence microscopy, and how to quantify the viability of electroporated cells. Data can be acquired at the single-cell or single-molecule level using fluorescence or FRET. The possibility of internalizing non-labeled molecules that trigger a physiological observable response in vivo is also presented. Finally, strategies of optimization of the protocol for specific biological systems are discussed.
Introduction
La plupart des études de fluorescence à l'intérieur des cellules vivantes dépendent de fusions de protéines avec des protéines fluorescentes (MF), comme une GFP. Ces études permettent marqueurs fluorescents du nombre de copies, motif de diffusion ou la localisation des protéines impliquées dans des processus tels que l'expression du gène ou du transport membranaire 2-7. MF offrent la spécificité d'étiquetage haute, la mise en œuvre facile, et sont disponibles dans un grand inventaire des variantes avec divers photophysique et les propriétés chimiques 1. Cependant, fluorophores organiques restent le premier choix pour des expériences in vitro en raison de leur plus grande photostabilité (jusqu'à 100 fois plus stable que MF) 8,9, de petite taille (jusqu'à 100 fois plus petit volume que MF) et la facilité d'étiquetage intramoléculaire (principalement par l'utilisation de résidus de cysteine). Tous ces facteurs sont particulièrement importants pour la fluorescence seule molécule et FRET 10 études.
Plusieurs méthodes d'internalisation Combincompris les avantages de l'étiquetage biologique et dans la détection in vivo ont été introduites au cours de la dernière décennie; Cependant, ces méthodes soit emploient relativement importantes polypeptides balises (tags par exemple, TMP, HALO, ou 20 kDa SNAP) 11-14, nécessitent l'utilisation d'acides aminés non naturels 15, ou sont limités à grandes cellules eucaryotes, seule membrane (par exemple. , gratter chargement, seringue chargement, la micro-injection) 16-19.
Ce protocole décrit un roman, simple et à haut débit méthode d'internalisation que les couples les avantages de fluorophores organiques avec l'observation in vivo. Pour développer cette technique, nous avons adapté le procédé d'électroporation couramment utilisé pour transformer des cellules avec l'ADN plasmidique 20,21 afin de charger les micro-organismes tels que E. coli ou S. cerevisiae avec des biomolécules organiquement étiquetés. Le protocole se compose de quatre étapes simples: l'incubation des cellules avec des biomolécules marquées,l'électroporation, la récupération des cellules, et le lavage des cellules pour éliminer les biomolécules non-internalisé. Ici, nous présentons ce protocole d'électroporation, ainsi que les procédés d'imagerie cellulaire et d'analyse de données pour étudier la fluorescence à base de cellules et une seule molécule et FRET signaux.
L'électroporation de décharge repose sur un champ électrique à haute tension à travers une faible ionique suspension de cellules de force pour former des pores de la membrane à travers laquelle transitoire biomolécules peuvent pénétrer dans les cellules (figure 1) 20,21. Tout comme avec la transformation de bactéries ou de la levure avec de l'ADN plasmidique, les cellules doivent être préparés avant l'électroporation pour assurer leur electrocompetency. Cette procédure, composé de plusieurs étapes de lavage avec de l'eau, augmente la perméabilité de la membrane et réduit la force ionique de la solution de cellules pour éviter un arc électrique dans la cuvette d'électroporation. Dans ce protocole, les cellules peuvent être préparés comme décrit ci-dessous (voir le protocole: 1.1) ou acheté d'un fournisseur commercials.

Figure 1: Représentation schématique du protocole d'internalisation De gauche à droite:. Ajouter quelques microlitres de biomolécules marquées à l'aliquote de cellules électrocompétentes (fragments d'ADN doublement marquée et les bactéries dans cet exemple); incuber 1 à 10 minutes sur de la glace et à transférer une cuvette d'électroporation de pré-refroidi; électroporer puis ajouter 0,5 à 1 ml de milieu riche pour les cellules immédiatement après; incuber à 37 ° C (ou la température requise par l'organisme, par exemple, 29 ° C pour les levures) pour laisser les cellules se rétablir; effectuer des 5 étapes de lavage pour enlever l'excédent non-internalisées molécules marquées; remettre en suspension le culot final dans 100-200 pi de tampon PBS et pipette 10 ul sur un tampon d'agarose; couvrir le pad avec une lamelle nettoyé et image sur un microscope à fluorescence (en mode grand champ ou en mode HILO).
cellules électrocompétentes sont incubées avec les biomolécules marqués juste avant électroporation, qui peuvent être effectuées à l'aide électroporateurs standards trouvés dans la plupart des laboratoires de biochimie. Immédiatement après l'électroporation, les cellules sont incubées dans un milieu riche qui permet leur récupération avant de les laver (Figure 1). L'excès de biomolécules marquées non-internalisées est d'abord éliminé par lavage dans un tampon contenant une concentration relativement élevée de sel et un peu de détergent (Voir PROTOCOLE: 3,3). La présence de sel perturbe interactions électrostatiques non spécifiques formés par des biomolécules marquées non internalisés qui autrement peuvent coller sur la membrane externe. De même, la présence de détergent dans le tampon de lavage perturbe interactions hydrophobes non spécifiques.
Alors que l'ADN internalisation est simple (Figure 2), des précautions doivent être prises lors de l'internalisation des protéines marquées à l'aide de l'électroporation. Tout d'abord, le stock échantillon de protéines organiquement marqué pourrait encore contenir un petit pourcentage de colorant libre. Molécules de colorant libres sont beaucoup plus petites que les protéines et pourraient donc être internalisés préférentiellement. Pour se assurer que la grande majorité des molécules fluorescentes internalisés observées correspondent à la protéine d'intérêt, l'échantillon de protéine initiale doit contenir moins de 2% de colorant ~ libre (Figure 5) 22. L'excès de protéines marquées non-internalisées peut également tenir à la membrane cellulaire externe après l'électroporation; ce phénomène est spécifique à la protéine et doit être vérifié pour chaque nouvelle protéine. Nous vous proposons plusieurs options qui permettent l'élimination des protéines non-intériorisée de l'échantillon de cellules chargé (Voir PROTOCOLE: 3.3.3).
Enfin, les cellules sont remises en suspension dans un petit volume de tampon phosphate et un tampon pipette sur agarose, ce qui permet leur image sur un microscope à fluorescence. Immobilisation sur des patins agarose est un simple et moyen efficace de l'imagerie des cellules sur une lamelle sans nuire à leur intégrité. Le tampon doit contenir un milieu de culture à faible fluorescence.
Imagerie cellulaire peut être effectuée soit en champ large, totale fluorescence à réflexion interne (FRBR) ou en utilisant HILO (fortement inclinées et laminé feuille optique) microscopie. Dans la configuration HILO, le faisceau laser pénètre plus profondément dans l'échantillon que dans TIRF, mais ne se allume pas tant que la totalité de l'échantillon de grand champ, ce qui permet un plus grand rapport 23 signal-sur-bruit. En fonction de la résolution de la puissance du laser et le temps utilisé, les biomolécules peuvent être internalisés comptées (à l'aide de l'analyse pas à pas-photoblanchiment, figure 3), localisées, suivies ou 24-28. L'internalisation des constructions doublement marquée avec une paire de fluorophores de FRET permet la quantification de FRET tant au unicellulaire ou niveaux seule molécule (figure 6).
Différents paramètres peuvent être modifiésen fonction de la sortie souhaitée et le système biologique étudié. Tout d'abord, la quantité de matière internalisé par la cellule peut être réglée en modifiant la concentration de biomolécules marquées ajouté aux cellules avant l'électroporation (figure 2). l'intensité de champ d'électroporation aura aussi une influence à la fois l'efficacité de chargement et la viabilité des cellules; comme prévu, tandis que l'efficacité de chargement augmente avec l'intensité du champ, la viabilité des cellules électroporées diminue (figure 4A). Les deux paramètres peuvent être quantifiés en enregistrant le pourcentage de charge et la division des cellules après l'électroporation. Ce dosage de la viabilité couplée à l'imagerie de fluorescence vérifie également l'observation des biomolécules internalisés dans les cellules vivantes et permet une observation continue sur plusieurs générations (figure 4B).
En résumé, ce protocole permet l'internalisation des marquées par fluorescence des molécules ADN et de protéines dansE. coli ou S. 26 cerevisiae. Les molécules individuelles marquées avec des fluorophores organiques peuvent être suivis avec la résolution spatio-temporelle élevée pour des échelles de temps un ordre de grandeur plus de médecins de famille. Enfin, cette méthode est compatible avec grand champ, la FRBR et la détection confocale, ainsi que des systèmes d'excitation pulsés, comme ALEX (alternant excitation laser 28,29).
Protocole
1. Préparation de la cellule
- Préparation de bactéries électrocompétentes laboratoire faits
- Préparer une préculture d'une nuit 5 à 10 ml d'une seule colonie de la souche de E. coli d'intérêt dans un milieu à faible fluorescence tel que M9 ou EZ Rich milieu défini.
- Dans la matinée, inoculer une nouvelle culture de 400 ml avec la préculture nuit tels que la DO 600 nm commence à 0,02. Ajouter 400 ml de la faible fluorescence moyenne 2,5 ml de 1 M MgSO 4 et 2,5 ml de 1 M de MgCl2.
- Croître à 37 ° C et 250 tours par minute jusqu'à ce que la DO 600 nm atteint 0,4 à 0,6.
- Arrêter la croissance par refroidissement de la culture dans un bain d'eau glacée pendant 10 à 15 min.
Remarque: A partir de maintenant, effectuer toutes les étapes à 4 ° C (sur la glace). - Centrifuger la culture 15 min à 1000 x g. Jeter le surnageant et remettre en suspension le culot cellulaire dans 250 ml réfrigéré distillée stérile et H 2 O.
- Répétez la centrifugation et remise en suspension étapes tWice, en diminuant le volume d'eau à 100 ml, puis 50 ml.
- Centrifuger la culture 10 min à 1000 x g. Jeter le surnageant et remettre en suspension le culot cellulaire dans 25 ml réfrigérée et stérile distillée H 2 O + 10% de glycerol.
- Répéter les étapes centrifugation et remise en suspension trois fois, ce qui diminue le volume de solution de glycerol à 10% à 10 ml, 5 ml, et enfin à 500 ul.
- Aliquoter les cellules dans des aliquotes de 20 ul chacune, flash gel dans de l'azote liquide et conserver à -80 ° C.
- Cellules bactériennes électrocompétentes commerciaux
- Diluer une aliquote de la cellule commerciale de 1: 1 avec de l'eau distillée glacée stérile. Assurez aliquotes 20 pi et conserver à -80 ° C.
- Préparer la levure électrocompétentes
Remarque: électrocompétentes S. cerevisiae cellules sont préparées avant chaque expérience d'électroporation et ne peuvent être stockés à -80 ° C comme pour E. coli.- Pour commencer, inoculer 5Moyenne 0 ml YPD avec une seule colonie de la souche d'intérêt souhaité.
- Incuber à 30 ° C et 250 tours par minute jusqu'à ce que la DO 600 nm atteint 0,6 à 0,8.
- Centrifuger les cellules à 1000 g pendant 5 min à 4 ° C.
- Reprendre le culot dans 25 ml réfrigérés et stériles distillée H 2 O.
- Répétez les étapes de lavage remet en suspension deux fois dans 25 ml d'eau et remet en suspension deux fois dans 2 ml d'une solution refroidie de sorbitol à 1 M.
- Remettre en suspension les cellules dans 250 pi de 1 M de sorbitol et de diviser les cellules en 50 aliquotes.
2. Agarose préparation du pad
- Pour supprimer fond particules fluorescentes, graver une lamelle dans un four à 500 ° C pendant 1 heure. "Clean brûlé" lamelles peuvent être stockées pendant des semaines à la température ambiante couvert de papier d'aluminium.
Note: D'autres méthodes de nettoyage courants tels que le nettoyage au plasma ou une solution de piranha pourraient être utilisés tant que le fondfluorescence des diapositives nettoyés reste quasi-nulle. - Préparer une solution d'agarose à faible fluorescence par fusion dans un four à micro-ondes un agarose à 2% - solution de l'eau distillée (70 ° C). Immédiatement ajouter 500 pi de la solution de gélose à 2% clair pour 500 pi de 2X milieu de culture de fluorescence faible et mélanger doucement.
- Avant il se refroidit et se durcissent, Pipette rapidement cette agarose - solution à moyen sur une lamelle de microscope (Non 1,5 d'épaisseur) afin de former un bloc d'environ 2 cm de diamètre et une hauteur de quelques millimètres. Éviter les bulles et les faire éclater avec une pipette conseils si nécessaire.
- Aplatissez le pad avec une deuxième lamelle "brûlé" (Pas d'épaisseur 1.5, voir la figure 1).
Remarque: Cette lamelle supérieure contribue à la formation d'un tampon homogène plane et le protéger de la poussière et de séchage alors que les cellules sont en cours de préparation. Milieu minimal M9 ou comme milieu riche comme EZ Rich milieu défini ont été testés pour leur faible fluorescence.
3. Electroporation
- Incubation
- Ajoutez jusqu'à 5 pi de molécules marquées stockées dans un tampon pauvre en sel (<50 mM de sel) à une seule aliquote de cellules compétentes (bactéries 20 pi ou 50 pi de levure) et incuber 10 min sur la glace.
Remarque: la concentration de molécules marquées par fluorescence dans les solutions mères et ainsi la quantité de molécules marquées ajoutées à la cellule avant l'électroporation est directement corrélée à l'efficacité de chargement (Figure 3, et discussion). Étant donné que certaines protéines sont moins compatibles avec la condition de faible sel, la concentration de sel dans le tampon de stockage peut être augmentée, mais le volume de molécules marquées ajouté aux cellules avant l'électroporation doit alors être diminué. - Transférer le mélange de cellules et de biomolécules marquées dans une cuvette d'électroporation de pré-refroidie (0,1 et 0,2 cm espacement pour les bactéries et les levures, respectivement). Tapoter doucement la cuvette sur le banc pour éliminer les bulles potentiels de la solution.
- Placez le cuvette dans le électroporateur et appliquer une impulsion électrique à haute tension à la solution (de 0,9 à 1,8 kV / cm, voir la discussion pour plus de détails sur le choix de tension). Une telle impulsion forme des pores transitoires dans les membranes cellulaires permettant biomolécules marquées de diffuser dans les cellules.
- Vérifiez que la constante de temps affiché sur le électroporateur est entre 4-6 ms. La baisse des constantes de temps sont souvent dues à la concentration de sel trop élevée et / ou la présence de bulles dans la cuve, et conduiront à très faible ou nulle chargement des cellules.
- Ajoutez jusqu'à 5 pi de molécules marquées stockées dans un tampon pauvre en sel (<50 mM de sel) à une seule aliquote de cellules compétentes (bactéries 20 pi ou 50 pi de levure) et incuber 10 min sur la glace.
- Récupération
- Immédiatement après l'électroporation, ajouter 500 pi de milieu riche comme le SOC, Moyen EZ Rich définies, YPD ou tout milieu riche aux cellules.
- Incuber l'échantillon à 37 ° C pour les bactéries et 29 ° C pour les levures pendant 2 à 10 min. Pour les mesures de viabilité, où l'utilisateur souhaite évaluer le pourcentage de cellules en croissance et en division après l'électroporation, en utilisant un temps de récupération plus longue (jusqu'à 1 heure) en we observer ces temps de latence avant la première division cellulaire.
- Les étapes de lavage
- Laver les cellules pour enlever les biomolécules non intériorisé par filage bas les cellules pendant 1 min à 3300 xg et 4 ° C. Jeter le surnageant et remettre en suspension les cellules dans 500 ul de PBS.
Remarque: pour chaque échantillon, préparer un témoin négatif de cellules incubées avec la même quantité de biomolécules marquées, mais pas électroporation et lavées de la même manière que l'échantillon principal. - Répétez les étapes précédentes 3 fois.
- Dans le cas de l'internalisation protéine, d'optimiser le processus de lavage en fonction des propriétés et le comportement de la protéine marquée d'intérêt. Les étapes suivantes sont des exemples d'optimisations possibles:
- Effectuer les trois premiers cycles de lavage utilisant du PBS contenant 100 mM de NaCl et 0,005% de Triton X100 pour éliminer les protéines non intériorisé qui pourrait se en tenir à la membrane externe de la cellule 22, 26.
- Filtrer les cellules électroporées avec un filtre de diamètre de pore de 0,22 um monté à l'intérieur d'un tube de microcentrifugation de 1,5 ml à la pipette les cellules électroporées dans le filtre. Isoler pendant 3 min à 800 xg et 4 ° C. Jeter le accréditives. Ajouter 500 ul nouvelle PBS sur les cellules et les tourner une fois de plus comme avant et répéter ces étapes une fois 22.
- Ajouter une petite quantité de la protease K (10 ng dans 500 ul de PBS) pendant le premier cycle de lavage pour permettre la digestion de toute protéine non-internalisé.
- Effectuer les trois premiers cycles de lavage utilisant du PBS contenant 100 mM de NaCl et 0,005% de Triton X100 pour éliminer les protéines non intériorisé qui pourrait se en tenir à la membrane externe de la cellule 22, 26.
- Isoler les cellules pendant 1 minute à 3300 xg et 4 ° C. Jeter le surnageant et remettre en suspension les cellules dans 150 pi de PBS.
- Répartir la solution de cellules chargées sur le tampon d'agarose en enlevant la lamelle supérieure et 10 ul d'étalement de la goutte de suspension cellulaire par gouttelettes. Remplacer une lamelle propre et inutilisé brûlée (Pas d'épaisseur 1,5, correspondant au microscope de spécification objectif) sur til le dessus du pavé et appuyez très doucement sur la diapositive.
- Protéger les cellules électroporées de la lumière en stockant les plaquettes dans une boîte opaque tandis imagerie différents échantillons.
- Laver les cellules pour enlever les biomolécules non intériorisé par filage bas les cellules pendant 1 min à 3300 xg et 4 ° C. Jeter le surnageant et remettre en suspension les cellules dans 500 ul de PBS.
Acquisition de données 4. Microscope
Remarque: Single-cellule et une seule molécule microscopie de fluorescence en microorganismes vivants peuvent être effectuées sur ne importe quel microscope de fluorescence approprié (ou commercial construit sur mesure).
- Paramètres
- Widefield ou HILO éclairage
- échantillons d'image avec ne importe quel microscope FRBR / une seule molécule.
Remarque: Par exemple, nous utilisons dans le laboratoire d'un microscope inversé personnalisé avec un set-up de la FRBR. Poutres d'un 532-nm et une diode laser 637-nm sont combinés et collimatés avant de se concentrer sur le plan focal arrière de l'objectif. La fluorescence de l'échantillon est collecté à travers le même objectif, séparée de la lumière d'excitation au moyen d'un passe-long et un filtre coupe-bande, et divisé en récanaux d et vert en utilisant un miroir dichroïque. Les deux canaux sont imagés sur moitiés séparées de la puce d'un (EM-CCD) d'électrons-multipliant dispositif à couplage de charge. Les vidéos sont enregistrées en utilisant le mode cinétique. Des images de la lumière blanche sont obtenus en utilisant une lampe à lumière blanche et d'un condenseur fixée au microscope comme une source d'éclairage. - Pour l'observation générale molécule unique, réglez le mode du microscope pour la FRBR ou HILO 23 éclairage (voir discussion pour plus de détails sur la FRBR contre imagerie HILO). Pour définir un mode Hilo, sur un microscope de la FRBR, diminuer légèrement l'angle d'incidence de la lumière d'excitation de mettre l'accent légèrement supérieure à la surface de la lamelle (image de l'intérieur de la cellule plutôt que sa membrane inférieure en contact avec la lamelle, voir 4.5.4 ).
- Pour l'analyse du niveau de la cellule, à long seule molécule suivi des expériences ou par étapes analyse de photoblanchiment, réglez le mode du microscope d'éclairage à un ensuri de mode grand champng l'observation continue de l'ensemble du volume de la cellule et donc de toutes les molécules marquées internalisés.
- échantillons d'image avec ne importe quel microscope FRBR / une seule molécule.
- Typiquement, les pouvoirs utilisation d'excitation autour de 0,5-3 MW (~ 50 à 400 W / cm 2).
Remarque: des puissances laser inférieures sont utiles pour réaliser l'observation de fluorescence à long terme et le suivi (plus de 1 minute), alors que des puissances laser élevées peuvent être nécessaires pour la résolution spatio-temporelle plus ou analyse de photoblanchiment par étapes. - Utilisez des temps d'exposition allant de 15 ms pour le suivi des expériences à 100 ms pour l'observation plus générale et la quantification de l'intensité. Note: D'autres taux et les modes châssis peuvent être utilisés tels que l'éclairage stroboscopique, en particulier pour l'étude des espèces diffusantes rapide 30.
- Dans le microscope TIRF, enregistrer le canal de fluorescence sur un CCD multiplication d'électrons (EMCCD) caméra à un grossissement résultant en une longueur de pixel d'environ 100 nm / pixel. La configuration de la FRBR est décrit plus en détail dans la référence 26.
- Widefield ou HILO éclairage
- Acquisition de données
- Couper ou bloquer la lumière laser jusqu'à ce que le début de l'expérience. Mettez le gain de la caméra EMCCD pour éviter tout dommage à l'appareil photo en raison de la surexposition.
- Placez le agarose pad en sandwich à l'envers sur la platine du microscope, avec le côté recouvert de cellules vers le bas, afin d'apporter la cellule proche de l'objectif. Définir le focus sur les cellules en lumière transmise mode de microscopie 28. Enregistrer une image de chaque cellule de vue dans l'imagerie de la lumière blanche afin de localiser la cellule donne un aperçu avant d'éteindre la lumière blanche.
- Protéger l'échantillon de la lumière ambiante laboratoire.
- Pour le mode d'excitation HILO, ajuster l'angle du faisceau d'excitation pour chaque rapport signal-sur-bruit en éclairant uniquement la partie de l'échantillon à proximité de la surface de la lamelle.
- Pour obtenir un éclairage Hilo, focaliser le faisceau laser dans le plan focal arrière d'un 100x NA 1.4 Objectif 28 (nume supérieurouvertures riques tels que 1,45 ou 1,49 conviennent également). En déplaçant la lentille de focalisation perpendiculaire à la poutre, la mise au point se déplace à l'écart du centre objectif pour que le faisceau sort de l'objectif avec un angle.
- Ajuster la position de la lentille afin de maximiser le rapport signal sur bruit, l'intensité de fluorescence intracellulaire extracellulaire par rapport à un signal de fond.
- Mettez le gain de la caméra et commencer à l'acquisition de données avant d'allumer le laser.
- Lors de l'enregistrement des données, acquérir une image en lumière blanche de chaque FOV avant ou après l'enregistrement des données de fluorescence; Cela permet d'identifier les limites des cellules dans les canaux de fluorescence.
- Pour la viabilité estimation
- Utilisez un milieu riche faible fluorescence dans le tampon d'agarose pour permettre aux cellules de se développer après l'électroporation.
- Équilibrer le microscope à la température optimale pour le microorganisme étudié (37 ° C pour E. coli, 29 ° C pour S. cerevisiae) Avec un système de chauffage objectif.
- Enregistrez deux blancs légers et de fluorescence images toutes les 30 minutes, en veillant à rester sur exactement le même champ de vision lors de l'enregistrement de données entière. Un décalage de ≈1 heures est généralement observé avant que les cellules commencent à se diviser.
- Compter le nombre de biomolécules intériorisées par cellule
- Réglez la puissance du laser à des valeurs élevées (2-3 MW) et à long temps d'exposition (100 ms).
- Réglez l'éclairage en mode widefield pour éclairer la totalité de la cellule.
- Enregistrez des vidéos comme décrit dans l'étape 4.2 en veillant à enregistrer des images supplémentaires (de 50 à 100 frames) après l'achèvement de la photoblanchiment de fluorescence.
5. Analyse des données
- Analyse générale
- Analyser les images enregistrées et des films, à la fois dans la lumière blanche et de fluorescence excitations, en utilisant un logiciel d'imagerie, comme le ImageJ du logiciel libre.
- Dans ImageJ, ouvrez les images ou les films enregistrés sur le microscope (format TIF) dans Fichier> Ouvrir> Votre emplacement du fichier.
- Pour comparer qualitativement intensités de fluorescence sur un écran d'ordinateur, assurez-vous que toutes les images de fluorescence sont affichés avec les mêmes luminosité et le contraste dans Image> Réglages> Luminosité / Contraste. Ajuster manuellement ou automatiquement les paramètres pour une image sélectionnée, appuyez sur le bouton "Set" et sélectionnez l'option "Propager à toutes les autres images".
- Réglez le type d'informations à extraire: Analyse> Définir mesures, et sélectionnez (au moins) "Zone", "écart-type", "Min et valeur de gris max" et "moyennes valeur de gris".
- Pour comparer les intensités de fluorescence de la cellule, sélectionnez la zone s d'intérêt en utilisant le bouton de sélection Freehand des ImageJ et extraire les intensités de cellules dans Analyser> Mesure. Le tableau qui en résulte contient les valeurs de mesure etpeuvent être enregistrés et / ou copié à d'autres logiciels. La valeur "moyenne" correspond à l'intensité moyenne par pixel dans la région sélectionnée et peut être directement comparée entre les cellules ou entre une cellule et le fond.
- Dans un échantillon de cellules électroporation, une cellule est considéré comme chargé si son intensité moyenne par pixel est plus grande que l'intensité moyenne par pixel du contrôle négatif plus 3 fois son écart-type (Av (je ai chargé la cellule)> Av (je -EP) + 3 * StdDev (je -EP)).
- Construire fausse-couleur des images et des films fluorescence superposition afin d'évaluer la qualité et le chargement des échantillons.
- Dans ImageJ, images recouvrement tels que l'image de la lumière blanche et l'image de fluorescence correspondant à la même FOV dans Image> Couleurs> Fusionner les canaux. Sélectionnez une couleur pour chaque image (C4 (gris) pour la lumière blanche, C1 (rouge) pour le canal rouge, C2 (vert pour le canal vert ... etc.).
- Chèquesur l'image de superposition que la fluorescence est situé dans les limites de la cellule (image de lumière blanche) et que la fluorescence de fond est faible et homogène (sans taches lumineuses à l'extérieur des limites de la cellule).
- Avant d'analyser un grand nombre de cellules, de vérifier qualitativement les images correspondant à la échantillon négatif sont semblables aux images de cellules vides et afficher intensités beaucoup plus faibles que les cellules électroporées.
- Pour les expériences de viabilité, compter manuellement les pourcentages de la division, non-séparation, mais visiblement intacte (identique) et (morts) cellules endommagées du même champ de vision de plus en plus au fil du temps (voir 4.2.6).
- Évaluer la viabilité d'au moins 200 cellules par échantillon (électroporation, contrôle négatif et cellules vides) afin de recueillir suffisamment de statistiques.
- Analyser les images enregistrées et des films, à la fois dans la lumière blanche et de fluorescence excitations, en utilisant un logiciel d'imagerie, comme le ImageJ du logiciel libre.
- Analyse basée sur des cellules
- Compter le nombre de biomolécules intériorisées par cellule par analyse de photoblanchiment par étapes
- cellules sectoriels par selecting la zone d'intérêt en utilisant le bouton de sélection Freehand des ImageJ et dessiner une forme entourant précisément la cellule (l'équivalent de la membrane cellulaire).
- Extraire les intensités de cellules au fil du temps dans Image> Piles> axe Z Terrain profil. Le graphe résultant représente l'intensité moyenne par pixel pour la zone à l'intérieur de la limite de la cellule par rapport à chaque image du film résultant dans une courbe de photoblanchiment pour cette cellule spécifique. Il contient une décroissance exponentielle de l'intensité initiale de la cellule atteignant une asymptote inférieure (arrière-plan de fluorescence). Les valeurs de mesure et peuvent être enregistrés et / ou copiés vers un autre logiciel en cliquant sur "Enregistrer" ou "Copier".
- Copiez et collez les valeurs de blanchiment dans une colonne de tableur (je crus).
- Calculer la autofluorescence moyenne par pixel restant après photoblanchiment (I auto) faisant la moyenne des valeurs brutes I obtenus pour les 50 à 100 dernières images (de asymptote inférieure).
- Subtract l'auto-fluorescence moyenne par pixel restant après photoblanchiment pour cette cellule de la courbe de photoblanchiment initiale: Je blanchiment = I première - je automobile.
- Utilisez référence soustrait photoblanchiment TimeTraces (je blanchiment par rapport cadre) montrant moins de 10 étapes quantifiés pour évaluer la taille de l'échelon moyen (intensité de fluorophore unitaire) en raison de la décoloration d'un seul fluorophore 26.
- Évaluer le nombre de molécules par cellule internalisés en divisant l'intensité de cellule de ligne de base soustraite initial (I blanchiment à t = 0) par l'intensité du fluorophore unitaire.
- L'efficacité de FRET seule cellule
- Mesurer l'intensité moyenne des cellules par pixel dans les deux canaux donneur et accepteur émission (sur donateurs excitation) et dans la limite de cellule pour chaque canal comme expliquer en 5.1.1.4.
- Mesurer l'intensité moyenne de pixel pour le fond dans chaque chAnnel d'une zone vide de la diapositive.
- Soustraire cette intensité de fond de l'intensité moyenne par pixel. Utilisez ces intensités de fluorescence fond soustrait pour calculer FRET pour chaque cellule en calculant l'intensité accepteur fond soustrait divisé par le total (+ accepteur donneur) intensité de fond soustrait lors de donateurs excitation: Je accepteur / (I + I accepteur donneur)
- Compter le nombre de biomolécules intériorisées par cellule par analyse de photoblanchiment par étapes
- Analyse une seule molécule
- Suivi une seule molécule et l'analyse de diffusion
Remarque: Le protocole de suivi de diffusion des molécules fluorescentes dans les cellules vivantes et d'évaluer leur coefficient apparent de diffusion a été décrite 26, 28.- En bref, adapter les images de fluorophores simples dans chaque image par une gaussienne elliptique 2D. Lien localisée molécules à une piste se ils sont apparus dans des trames consécutives dans une fenêtre de 5-7 pixels (de 0,48 à 0,67 um). Utilisez un memory paramètre de 1 cadre pour tenir compte de la disparition transitoire d'un fluorophore en raison de clignotant ou la localisation manquer.
- Analyse in vivo smFRET
- Identifier manuellement molécules localisées à l'intérieur de cellules à partir de films en passant par un film dans ImageJ et identifier des molécules immobiles (ou assez immobiles) dans le (accepteur) canal de FRET.
- Pour extraire les intensités dans le canal accepteur et donneur correspondant à une molécule immobile, sélectionnez la zone autour de la molécule dans chaque canal en utilisant le bouton de sélection "Oval" de ImageJ (cercle autour de chaque fluorophore unique, en utilisant un rayon ~3-pixel) et extraire les intensités de molécules dans Analyser> Mesure. Le tableau qui en résulte contient les valeurs de mesure et peut être sauvegardé et / ou copié à d'autres logiciels.
- Calculer les valeurs de fond par canal de l'intensité moyenne de pixel d'un cercle de la même taille dans une zone vide de la diapositive sur toutes les trames analysées.
- Utilisez des valeurs de fluorescence fond soustrait dans les canaux de donneur et accepteur (sur donateurs excitation) pour la fluorescence et FRET traces de temps, comme dans le seul cellulaire FRET cas (voir 5.2.1.7).
Remarque: l'analyse et des algorithmes automatisés et robuste ont été décrits dans les références 26 à 28, 31.
- Suivi une seule molécule et l'analyse de diffusion
Résultats
La préparation des échantillons
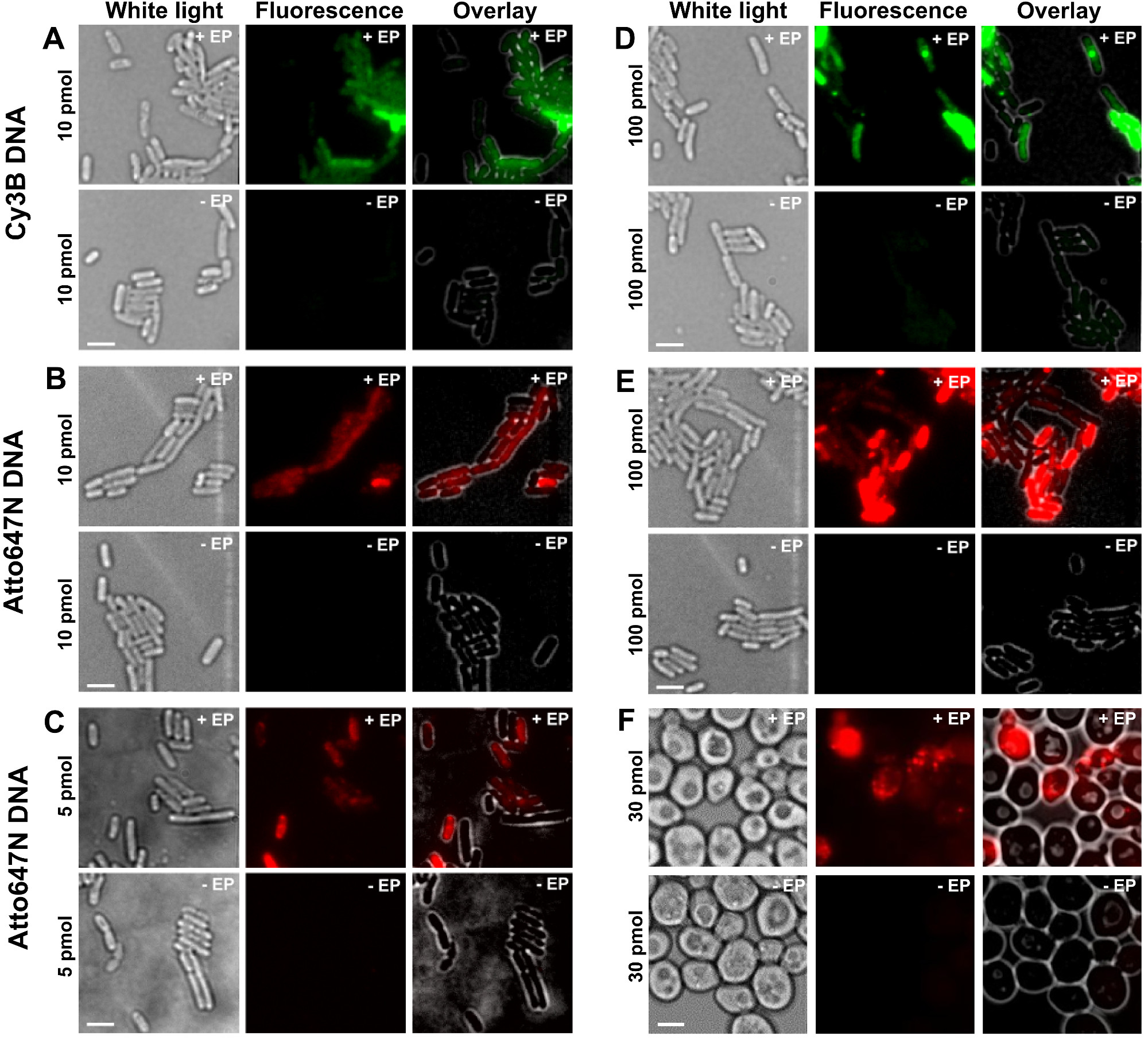
Les différentes étapes du protocole sont présentés comme des schémas de la figure 1. Par exemple, nous avons représenté le chargement des bactéries avec doublement marquée (donneur et colorants accepteurs) fragments d'ADN. Les résultats représentatifs pour l'internalisation ADN sont présentés à la figure 2. Pour chaque échantillon, une électroporation, les données pour les cellules vides et les cellules non-électroporées ont également été enregistrées (figure 2). "Cellules vides" correspondent aux cellules électrocompétentes ni incubées avec des biomolécules fluorescentes ni électroporation; leur intensité dans le canal de fluorescence reflète le niveau d'autofluorescence dans des conditions expérimentales identiques (puissance du laser, la résolution de temps, température, etc.). "cellules non électroporés" (également appelé -EP, ce est à dire, moins EP) correspondent à un témoin négatif dans lequel les cellules électrocompétentes ont été incubées avec la biomolec fluorescentules mais pas électroporation. Ces cellules non électroporés doivent présenter un niveau de fluorescence proche de l'autofluorescence des cellules vides et significativement plus faible que l'intensité de fluorescence affichée par les cellules chargées, électroporation. Cela confirme l'élimination de tous les biomolécules marquées non internalisés qui aurait adhéré à la membrane cellulaire externe.

Figure 2: Les résultats représentatifs pour l'internalisation des ADNdb marquées avec des fluorophores à différentes concentrations en bactéries (AE) et de levure (F) De gauche à droite:. Lumière blanche, fluorescence et de superposer des images. - / + EP désigne incubation sans / avec électroporation. Barres d'échelle: 3 pm. A. CY3B ADN double brin, 10 pmol, E. coli. B. ATTO647N ADN double brin, 10 pmol, E. coli. C. Alexa647 ADNdb, 5 pmol, E. coli. D. CY3B ADNdb, 100 pmol, E. coli. E. ds ATTO647NADN, 100 pmoles, E. coli. F. ATTO647N ADN double brin, 30 pmol, levure. Ce chiffre a été modifié de la référence 26. Se il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
Compter le nombre de biomolécules intériorisées par cellule
La procédure d'estimer le nombre de biomolécules marquées internalisées par cellule en utilisant une analyse photoblanchiment est présentée en figure 3 et deux films supplémentaires, ainsi que des résultats représentatifs obtenus avec différentes concentrations d'ADN marqué. Chargement des cellules efficacité augmente avec le montant initial de l'ADN marqué incubés, permettant à l'utilisateur de régler le nombre de molécules marquées par cellule d'un niveau "molécule unique" (<10, complémentaire Film 2B) à un niveau "d'ensemble" (> 10 , 2A Movie supplémentaire). Une méthode robuste d'estimation du pourcentage de cellules chargées est to compter le nombre de cellules électroporées présentant une intensité de cellule au-dessus de l'intensité moyenne de cellule des cellules non électroporés plus 3 fois l'écart de leur type (à savoir, Av (I-EP) + 3 * StdDev (I-EP) où Av = moyenne, I = intensité par pixel, Std. Dev. = écart type et -EP = non électroporés), comme illustré sur la figure 3.

Figure 3:. Compter le nombre de molécules internalisées en utilisant l'analyse photoblanchiment (A) unicellulaire photoblanchiment analyse. Des exemples de TimeTraces d'intensité de fluorescence (bleu: données brutes; rouge: ajustement; encarts: WL et images de fluorescence de E. coli chargé avec ATTO647N marqué ADNdb avant et après le blanchiment). Haut: une seule étape événement de blanchiment. Moyen: cellule contenant ± 3 molécules montrant blanchimentet clignote. Bottom: cellule contenant> 10 étapes correspondant à au moins 10 molécules (B) de l'histogramme des intensités seule étape de hauteur à partir d'un algorithme d'ajustement de l'étape-automatisé à partir de 57 cellules contenant moins de six étapes distinctes.. Single-forme gaussienne est centrée à 11 ± 3 au, correspondant à une intensité de fluorophore unitaire de 8100 photons par seconde. L'astérisque marque la collecte toutes les hauteurs étape supérieure ou égale au 50 (C) histogramme des molécules internalisées par cellule par électroporation avec différentes quantités d'ADN double brin ATTO647N, calculées après divisant l'intensité de fluorescence initiale par l'intensité de fluorophore unitaire bin. De haut en bas: les cellules vides (ce est à dire, non incubées avec des molécules fluorescentes et non électroporés), non électroporés (mais incubées avec des molécules fluorescentes, nommé -ep), et incubées avec des cellules électroporées 10 et 100 pmol ADN double brin (appelé + EP). Les cellules vides et non-électroporation correspondentà autofluorescence, tandis que les cellules électroporées montrent une large distribution des molécules internalisées, avec une proportion plus élevée de cellules hautement chargés à 100 pmol (≥ 4 molécules, voir bin astérisque marqué). l'efficacité d'internalisation (fraction de cellules avec Int.> moyenne + 3x Std. Dev. Extrait de non -EP) pour les échantillons 10 et 100 pmol était de 94% et 90%, respectivement. 121 ± 106 molécules pour 10 pmol ADN double brin, et 176 ± 187 molécules pour 100 pmol ADNdb: nombre de molécules internalisées par cellule moyenne. Réglages: 100 ms exposition, éclairage grand champ. Barres d'échelle: 1 um. Ce chiffre a été modifié de la référence 26. Se il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
Chargement des cellules et la viabilité
En plus de modifier la quantité de biomolécules marquées ajouté aux cellules avant l'électroporation, lal'utilisateur peut régler la quantité de molécules internalisées en choisissant différentes forces sur le terrain lors de l'électroporation (Figure 4, Film supplémentaire 1). Une plus grande force sur le terrain conduisent à une plus grande efficacité d'internalisation, mais conduisent à une légère diminution de la viabilité cellulaire. Pour les protéines internalisation, l'utilisation d'une étape de filtration peut aider à éliminer les non-internalisées protéines marquées (voir 3.3.3.1.1). Dans de tels cas, la filtration de la cellule se assure que les protéines fluorescentes sont observées en effet internalisé à l'intérieur du cytoplasme bactérien; Nous notons, toutefois, que la filtration a également un impact négatif sur la viabilité cellulaire (pour plus de détails, voir REF 22).

Figure 4: Influence de la tension d'électroporation lors du chargement et la viabilité cellulaire (A) Diagramme à barres représentant l'effet du champ d'électroporation sur le chargement effic.iency (barres rouges) et la viabilité des cellules (barres vertes). 84 ± 8% de la cellule non-électroporation (0 kV / cm) fracture après 1 h sur un tampon d'agarose à 37 ° C. Dans les mêmes conditions, 78 ± 3% et 49 ± 3% des cellules électroporées à 0,9 kV / cm et 1,8 kV / cm divisent respectivement après 1 h. Pour une efficacité de chargement, 73 ± 8% des cellules sont chargées à 0,9 kV / cm, tandis que 92 ± 6% des cellules sont chargées à 1,8 kV / cm. Les barres d'erreur représentent l'écart-type calculé à partir de trois mesures indépendantes; plus de 200 cellules ont été analysées pour chaque échantillon et chaque répétition. Augmentation globale de l'efficacité de chargement avec une tension d'électroporation de la légère détriment de la viabilité des cellules. (B) des mesures de fluorescence à base de cellules sur plusieurs générations montrent que l'intensité globale de fluorescence est partagé à parts égales entre les deux cellules filles. Cellule 1 et 2 se réfère au nombre de cellules dans l'image de la lumière blanche (à gauche) et l'image de fluorescence à t = 0. Echellebar: 1 um). Ce chiffre a été modifié de la référence 26.
Protein internalisation
Les résultats représentatifs pour la protéine internalisation sont les figures 5A & B. Il est particulièrement important d'éliminer autant du colorant libre (ne ayant pas réagi) restant de l'échantillon de protéine que possible avant l'électroporation. Dans les exemples des figures 5A et B, l'échantillon marqué CY3B fragment de Klenow (CY3B-KF, où KF est le fragment de Klenow de l'ADN de E. coli polymerase I, 66 kDa) ne contient que 1% de colorant libre; cette contribution de teinture à la charge globale de la cellule est négligeable. La comparaison de l'échantillon d'intérêt avec une électroporation à la fois les cellules non électroporés (incubées avec la même quantité de protéine marquée) ainsi que les cellules électroporées avec la quantité équivalente de colorant libre constituent deux contrôles nécessaires pour se assurer que les molécules fluorescentes observéessont bien intériorisé protéines marquées.

Figure 5: Protein internalisation de bactéries vivantes (A) représentant sur le terrain de vues de recouvrement de fluorescence.. Électroporation des cellules à la tension 1,4 kV avec la sous-unité de 50 pmol RNAP partir d'une solution de protéine de stock qui ne contenait que 1% sans colorant CY3B. Non électroporés (non -EP) et des cellules vides sont définis comme précédemment. Colorant libre a été intériorisée à la même concentration que dans l'échantillon électroporation l'RNAP de. Imagerie en mode champ large, 532-nm excitation à 1 mW, 50 ms exposition. (B) Répartition des intensités moyenne de cellules non corrigées pour les échantillons (A), donnée en proportion du nombre total de cellules. Plus de 400 cellules par échantillon ont été segmentés. Ce chiffre a été modifié de la référence 22. (C) L'internalisation des UNLAbeled l'ARN polymerase T7 (T7 RNAP, 98 kDa) dans électrocompétentes DH5a portant le plasmide codant pour la GFP émeraude pRSET-EmGFP (EmGFP) sous le contrôle d'un promoteur de T7. Gauche: Schéma d'essai. Moyen: superposition fluorescence. Droite: histogrammes des intensités de fluorescence à base de cellules pour l'échantillon non-électroporation (en haut) et les cellules incubées et électroporation avec T7 RNAP (en bas); environ 11% des cellules électroporées montrent l'intensité de fluorescence élevé (fraction de cellules avec Int.> signifie + 3x Std. Dev. de non échantillon de -EP) indiquant expression de EmGFP. Les astérisques indiquent bacs recueillir toutes les intensités supérieure ou égale barre d'échelle de 1100 au: 3 um. Ce chiffre a été modifié de la référence 26.
La figure 5C présente une autre demande de protéine électroporation. Ici, la protéine électroporation est non marqué, mais son internalisation déclenche une réponse fluorescente observable. Cette expérience confirme la présence et la fonctionnalité des protéines électroporées dans le cytoplasme de la cellule. T7 ARN polymérase non marqué (98 kDa) a été internalisé dans E. coli souche DH5a contenant un plasmide codant pour la protéine fluorescente EmGFP sous le contrôle d'un 26 promoteur T7. Comme le gène de l'ARN polymerase T7 est absent dans DH5a, EmGFP expression dans nos expériences nécessite que fonctionnelle de l'ARN polymerase de T7 est introduit dans les cellules par électroporation (figure 5C). Après l'électroporation avec 1 pmol T7 RNAP,> 11% des cellules (barre bleue, figure 5C) présentait une fluorescence plus élevé que le contrôle négatif (incubées avec la même quantité de T7 RNAP, mais pas électroporation). Ce résultat démontre que la proportion des molécules T7 RNAP internalisées par électroporation conservent leur intégrité et in vivo peuvent remplir leurs fonctions prévues dans le cytoplasme de la cellule.
In vivo FRET à la seule moléculeet les niveaux unicellulaires
Enfin, l'internalisation et l'analyse des espèces doublement marquée dans les bactéries vivant est présenté dans la figure 6 et Movie supplémentaire 3. fusions de protéines fluorescentes Comme ne sont pas idéales pour les études in vivo smFRET, la capacité de livrer des biomolécules doublement marquée dans des cellules vivantes utilisant l'électroporation est une des grands atouts de cette méthode. Figure 6A présente une seule cellule FRET analyse des bactéries chargées de différentes normes FRET ADN (utilisant CY3B et fluorophores Atto647N comme paire donneur-accepteur de FRET). Les cellules sont électroporées avec 20 pmoles d'ADNdb trois normes FRET courts doublement marquées avec l'efficacité de FRET apparentes (E *) de 0,17, 0,48, 0,86 et in vitro (préalablement déterminées 26). Tous ADN pénètrent dans les cellules de manière efficace (figure 6A, à gauche) et le pic principal de chaque E distribution d'une seule cellule de * accorde bien avec les résultats in vitro (figure 6A, À droite). Dans les échantillons intermédiaire et élevée FRET, les populations cellulaires avec inférieure E * que prévu sont observées, probablement en raison d'une combinaison d'accepteur blanchiment et l'inactivité photophysique, la cellule variable chargement (ainsi, à rapport variable signal-bruit) et dégradation de l'ADN .

Figure 6:.. Les résultats représentatifs pour une seule cellule et une seule molécule FRET observation dans des bactéries vivant études Ensemble et smFRET dans les bactéries simples (A) Analyse des cellules chargées avec 20 pmol de chacune des trois normes ADN FRET présentant faible (~ 0,17), intermédiaire (~ 0,48), et élevé (~ 0,86) FRET (mesurée en utilisant des mesures in vitro une seule molécule, voir REF 26). Gauche: la lumière blanche et rouge vert / images (FRET) fluorescence de superposition (échelle de bar: 3 pm). Des exemples de valeurs de FRET provenant de cellules différentes sont indiquées (blanc). Gréement ht (de haut en bas):. non corrigée FRET à base de cellules (E *) histogrammes pour les donateurs seulement (vert foncé), faible (vert clair), intermédiaire (jaune) et haute (rouge) FRET normes d'ADN (B-D) Dans smFRET vivo. Les cellules sont chargées avec FRET intermédiaire ADN 0,25 pmol (panneau B), 0,25 pmol haute-FRET ADN (groupe C), et 5 pmol doublement marquée KF (panneau D). Colonne de gauche: rouge vert fluorescence superposition / d'une seule image avant et après accepteur photoblanchiment. Moyen colonne: traces de temps correspondant à la molécule dans le cercle jaune. FRET efficacité, l'intensité des émissions des donateurs, et des intensités accepteur d'émissions sont affichés en bleu, vert et rouge, respectivement. Colonne de droite: histogrammes de FRET de donateurs que des molécules (vert) et des molécules donneur-accepteur (jaune, rouge, et gris) à partir de 20 traces de temps pour chaque échantillon. Barres d'échelle: 3 pm A, 1 pm B-D. Ce chiffre a été modifié de la référence 26.208fig6large.jpg "target =" _ blank "> Se il vous plaît cliquer ici pour voir une version plus grande de cette figure.
Pour observer smFRET in vivo pour échantillons ADN ou de protéines, de faibles quantités (0,25 pmol) de normes ADN intermédiaire et élevée FRET (figure 6B, C) ou 5 pmol de fluorophores doublement marquées KF (Alexa647 / CY3B que paire de FRET, Figure 6D) sont soumis à une électroporation dans E. coli. Ces concentrations ont conduit à de nombreuses cellules chargées avec quelques (n = 1-10) molécules marquées, permettant la localisation directe, de suivi et de surveillance pour FRET molécules simples. Certaines molécules diffusent librement, tandis que d'autres semblent immobile ou diffusent lentement (Film supplémentaire 3). TimeTraces de biomolécules immobilisées doublement marquée (Figure 6, milieu) durent de 1 à 30 s et de montrer les caractéristiques de smFRET: changements anticorrelated dans la fluorescence du donneur et accepteur sur accepteur blanchiment (par exemple, t ~ 16 sec; la figure 6B, milieu ), suivi par simple-Étape blanchiment donneur (par exemple, ~ t; 19 sec; Figure 6B). FRET distributions générés par ces TimeTraces (figure 6, à droite) résultat dans une valeur moyenne qui est en excellent accord avec publié des études in vitro 26,31,32. Ces résultats établissent la capacité pour les études de smFRET quantitatives sur les ADN et les protéines intériorisées, et suggèrent que les protéines conservent leur intégrité et de la structure lors de l'électroporation et l'internalisation (soutenue par les expériences T7 RNAP d'internalisation).
Film supplémentaire 1:. La viabilité cellulaire Gauche: images lumière blanche. Images de fluorescence: Droit. GIF animé d'animation montrant la division après l'électroporation (1,8 kV / cm) de bactéries chargées de l'ADN marqué 10 pmol de Atto647. La diminution apparente globale de fluorescence est dû à la dilution de l'ADN marqué à la division cellulaire et aussi partiellement au photoblanchiment qui se produit pendant chaquemesure.
Film complémentaire 2: études de photoblanchiment à base de cellules A.. Un exemple représentatif d'une cellule lourdement chargé (contenant> 100 molécules d'ADN Atto647N marqué). En haut à gauche, une image blanche de lumière de la cellule d'intérêt (rectangle rouge). En haut à droite, le film des cellules chargées montrant leur décroissance de la fluorescence sur plusieurs minutes. En bas, trace de temps de la décroissance globale de l'intensité de fluorescence de la cellule d'intérêt. Fluorophores organiques peuvent présenter photoblanchiment vies deux ordres de grandeur plus élevé que les médecins de famille (ici, ~ 41 sec pour Atto647N). B. Exemple représentatif d'une cellule chargée de moins de 10 molécules marquées (trois dans ce cas). Haut, même que le panneau A. bas, trace de temps de l'intensité de fluorescence globale de la cellule d'intérêt montrant simple blanchiment de l'étape et / ou clignotant correspondant à des fluorophores organiques simples. La hauteur moyenne de ces étapes correspond à l'intensité unitaire seule molécule (ici ~12 au) utilisée pour estimer le nombre initial de molécules internalisées par cellule. Films sous excitation continue laser rouge à une puissance de 300 mW et 100 ms par image.
Film supplémentaire 3: Je n vivo seule molécule FRET Haut:. Les cellules chargées avec 0,25 pmol haute-FRET ADN (comme dans la figure 6C) surveillé en continu à 50 ms par image sous éclairage nTIRF utilisant 1 mW vert (532 nm) laser. Chaque cadre est d'un vert rouge (FRET) fluorescence superposition / de chaque canal. Diffusion et rouge immobile (intact) et vert (seule étiquette actif) molécules d'ADN peuvent être observées. Bottom: Temps trace correspondant à la molécule dans le cercle jaune. FRET efficacité, l'intensité des émissions des donateurs, et des intensités accepteur d'émissions sont affichés en bleu, vert et rouge, respectivement. Événement anti-corrélé blanchiment accepteur (transitions rouge au vert) correspond à la signature d'une seule molécule FRET.
Discussion
De nombreux paramètres peuvent être modifiés pendant l'électroporation des cellules et d'acquisition de données selon le système biologique d'intérêt et la nature précise de l'expérience (au niveau de la cellule ou molécule unique analyse). Par exemple, lorsque l'ADN par électroporation dans des bactéries, 0,25 à 5 pmoles de fragments d'ADN double brin marqués conduit à une faible efficacité d'internalisation, ce qui permet la détection directe seule molécule (par exemple., Sans avoir besoin de photoblanchiment à l'avance). Au-dessus de 5 pmol ADN double brin, les cellules ont tendance à être lourdement chargé, un régime mieux adapté pour l'analyse unicellulaire. Tous les ADN marqués devraient également être préalablement purifié sur gel pour éliminer toute trace de colorant libre (fluorophore ne ayant pas réagi) de la solution ADN stock. En outre, les problèmes potentiels avec la dégradation de l'ADN, en particulier pour des expériences smFRET, peuvent être réglés par des ADN avec des acides nucléiques non naturels, ou des motifs qui protègent exonucléase accessible terminaisons tels que des boucles en épingle à cheveux.
Un autre adjustable paramètre dans électroporation est l'intensité du champ appliqué pendant l'électroporation. Faible intensité de champ (~ 1 kV / cm) conduira à une faible efficacité de chargement approprié pour les études à molécule unique. Intensités de champ plus élevée (jusqu'à 1,8 kV / cm) augmenteront l'efficacité de chargement; Cependant, il existe une corrélation inverse entre l'intensité du champ et la viabilité des cellules après l'électroporation (voir figure 4). Pour référence, une intensité de champ normal utilisé pour les bactéries et la levure est une électroporation à environ 1,5 kV / cm. La constante de temps représentant la durée de cette décomposition, est un paramètre pratique à suivre, car les gouttes de constante de temps dès que tout phénomène d'arc se produit dans la cuvette. D'après les réglages normaux, la constante de temps doit être supérieure à 4 ms; des valeurs inférieures conduiront à une faible efficacité de chargement ou de cellules endommagées, même non-chargées. La plupart des électroporateurs offrent d'autres degrés de liberté (comme "troncature d'impulsion» ou «forme des impulsions") qui peut être modifiée pour accorder à la foisle chargement et la viabilité cellulaire. Nous avons appliqué cette méthode à la fois les bactéries et les levures, cependant procédures similaires devraient également permettre l'internalisation des biomolécules marquées dans les cellules de mammifère en utilisant les paramètres de électroporateur appropriées depuis leur membrane est en fait moins complexe (unique bicouche lipidique) et depuis l'électroporation a déjà été utilisé avec de telles cellules 21.
Lorsque l'internalisation des protéines marquées, tout colorant libre doit être retiré de la solution de protéine marquée des stocks électroporation préalable. Molécules de colorant libres, en raison de leur petite taille, peuvent être internalisés préférentiellement au cours des protéines d'intérêt, et sont difficiles à distinguer lors de l'analyse des données (malgré leur diffusion devrait plus rapide). A titre indicatif, pour un échantillon biologique de la protéine marquée pour être adapté à l'électroporation, la quantité de colorant restant libre doit être inférieure à 2% (balayage à fluorescence détectée à l'aide d'une SDS-PAGE) 22. Ce procédé est particulièrement importante,que certaines molécules pourraient tenir à des membranes externes des bactéries ou des levures par électroporation. À cet égard, l'échantillon de contrôle négatif doit afficher intensité de fluorescence par cellule nettement plus faible que les cellules électroporées, idéalement aussi bas que le niveau de cellules vides (cellules qui ne ont pas été incubées avec des biomolécules marquées par fluorescence, ni électroporation, figure 2) autofluorescence.
Comme avec l'ADNdb, l'efficacité d'internalisation des protéines marquées est liée à la quantité de biomolécules ajouté aux cellules avant l'électroporation. Cependant, d'autres paramètres, comme la taille et la charge, jouent un rôle dans l'internalisation. Les petites protéines présentent des rendements élevés d'internalisation, tandis que les protéines plus grandes (jusqu'à 98 kDa) peut être internalisé avec succès, mais avec une efficacité plus faible (figure 5) 26. Le point isoélectrique de la protéine, les interactions potentielles avec la membrane cellulaire et d'autres paramètres physico-chimiques aussiinfluence chargement cellulaire pendant électroporation. En conséquence, les utilisateurs ont besoin pour optimiser les expériences de leur propre système, sachant qu'une forte concentration initiale de protéine marquée (> 50 M) donnera la meilleure chance pour le chargement réussi. Électroporation propose également un nouvel outil de perturber et d'analyser la fonction cellulaire en introduisant des protéines et d'autres biomolécules dans les cellules (soit marqué ou non marqué). Les expériences de la T7 ARN polymerase (Figure 5C) présentent une telle exemples d'une expérience dans laquelle on peut introduire une biomolécule qui peut modifier l'expression de gènes in vivo en utilisant l'électroporation.
Lorsque la réalisation d'expériences de fluorescence seule molécule, illumination TIR est généralement privilégiée par rapport à d'autres modes d'éclairage car il offre le meilleur rapport signal-sur-bruit en excitant seulement fluorophores dans une section mince dessus de la surface de lamelle (~ 100 nm). Cependant imagerie de diffusion à l'intérieur des biomolécules marquées micro-organismes vivants peuvent reexiger un éclairage plus profond (jusqu'à 0,8 um pour E. coli). Deeper illumination est réalisée en mode Hilo, tout en préservant un rapport signal-bruit. D'autre part, l'imagerie à grand champ est particulièrement important pour l'analyse de photoblanchiment étapes, où l'utilisateur est l'estimation du nombre de molécules internalisées par photoblanchiment toute une cellule chargée avec une puissance laser élevée et en divisant l'intensité initiale de la fluorescence de la cellule par l'intensité unitaire produite par une seule molécule (l'étape de photoblanchiment unique, figure 3). Imagerie Widefield est également nécessaire pour le suivi de la molécule à long terme afin de localiser les molécules diffusantes d'intérêt même si leurs trajectoires couvrent le volume de cellule entière.
Dans ce protocole, nous présentons comment électroporation, une technique standard pour les biologistes et de biochimistes pour délivrer des acides nucléiques dans les cellules, constitue une méthode simple pour délivrer biomolécules fluorescentes dans divers types de cellules. Thest nouvelle technique à haut débit offre un outil unique d'observer des molécules marquées dans leur environnement natif. En d'addition biomolécules marquées avec des fluorophores couvrant une large gamme de longueurs d'onde, l'électroporation peut délivrer des molécules modifiées par de nombreux groupes chimiques, tels que des nucleotides et des acides aminés non naturels, des chélateurs de métaux, des agents de réticulation, et les groupes de mise en cage. Si le système biologique d'intérêt ne est pas indispensable au développement de la cellule, le gène codant pour la protéine cible peut également être supprimée (ou knocked-down), se assurer que les protéines observées après internalisation représentent tous (ou presque) de la piscine de protéine intracellulaire . En substance, l'électroporation peut "greffe" la flexibilité de bioconjugués in vitro dans des cellules vivantes et donc bénéficier des efforts de la biologie synthétique, la biologie des systèmes, et dans la détection de molécule unique vivo.
Déclarations de divulgation
The authors have nothing to disclose.
Remerciements
We thank Stephan Uphoff for discussions.
R.C. was supported by Linacre College, Oxford University. A.P. was supported by the German Academic Exchange Service (DAAD), the German National Academic Foundation and EPSRC. M.S. was supported by the Wellcome Trust. A.N.K. was supported by a UK BBSRC grant (BB/H01795X/1), and a European Research Council Starter grant (261227).
matériels
| Name | Company | Catalog Number | Comments |
| ElectroMax DH5-alpha Comptent cells | Invitrogen | 11319-019 | or any other commercial or lab-mage electrocompetant bacteria or yeast. |
| EZ Rich Defined Madia | Teknova | M2105 | low fluorescence rich media |
| MicroPulser Electroporation Apparatus | Biorad | 165-2100 | or any classical electroporator for microorganism transformation |
| Certified Molecular Biology agarose | Biorad | 161-3100 | low fluorescence agarose for agarose pad |
| Microscope coverslips No 1.5 thickness | Menzel | BB024060SC | remove background particles by heating slides in furnace at 500 °C for 1h |
| Single-molecule fluorescence microscope | Home-built | described in REFs | |
| Localization software | Custom-written, available online | MATLAB and C++ software package that can be adapted for localization analysis. | |
| Tracking software | Available online | MATLAB implementation by Blair and Dufresne. |
Références
- Tsien, R. Y. The green fluorescent protein. Annu Rev Biochem. 67, 509-544 (1998).
- Leake, M. C., et al. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature. 443, 355-358 (2006).
- Taniguchi, Y., Kawakami, M. Application of HaloTag protein to covalent immobilization of recombinant proteins for single molecule force spectroscopy. Langmuir. 26, 10433-10436 (2010).
- Xie, X. S., Choi, P. J., Li, G. W., Lee, N. K., Lia, G. Single-molecule approach to molecular biology in living bacterial cells. Annual review of biophysics. 37, 417-444 (2008).
- Lee, J. H., et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 343, 1360-1363 (2014).
- Miesenbock, G., De Angelis, D. A., Rothman, J. E. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 394, 192-195 (1998).
- Sauer, M. Localization microscopy coming of age: from concepts to biological impact. J Cell Sci. 126, 3505-3513 (2013).
- Dempsey, G. T., Vaughan, J. C., Hao Chen, K., Zhuang, X. Evaluation of fluorophores for optimal performance in localizationbased super-resolution imaging. Nat Meth. 8, 1027-1041 (2011).
- Shaner, N. C., Steinbach, P. A., Tsien, R. Y. A guide to choosing fluorescent proteins. Nat Meth. 2, 905-909 (2005).
- Landgraf, D., Okumus, B., Chien, P., Baker, T. A., Paulsson, J. Segregation of molecules at cell division reveals native protein localization. Nat. Methods. 9, 480-482 (2012).
- Jaitin, D. A., et al. Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science. 343, 776-779 (2014).
- Aldridge, S., et al. AHT-ChIP-seq: a completely automated robotic protocol for high-throughput chromatin immunoprecipitation. Genome Biol. 14, R124 (2013).
- Keppler, A., et al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 21, 86-89 (2003).
- Wombacher, R., et al. Live-cell super-resolution imaging with trimethoprim conjugates. Nat. Methods. 7, 717-719 (2010).
- Zhang, Z., et al. A new strategy for the site-specific modification of proteins in vivo. Biochemistry. 42, 6735-6746 (2003).
- McNeil, P. L., Murphy, R. F., Lanni, F., Taylor, D. L. A method for incorporating macromolecules into adherent cells. J Cell Biol. 98, 1556-1564 (1984).
- Clarke, M. S., McNeil, P. L. Syringe loading introduces macromolecules into living mammalian cell cytosol. J Cell Sci. 102, 533-541 (1992).
- Sakon, J. J., Weninger, K. R. Detecting the conformation of individual proteins in live cells. Nat. Methods. 7, 203-205 (2010).
- Taylor, L. S. Electromagnetic syringe. IEEE Trans. Biomed. Eng. 25, 303-304 (1978).
- Dower, W. J., Miller, J. F., Ragsdale, C. W. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 16, 6127-6145 (1988).
- Neumann, E., Schaefer-Ridder, M., Wang, Y., Hofschneider, P. H. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J. 1, 841-845 (1982).
- Sustarsic, M., et al. Optimized delivery of fluorescently labeled proteins in live bacteria using electroporation. Histochem Cell Biol. , (2014).
- Tokunaga, M., Imamoto, N., Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods. 5, 159-161 (2008).
- Sinha, A., et al. A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature. 000, 1-5 (2014).
- English, B. P., et al. Single-molecule investigations of the stringent response machinery in living bacterial cells. Proc Natl Acad Sci U S A. 108, E365-E373 (2011).
- Crawford, R., et al. Long-lived intracellular single-molecule fluorescence using electroporated molecules. Biophys J. 105, 2439-2450 (2013).
- Uphoff, S., Reyes-Lamothe, R., Garza de Leon, F., Sherratt, D. J., Kapanidis, A. N. Single-molecule DNA repair in live bacteria. Proc Natl Acad Sci U S A. 110, 8063-8068 (2013).
- Uphoff, S., Sherratt, D. J., Kapanidis, A. N. Visualizing Protein-DNA Interactions in Live Bacterial Cells Using Photoactivated Single-molecule Tracking. J Vis Exp. , (2014).
- Hohlbein, J., Gryte, K., Heilemann, M., Kapanidis, A. N. Surfing on a new wave of single-molecule fluorescence methods. Phys Biol. 7, 031001 (2010).
- Xie, X. S., Yu, J., Yang, W. Y. Perspective - Living cells as test tubes. Science. 312, 228-230 (2006).
- Santoso, Y., et al. Conformational transitions in DNA polymerase I revealed by single-molecule FRET. Proc Natl Acad Sci U S A. 107, 715-720 (2010).
- Hohlbein, J., et al. Conformational landscapes of DNA polymerase I and mutator derivatives establish fidelity checkpoints for nucleotide insertion. Nature communications. 4, 2131 (2013).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.