
Method Article
Siguiente Generación de secuenciación para la detección de mutaciones procesable en tumores sólidos y líquidos
En este artículo
Resumen
This manuscript describes clinical protocols for two next-generation sequencing panels. One panel interrogates hematologic malignancies while the other panel targets genes commonly mutated in solid tumors. Molecular classification of driver mutations in human malignancies offers valuable prognostic and predictive information.
Resumen
As our understanding of the driver mutations necessary for initiation and progression of cancers improves, we gain critical information on how specific molecular profiles of a tumor may predict responsiveness to therapeutic agents or provide knowledge about prognosis. At our institution a tumor genotyping program was established as part of routine clinical care, screening both hematologic and solid tumors for a wide spectrum of mutations using two next-generation sequencing (NGS) panels: a custom, 33 gene hematological malignancies panel for use with peripheral blood and bone marrow, and a commercially produced solid tumor panel for use with formalin-fixed paraffin-embedded tissue that targets 47 genes commonly mutated in cancer. Our workflow includes a pathologist review of the biopsy to ensure there is adequate amount of tumor for the assay followed by customized DNA extraction is performed on the specimen. Quality control of the specimen includes steps for quantity, quality and integrity and only after the extracted DNA passes these metrics an amplicon library is generated and sequenced. The resulting data is analyzed through an in-house bioinformatics pipeline and the variants are reviewed and interpreted for pathogenicity. Here we provide a snapshot of the utility of each panel using two clinical cases to provide insight into how a well-designed NGS workflow can contribute to optimizing clinical outcomes.
Introducción
secuenciación de próxima generación (NGS) de las muestras de oncología clínica se ha vuelto más ampliamente disponibles en los últimos años como el crecimiento puntos literatura científica para la importancia de identificar los cambios genéticos dirigibles y los marcadores moleculares predictivos / pronósticos. Panel de genes múltiples análisis y estudios de secuenciación del exoma enteros en ambos epiteliales 1,2 y 3 tumores malignos hematológicos han solidificado el concepto de heterogeneidad tumoral y la evolución clonal como la enfermedad progresa y recaídas. Además, a diferencia de las tecnologías competidoras como la reacción en cadena de la polimerasa (PCR) o la secuenciación de Sanger, NGS puede detectar la mayoría de las alteraciones genómicas en todos los genes del cáncer clínicamente relevantes en un único ensayo 4.
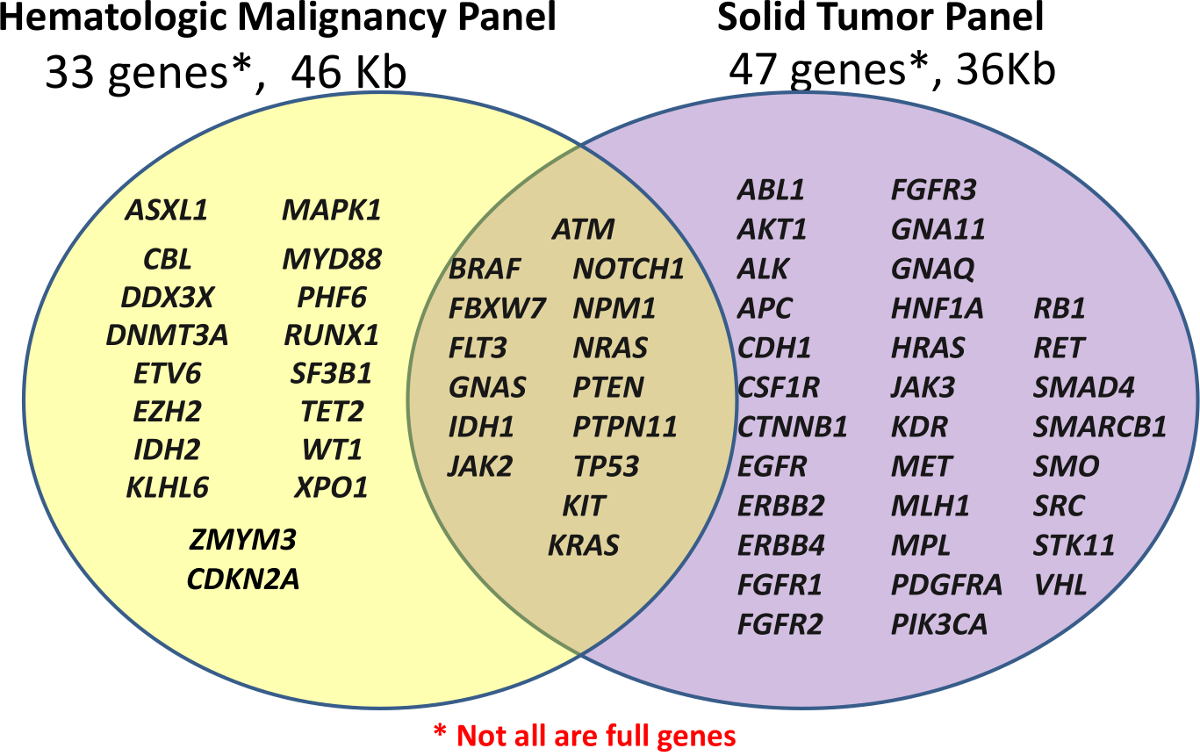
El Centro de Diagnóstico personalizado lanzó inicialmente con dos paneles clínicos NGS, costumbre panel hematológico (Hemo-NGS Panel) y un off-the-shelf Cancer Panel de muestras FFPE (Solid-NGS panel) (véase Figura 1). Estos paneles cubren las regiones de interés clínicamente relevantes o altos de los genes seleccionados; no todos los genes o exones están totalmente cubiertos. Los amplicones son generadas por hibridación de la sonda, seguido por extensión y ligación. Las regiones seleccionadas se amplifican aún más el uso de PCR con cebadores universales de doble indexados, lo que permite un máximo de 96 muestras que deben mezclarse para la secuenciación.

Figura 1:. Lista de genes cubiertos en los paneles Biblioteca preparación se lleva a cabo utilizando ya sea la costumbre panel hematológico (Grupo Hemo-NGS) de 33 genes o el off-the-shelf Cancer Panel de amplicón (Solid-NGS) de 47 genes. No todos los genes o exones están cubiertos en su totalidad, ya que algunos amplicones sólo podrán cubrir ciertos puntos de acceso. Por favor, haga clic aquí para ver una versión más grande de esta figura. </ A>
{kind=link}
El contenido del Panel de hemo-NGS se deriva de múltiples fuentes, pero se centra alrededor de 16 genes mutados en la leucemia mieloide aguda (LMA) descrito anteriormente como demuestra un alto nivel de utilidad clínica 5. El Grupo Solid-NGS se produce comercialmente con las regiones seleccionadas sobre la base de los genes mutados en el cáncer comúnmente como se informa en el Catálogo de mutaciones somáticas en el Cáncer (cósmica) de base de datos 6.
Varios pasos clave caracterizan el flujo de trabajo general para NGS clínicos. Después de que el médico ordena la prueba, un patólogo determina la adecuación de la muestra de análisis para el porcentaje de tumor y el volumen de muestra siguientes. En nuestra institución, requerimos% del tumor, al menos, 10 debido a la tasa de error de secuenciación de fondo ( "ruido") de la tecnología y la eficiencia del enfoque específico. Si el tejido es adecuado para las pruebas, se extrae ADN genómico. Este ADN se somete a control múltiple de la calidad (QC) pasos. Si el ADN pasa QC, se genera y se secuenció una biblioteca de amplicón. Los datos resultantes se analizaron a través de una tubería de la bioinformática en el local. Tras el análisis de la bioinformática, las variantes son revisados manualmente y se interpretan para la patogenicidad antes de su incorporación en un informe clínico. A continuación se describen dos casos que pasaron por este flujo de trabajo riguroso y en última instancia condujo a cambios en la gestión clínica.
Caso 1 - Leucemia mieloide aguda
Una biopsia de la médula ósea de los pacientes fue un diagnóstico de la LMA, sin maduración. Los estudios citogenéticos fueron enviados en la muestra de médula ósea y demostraron un cariotipo femenino normal. Hubo 95% de blastos circulantes presentes, por lo que una muestra de sangre periférica se envían para las pruebas de diagnóstico personalizado en el panel de hemo-NGS.
leucemia mieloide aguda (LMA) es un tumor maligno hematológico del linaje mieloide de las células blancas de la sangre. La detecciónde las mutaciones genéticas en la LMA se ha convertido cada vez más importante para el pronóstico y tratamiento, con mutaciones genéticas recurrentes reconocen como importante en la patogénesis y el pronóstico 7. Las mutaciones en NPM1 y CEBPA se han asociado con un riesgo de pronóstico favorable, mientras que las duplicaciones en tándem internas (DTI) en FLT3 se han asociado con un resultado menos favorable 8. Un creciente cuerpo de evidencia apoya un papel patogénico de estas y otras mutaciones en la LMA 9.
Caso 2 - Pulmón Adenocarcinoma
Una biopsia de una masa supraclavicular izquierdo de paciente B demostró adenocarcinoma pulmonar. El material de biopsia de la (FFPE) de masa de los ganglios linfáticos fijado en formalina incluido en parafina fue enviado para la prueba genómico (Panel Solid-NGS) como rollos / rizos con% tumor mayor que 50, para identificar si una mutación estaba presente para la intervención terapéutica dirigida.
Canc de pulmóner es la principal causa de mortalidad relacionada con el cáncer en los Estados Unidos y se divide en dos tipos principales, cáncer de pulmón no microcítico (CPNM) y cáncer de pulmón de células pequeñas (SCLC). NSCLC puede definirse adicionalmente como sea adenocarcinoma o carcinoma de células escamosas, basado en la histología de la lesión. Adenocarcinoma de pulmón es el subtipo más común de cáncer de pulmón, visto tanto en fumadores y no fumadores, y es la forma más común de cáncer de pulmón en los no fumadores 10. Los estudios moleculares de los adenocarcinomas de pulmón han identificado mutaciones en múltiples oncogenes 11. Las mutaciones del conductor más comunes identificadas en los fumadores tienen mutaciones en KRAS y BRAF. Las mutaciones más comunes en los no fumadores son mutaciones en EGFR, y reordenamientos de los genes ALK, RET y ROS1. Los tumores de pulmón se han descrito con un exón 20 de inserción en marco en el gen ErbB2 (HER2 / neu). La anomalía más frecuente en la ESR2 / neu es una amplificación de este locus en el cáncer de mama para que una terapia dirigida está disponible (trastuzumab: un anticuerpo monoclonal humanizado contra HER2 / neu). El HER2 / neu exón 20 de inserción que se observa en 2-4% de pulmón adenocarcimomas 12 ha mostrado una respuesta parcial a la terapia de combinación con HER2 / neu y los inhibidores de mTOR (neratinib y temsirolimus, respectivamente) 13.
Protocolo
Este protocolo comprende las etapas más destacadas de dos pruebas de laboratorio desarrollado validados para el perfil genómico de tumores sólidos y líquidos, respectivamente. Las pruebas realizadas en el laboratorio se realiza de acuerdo con los requisitos de las Enmiendas de Mejoras de Laboratorio Clínico (CLIA) de 1988.
1. Extracción de ADN a partir de sangre periférica o de médula ósea
- Determinar la cantidad de sangre o de médula ósea para tomar en función de la Tabla 1.
| Muestra / CMB | La cantidad a ser tratada como 1 ml de sangre |
| Médula ósea | 250 l |
| Sangre WBC 12.000 - 50.000 | 1 ml |
| Sangre WBC 50.000 - 100.000 | 500 l |
| WBC de sangre 100.000 - 200.000 | 200 l |
| Sangre WBC> 200.000 | 100 l |
| * Para la sangre WBC <12.000, tomar 2 ml de sangre | |
Tabla 1:. / Médula Ósea volumen de sangre que use la tabla Puesto que el recuento de glóbulos blancos puede variar de una muestra a otra, es difícil especificar un volumen específico de sangre para su uso. Por lo tanto, la cantidad de sangre a usar para el ensayo debe ser determinada observando el recuento de glóbulos blancos (WBC) antes de empezar el ensayo. Aunque se utiliza menos sangre, todavía debe ser tratado como si sus 1 ml ya que el volumen de sangre que se utiliza se reduce debido a que el número de células presentes es mayor de lo normal.
- Siga el protocolo del kit disponible comercialmente para aislar el ADN genómico.
2. Extracción de ADN de tejido incluidas en parafina (FFPE) fijado en formalina
- Residencia enla región del tumor el patólogo con un círculo en la diapositiva H & E, se alinean las diapositivas no teñidas con la guía de H & E diapositivas y delinear un área similar para su extracción. Por macro-disección, proceso de un solo espécimen / set del paciente de diapositivas a la vez.
- Se calientan los portas en un bloque de calor a 45 ° C para fundir la parafina ligeramente. Raspe con cuidado el tejido dentro de las líneas que se marcan en la diapositiva, utilizando un bisturí nueva para cada muestra a extraer. Coloque los raspados de cera en el tubo debidamente etiquetado 1,5 ml. Tenga cuidado porque la cera es muy raspada electrostática y puede saltar fuera del tubo.
- Añadir 320 l de solución de desparafinación para cada cinco a seis 5 micras secciones (25 - 30 micras en total). Por ejemplo, si un tubo que contiene 3 secciones de un 10 micras rollo / enrollamiento se va a procesar, a continuación, utilizar 320 l, pero si se obtuvieron 5 secciones en el mismo espesor a continuación, utilizar 640 l.
- Agitar vigorosamente durante al menos 10 segundos y realizar aqgiro uick en una microcentrífuga para extirpar el tejido / cera de los lados y la tapa y en la solución. Incubar a 56 ° C durante 3 min, y luego se incuba a temperatura ambiente durante 5 -. 10 min.
- Después de la incubación RT, añadir 180 l de tampón ATL por cada 320 l de solución de desparafinación añadido. Picar el tejido diez veces usando un mini-mano de mortero estéril usando una nueva mano de mortero para cada espécimen. Asegúrese de que no hay tejido pegado a la mano del mortero, ya que puede ser muy pegajosa. Vortex vigorosamente la suspensión durante 3 segundos y centrifugar a máxima velocidad durante 1 minuto.
- Añadir 10 l de proteinasa K a la fase clara inferior. Mezclar suavemente pipeteando arriba y hacia abajo para asegurar que se volvió a suspender el tejido. NO hacer vórtice. Incubar a 56 ° C durante la noche con agitación a 400-500 rpm.
- A la mañana siguiente, compruebe si el tejido se haya disuelto completamente (esto ha ocurrido si la solución es clara inferior). Si la solución inferior no está claro, a continuación, agitar vigorosamente durante 3 segundos y centrifugar a máxima velocidad for 1 min. Añadir un extra de 5 - 10 l de proteinasa K y se incuba a 56 ° C durante 30 - 60 min.
- Se incuba a 90 ° C durante 1 hora para ayudar a revertir el formaldehído entrecruzamiento. Dejar que las muestras se enfríen a temperatura ambiente durante 5 - 10 minutos a centrifugar brevemente y luego cada tubo para consolidar el líquido.
- Pasar la fase clara inferior en un tubo etiquetado 1,5 ml. Si hay varios tubos de la misma muestra de pacientes (por ejemplo, el caso si se utilizan múltiples rollos), recombinar las fases inferiores en un tubo en este punto. Nota: La transferencia de pequeñas cantidades de la solución de desparafinación no debe interferir con el procedimiento de purificación, pero existe un riesgo si una gran cantidad se transfiere.
- Añadir 2 l de solución de RNasa A. Vórtice suavemente o invierte 25 veces y vuelta rápida en una microcentrífuga. Incubar a temperatura ambiente durante 5 min.
- Añadir 200 l de solución de proteína Precipitación. Si al hacer una o dos rollos, utilizar 200 l. Si hace tres rodilloss de una vez, a continuación, utilizar 400 l. Vortex vigorosamente a alta velocidad durante 30 segundos para mezclar uniformemente los tampones de lisis. Incubar en hielo durante 5 min o las muestras pueden permanecer en el hielo durante un máximo de una hora.
- Se centrifuga a 5.000 g durante 5 min. La proteína precipitada debe formar un apretado, sedimento blanco. Verter el sobrenadante en un tubo etiquetado 1,5 ml, y a continuación incubar las muestras en hielo durante al menos 3 min. Se centrifuga a 5.000 g durante 3 min.
- Añadir 200 l de 2-propanol (isopropanol) para cada 180 l de tampón ATL añaden antes a un tubo etiquetado 1,5 ml (que podría ser necesario el uso de un tubo de 2 ml). Por ejemplo, si al hacerlo tres rollos añaden 600 l de isopropanol. Añadir 1 l de glucógeno por cada 180 l de tampón ATL añadido anteriormente a la isopropanol e invertir el tubo varias veces para mezclar.
- añadir con cuidado el sobrenadante de la etapa de precipitación de la proteína en la mezcla de isopropanol. Mezclar el tubo invirtiendo suavemente al menos 50 veces.
- Centrifugar a máxima spEED durante 3 min. El ADN será visible tan pequeño sedimento blanco en la parte inferior del tubo.
- Vierta o aspirar el sobrenadante en el tubo de recuperación apropiado. Mantenga un tubo de 1,5 ml de residuos por separado para cada muestra en caso de que el pellet viene desalojado por lo que no se perderá o se mezcla con los residuos de otros especímenes. Escurrir el tubo sobre una toalla de papel y compruebe se elimina la mayor parte del isopropanol.
- Añadir 300 l de etanol recién hecho 70%. Invertir el tubo suavemente varias veces para lavar el sedimento. Tratar de asegurar la pastilla viene desalojada para asegurar una limpieza más profunda.
- Centrifugar a velocidad máxima durante 5 min. y luego retirar con cuidado el etanol. El sedimento puede estar suelto, por lo que vierta o aspirado lentamente y ver el sedimento. Eliminar el exceso de etanol a partir de la parte interior del tubo, sin tocar el pellet. Permitir que las muestras se sequen al aire durante 5 - 15 min, teniendo cuidado de no sobre secar la muestra.
- Añadir entre 25 - 100 l de solución de hidratación de ADN al correomuestra ACH basado en el tamaño del sedimento de ADN y la cantidad a partir de tejido. Vortex los tubos vigorosamente y girar brevemente en una microcentrífuga. Incubar durante 1 hora a 65 ° C para rehidratar completamente el ADN.
3. Control de Calidad de ADN genómico
Nota: Hay tres pasos independientes para el control de la calidad del ADN (QC). Véase la Tabla 2 para mayor explicación de por qué se lleva a cabo cada paso del control de calidad.
| Instrumento | Resultado | Indicación | Rango Ideal |
| DropSense96 | A260 / A230 | Identificación de contaminantes químicos (por ejemplo, etanol) | 1.50 - 2.2 |
| DropSense96 | A260 / A280 | Identificación de contaminantes proteicos | 1.60 - 2.2 |
| DropSense96 | Concentración | la cuantificación de ADN | > 1 ng / l |
| TapeStation | ADN Smear | Determinación de la integridad del ADN (por ejemplo, la degradación / fragmentación de ADN extraído) | 50%> 1.000 pb |
| 2.0 qubit | Concentración | Más precisa cuantificación de ADN | > 1 ng / l |
Tabla 2:. QC ADN Resultados esperados Todos estos valores se tienen en cuenta antes de ejecutar una muestra que permite proceder a la etapa de preparación de la biblioteca.
- Siguiendo el protocolo del fabricante, ejecute 2 l de ADN extraído en un fluorómetro para obtener la concentración de trabajo (ng / l) de la muestra.
- Ejecutar 1 - 2 l de cada muestra en un espectrofotómetro UV / VIS para comprobar la calidad de la muestra (A260 / A230 y A260 rata / A280ios) según las instrucciones del fabricante.
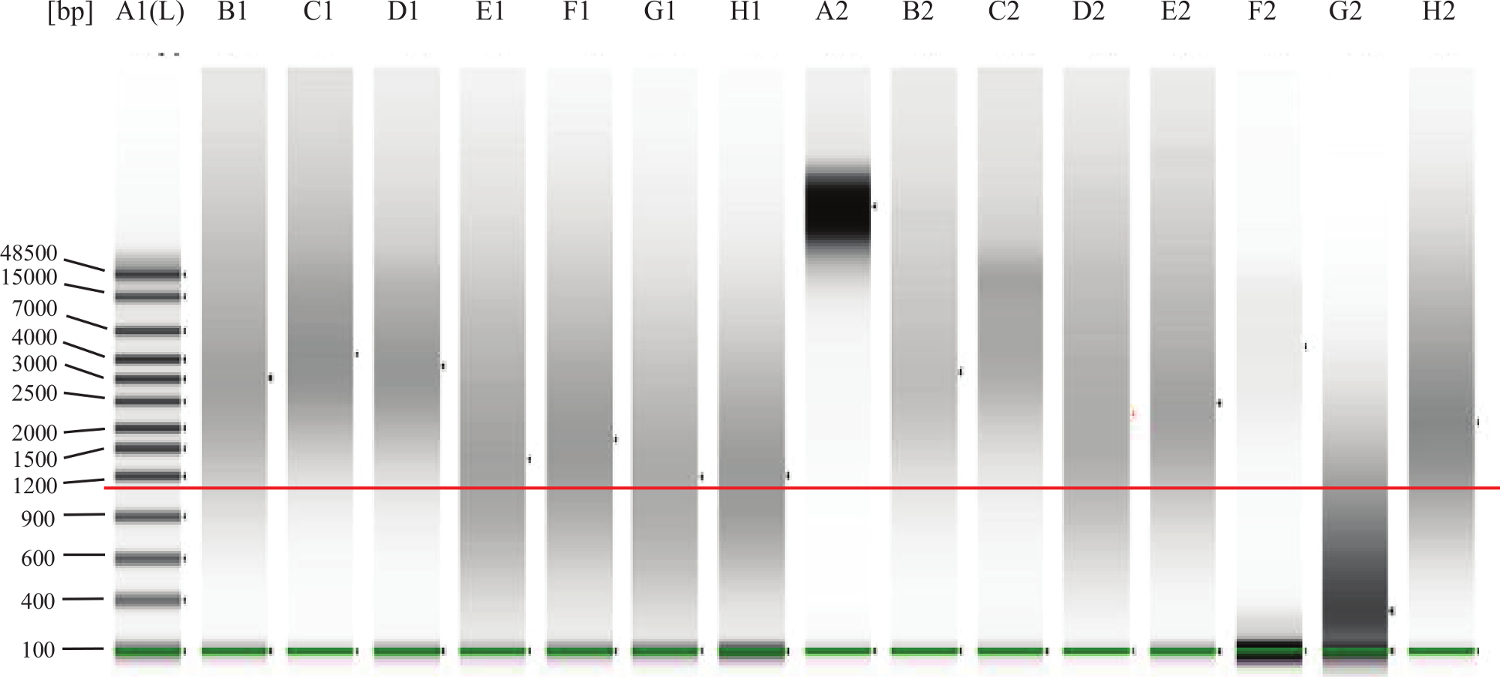
- Para las muestras FFPE: Siguiendo las instrucciones del fabricante, ejecutar 1 l de cada muestra en un sistema de electroforesis en gel de microfluidos para evaluar la degradación / fragmentación del ADN. Vea la Figura 2 para ver ejemplos.

Figura 2:. El ADN genómico del control de calidad del gel Ejemplo La línea verde sobre las bandas más bajas es indicar el marcador más bajo. La línea roja añadido-in es para indicar aproximadamente 1.000 pb. Carril A2 representa el ADN intacto, como uno esperaría de Sue tis fresco (por ejemplo, sangre periférica o médula ósea). Lanes B1, C1, D1, B2, C2, E2 son ejemplos de buenas FFPE de ADN, intacta. El ADN en F2 carril parece estar intacta, pero en una concentración demasiado baja. El ADN en G2 carril es degradada o fragmentada y no funcionará en el ensayo. Lanes E1, F1, G1, H1, D2,H2 representan ADN que cae dentro de la "zona gris" lo que significa que el ensayo puede funcionar bien, pero algunos de los ADN podría ser demasiado dañado o reticulado y por lo tanto no va a funcionar bien en el ensayo. Haga clic aquí para ver una más grande versión de esta figura.
{kind=link}
4. Preparación de amplicones Biblioteca
- La hibridación de oligo-piscina y Extensión La ligadura de oligos Bound
- En la sala de Pre-PCR, añadir el volumen necesario de bajo EDTA TE, 5 l de tubo Oligo del Panel (por ejemplo, Solid-NGS Panel o hemo-NGS Panel), y 100 a 250 ng de ADN genómico de cada uno correspondiente bien en una placa de 96 pocillos semi-bordeado etiquetado como la placa de hibridación (HYB).
- Añadir 40 l de oligo hibridación para la secuenciación del reactivo 1 (OHS1) a cada muestra en la placa HYB. pipetear suavemente hacia arriba y hacia abajo al menos 5 - 6 veces para mezclar. Cambie las puntas después de cada columna para evitar quecontaminación cruzada.
- Sellar la placa con papel de aluminio HYB adhesivo y centrifugar a 1000 xg durante 30 seg.
- Incubar la placa HYB en la incubadora de hibridación precalentado a 95 ° C durante 1 min. Ajuste la temperatura de la incubadora de hibridación a 40 ° C. Continuar la incubación el tiempo que se tarda la incubadora a disminuir desde 95 ° C a 40 ° C (~ 90 min). Nota: Este enfriamiento gradual es crítica para la hibridación adecuada.
- Durante el último 15 - 20 min de incubación de la hibridación, lavado previo de la unidad de placa de filtro (FPU). Prepare sólo los pocillos para ser utilizados en el ensayo actual, es decir, sólo utilice pozos frescos / no utilizadas de una placa de filtro previamente abierta, pero nunca volver a utilizar los pozos que se han utilizado. Nota: Esto debe ser clara basada en el número de juego en la placa de filtro, las marcas de la placa de filtro, y el sellador utilizado en toda la placa.
- Usando una pipeta multicanal, añadir 45 l de lavado astringente 1 (SW1) a cada pocillo.
PRECAUCIÓN: Contiene formamida. Cubierta y centrifugar la FPU a 2.250 xg durante 3 min a 20 ° C. Compruebe cada bien para líquido residual (> 15 l / pocillo). - Si existe líquido residual, girar el ° FPU 180 y repetir el paso de centrifugación de nuevo para una 3 min adicionales. Si el líquido residual hace girar a través de la segunda vez, a continuación, proceder al siguiente paso, si no, entonces puede haber un defecto en la placa de filtro y tendrá que ser reemplazado la placa actual.
- Usando una pipeta multicanal, añadir 45 l de lavado astringente 1 (SW1) a cada pocillo.
- Una vez que la incubadora de hibridación se ha enfriado a 40 ° C, se centrifuga la placa a 1000 xg durante al menos 30 segundos a 20 ° C para recoger la condensación. Transferir todo el volumen de cada muestra de la placa de HYB sobre el centro de los pocillos correspondientes prelavada de la FPU. Cambiar las puntas después de cada columna para evitar la contaminación cruzada. Cubrir la FPU y se centrifuga a 2.250 g durante 3 min a 20 ° C.
- Añadir 45 l de SW1 y centrifugar a 2250 xg durante 3 min a 20 ° C. Repita para un total de dos lavados. Gire el FPU 180 ° y se centrifuga de nuevo a 2.250 xg durante 3 minutos para eliminar por completo todo el SW1.
- Disimular la FPU. Desechar todo el flujo a través (que contiene formamida) en el contenedor de residuos adecuado. Volver a montar la FPU utilizando una placa de recolección de residuos MIDI diferente. Añadir 45 l de tampón universal 1 (UB1) a cada pocillo de muestra y se centrifuga a 2250 xg durante 3 min a 20 ° C. PRECAUCIÓN: Contiene formamida.
- Añadir 45 l de Extensión ligadura Mix 3 (ELM3) a cada pocillo de muestra en la placa de FPU y la pipeta hacia arriba y abajo 3 veces para mezclar. Nota: La reacción de extensión-ligación se lleva a cabo en la membrana de placa filtrante.
- Sellar la placa con papel de aluminio FPU adhesivo y se incuba el conjunto completo FPU en un pre-calienta 37 ° C incubadora durante 45 min.
- La indexación y la amplificación por PCR
- Alícuota de los índices que se utilizan en los pocillos correspondientes en el indexado de amplificación Plcomió (IAP) disponiendo los cebadores en el Aparato índice de placa, dispuestos de la siguiente manera: Tubos de cebadores i5 (gorras blancas, solución clara) verticalmente, alineados con las filas A a H, tubos de cebadores i7 (tapas naranjas, solución amarilla) horizontalmente , alineados con las columnas 1 a 12. el uso de un p10 pipeta multicanal, añadir 4 l de cebadores i7 (solución amarilla) a cada fila de la IAP y añadir 4 l de cebadores i5 (solución clara) a cada columna de la IAP.
- En el hielo o un bloque de refrigeración, preparar la mezcla maestra de PCR mediante la adición de 0,5 l de ADN polimerasa 1 (TDP1) a 25 l de PCR Master Mix 2 (PMM2) por muestra. Invertir, rápidamente vórtice, y centrifugar brevemente la PCR Master Mix para mezclar.
- Añadir 22 l de la mezcla maestra de PCR a cada pocillo de la IAP y la pipeta hacia arriba y abajo 3 veces para mezclar. Cambiar las puntas entre los pozos. Mantenga la IAP a 4 ° C.
- Después de los 45 min de reacción de extensión-ligación (paso 4.1.10), retire la FPU de la incubadora y retirar con cuidado la columinum sello de aluminio. Cubrir con la tapa y se centrifuga a 2.250 g durante 3 min a 20 ° C.
- Añadir 25 l de NaOH 50 mM a cada pocillo de muestra en la FPU. Pipetear arriba y abajo de al menos 6 veces; asegurar las puntas de pipeta entrar en contacto con la membrana. Cambie las puntas después de cada columna. Incubar a temperatura ambiente durante 5 min.
- Transferir las muestras eluyeron de la FPU a la IAP como sigue:
- Usando una pipeta multicanal p100 ajustado a 20 l, pipetear el NaOH en la placa de FPU arriba y hacia abajo al menos 6 veces. Incline ligeramente la placa FPU para garantizar la aspiración completa de la placa.
- Transferencia de 20 l de la FPU a la columna correspondiente de la IAP. pipeta suavemente hacia arriba y hacia abajo al menos 6 veces para combinan a fondo el ADN con la mezcla maestra de PCR. Sellar la IAP con película adhesiva y se centrifuga a 1.000 xg durante 1 min a 20 ° C.
- Llevar la placa de PCR en la sala de post-PCR y la carga de la placa en un termociclador. Ejecutar el PCR programa que consiste en un paso de desnaturalización de calor a 95 ° C durante 3 min; seguido por 25 ciclos de 95 ° C durante 30 segundos, 62 ° C durante 30 seg, 72 ° C durante 60 seg; seguido de una extensión final de 72 ° C durante 5 min; terminando con un mantenimiento a 10 ° C. Si no proceder a la siguiente etapa después de la finalización de la PCR, la placa puede permanecer en el ciclador térmico durante la noche, o puede ser almacenado a 2-8 ° C hasta dos días.
- La purificación de PCR y Normalización basado en perlas
- Retire las perlas magnéticas, de purificación de tampón de elución (EBT), y reactivos de la electroforesis en gel del 4 ° C nevera y lugar a temperatura ambiente al menos 20 min antes de la siguiente etapa.
- Una vez que la PCR se haya completado, se centrifuga a 1.000 xg durante 1 min a 20 ° C para recoger la condensación. Transferencia de 1 l de cada reacción de PCR para la tira de tubos / pocillos de las placas que contienen 4 l de agua para diluir las muestras 1/5. Pipetear arriba y abajo para mezclar.
- Añadir 2 l de ladiluido muestras PCRed a 2 l de tampón de microfluidos-gel. Sellar las tiras / placa. Se agita a 1800 rpm durante al menos 30 segundos y se centrifuga a 1000 xg durante 30 seg. Siguiendo el protocolo del fabricante, ejecute la mezcla en un gel de microfluidos para evaluar si la preparación de la biblioteca produjo una biblioteca aceptable (véase la Figura 3).
- Vórtice de purificación de las perlas magnéticas hasta que estén bien suspendidas y el color aparece homogénea. Añadir 45 l de las perlas para cada muestra.
- Sellar la placa con película adhesiva transparente y agitar la placa a 1.800 rpm durante 2 minutos. Se incuba a temperatura ambiente sin agitación durante 10 min.
- Coloque la fuente sobre un soporte magnético. Una vez que el sobrenadante se haya resuelto, retirar con cuidado y desechar el sobrenadante. Si ningún perlas son aspiradas inadvertidamente en las puntas, prescindir de las perlas de nuevo a la placa y dejar que la placa de apoyo en el imán durante 2 minutos y confirme que el sobrenadante se haya resuelto.
- Con la placa de tque soporte magnético, añadir 200 l de recién preparada etanol al 80% a cada pocillo de muestra. Mover la placa de ida y vuelta un par de veces. Se incuba la placa en el soporte magnético durante 30 segundos. Con cuidado, retirar y desechar el sobrenadante. Repita para un total de dos lavados.
- Eliminar el exceso de etanol por centrifugación brevemente la placa a 1000 xg para reducir cualquier etanol en los lados del tubo, la colocación de la placa posterior en el soporte magnético, y el uso de una pipeta de p10 de múltiples canales ajustado a 10 l para eliminar el etanol. Permitir que las perlas se sequen al aire durante 5 - 8 minutos.
- Usando una pipeta multicanal p100, añadir 30 l de EBT a cada pocillo. Pipetear arriba y abajo varias veces para asegurar las perlas vienen del lado del tubo. Sellar la placa con película adhesiva transparente y agitar la placa a 1.800 rpm durante 2 minutos.
- Se incuba a temperatura ambiente sin agitación durante 3 min. Si hay muestras en las que los granos no se resuspendió completamente, suavemente la pipeta hacia arriba y abajo para volver a suspender las perlas.
- Colocar la placa en un soporte magnético. Usando una pipeta multicanal p100, la transferencia de 20 l del sobrenadante a una placa nueva llamada la placa Biblioteca Normalización (LNP). La transferencia de los restantes 10 ~ l de las bibliotecas de secuenciación individuales de una placa separada llamada la restante plano limpio Biblioteca Plate (RCLP). Almacenar esta placa junto con la preparación de la biblioteca final, ya que se puede utilizar como una reserva para una segunda carrera de secuenciación, si es necesario. Si no proceder a la siguiente etapa, la LNP y RCLP se pueden almacenar a -15 a -25 ° C.
- Vigorosamente vórtice y volver a suspender la biblioteca de los granos de normalización 1 (LNB 1). Es crítico para resuspender completamente el sedimento LNB1 talón en la parte inferior del tubo. Preparar la mezcla de normalización, mediante la mezcla de 8 l de LNB1 con 44 l de Normalización Biblioteca Aditivos 1 (LNA1) por muestra. Vigorosamente la mezcla vórtice Normalización durante 10 - 20 segundos.
PRECAUCIÓN: LNA1 contiene formamida. - Con una inversión de intermitentend agitación de la mezcla de Normalización, añadir 45 l de cada muestra de la LNP. Sellar la placa con película adhesiva transparente y agitar la placa a 1.800 rpm durante 30 min. Este 30 min de incubación es crítica para la correcta normalización de la biblioteca como incubaciones de mayor o menor que 30 min puedan afectar a la representación de la biblioteca y la densidad de clúster.
- Durante los 30 minutos de incubación, preparar los reactivos para secuenciación descongelando el cartucho de reactivos y Hyb Buffer (HT1) [PRECAUCIÓN: Ambos contienen formamida]. Además, conseguir hielo para un paso posterior y garantizar un bloque de calor adecuado para tubos de 1,5 ml de centrífuga se establece en 96 ° C.
- Cuando la etapa de mezcla 30 min se ha completado, colocar la LNP en un soporte magnético. Una vez que el sobrenadante se haya resuelto, utilizar una pipeta multicanal para retirar cuidadosamente y desechar el sobrenadante en el contenedor de residuos adecuado.
- Eliminar la LNP del soporte magnético y lavar las perlas con la biblioteca de Normalización de lavado 1 (LNW1) como sigue:
- Añadir 45l de LNW1 a cada pocillo de muestra.
PRECAUCIÓN: Contiene formamida. Sellar la placa con película adhesiva transparente y agitar la placa a 1.800 rpm durante 5 min. Repita para un total de dos lavados. Asegúrese de eliminar todo el LNW1 después del segundo lavado.
- Añadir 45l de LNW1 a cada pocillo de muestra.
- Eliminar la LNP del soporte magnético y, usando una pipeta multicanal, añadir 30 l de NaOH 0,1 N (menos de una semana de edad) a cada pocillo para eluir la muestra. Sellar la placa con película adhesiva transparente y agitar la placa a 1.800 rpm durante 5 min.
- Durante el 5 min de elución, añadir 30 l de biblioteca de Normalización Storage Buffer 1 (LNS1) a cada pocillo para ser usado en una nueva placa llamada la placa de almacenamiento (SGP).
- Coloque la LNP en el soporte magnético. Una vez que el sobrenadante se haya resuelto, la transferencia de la elución de 30 l para la LNS1 en el PEC. Cambiar las puntas entre muestras para evitar la contaminación cruzada.
- Sellar la placa con película adhesiva y se centrifuga a 1000 xg durante al menos 30 seg.
- Añadir 51; l de cada muestra que se secuenció para un amplicón Biblioteca Pooled marcado (PAL) tubo de 1,5 ml. Vórtice del PAL para mezclar y centrifugar brevemente en una microcentrífuga.
- Dependiendo de qué química de secuenciación se esté utilizando (V2 o V3) añadir 4 - 10 l de la PAL de 590 - 596 l de HT1. En general, añadir 5,8 l de PAL a 595 l de HT1 para la química V2 y 8,5 l de PAL a 592 l de HT1 para la química V3. Etiqueta este tubo a medida que el tubo diluido amplicón Biblioteca (DAL).
- Vórtice del DAL y brevemente de girar en una microcentrífuga. Incubar la DAL a 96 ° C durante 2 min. Invertir el tubo DAL 3 veces y poner el DAL en hielo durante al menos 5 min, mientras se prepara el secuenciador para la secuenciación. Después de la 5 min, la DAL está listo para ser cargado.
- Si terminado con el PEC, sellar la placa con papel de aluminio película adhesiva y la etiqueta con la fecha y la placa de identificación. Almacenar el PEC sellada y PAL de -15 a -25 ° C.

Figura 3:. Biblioteca de preparación del control de calidad del gel Ejemplo La línea verde sobre las bandas más bajas es indicar el marcador inferior y la línea púrpura sobre las bandas superiores es para indicar el marcador más alto. Todo funcionaba bien para los carriles H1, A2, B2, C2, E2, y G2. La preparación de la biblioteca no funciona de manera óptima, por carriles D2, F2 y H2, pero los resultados todavía será obtenida apenas puede ser que no tenga una cobertura adecuada. Para A3, la preparación de la biblioteca apenas funcionaba y lo más probable es que esta muestra de ADN no era adecuado para el ensayo. Las bandas más bajas por encima de la marca más baja son los cebadores no utilizados, debido a que la muestra se toma directamente de la PCRed bien. La muestra NTC sólo debe tener la banda no utilizado de imprimación, y nada más. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
5. Secuenciación
- Asegurar una correcta SampleSheet.csv se ha hecho para la carrera. Ver Figura 1 complementario para un ejemplo.
- Enjuague y seque la celda de flujo y añadir 600 l de la DAL para el cartucho de reactivos descongelado.
- Prepare el secuenciador para la secuenciación siguiendo las instrucciones que aparecen en pantalla.
Análisis 6. Datos
- Ejecutar el gasoducto bioinformática. Utilizar la tubería bioinformático en la empresa, diseñado a medida para identificar las mutaciones, inserciones, supresiones y amplificaciones 18.
- Después de la tubería ha completado, compruebe los archivos de registro de errores / advertencia, ya que cualquier ayuda significativa errores / advertencias en el control de calidad del procesamiento en paralelo.
- Analizar las estadísticas de ejecución (Tabla 3) para asegurar la biblioteca secuenciado ha pasado el laboratorio determina las métricas de control de calidad (Figura 4). Revisar manualmente cada variante mediante la visualización de los archivos .bam en un visor de datos genómica (por ejemplo, el Integrativa Genómica Vijarro 16 (IGV)).
NOTA: Sólo las variantes dentro de la gama de frecuencias de alelos y validado por encima de la profundidad mínima de cobertura después de la filtración de calidad se dan (usando la nomenclatura del Genoma Humano Variación Sociedad (vehículos pesados)). Para informes finales, cada variante exonic se clasifican en: patógena, probablemente enfermedad asociada, variante de significado desconocido (VUS), probablemente benigna, y benignos. Todas las variantes anteriores, los criterios de notificación de 5% la frecuencia de alelos, con excepción de aquellas variantes consideradas benignas y sinónimo cambios, se informó.

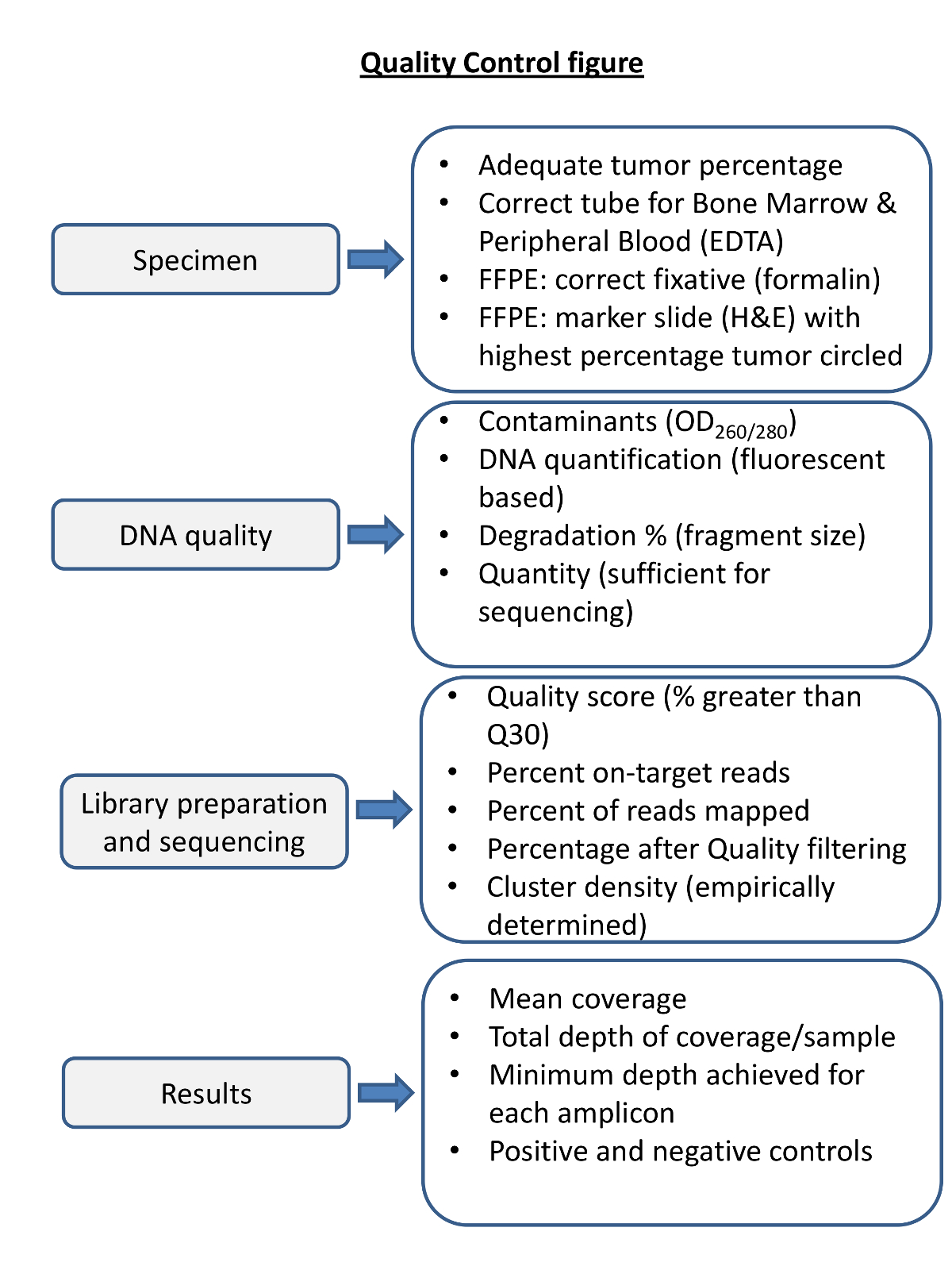
Figura 4. Vista general de Control de Calidad Pasos para NGS. El control de calidad de cada paso en el proceso es necesario para asegurar la secuenciación dará resultados tales que las métricas de secuenciación de pre y post son considerados. tratamiento espécimen apropiada es esencial para el ADN de alta calidad. sangre ymédula ósea en fijadores inapropiados puede producir ADN de baja calidad. La fijación inadecuada de las muestras de tumor sólido puede degradar el ADN (por ejemplo, la fijación en B5). la calidad del ADN se debe evaluar la contaminación de proteínas y ARN por medio de espectrofotometría, y evaluarse con precisión la cantidad y la integridad del ADN. Las métricas de secuenciación deben ser determinados empíricamente en el laboratorio secuenciación y seguido para cada reacción de secuenciación y cada espécimen. Antes de informar sobre los resultados de secuenciación para cada muestra deberá determinar, en la cobertura, la profundidad y el desempeño adecuado de controles positivos y negativos. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Resultados
Caso 1 - Panel de hemo-NGS
El ADN extraído de sangre periférica leucémica era de suficiente calidad y cantidad (176 ng / l) para el Grupo Hemo-NGS. La profundidad media global de la cobertura fue 4,933x (por encima de la profundidad media mínima de cobertura de 1.000 x). Estadísticas de ejecución adicionales están en la Tabla 3. De las 8 regiones por debajo cobertura 250x, 3 se debieron a corte incorrecto de los cebadores (es decir, la secuencia del cebador no se ha eliminado correctamente debido a errores de secuenciación), 1 fue un artefacto conocido del ensayo, y el otro 4 eran regiones parciales de exones de genes diferentes sin variantes notificables. Aunque nuestro protocolo clínico sólo se incluye la presentación de informes variantes cubiertas por al menos 250 lecturas, todos los datos con al menos 100 lee se importan en la base de datos para su revisión.
Procesamiento de datos from la tubería bioinformática detectado tres mutaciones reportables, asociadas a la enfermedad; una mutación sin sentido en FLT3, una mutación sin sentido en IDH2, y una mutación de desplazamiento de marco en NPM1. Las variantes exónicas con sus frecuencias de los alelos se describen en la Tabla 4. Una mutación común en FLT3 es el FLT3 -interna duplicación en tándem (ITD) que no se llama automáticamente por nuestro tubería y requiere una inspección visual del exón 14. La inspección visual del exón 14 de FLT3 no mostró la inserción o la duplicación en la muestra presentados. El FLT3 I836del estaba en el 1% y frecuencia de los alelos no se incluyó en el informe final, ya que cayó por debajo de la frecuencia de los alelos mínimo validado de 5%. Esta mutación no está en la misma molécula de ADN como el cambio FLT3 D835Y (es decir, observado en la misma región del amplicón, pero no en "cis" en cualquiera de secuenciación lecturas) y sólo se observó por la revisión manual de la fi .bamles al verificar el cambio p.D835Y. Las frecuencias de los alelos más bajas de las dos mutaciones de FLT3 sugiere estas mutaciones pueden representar heterogeneidad y / o evolución clonal; sin embargo, la diferencia podría ser debido a la realización del ensayo para que amplicón o un polimorfismo de un solo nucleótido (SNP) o cerca de la superposición de la secuencia del cebador que afectó a la amplificación de este alelo.
Los resultados del panel de hemo-NGS para el caso 1 con mutaciones AML identificadas en FLT3, IDH2, y NPM1, tres genes frecuentemente mutados en la LMA. Las mutaciones en FLT3 se observan en ~ 25% de los pacientes adultos con LMA (base de datos de COSMIC 17) y son bien duplicaciones en tándem interna (ITD) o mutaciones de aminoácidos en el dominio tirosina quinasa. Los FLT3-ITD son la mutación más común y se asocian con mala respuesta a la quimioterapia estándar, mientras que la importancia pronóstica de la quinasa FLT3 dmutaciones puntuales Omain, como se ve en este paciente AML, tiene un impacto claro en el pronóstico 14. Isocitrato deshidrogenasa 2 (NADP +), mitocondrial (IDH2) es un gen que codifica un modificador de la epigenética que es comúnmente mutado en AML. Las mutaciones en modificadores epigenéticos son relativamente comunes en AML, con mutaciones en IDH1 y DNMT3A que representan otras mutaciones en esta clase de genes que conducen a la desregulación de genes. Las mutaciones en el gen de la nucleofosmina (NPM1) son una de las mutaciones adquiridas más comunes en AML y en general se considera que son un buen factor pronóstico (en ausencia de un FLT3 - ITD).
Co-mutaciones de NPM1 y la IDH2 se han descrito en la bibliografía como un indicador de pronóstico favorable 5, con una supervivencia global a los 3 años de 89%. Esto representa un beneficio significativo en la supervivencia en comparación con la supervivencia global a los 3 años deNPM1 tipo salvaje y IDH2 del 31%. Por ejemplo, el estándar de la práctica de atención incluye el análisis de mutaciones para NGP 1 y las mutaciones de FLT3-ITD. En este escenario, la detección de solamente una mutación NPM1 dejaría de manera apropiada paciente estratificar por el riesgo, como las mutaciones secundarias pueden ser favorable (por ejemplo, IDH2) o desfavorable (por ejemplo, TET2), reduciendo de confianza para el alivio de un trasplante de médula ósea.
Caso 2 - Grupo Solid-NGS
ADN extraído de los tejidos FFPE era de calidad y cantidad suficiente para el Solid-NGS panel, con una concentración de 252 ng / l, y sólo el 4% del ADN por debajo de 1.000 pares de bases (pb). Tras el análisis de datos de la profundidad media de cobertura fue de 9.167 lecturas (muy por encima de nuestro límite de profundidad mínima de 1.000 lecturas) sin regiones por debajo de 250 profundidad de lectura. métricas de control de calidad adicionales son shown en la Tabla 3.
Los datos procesados a través del gasoducto bioinformática detectaron dos mutaciones asociadas a la enfermedad: una inserción en marco en el exón 20 de ErbB2 (HER2 / neu) y una mutación sin sentido en TP53. Todas las variantes exonic con sus frecuencias alélicas están representados en la Tabla 5. La inserción en ErbB2 en realidad representa un tándem duplicado (Her2 / neu) secuencia, como se refleja en la nomenclatura. Identificación y confirmación de la revisión manual en marco de inserción requerida de la secuenciación lee a través de IGV. La detección de las frecuencias de mutación de mayor que 50%, como se ve, tanto para el TP53 y Her2 / neu mutaciones es sugerente de una pérdida de heterocigosidad evento (LOH) (véase la discusión).
Los resultados panel sólido-NGS para el caso 2 con mutaciones detectadas i adenocarcinoma de pulmónn ErbB2 (HER2 / neu) y TP53, dos genes no probados comúnmente para como parte de la corriente de estándar de atención para pacientes con cáncer de pulmón. HER2 / neu codifica un receptor de la tirosina quinasa similar a otro gen comúnmente mutado en el cáncer de pulmón, EGFR . La activación de HER2 exón / neu 20 inserciones se observan en 2-4% de los adenocarcinomas de pulmón, dar cuenta de la mayoría de las mutaciones HER2 / neu observadas en el cáncer de pulmón, y se ven típicamente en los tumores sin mutaciones en otros genes del controlador como EGFR y ALK 12 . Hay varias líneas de evidencia que muestra potencial para las diferentes opciones de tratamiento para los pacientes con la activación de las inserciones de HER2 / neu, incluyendo la respuesta parcial a la terapia de combinación con HER2 / neu y los inhibidores de mTOR 13 y el control de la enfermedad significativa con el anticuerpo monoclonal trastuzumab en combinación con quimioterapia 15. El descubrimiento de un cambio TP53 no es uncoMMON en el cáncer, pero en este momento no existen terapias viables.
| Caso 1 | caso 2 | |
| Arranque Total de Lecturas | 2,215,926 | 2,129,110 |
| Porcentaje de Lee Mapping | 98.42% | 98.29% |
| Porcentaje de Lee en el blanco | 99.01% | 97.29% |
| Porcentaje de Lee en el blanco Después de filtro | 97.60% | 95.45% |
| Porcentaje de útil Lee | 94.87% | 91.79% |
| Porcentaje de bases por encima de Cobertura 250x | 98.40% | 100% |
| Porcentaje de bases ACobertura Bove 1000x | 95.90% | 99,70% |
| A continuación cobertura 250x - Número de amplicón | 8 | 0 |
Tabla 3:. Secuenciación Run QC Métrica Este es un resumen de las estadísticas más importantes de ejecución, sin incluir la cobertura media, que se utilizan para la revisión de datos para determinar si una muestra de la biblioteca de preparación ha pasado del control de calidad. Todo el proceso es exitoso si todos los porcentajes están por encima de 90%, pero es posible, con el arrastre de SW1 o UB1 en la etapa de lavado FPU o cebador diafonía, a tener menor 'Porcentaje en Target' en el intervalo de 80 - 90%. Si el 'Porcentaje Asignado' es demasiado bajo, que indicaría una contaminación de bacterias u otro ADN, ya que todas las muestras deben estar alineados a humano. Cuando cualquiera de estas especificaciones se encuentran por debajo del 80%, la muestra se encuentra en posición para una revisión adicional para ayudar a determinar cómo proceed y mejorar el proceso.

Tabla 4:.. Caso 1 resultados detectados patógena, enfermedad asociada, variantes de significado incierto (VUS), y variantes exonic probablemente benignas por encima de los criterios de notificación de la frecuencia de los alelos 5% se enumeran Por favor, haga clic aquí para descargar la tabla.

Tabla 5:.. Caso 2 resultados detectados patógena, enfermedad asociada, variante de significado desconocido (VUS), y exonic variantes probablemente benignas por encima de los criterios de notificación de 5% frecuencia de los alelos están enumerados Haga clic aquí para descargar esta tabla.
Adicional Figura 1: Un ejemplo de un SampleSheet.csv a base de amplicón. Esta hoja transmite al secuenciador lo que la química para funcionar (en este caso amplicón), lo que el flujo de trabajo (por ejemplo, GenerateFastq), lo que Aplicación y ensayo (por ejemplo, FastqOnly), el número de bases (o lee) a la secuencia (en este caso 186 pb x 186 pb), y por último lo muestras se asocian con ciertos índices (en este caso doble indexación). Las partes que están resaltados en amarillo pueden ser cambiados a lo que el experimentador quiere, pero en este caso el laboratorio utiliza estos parámetros. Por favor, haga clic aquí para descargar esta figura.
Discusión
Como las dos pruebas NGS descritos en este manuscrito se ofrecen clínicamente, la consideración práctica más importante es el control de calidad. En concreto, cerca de la consideración se debe pagar a la calidad y cantidad de ADN extraído. Esto es especialmente importante para las muestras FFPE que a menudo son muy degradada con rendimiento de ADN variable. Un método de precipitación con isopropanol se desarrolló con el fin de maximizar el rendimiento de ADN a partir de muestras FFPE como se encontraron los métodos basados en las columnas para dar lugar a veces a cizallamiento DNA con volúmenes de elución limitados. Por lo tanto, la mayoría de las veces cuando un espécimen produce demasiado baja concentración o es demasiado degradada para el ensayo, lo más probable debido al tamaño del tejido, el tipo o la fijación y no el proceso de extracción. Para las muestras de sangre / médula ósea, si hay un error de extracción, por lo general es debido a un ser hemodilute muestra (es decir, no tener suficiente número de glóbulos blancos o tumorales en ese sorteo) o quimioterapia ablación.
. Nt "> Durante la validación, se debería establecer puntos de corte para la aceptabilidad de la calidad y cantidad de ADN La entrada recomendada de los 100 - 250 ng se utiliza a menudo en el ensayo, sin embargo, si la calidad del ADN es buena, a continuación, cantidades inferiores de los insumos pueden tener éxito. Además, si la calidad del ADN es pobre (es decir, la cantidad de ADN amplificable es de menos de 100 a 250 ng) a continuación, las cantidades de entrada más altas pueden mejorar la calidad de los resultados de secuenciación (ya que la cantidad de ADN amplificable se llega a la entrada recomendado) . las métricas de calidad y cantidad del ADN se debe aplicar a cada muestra antes de avanzar el ADN en la preparación de la biblioteca. Esas muestras en una "zona gris" (véase la Figura 2) se debe ejecutar a discreción del director del laboratorio o la persona designada. en la actualidad el mejor manera de predecir si el ADN no llevará a cabo bien durante la secuenciación es realizar un ensayo basado en qPCR que permite la evaluación de la cuantificación y la calidad de ADN de entrada. Este enfoque se refiere a la bioavailability de diferentes fragmentos de tamaño en la muestra, a través de la amplificación de fragmentos de tamaño diferentes (por ejemplo, 100 pb, 150 pb, 200 pb y 300 pb) y la comparación de rendimientos.En la actualidad, la preparación de la biblioteca consiste en un gran número de pasos manuales, donde un paso en falso en una de las diversas coyunturas puede causar la biblioteca para fallar o ser de mala calidad. El análisis en gel de microfluidos es el único paso de control de calidad para comprobar una cuestión biblioteca de preparación antes de la secuenciación. En consecuencia, hay varios pasos críticos en los que la atención extra puede aumentar la probabilidad de una reacción exitosa. Es imprescindible para garantizar el correcto de la muestra y de oligonucleótidos se utilizan para cada espécimen. Asegurar y registrar adecuadamente que cada muestra contiene una de las 96 combinaciones únicas de pares de cebadores de PCR-duales indexadas reduce la posibilidad de una muestra de confusión. También, es importante asegurar la placa de filtro (FPU) drena correctamente; si no drena adecuadamente esto puede provocar que el extepaso nsion-ligadura de la preparación de la biblioteca para realizar subóptima y conducir a la secuencia de datos de mala calidad. Después de biblioteca QC, es crítico para asegurar que las perlas LNB1 están completamente resuspendieron y que la solución LNB1 / LNA1 se mezcla bien antes de añadir a las muestras como la concentración de esta mezcla se utiliza para determinar la molaridad de la biblioteca. Finalmente, si el paso del talón de elución da lugar a una cantidad subóptima de biblioteca eluyendo de las perlas que disminuirá la densidad de agrupamiento y, posiblemente, causar la biblioteca para no obtener una cobertura media adecuada. Por el contrario, un exceso de biblioteca dará lugar a una peor calidad lee. Por lo tanto, es importante ser constante en la etapa de normalización basado en perlas para asegurar la puesta en común óptima y la agrupación de las bibliotecas en el secuenciador.
Además de la preparación de la biblioteca, es fundamental para validar un gasoducto bioinformática que producirá llamadas mutaciones exactas de los archivos RAW, desmultiplexadas FASTQ. la elección de unasolución personalizada puede llevar mucho tiempo, ya que hay muchos alineadores de código abierto y comercialmente disponibles, las personas que llaman variantes y paquetes de software NGS que uno tiene que tamizar a través. tendrán que ser diseñado algoritmos personalizados para extraer estadísticas de rendimiento esenciales, identificar las mutaciones recurrentes únicos que eludió herramientas de código más abiertas, y determinar la condición de número de copia sobre cada uno de los loci. Durante el proceso de validación de un gasoducto bioinformática, es importante para determinar los puntos de corte de notificación obligatoria para las variantes que cumplan o superen tanto una profundidad mínima de cobertura después de la filtración de la calidad (por ejemplo, un mínimo de 250 lecturas) y una frecuencia mínima alelo (por ejemplo, 4 %). Dado que este un ensayo basado en amplificación de multiplexado, es importante para determinar la longitud media mínima profundidad de la cobertura (por ejemplo, 1,000x) de que la biblioteca tiene que obtener para ser capaz de obtener el amplicón de menor nivel de rendimiento a la profundidad mínima de lecturas. Además, la naturaleza multiplexada del ensayo hace causa desactivar los efectos de objetivo y estos artefactos '' tendrá que ser descubierto y totalmente examinados antes de su lanzamiento. Otra limitación importante para el ensayo descrito es una necesidad para las muestras que contienen más de 10% de tumor con el fin de lograr la frecuencia del alelo mínimo validado.
La detección de baja frecuencia, 1%, inserciones FLT3 es evidencia de que la revisión manual sigue siendo deseable en este proceso. Incluso con un alelo frecuencia de corte del 5%, puede que algunas mutaciones importantes perdidas y por lo tanto la revisión manual serán esenciales para determinar estas variantes. Para FLT3-ITD, la inspección visual del exón 14 se lleva a cabo para todos los pacientes con LMA para asegurar un nivel bajo o grandes inserción / duplicación no pasa desapercibida. Además, HER2 exón 20 inserciones que son comúnmente al lado de la secuencia del cebador, necesitan intervención manual. A pesar de tener un gasoducto bioinformática robusta, algunas variantes podrían pasarse por alto que es sólo la naturaleza de tener un corte duroapagado para la mayoría de las estadísticas mencionadas anteriormente. se necesitan mejores bioinformática para ayudar a aliviar este problema, al igual que una mejor preparación de la biblioteca y / o secuenciación metodologías, ya que es más ventajoso disponer de datos de buena calidad en los puntos de corte aún más bajos que contienen un menor número de artefactos y falsos positivos.
La detección e interpretación de frecuencias de los alelos puede ser difícil debido a la dificultad en la determinación del porcentaje de tumor y el sesgo de la amplificación de algunas regiones del genoma. Además, frecuencias de los alelos más de 50% se pueden detectar, como se observa en el caso 2. Esto se interpreta como una pérdida de heterocigosidad (LOH) evento, ya sea debido a la pérdida del alelo normal, lo que lleva a la aparente aumento del mutante lee, una ganancia del alelo mutante (por ejemplo, 2 mutante y una copia normal) u otros mecanismos. Estos mecanismos pueden ser dilucidado mediante la utilización de la gama de hibridación genómica comparativa (aCGH 19) y / o una matriz de genotipado SNP. 20.
Las metodologías de enriquecimiento de destino actuales se basan en procedimientos de día completo de cualquiera de las técnicas de PCR multiplex de captura de híbridos ineficientes o que dan lugar a la necesidad de una mayor cobertura de la secuenciación de una sola muestra y más fuera de la secuencia de destino lee. Las aplicaciones adicionales para oncología molecular NGS se espera en un futuro próximo incluirán métodos de preparación de bibliotecas fáciles que pueden ser completamente automatizados y ser capaz de procesar las muestras con cantidades muy pequeñas de ADN de entrada (es decir, menos de 1 ng), así como las muestras con alto grado de degradación DNA. Para hacer frente a estos retos, la mayoría de los métodos estará probablemente basado en la PCR, o bien ser un enfoque de PCR de varios pasos o un enfoque de PCR singleplex masivamente paralelas. Además, los códigos de barras molecular de amplicones individuales se ha demostrado que reduce drásticamente el ruido de fondo de secuenciación y permitirá a las pruebas de las muestras con proporciones más bajas de las células tumorales para lograr menores frecuencias de los alelos y avanzar hacia la captura de circulanteing células tumorales.
La detección de mutaciones asociadas a la enfermedad en muestras de cáncer ha sido norma de atención durante décadas. Históricamente, los genes se prueban a menudo en secuencia, un gen / exón a la vez, con la identificación de una mutación que conduce a la final de la secuencia de prueba. El advenimiento de la NGS ha permitido un enfoque menos sesgada a la secuenciación de múltiples genes asociados con muchos tipos de cáncer en paralelo que conducen a la identificación de múltiples mutaciones que están asociadas con la neoplasia. La utilidad clínica de NGS para la detección de mutaciones somáticas en el cáncer es cada vez más evidente. De hecho, el análisis basado en NGS de muestras tumorales representa un nuevo paradigma que desafía las pruebas genéticas tradicionales, sola, pero la utilidad clínica es muy clara. Los laboratorios clínicos actuales tienen la gran oportunidad de casarse con la validación del método de ensayo e interpretación cuidadosa con la aplicación de esta tecnología de gran alcance.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Los autores desean agradecer la ayuda de Daniel salvaje para la lectura del manuscrito y la asistencia en la producción.
Materiales
| Name | Company | Catalog Number | Comments |
| Genomic DNA ScreenTape | Agilent Technology | 5067-5365 | |
| Genomic DNA Reagents | Agilent Technology | 5067-5366 | |
| High Sensitivity D1000 ScreenTape | Agilent Technology | 5067-5584 | |
| High Sensitivity D1000 Reagents | Agilent Technology | 5067-5585 | |
| TapeStation 2200 | Agilent Technology | G2965A | |
| TapeStation Analysis Software | Agilent Technology | A.01.04 or higher | |
| 96-well Tube Storage Racks | Any Vendor | ||
| 15/50 ml Tube Rack | Any Vendor | ||
| 96-well Plate Rack | Any Vendor | ||
| Pipette, single-channel, 0.5–2.5 μl | Any Vendor | ||
| Pipette, single-channel, 1–10 μl | Any Vendor | ||
| Pipette, single-channel, 2–20 μl | Any Vendor | ||
| Pipette, single-channel, 10–100 μl | Any Vendor | ||
| Pipette, single-channel, 20–200 μl | Any Vendor | ||
| Pipette, single-channel, 100–1,000 μl | Any Vendor | ||
| Serological Pipettor | Any Vendor | ||
| Vortexer | Any Vendor | ||
| Ice bucket | Any Vendor | ||
| Microcentrifuge (for tubes and strip tubes) | Any Vendor | ||
| Freezer, -20 °C | Any Vendor | ||
| 4 °C Refrigerator | Any Vendor | ||
| Water or Bead Bath | Any Vendor | ||
| Incubator (37 °C) | Any Vendor | ||
| Serological Pipettes, 1 ml | Any Vendor | ||
| Serological Pipettes, 5 ml | Any Vendor | ||
| Serological Pipettes, 10 ml | Any Vendor | ||
| Serological Pipettes, 25 ml | Any Vendor | ||
| Gloves | Any Vendor | ||
| Razor Blades/Scaples | Any Vendor | ||
| KimWipes | Any Vendor | ||
| 15 ml Conical Tube | Any Vendor | ||
| 50 ml Conical Tube | Any Vendor | ||
| Paper Towels | Any Vendor | ||
| 200 proof Ethanol | Any Vendor | Store in Flammable Cabinet | |
| 2-Propanol (Isopropanol) | Any Vendor | Store in Flammable Cabinet | |
| 25 ml Reservoirs | Any Vendor | ||
| 10 N NaOH | Any Vendor | ||
| Pipette, 8-channel, 1 – 10 μl | Any Vendor | ||
| Pipette, 8-channel, 10 – 100 μl | Any Vendor | ||
| Pipette, 8-channel, 20 – 300 μl | Any Vendor | ||
| Ice Bucket | Any Vendor | ||
| Water Squirt Bottle | Any Vendor | ||
| Alcohol Squirt Bottle | Any Vendor | ||
| Lens Cleaning Paper | Any Vendor | ||
| Plates, 96-well PCR, Semi-Skirted | Any Vendor | ||
| Tube strips, 8-well, 0.2 ml | Any Vendor | ||
| Agencourt AMPure XP Beads | Beckman Coulter | A63881 | |
| BioShake IQ or 3000-T elm | Bulldog Bio/Q.Instruments | 1808-0506/ 1808-0517 | |
| DropPlate96 S - LabChipDS | Caliper | 128876 | |
| DropPlate96 D - LabChipDS | Caliper | 132848 | |
| DropSense96 | Caliper (Trinean) | ||
| DropQuant Software | Caliper (Trinean) | ||
| Plate Sealing Film | Denville | B1212-5S | |
| Aluminum Seal Foil | Denville | B1212-6S | |
| Nuclease-Free, Pure Water System | EMD Millipore | ||
| 5424 centrifuge | Eppendorf | 22621408 | |
| 5804R centrifuge | Eppendorf | 22623508 | Both 15 ml tube and plate rotators, preferably a centrifuge that can go up to 2,500 x g. |
| Safe-Lock Tube 1.5 ml, Natural | Eppendorf | 22431021 | |
| 5 ml Tube, DNA LoBind Tube | Eppendorf | 30108310 | |
| 5430R Centrifuge | Eppendorf | 022620645 | Any plate rotator centrifuge will work |
| Hybex Microsample Incubator | Fisher Scientific | 1057-30-0 | |
| Hybex 0.2 ml Tube Block | Fisher Scientific | 1057-31-0 | |
| TruSeq Amplicon – Cancer Panel | Illumina | FC-130-1008 | 96 reactions |
| TruSeq Custom Amplicon | Illumina | PE-940-1011 | 96 reactions |
| TruSeq Custom Amplicon Index Kit | Illumina | FC-130-1003 | 96 Indices, 384 Samples |
| MiSeq Reagent Kit v3, 500 Cycles | Illumina | MS-102-3003 | |
| MiSeq Reagent Kit v2, 300 Cycles | Illumina | MS-102-2002 | |
| MiSeq Reagent Kit v2, 500 Cycles | Illumina | MS-102-2003 | |
| Experiment Manager | Illumina | 1.3 or higher | |
| MiSeq Reporter | Illumina | 2.0 or higher | |
| Sequencing Analysis Viewer | Illumina | 1.8 or higher | |
| TruSeq Index Plate Fixture and Collar Kit | Illumina | FC-130-1007 | |
| MiSeq v2 | Illumina | SY-410-1003 | |
| TruSeq Custom Amplicon Filter Plate | Illumina | FC-130-1006 | |
| Index Adapter Replacement Caps | Illumina | 11294657 | |
| Qubit 2.0 | Invitrogen | Q32866 | |
| Qubit 0.5 ml Tubes | Invitrogen | Q32856 | |
| Qubit dsDNA Broad Range Assay Kit | Invitrogen | Q32853 | |
| DynaMa6-96 Magnetic Stand, Side Skirted | Invitrogen | 120.27 | |
| GeneAmp PCR System 9700 (gold/silver block) | Life Technologies | N8050200 | |
| Gentra Puregene Blood Kit | Qiagen | 158489 | |
| Deparaffinization Solution (16 ml) | Qiagen | 19093 | |
| Buffer ATL (4 x 50ml) | Qiagen | 939011 | |
| Protein Precipitation Solution (50 ml) | Qiagen | 158910 | |
| DNA Hydration Solution (100 ml) | Qiagen | 158914 | |
| Glycogen Solution (500 μl) | Qiagen | 158930 | |
| Qiagen Proteinase K | Qiagen | 19133 | |
| Rnase (5 ml) | Qiagen | 158924 | |
| Nuclease-Free Water (10 x 50 ml) | Qiagen | 129114 | |
| Pestles | USA Scientific | 1415-5390 | |
| TipOne RPT 10 µl elongated filter pipet tips in sterilized racks, 10 racks of 96 tips (960 tips). | USA Scientific | 1180-3810 | |
| TipOne RPT 100 µl natural, beveled filter pipet tips in sterilized racks, 10 racks of 96 tips (960 tips) | USA Scientific | 1180-1840 | |
| TipOne RPT 200 μl natural, beveled filter pipet tips in racks, sterilized racks, 10 racks of 96 tips (960 tips) | USA Scientific | 1180-8810 | |
| TipOne RPT 20 μl natural, beveled filter pipet tips in racks, sterilized racks, 10 racks of 96 tips (960 tips) | USA Scientific | 1180-1810 | |
| TipOne RPT 1,000 μl natural, graduated XL filter pipet tips in | USA Scientific | 1182-1830 |
Referencias
- Gerlinger, M., et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 366 (10), 883-892 (2012).
- Campbell, P. J., et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 467 (7319), 1109-1113 (2010).
- Ding, L., et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 481 (7382), 506-509 (2012).
- Frampton, G. M., et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nature Biotechnol. 31 (11), 1023-1031 (2013).
- Patel, J. P., et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 366 (12), 1079-1089 (2012).
- Forbes, S. A., et al. COSMIC (the Catalogue of Somatic Mutations in Cancer ): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 38 (Database Issue), 652-657 (2010).
- Shih, A. H., Abdel-wahab, O., Patel, J. P., Levine, R. L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 12 (9), 599-612 (2012).
- Liersch, R., Müller-Tidow, C., Berdel, W. E., Krug, U. Prognostic factors for acute myeloid leukaemia in adults - biological significance and clinical use. Br J Haematol. 165 (1), 17-38 (2014).
- Bacher, U., Schnittger, S., Haferlach, T. Molecular genetics in acute myeloid leukemia. Curr Opin Oncol. 22 (6), 646-655 (2010).
- Subramanian, J., Govindan, R. Lung cancer in "Never-smokers": a unique entity. Oncology (Williston Park). 24 (1), 29-35 (2010).
- Sakashita, S., Sakashita, M., Tsao, M. S. Genes and pathology of non-small cell lung carcinoma. Semin Oncol. 41 (1), 28-39 (2014).
- Arcila, M. E., Chaft, J. E., Nafa, K. Prevalence clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin Cancer Res. 18 (18), (2012).
- Gandhi, L., et al. Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2-dependent and other solid tumors. J Clin Oncol. 32 (2), 68-75 (2014).
- Sheikhha, M. H., Awan, A., Tobal, K., Liu Yin, J. A. Prognostic significance of FLT3 ITD and D835 mutations in AML patients. Hematol J. 4 (1), 41-46 (2003).
- Mazières, J., et al. Lung cancer that harbors an HER2 mutation epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 31 (16), 1-8 (2014).
- Robinson, J. T., et al. Integrative Genomics Viewer. Nat Biotechnol. 29 (1), 495-500 (2011).
- Forbes, S. A., et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 43 (Database issue), D805-D811 (2014).
- Daber, R., Sukhadia, S., Morrissette, J. J. Understanding the limitations of next generation sequencing informatics, an approach to clinical pipeline validation using artificial data sets. Cancer Genetics. 206 (12), 441-448 (2013).
- Haraksingh, R. R., et al. Genome-Wide Mapping of Copy Number Variation in Humans: Comparative Analysis of High Resolution Array Platforms. PLoS ONE. 6 (11), e27859(2011).
- de Leeuw, N., et al. SNP Array Analysis in Constitutional and Cancer Genome Diagnostics - Copy Number Variants, Genotyping and Quality Control. Cytogenet Genome Res. 135 (3-4), 212-221 (2011).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados