
Method Article
Successivo sequenziamento di nuova generazione per il rilevamento di Actionable mutazioni nei tumori solidi e liquidi
In questo articolo
Riepilogo
This manuscript describes clinical protocols for two next-generation sequencing panels. One panel interrogates hematologic malignancies while the other panel targets genes commonly mutated in solid tumors. Molecular classification of driver mutations in human malignancies offers valuable prognostic and predictive information.
Abstract
As our understanding of the driver mutations necessary for initiation and progression of cancers improves, we gain critical information on how specific molecular profiles of a tumor may predict responsiveness to therapeutic agents or provide knowledge about prognosis. At our institution a tumor genotyping program was established as part of routine clinical care, screening both hematologic and solid tumors for a wide spectrum of mutations using two next-generation sequencing (NGS) panels: a custom, 33 gene hematological malignancies panel for use with peripheral blood and bone marrow, and a commercially produced solid tumor panel for use with formalin-fixed paraffin-embedded tissue that targets 47 genes commonly mutated in cancer. Our workflow includes a pathologist review of the biopsy to ensure there is adequate amount of tumor for the assay followed by customized DNA extraction is performed on the specimen. Quality control of the specimen includes steps for quantity, quality and integrity and only after the extracted DNA passes these metrics an amplicon library is generated and sequenced. The resulting data is analyzed through an in-house bioinformatics pipeline and the variants are reviewed and interpreted for pathogenicity. Here we provide a snapshot of the utility of each panel using two clinical cases to provide insight into how a well-designed NGS workflow can contribute to optimizing clinical outcomes.
Introduzione
sequenziamento di prossima generazione (NGS) di campioni oncologia clinica è diventato più ampiamente disponibile nel corso degli ultimi anni, come sempre più punti di letteratura scientifica l'importanza di individuare i cambiamenti genetici per il targeting e marcatori molecolari predittivi / prognostici. Pannello Multi-gene analisi e interi studi sequenziamento in entrambi epiteliali 1,2 e 3 ematologiche maligne hanno solidificato il concetto di eterogeneità del tumore e l'evoluzione clonale come malattia progredisce e ricadute. Inoltre, a differenza delle tecnologie concorrenti come la reazione a catena della polimerasi (PCR) o sequenziamento Sanger, NGS in grado di rilevare la maggior parte delle alterazioni genomiche in tutti i geni del cancro clinicamente rilevanti in un singolo test 4.
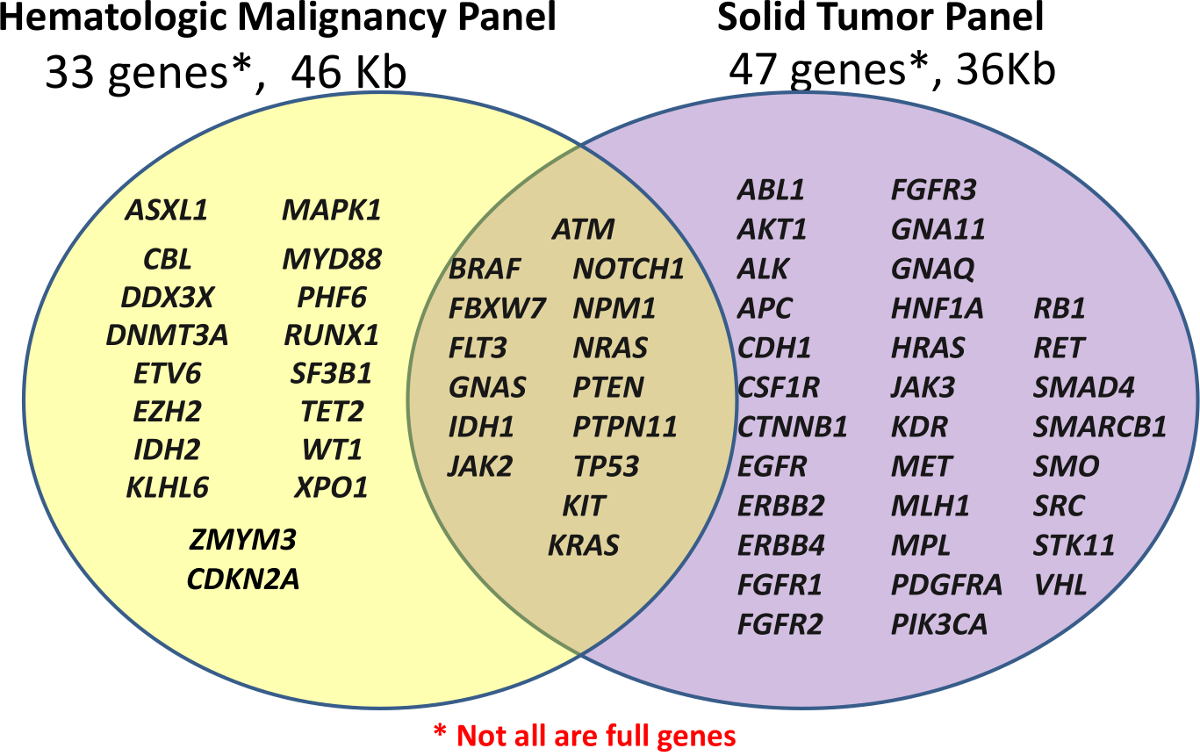
Il Centro per la diagnostica personalizzati inizialmente lanciato con due NGS pannelli clinici, una consuetudine pannello ematologica (eme-NGS Panel) e un off-the-shelf Cancer Panel per i campioni FFPE (Solid-NGS Panel) (vedi Figura 1). Questi pannelli coprono le regioni di interesse clinicamente rilevanti o elevati di geni selezionati; Non tutti i geni o esoni sono completamente coperti. Ampliconi sono generati da ibridazione sonda seguita per estensione e legatura. Le regioni interessate sono ulteriormente amplificati con PCR con primer dual-indicizzato universale, consentendo fino a 96 campioni devono essere aggregate per il sequenziamento.

Figura 1:. Elenco dei geni coperti nei pannelli preparazione Biblioteca viene eseguita utilizzando sia l'usanza ematologiche Panel (Pannello di eme-NGS) di 33 geni o off-the-shelf Amplicon Cancer Panel (Solid-NGS) di 47 geni. Non tutti i geni o esoni sono coperti per intero, come alcuni ampliconi può riguardare soltanto alcune hotspot. Clicca qui per vedere una versione più grande di questa figura. </ A>
{kind=link}
Il contenuto per il pannello di eme-NGS è stato derivato da più fonti, ma centra circa 16 geni mutati nella leucemia mieloide acuta (AML) precedentemente descritto come dimostrazione di un alto livello di utilità clinica 5. Il-NGS solido pannello è prodotto commercialmente con le regioni mirate basate su geni comunemente mutato nel tumore come riportato nel catalogo di mutazioni somatiche in Cancro (COSMIC) del database 6.
Diversi passaggi chiave che caratterizzano i flussi di lavoro per NGS clinici. Dopo il medico ordina il test, un patologo determina l'adeguatezza del campione seguenti analisi per la percentuale del tumore e volume del campione. Nella nostra istituzione, si richiede almeno il 10% del tumore a causa del tasso sfondo sequenziamento di errore ( "disturbo") della tecnologia e l'efficienza del approccio mirato. Se il tessuto è adeguata per il test, il DNA genomico è estratto. Questo DNA viene poi sottoposto a controllo multiplo qualità (QC) passi. Se il DNA passa QC, una libreria amplicone viene generato e sequenziato. I dati risultanti viene analizzata attraverso un bioinformatica pipeline in-house. A seguito di analisi bioinformatica, le varianti sono rivisti manualmente ed interpretati per la patogenicità prima dell'incorporazione in una relazione clinica. Qui di seguito vengono descritti due casi che ha attraversato questo rigorosa del flusso di lavoro e, infine, ha portato a cambiamenti nella gestione clinica.
Caso 1 - leucemia mieloide acuta
Una biopsia del midollo osseo dal paziente A era diagnostico per AML, senza la maturazione. Studi citogenetici sono stati inviati sul campione di midollo osseo e hanno dimostrato una normale cariotipo femminile. Ci sono stati 95% di blasti circolanti presenti, in modo da una periferica campione di sangue è stato inviato per l'esame diagnostico personalizzato sul pannello eme-NGS.
Leucemia mieloide acuta (LMA) è un tumore maligno ematologica della linea mieloide dei globuli bianchi. La rilevazionedi mutazioni geniche in LMA è diventato sempre più importante per la prognosi e il trattamento, con mutazioni genetiche ricorrenti riconosciuti come importante nella patogenesi e la prognosi 7. Le mutazioni in NPM1 e CEBPA sono stati associati con un rischio prognostico favorevole, mentre duplicazioni tandem interni (DTI) a FLT3 sono stati associati con un esito meno favorevole 8. Un crescente corpo di evidenze supporta un ruolo patogenetico di queste e altre mutazioni in AML 9.
Caso 2 - Polmone Adenocarcinoma
Una biopsia di una massa sopraclaveare sinistro da paziente B ha dimostrato adenocarcinoma polmonare. materiale bioptico dal incluso in paraffina (FFPE) linfonodo fissato in formalina è stato inviato per il test genomico (pannello solido-NGS), come rotoli / riccioli con più del 50% del tumore, per identificare se una mutazione era presente per un intervento terapeutico mirato.
Lung cancER è la principale causa di mortalità per cancro correlati negli Stati Uniti ed è diviso in due tipi principali, non a piccole cellule del polmone (NSCLC) e carcinoma polmonare a piccole cellule (SCLC). NSCLC può essere ulteriormente definita come adenocarcinoma o carcinoma a cellule squamose, sulla base l'istologia della lesione. Polmone adenocarcinoma è il sottotipo più comune di cancro del polmone, visto in fumatori e non fumatori, ed è la forma più comune di cancro al polmone per i non fumatori 10. Studi molecolari degli adenocarcinomi del polmone hanno identificato mutazioni in diversi oncogeni 11. Le mutazioni del driver più comuni individuate nei fumatori sono mutazioni nel gene KRAS e BRAF. Le mutazioni più comuni nei non fumatori sono mutazioni in EGFR, e riarrangiamenti che coinvolgono i geni ALK, RET e ROS1. Tumori del polmone sono stati descritti con un esone 20 inserimento in-frame nel ERBB2 gene (HER2 / neu). L'anomalia più comune in HER2 / neu è un ampliamento di questo locus nel carcinoma mammario per le quali una terapia mirata è disponibile (trastuzumab: un anticorpo monoclonale umanizzato contro HER2 / neu). Il 20 inserimento HER2 / neu esone che si osserva nel 2-4% del polmone adenocarcimomas 12 ha mostrato risposta parziale alla terapia di combinazione con HER2 / neu e inibitori di mTOR (neratinib e temsirolimus, rispettivamente) 13.
Protocollo
Questo protocollo comprende le fasi salienti di due prove di laboratorio sviluppato convalidate per il profilo genomico di tumori solidi e liquidi, rispettivamente. Il test eseguito in laboratorio è fatto in conformità con i requisiti dei Clinical Laboratory Improvement emendamenti (CLIA) del 1988.
1. Estrazione del DNA dal sangue periferico o midollo osseo
- Determinare la quantità di sangue o midollo osseo a prendere basa sulla Tabella 1.
| Esempio / WBC | Importo di essere trattato come 1 ml di sangue |
| Bone Marrow | 250 microlitri |
| WBC Sangue 12.000 - 50.000 | 1 ml |
| WBC Sangue 50,000 - 100,000 | 500 microlitri |
| WBC Sangue 100.000 - 200.000 | 200 l |
| WBC Sangue> 200.000 | 100 pl |
| * For Blood WBC <12.000, prendere 2 ml di sangue | |
Tabella 1:. Il sangue / midollo osseo Volume per Usa mappa Dal momento che il numero di globuli bianchi può variare da campione a campione, è difficile indicare un volume specifico di sangue da utilizzare. Pertanto, la quantità di sangue da utilizzare per il saggio deve essere determinata osservando il numero dei globuli bianchi (WBC) prima di iniziare il dosaggio. Sebbene meno sangue viene utilizzato, esso dovrebbe essere trattata come se le sue 1 ml Poiché il volume di sangue utilizzato è ridotta perché il numero di cellule presenti è maggiore del normale.
- Seguire il protocollo del kit disponibile in commercio per isolare il DNA genomico.
2. DNA Estrazione da paraffina-embedded (FFPE) tessuto fissato in formalina
- Basato sula regione del tumore il patologo cerchiata sul vetrino H & E, allineare i vetrini non colorati con la guida di H & E scivolo e delineare una zona simile per l'estrazione. Per macro-dissezione, processo di un solo esemplare / set del paziente di diapositive alla volta.
- Scaldare i vetrini su un blocco C di calore di 45 ° o leggermente fondere la paraffina. raschiare delicatamente il tessuto all'interno delle linee che sono contrassegnati sul vetrino, con un nuovo bisturi per ogni campione da estrarre. Posizionare la raschiatura cera nel tubo adeguatamente etichettati 1,5 ml. Bisogna fare attenzione perché la cera è molto raschiato elettrostatico e può saltare fuori dal tubo.
- Aggiungere 320 ml di Sparaffinatura soluzione per ogni cinque o sei 5 sezioni micron (25 - 30 micron totali). Ad esempio, se un tubo contenente 3 sezioni di 10 micron rotolo / arricciatura sta per essere trasformati, quindi utilizzare 320 microlitri, ma se sono stati ottenuti 5 sezioni allo stesso spessore quindi utilizzare 640 microlitri.
- Vortex vigorosamente per almeno 10 secondi ed eseguire aqrotazione uick in una microcentrifuga per rimuovere il tessuto / cera dai lati e cappuccio e nella soluzione. Incubare a 56 ° C per 3 min, e incubare a temperatura ambiente per 5 -. 10 min.
- Dopo l'incubazione RT, aggiungere 180 ml di tampone ATL per ogni 320 ml di soluzione Sparaffinatura aggiunto. Tritare il tessuto dieci volte con un mini-pestello sterile utilizzando una nuova pestello per ogni campione. Assicurarsi che vi sia nessun tessuto attaccato al pestello, come può essere molto appiccicoso. Vortex la sospensione vigorosamente per 3 secondi, quindi si centrifuga alla massima velocità per 1 min.
- Aggiungere 10 ml di proteinasi K alla fase chiara inferiore. Mescolare delicatamente pipettando su e giù per garantire il tessuto viene risospeso. Non lo fanno vortice. Incubare a 56 ° C per una notte con agitazione a 400-500 giri al minuto.
- La mattina successiva, verificare se il tessuto è completamente sciolto (questo si è verificato se la soluzione più bassa è chiaro). Se la soluzione inferiore non è chiaro, quindi vortice vigorosamente per 3 secondi e centrifugare a massima velocità for 1 min. Aggiungere un extra 5 - 10 ml di proteinasi K e incubare a 56 ° C per altri 30 - 60 min.
- Incubare a 90 ° C per 1 ora per contribuire a invertire la formaldeide reticolazione. Lasciare che i campioni raffreddare a temperatura ambiente per 5 - 10 minuti e poi centrifugare brevemente ciascun tubo per consolidare il liquido.
- Trasferire la fase chiara in basso in una provetta etichettata 1,5 ml. Se vi sono più tubi di stesso campione del paziente (ad esempio il caso se vengono utilizzate più rotoli), ricombinare le fasi inferiori in un tubo a questo punto. Nota: Trasferimento di piccole quantità di soluzione Deparaffinizzazione non dovrebbe interferire con la procedura di purificazione, ma vi è un rischio se una grande quantità viene trasferito.
- Aggiungere 2 ml di soluzione di RNase A. Vortex delicatamente o invertire 25 volte e rotazione rapida in una microcentrifuga. Incubare a temperatura ambiente per 5 min.
- Aggiungere 200 ml di proteine precipitazioni Solution. Se facendo uno o due panini, usare 200 ml. Se facendo tre rullis in una sola volta, quindi utilizzare 400 ml. Vortex vigorosamente ad alta velocità per 30 secondi per mescolare in modo uniforme i buffer di lisi. Incubare in ghiaccio per 5 minuti o campioni possono rimanere sul ghiaccio per un massimo di un ora.
- Centrifugare a 5000 xg per 5 min. La proteina precipitata dovrebbe formare una stretta, pellet bianco. Versare il supernatante in una provetta etichettata 1,5 ml, e poi incubare i campioni in ghiaccio per almeno 3 minuti. Centrifugare a 5.000 xg per 3 min.
- Aggiungere 200 ml di 2-propanolo (isopropanolo) per ogni 180 ml di tampone ATL inseriti anteriormente alla provetta da 1,5 ml (potrebbe essere necessario utilizzare un tubo 2 ml). Ad esempio, se facendo tre rotoli aggiungono 600 ml di isopropanolo. Aggiungere 1 ml di glicogeno per ogni 180 ml di tampone ATL aggiunto in precedenza per l'isopropanolo e capovolgere il tubo più volte per mescolare.
- Aggiungere con cautela il surnatante dal punto di proteine Precipitazione nella miscela isopropanolo. Mescolare il tubo capovolgendo delicatamente almeno 50 volte.
- Centrifugare a massima speed per 3 min. Il DNA sarà visibile come piccoli pellet bianco sul fondo della provetta.
- Versare o aspirare il surnatante nel tubo dei rifiuti. Mantenere una provetta da 1,5 ml differenziata per ciascun campione nel caso in cui il pellet viene sloggiato in modo da non essere perso o mescolato con lo spreco di altri esemplari. Scolare il tubo su un tovagliolo di carta e garantire la maggioranza della isopropanolo viene rimosso.
- Aggiungere 300 ml di appena fatto il 70% di etanolo. Capovolgere la provetta delicatamente più volte per lavare il pellet. Cercare di garantire il pellet viene sloggiato per garantire una pulizia più accurata.
- Centrifugare a massima velocità per 5 min. e poi rimuovere con attenzione l'etanolo. Il pellet può essere sciolto, in modo da versare o aspirare lentamente e guardare il pellet. Rimuovere l'eccesso di etanolo dall'interno del tubo, senza toccare il pellet. Consentire ai campioni di aria secca per 5 - 15 minuti, facendo attenzione a non più di asciugare il campione.
- Aggiungere tra 25-100 ml di DNA idratazione soluzione di ecampione gni base alle dimensioni del pellet di DNA e l'importo iniziale di tessuto. Vortex tubi energicamente e brevemente girare in una microcentrifuga. Incubare per 1 ora a 65 ° C per reidratare pienamente il DNA.
3. Controllo Qualità DNA genomico
Nota: Ci sono tre fasi indipendenti per il controllo della qualità del DNA (QC). Vedere la Tabella 2 per ulteriori spiegazioni del motivo per cui viene eseguito ogni passaggio di controllo di qualità.
| Strumento | Risultato | Indicazione | gamma ideale |
| DropSense96 | A260 / A230 | L'identificazione di contaminanti chimici (per esempio, etanolo) | 1,50-2,2 |
| DropSense96 | A260 / A280 | Identificazione di contaminanti proteici | 1,60-2,2 |
| DropSense96 | Concentrazione | la quantificazione del DNA | > 1 ng / ml |
| TapeStation | DNA Smear | Determinazione della integrità del DNA (ad esempio, la degradazione / frammentazione del DNA estratto) | 50%> 1000 bp |
| qubit 2.0 | Concentrazione | Più precisa quantificazione del DNA | > 1 ng / ml |
Tabella 2:. DNA QC Risultati attesi Tutti questi valori sono presi in considerazione prima di eseguire permettendo un campione di procedere alla fase di preparazione biblioteca.
- Seguendo il protocollo del produttore, eseguire 2 ml di DNA estratto su un fluorimetro per ottenere la concentrazione di lavoro (ng / ml) del campione.
- Eseguire 1 - 2 ml di ogni campione su un UV / VIS spettrofotometro per controllare la qualità del campione (A260 / A230 e A260 / A280 rattoios) secondo le istruzioni del produttore.
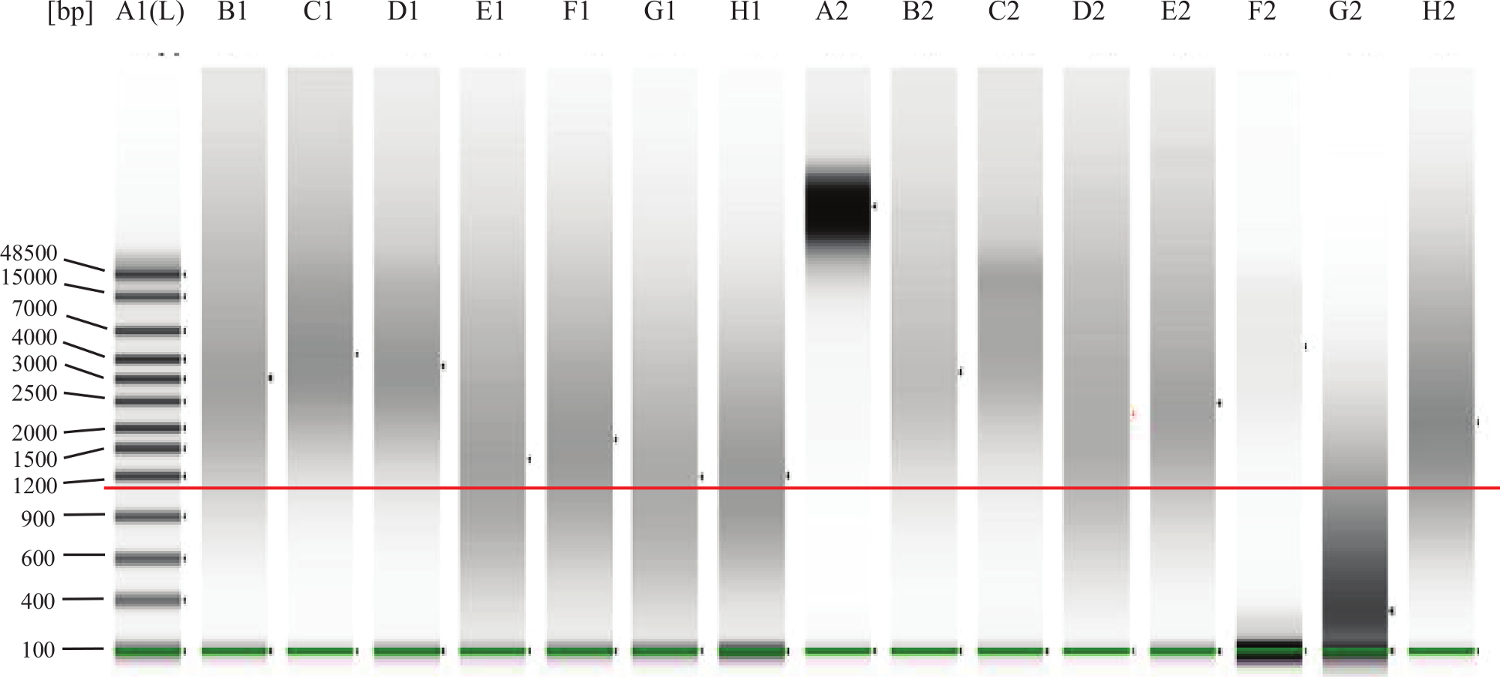
- Per FFPE campioni: Seguendo le istruzioni del produttore, eseguire 1 ml di ogni campione su un sistema di gel-elettroforesi microfluidica per valutare il degrado / frammentazione del DNA. Vedere la Figura 2 per gli esempi.

Figura 2:. Il DNA genomico QC Gel Esempio La linea verde sopra le fasce più basse è quello di indicare il marcatore più bassa. La linea rossa aggiunto-in è quello di indicare circa 1.000 bp. Corsia A2 rappresenta DNA completamente intatto, come ci si aspetterebbe da Sue tis fresco (ad esempio, sangue periferico o midollo osseo). Lanes B1, C1, D1, B2, C2, E2 sono esempi di buona, DNA FFPE intatto. Il DNA in corsia F2 sembra essere intatto, ma ad una concentrazione troppo bassa. Il DNA in corsia G2 è degradato o frammentato e non funzionerà nel saggio. Corsie E1, F1, G1, H1, D2,H2 rappresentano il DNA che rientra nella 'zona grigia' il che significa che il test potrebbe funzionare bene, ma un po 'del DNA potrebbe essere troppo danneggiato o reticolato e quindi non si esibirà anche nel dosaggio. Cliccate qui per visualizzare un più grande versione di questa figura.
{kind=link}
4. Amplicon Biblioteca Preparazione
- Ibridazione di Oligo Pool e Extension-legatura di Oligos Bound
- Nella sala pre-PCR, aggiungere il volume necessario di Low EDTA TE, 5 ml di tubo Oligo del pannello (ad esempio, Solid-NGS Panel o eme-NGS Panel), e 100-250 ng di DNA genomico per ciascuna corrispondente bene in un 96 ben piatto semi-gonna etichettato come il (HYB) piastra di ibridazione.
- Aggiungere 40 ml di oligo Ibridazione per il sequenziamento reagente 1 (OHS1) per ogni campione nella piastra HYB. Delicatamente pipetta su e giù almeno 5 - 6 volte per mescolare. Modificare suggerimenti dopo ogni colonna per evitarecontaminazione incrociata.
- Sigillare la piastra HYB con un foglio di alluminio adesivo e centrifugare a 1000 xg per 30 sec.
- Incubare la piastra HYB nella ibridazione incubatore pre-riscaldato a 95 ° C per 1 min. Impostare la temperatura dell'incubatore ibridazione a 40 ° C. Continuare incubazione finché prende l'incubatrice diminuire da 95 ° C a 40 ° C (~ 90 min). Nota: Questo raffreddamento graduale è fondamentale per il corretto ibridazione.
- Durante gli ultimi 15 - 20 min di incubazione ibridazione, pre-lavare il gruppo filtro piastra (FPU). Preparare solo i pozzetti da utilizzare nel saggio corrente, cioè, utilizzare solo / pozzetti non utilizzati freschi di una piastra filtro aperto in precedenza, ma mai riutilizzare pozzetti che sono stati utilizzati. Nota: Questo dovrebbe essere chiaro in base al numero kit sulla piastra di filtraggio, i segni sulla piastra di filtraggio, e il sigillante usato tutta la piastra.
- Usando una pipetta multicanale, aggiungere 45 ml di lavaggio stringente 1 (SW1) in ciascun pozzetto.
ATTENZIONE: Contiene formammide. Coprire e centrifugare la FPU a 2.250 xg per 3 minuti a 20 ° C. Controllare ogni bene per liquido residuo (> 15 ul / pozzetto). - Se esiste residui di liquido, ruotare la FPU a 180 ° e ripetere il passo centrifugare per un ulteriore 3 min. Se il liquido residuo gira attraverso la seconda volta, quindi procedere al passaggio successivo, se poi non ci può essere un difetto nella piastra di filtraggio e dovrà essere sostituito il piatto corrente.
- Usando una pipetta multicanale, aggiungere 45 ml di lavaggio stringente 1 (SW1) in ciascun pozzetto.
- Una volta che l'incubatore ibridazione è stata raffreddata a 40 ° C, centrifugare la piastra a 1.000 xg per almeno 30 sec a 20 ° C per raccogliere condensa. Trasferire l'intero volume di ogni campione dalla piastra HYB sul centro dei corrispondenti pozzetti pre-lavati della FPU. Cambiare suggerimenti dopo ogni colonna per evitare la contaminazione incrociata. Coprire la FPU e centrifugare a 2.250 xg per 3 minuti a 20 ° C.
- Aggiungere 45 ml di SW1 e centrifugare a 2.250 xg per 3 min a 20 ° C. Ripetere per un totale di due lavaggi. Ruotare il FPU a 180 ° e centrifugare di nuovo a 2.250 xg per 3 min per rimuovere completamente tutte le SW1.
- Dissimulare la FPU. Eliminare tutto il flow-through (contenente formammide) nel contenitore per rifiuti corretta. Rimontare la FPU utilizzando un diverso piatto MIDI raccolta dei rifiuti. Aggiungere 45 ml di tampone universale 1 (UB1) a ciascun pozzetto e centrifugare a 2.250 xg per 3 min a 20 ° C. ATTENZIONE: Contiene formammide.
- Aggiungere 45 ml di estensione legatura Mix 3 (ELM3) per ciascun pozzetto sulla piastra FPU e pipetta su e giù per 3 volte per mescolare. Nota: La reazione di estensione-legatura avviene sulla membrana piastra di filtraggio.
- Sigillare la piastra FPU con foglio di alluminio adesivo ed incubare l'intero gruppo FPU in un pre-riscaldato a 37 ° C incubatore per 45 min.
- Indicizzazione e PCR Amplification
- Un'aliquota gli indici utilizzati per i pozzi corrispondenti nella indicizzato Amplification Plate (IAP) disponendo i primer nel dispositivo Index Piastra, disposti nel modo seguente: i tubi di primer i5 (cappucci bianchi, soluzione limpida) in verticale, in linea con le righe da A a H, tubi i7 di primer (tappi arancione, soluzione gialla) in senso orizzontale , in linea con le colonne da 1 a 12. Utilizzando un p10 multicanale pipetta, aggiungere 4 ml di primer i7 (soluzione gialla) a ogni riga della IAP e aggiungere 4 ml di primer i5 (soluzione chiara) per ogni colonna della IAP.
- Su ghiaccio o un blocco di raffreddamento, preparare la PCR Master Mix con l'aggiunta di 0,5 ml di DNA polimerasi 1 (TDP1) a 25 ml di PCR Master Mix 2 (PMM2) per campione. Inverti, in modo rapido vortice, e centrifugare brevemente la PCR Master Mix per mescolare.
- Aggiungere 22 ml di PCR Master Mix per ogni pozzetto della IAP e pipetta su e giù per 3 volte per mescolare. Cambiare punte tra i pozzi. Mantenere la IAP a 4 ° C.
- Dopo il 45 min Extension-legatura di reazione (fase 4.1.10), rimuovere la FPU dal termostato e rimuovere con attenzione di aluminum sigillo di alluminio. Coprire con il coperchio e centrifugare a 2.250 xg per 3 min a 20 ° C.
- Aggiungere 25 ml di 50 mM NaOH a ciascun campione sulla buona FPU. Pipettare su e giù almeno 6 volte; garantire che i puntali delle pipette venire a contatto con la membrana. Modificare suggerimenti dopo ogni colonna. Incubare a temperatura ambiente per 5 min.
- campioni di trasferimento eluiti dalla FPU per la IAP come segue:
- Utilizzando un p100 pipetta multicanale impostato a 20 ml, pipetta la NaOH sulla piastra FPU su e giù per almeno 6 volte. Inclinare leggermente la piastra FPU per garantire l'aspirazione totale fuori la piastra.
- Trasferimento 20 microlitri dalla FPU al corrispondente colonna della IAP. Delicatamente pipetta su e giù per almeno 6 volte di combinare accuratamente il DNA con la PCR Master Mix. Sigillare la IAP con pellicola adesiva e centrifugare a 1000 xg per 1 minuto a 20 ° C.
- Portare la piastra PCR nella camera post-PCR e caricare la piastra su un termociclatore. Eseguire la PCR program costituito da un calore denaturazione fase a 95 ° C per 3 min; seguita da 25 cicli di 95 ° C per 30 sec, 62 ° C per 30 sec, 72 ° C per 60 sec; seguita da una estensione finale di 72 ° C per 5 min; terminando con una sospensione a 10 ° C. Se non procedere alla fase successiva dopo il completamento della PCR, la piastra può rimanere sul termociclatore overnight, oppure può essere conservato a 2 e 8 ° C fino a due giorni.
- PCR Purificazione e normalizzazione Bead-Based
- Rimuovere le sfere di purificazione magnetiche, tampone di eluizione (EBT), e reagenti gel elettroforesi dalla C frigorifero 4 ° e posto a temperatura ambiente almeno 20 minuti prima della fase successiva.
- Una volta che la PCR è completa, centrifugare a 1000 xg per 1 min a 20 ° C per raccogliere condensa. Trasferire 1 ml di ogni reazione PCR a striscia tubi / pozzetti della piastra contenente 4 ml di acqua per diluire i campioni 1/5. Pipetta su e giù per mescolare.
- Aggiungere 2 ml didiluito campioni PCRed a 2 ml di buffer di microfluidica-gel. Sigillare la strisce / piastra. Agitare a 1.800 rpm per almeno 30 secondi e centrifugare a 1000 xg per 30 sec. Seguendo il protocollo del produttore, eseguire il composto su un gel microfluidica per valutare se la preparazione biblioteca prodotto una libreria accettabile (vedere Figura 3).
- Vortice le sfere di purificazione magnetica fino a quando sono ben sospesi e il colore appare omogeneo. Aggiungere 45 ml di perline per ogni campione.
- Sigillare la piastra con pellicola adesiva trasparente e agitare la piastra a 1.800 rpm per 2 minuti. Incubare a temperatura ambiente, senza agitazione per 10 min.
- Porre la piastra su un supporto magnetico. Una volta che il surnatante ha autorizzato, rimuovere con attenzione e scartare il surnatante. Se qualche perle sono inavvertitamente aspirati nelle punte, dispensare le perle di nuovo alla piastra e lasciare che il resto piatto sul magnete per 2 minuti e confermare che il surnatante ha eliminato.
- Con la piastra su tegli supporto magnetico, aggiungere 200 ml di preparati al momento 80% di etanolo per ogni campione. Spostare la piastra in avanti e indietro un paio di volte. Incubare la piastra sul supporto magnetico per 30 sec. Rimuovere con cautela e scartare il surnatante. Ripetere per un totale di due lavaggi.
- Rimuovere l'eccesso di etanolo mediante centrifugazione brevemente la piastra a 1.000 xg per ridurre qualsiasi etanolo sui lati del tubo, ponendo la piastra posteriore sul supporto magnetico e usando una pipetta multicanale p10 impostata a 10 microlitri per rimuovere l'etanolo. Lasciare le perline per asciugare per 5-8 min.
- Utilizzando un p100 pipetta multicanale, aggiungere 30 ml di EBT a ciascun pozzetto. Pipetta su e giù un paio di volte per garantire le perline venire fuori il lato del tubo. Sigillare la piastra con pellicola adesiva trasparente e agitare la piastra a 1.800 rpm per 2 minuti.
- Incubare a temperatura ambiente senza agitare per 3 min. Se ci sono esempi in cui le sfere non sono completamente risospese, delicatamente pipetta su e giù per risospendere le sfere.
- Posizionare la piastra su un supporto magnetico. Utilizzando un p100 pipetta multicanale, trasferire 20 ml del surnatante in un piatto completamente nuovo chiamato la piastra Biblioteca Normalizzazione (LNP). Trasferire i restanti ~ 10 ml di singole biblioteche di sequenziamento di un piatto a parte chiamato il rimanente ripulito Biblioteca Piastra (RCLP). Conservare questa piastra con il prep libreria finale, in quanto può essere utilizzato come riserva per una seconda corsa di sequenziamento, se necessario. In caso contrario procedere alla fase successiva, la LNP e RCLP possono essere conservati a -15 a -25 ° C.
- Vigorosamente vortice e risospendere la Biblioteca Beads normalizzazione 1 (LNB1). È fondamentale per risospendere completamente i pellet LNB1 tallone sul fondo della provetta. Preparare la normalizzazione Mix, mescolando 8 ml di LNB1 con 44 ml di Biblioteca Normalizzazione Additivi 1 (LNA1) per campione. Vigorosamente vortice la normalizzazione Mix per 10-20 sec.
ATTENZIONE: LNA1 contiene formammide. - Con intermittente inversione unND vortex della normalizzazione Mix, aggiungere 45 ml di ogni campione della LNP. Sigillare la piastra con pellicola adesiva trasparente e agitare la piastra a 1.800 rpm per 30 min. Questo min di incubazione 30 è fondamentale per il corretto normalizzazione libreria come incubazioni più o meno di 30 min possono influenzare rappresentazione biblioteca e densità di cluster.
- Durante il 30 min di incubazione, preparare i reagenti per il sequenziamento da scongelamento la cartuccia del reagente e ibridazione Buffer (HT1) [ATTENZIONE: Entrambi contengono formammide]. Inoltre, ottenere ghiaccio per un passaggio successivo e garantire un blocco di calore adatto per provette da 1,5 ml centrifuga è impostato a 96 ° C.
- Quando la fase di miscelazione 30 min è completa, posizionare il LNP su un supporto magnetico. Una volta che il surnatante ha autorizzato, utilizzare una pipetta multicanale per rimuovere con attenzione e scartare il surnatante nel contenitore dei rifiuti.
- Rimuovere la LNP dal supporto magnetico e lavare le perline con Biblioteca Normalizzazione Wash 1 (LNW1) come segue:
- Aggiungere 45pl di LNW1 per ciascun campione.
ATTENZIONE: Contiene formammide. Sigillare la piastra con pellicola adesiva trasparente e agitare la piastra a 1.800 rpm per 5 min. Ripetere per un totale di due lavaggi. Assicurarsi di rimuovere tutti i LNW1 dopo il secondo lavaggio.
- Aggiungere 45pl di LNW1 per ciascun campione.
- Rimuovere la LNP dal supporto magnetico e, usando una pipetta multicanale, aggiungere 30 ml di 0,1 N NaOH (meno di una settimana) a ciascun pozzetto per eluire il campione. Sigillare la piastra con pellicola adesiva trasparente e agitare la piastra a 1.800 rpm per 5 min.
- Durante la 5 min eluizione, aggiungere 30 ml di Biblioteca Normalizzazione bagagli Buffer 1 (LNS1) in ciascun pozzetto per essere utilizzato in una nuova piastra di nome della piastra di stoccaggio (PSC).
- Posizionare la LNP sul supporto magnetico. Una volta che il surnatante ha autorizzato, trasferire l'eluizione 30 microlitri al LNS1 nel PSC. Cambiare punte tra i campioni per evitare la contaminazione incrociata.
- Sigillare la piastra con pellicola adesiva e centrifugare a 1000 xg per almeno 30 sec.
- Aggiungere 51; l di ciascun campione da sequenziato a un pool Amplicon libreria etichettato (PAL) provetta da 1,5 ml. Agitare il PAL per mescolare e centrifugare in una microcentrifuga brevemente.
- A seconda di ciò chimica sequenziamento utilizzato (V2 o V3) aggiungere 4 - 10 ml di PAL a 590 - 596 ml di HT1. Generalmente, aggiungere 5,8 ml di PAL a 595 ml di HT1 per la chimica V2 e 8,5 ml di PAL a 592 ml di HT1 per V3 chimica. Etichettare questo tubo come il tubo diluito Amplicon Library (DAL).
- Agitare il DAL e brevemente centrifugare in una microcentrifuga. Incubare la DAL a 96 ° C per 2 min. Invertire il tubo DAL 3 volte e mettere il DAL in ghiaccio per almeno 5 minuti, mentre si prepara il sequencer per il sequenziamento. Dopo il 5 min, il DAL è pronto per essere caricato.
- Se finito con il PSC, sigillare la piastra in alluminio adesiva pellicola stagnola ed etichettarlo con la data e targhetta di identificazione. Conservare il PSC sigillato e PAL a -15 a -25 ° C.

Figura 3:. Libreria Prep QC Gel Esempio La linea verde su bande più basse è quello di indicare il marker inferiore e la linea viola sulle bande superiori è quello di indicare il marcatore più alto. Tutto ha funzionato bene per corsie H1, A2, B2, C2, E2, e G2. La preparazione biblioteca non ha funzionato in modo ottimale, per corsie D2, F2, e H2, ma i risultati saranno ancora ottenuto solo che potrebbero non avere una copertura adeguata. Per A3, la preparazione biblioteca funzionava a malapena e molto probabilmente questo campione di DNA non era adeguata per il test. Le fasce più basse sopra il marcatore più basso sono i primer non utilizzati, perché l'aliquota è presa direttamente dal PCRed bene. Il campione NTC dovrebbe avere solo la banda di primer non utilizzati, e nient'altro. Cliccate qui per vedere una versione più grande di questa figura.
{kind=link}
5. Sequencing
- Assicurare una corretta SampleSheet.csv è stato fatto per la corsa. Vedere la Figura 1 supplementare per un esempio.
- Risciacquare e asciugare la cella di flusso e aggiungere 600 ml di DAL alla cartuccia reagenti scongelati.
- Preparare il sequencer per il sequenziamento seguendo le istruzioni sullo schermo.
Analisi 6. Dati
- Eseguire il gasdotto bioinformatica. Utilizzare il gasdotto bioinformatica in-house, progettato su misura per identificare mutazioni, inserzioni, delezioni e amplificazioni 18.
- Dopo che l'oleodotto ha completato, controllare i file di log per gli errori / warning, come ogni significativo aiuto errori / avvertenze nel controllo di qualità del trattamento pipeline.
- Analizzare le statistiche run (Tabella 3) per garantire la biblioteca sequenziato ha superato il laboratorio determinato metriche QC (Figura 4). Rivedere manualmente ogni variante visualizzando i file .bam in un visualizzatore di dati genomici (ad esempio, il Integrativa Genomica Vibrocca 16 (IGV)).
NOTA: solo le varianti all'interno della gamma allele frequenze convalidato e al di sopra della profondità minima di copertura dopo la filtrazione di qualità sono riportati (utilizzando i mezzi pesanti la nomenclatura di Human Genome Variation Society ()). Per la segnalazione finale, ogni variante exonic è stato classificato in: patogeno, probabilmente la malattia associata, variante sconosciuta rilevanza (VUS), probabilmente benigni, e benigna. Tutte le varianti sopra i criteri di rilevazione del 5% allele frequenza, ad eccezione di quelle varianti benigne ritenuti e cambiamenti sinonimo, sono stati segnalati.

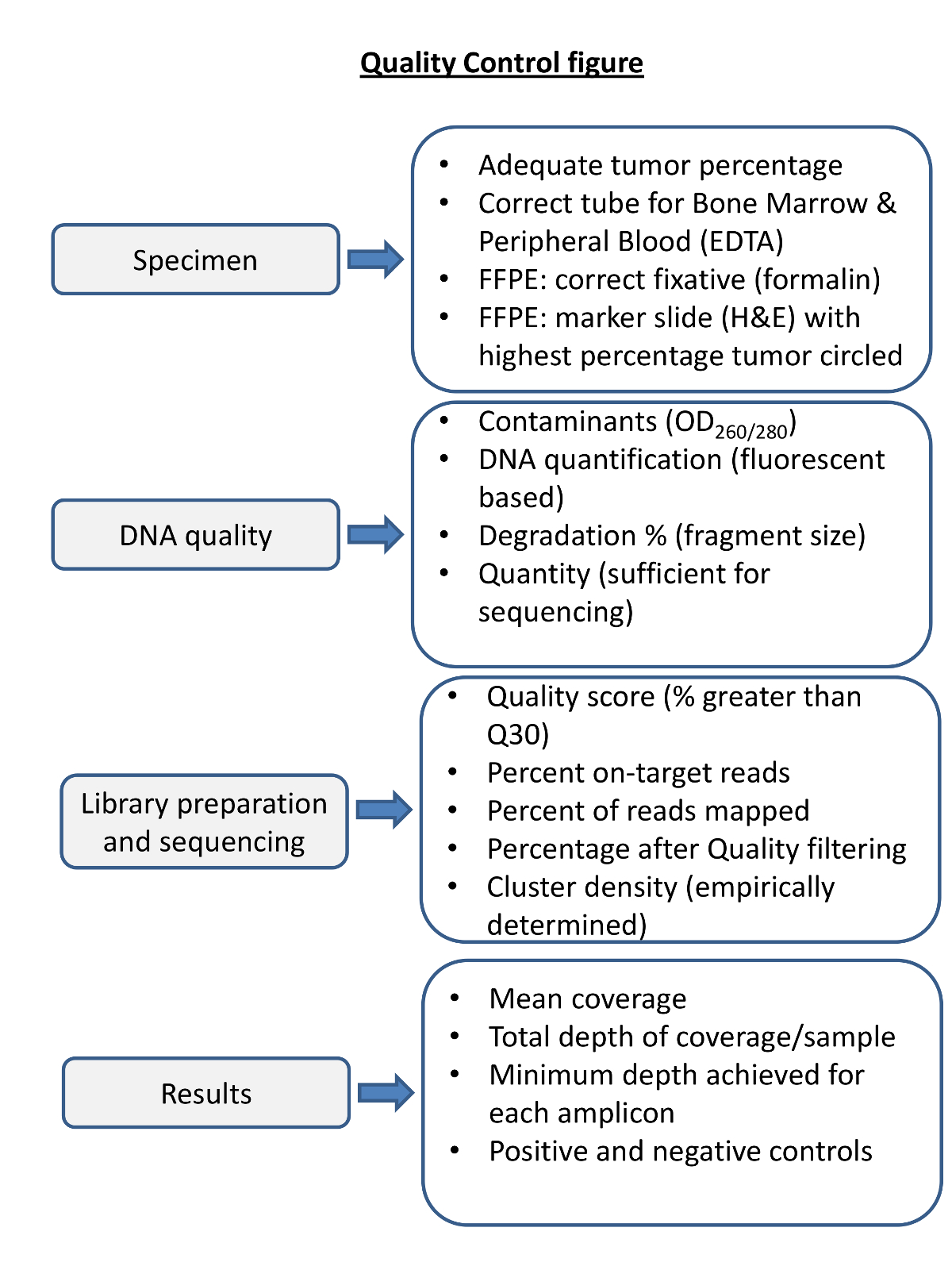
Figura 4. Presentazione di Quality gradini di per NGS. Il controllo di qualità di ogni fase del processo è necessario per garantire il sequenziamento produrrà risultati tali che i parametri di sequenziamento pre e post sono considerati. trattamento del campione appropriato è essenziale per DNA di alta qualità. sangue emidollo osseo in fissativi inappropriate può produrre DNA di bassa qualità. La fissazione inadeguato di campioni tumorali solide può degradare il DNA (ad esempio, fissazione in B5). qualità del DNA dovrebbe essere valutata per le proteine e la contaminazione di RNA con mezzi spettrofotometriche, e accuratamente valutato per la quantità e l'integrità del DNA. Le metriche di sequenziamento devono essere determinati empiricamente in laboratorio sequenziamento e seguiti per ciascuna reazione di sequenziamento e ogni campione. Prima di riportare i risultati di sequenziamento per ogni campione dovrebbero essere valutati per la copertura, la profondità, e adeguata esecuzione dei controlli positivi e negativi. Cliccate qui per vedere una versione più grande di questa figura.
{kind=link}
Risultati
Caso 1 - eme-NGS Panel
Il DNA estratto da sangue periferico leucemica era di qualità sufficiente e la quantità (176 ng / ml) per il pannello di eme-NGS. La profondità media complessiva di copertura è 4,933x (al di sopra della profondità media minima di copertura dei 1,000x). Ulteriori statistiche run sono in Tabella 3. Degli 8 regioni sottostanti copertura 250x, 3 erano dovuti a taglio improprio dei primer (cioè, la sequenza di innesco non è stato rimosso correttamente a causa di errori di sequenziamento), 1 era un artefatto conosciuto del dosaggio, e gli altri 4 erano zone parziali di esoni di geni diversi senza varianti informativa. Anche se il nostro protocollo clinico comprende solo la segnalazione varianti coperte da almeno 250 legge, tutti i dati con almeno 100 letture vengono importati nel database per la revisione.
Il trattamento dei dati from il gasdotto bioinformatica rilevato tre informativa, malattia mutazioni associate; una mutazione missense in FLT3, una mutazione missense nel IDH2, e una mutazione frameshift in NPM1. Le varianti exonic con le loro frequenze alleliche sono descritti nella Tabella 4. Una mutazione comune nel FLT3 è il FLT3 -Internal tandem duplicazione (ITD) che non viene chiamato automaticamente dalla nostra pipeline e richiede un controllo visivo dell'esone 14. Controllo visivo dell'esone 14 di FLT3 non ha mostrato alcuna inserimento o la duplicazione nel campione presentati. Il FLT3 I836del era al 1% frequenza allelica e non è stata inclusa nella relazione finale perché è caduto sotto la frequenza minima allele convalidata del 5%. Questa mutazione non è sulla stessa molecola di DNA come il cambiamento FLT3 D835Y (cioè, osservato nella stessa regione amplicone, ma non in "cis" di qualsiasi sequenza letture) ed è stata osservata solamente con una revisione manuale del fi .bamLes quando si verifica la variazione p.D835Y. Le frequenze alleliche inferiori delle due mutazioni FLT3 suggerisce queste mutazioni possono rappresentare l'eterogeneità e / o evoluzione clonale; tuttavia, la differenza potrebbe essere dovuta a prestazioni del test per quella amplicone o un polimorfismo a singolo nucleotide (SNP) vicino o sovrapposizione la sequenza di innesco che ha colpito l'amplificazione di questo allele.
I risultati del pannello eme-NGS per Caso 1 con leucemia mieloide acuta identificato mutazioni in FLT3, IDH2, e NPM1, tre geni comunemente mutati in AML. Le mutazioni in FLT3 si osservano in ~ 25% dei pazienti adulti con leucemia mieloide acuta (database COSMIC 17) e sono o duplicazioni tandem interno (DTI) o mutazioni missense nel dominio della tirosin-chinasi. I FLT3-DTI sono la mutazione più comune e sono associati con scarsa risposta alla chemioterapia standard, mentre il significato prognostico di FLT3 chinasi dmutazioni puntiformi OMain, come si vede in questo paziente AML, ha un impatto sulla prognosi chiaro 14. Isocitrate deidrogenasi 2 (NADP +), mitocondriale (IDH2) è un gene che codifica un modificatore di epigenetica che viene comunemente mutato in AML. Mutazioni nel modificatori epigenetici sono relativamente comuni nella LAM, con mutazioni nel IDH1 e dnmt3a che rappresentano altre mutazioni in questa classe di geni che portano a geni disregolazione. Le mutazioni nel gene per nucleophosmin (NPM1) sono una delle mutazioni acquisite più comuni in AML e sono generalmente considerati di essere un buon fattore prognostico (in assenza di un FLT3 - ITD).
Co-mutazioni NPM1 e IDH2 sono stati descritti in letteratura come indicatore prognostico favorevole 5, con una sopravvivenza globale a 3 anni del 89%. Ciò rappresenta un vantaggio significativo di sopravvivenza rispetto alla sopravvivenza globale a 3 anni diwild type NPM1 e IDH2 del 31%. Ad esempio, standard di pratica di assistenza include l'analisi di mutazione per NPM 1 e mutazioni FLT3-ITD. In questo scenario, l'individuazione di una mutazione unica NPM1 avrebbe mancato di opportunamente paziente stratificare per il rischio, come le mutazioni secondarie possono essere favorevoli (ad esempio, IDH2) o sfavorevole (ad esempio, TET2), riducendo la fiducia per alleviare un trapianto di midollo osseo.
Caso 2 - pannello solido-NGS
DNA estratto dal tessuto FFPE era di qualità sufficiente e quantità per Solid-NGS Panel, con una concentrazione di 252 ng / ml e solo il 4% del DNA inferiore a 1.000 paia di basi (bp). Dopo l'analisi dei dati la profondità media di copertura era 9.167 legge (ben al di sopra la nostra profondità di taglio minimo di 1.000 legge), senza regioni al di sotto di 250 profondità di lettura. Ulteriori metriche QC sono shown in Tabella 3.
I dati trattati attraverso il gasdotto bioinformatica rilevati due associata a malattia mutazioni: un inserimento in-frame in esone 20 di ERBB2 (HER2 / neu) e una mutazione missense nel TP53. Tutte le varianti exonic con le loro frequenze alleliche sono rappresentati nella Tabella 5. L'inserimento in ERBB2 in realtà rappresenta un tandem duplicato (HER2 / neu) sequenza, che si riflette nella nomenclatura. L'identificazione e la conferma della revisione manuale in-frame inserimento richiesta del sequenziamento legge attraverso IGV. Il rilevamento di frequenze di mutazione superiori al 50%, come visto sia per il TP53 e HER2 / neu mutazioni è indicativo di una perdita di eterozigosi evento (LOH) (vedi la discussione).
I risultati Solid-NGS pannello per Caso 2 con mutazioni adenocarcinoma del polmone rilevati in ERBB2 (HER2 / neu) e TP53, due geni non comunemente testati per come parte della corrente standard di cura per i pazienti affetti da cancro del polmone. HER2 / neu codifica per un recettore tirosin-chinasi simile ad un altro gene comunemente mutato nel cancro del polmone, EGFR . Attivazione HER2 esone / neu sono osservati 20 inserzioni in 2-4% degli adenocarcinomi del polmone, rappresentano la maggior parte dei HER2 mutazioni / neu osservate nel cancro del polmone, e sono tipicamente visto in tumori senza mutazioni in altri geni del driver, come EGFR e ALK 12 . Ci sono diverse linee di prove che dimostrano il potenziale per le varie opzioni di trattamento per i pazienti con l'attivazione di inserzioni HER2 / neu, tra cui risposta parziale alla terapia di combinazione con HER2 / neu e inibitori di mTOR 13 e controllo delle malattie significativa con l'anticorpo monoclonale trastuzumab in combinazione con la chemioterapia 15. Alla scoperta di un cambiamento TP53 non è uncoMMON nel cancro, ma in questo momento non esistono terapie attuabili.
| Caso 1 | caso 2 | |
| Totale a partire Legge | 2.215.926 | 2.129.110 |
| Percentuale di Legge Mapping | 98.42% | 98.29% |
| Percentuale di Legge su Target | 99.01% | 97.29% |
| Percentuale di Legge sulla destinazione dopo Filter | 97.60% | 95.45% |
| Percentuale di utilizzabile Legge | 94.87% | 91.79% |
| Percentuale delle basi sopra copertura 250x | 98.40% | 100% |
| Percentuale delle basi unLa copertura Bove 1000x | 95.90% | 99.70% |
| La copertura Sotto 250x - Amplicon Numero | 8 | 0 |
Tabella 3:. Sequencing Run QC Metrics Questo è un riassunto delle più importanti statistiche di esecuzione, che non includono la copertura media, che vengono utilizzati per la revisione dei dati per determinare se un campione biblioteca preparazione è passato controllo di qualità. L'intero processo è successo se tutte le percentuali sono superiori al 90%, ma è possibile, con riporto di SW1 o UB1 nel passo FPU lavaggio o Primer cross-talk, ad avere minore 'Percent su Target' nell'intervallo 80 - 90%. Se il 'Percent Mapped' è troppo bassa, che indicano una contaminazione di batteri o altro DNA, come tutti i campioni devono allinearsi umana. Quando una di queste specifiche sono al di sotto dell'80%, il campione è in posizione di ulteriore revisione per aiutare a determinare come proceed e migliorare il processo.

Tabella 4:.. Caso 1 Risultati rilevato patogeni, malattia associata, variante di significato sconosciuto (VUS), e varianti exonic probabilmente benigni sopra i criteri di rilevazione del 5% frequenza dell'allele sono elencate Cliccate qui per scaricare questa tabella.

Tabella 5:.. Caso 2 Risultati rilevato patogeni, malattie associate, variante di significato sconosciuto (VUS), e le varianti exonic probabilmente benigni sopra i criteri di rilevazione del 5% frequenza dell'allele sono elencati Cliccate qui per scaricare questa tabella.
Supplemento Figura 1: Un esempio di SampleSheet.csv amplicone-based. Questa scheda trasmette al sequencer ciò chimica per eseguire (in questo caso Amplicon), ciò workflow (ad esempio, GenerateFastq), ciò Applicazione e Assay (ad esempio, FastqOnly), quante basi (o legge) a sequenza (in questo caso 186 bp x 186 bp), ed infine quali campioni sono associate con alcuni indici (in questo caso doppia indicizzazione). Le parti che sono evidenziati in giallo possono essere modificati a tutto ciò che lo sperimentatore vuole, ma in questo caso il laboratorio utilizza questi parametri. Cliccate qui per scaricare questa cifra.
Discussione
Poiché le due prove NGS descritti in questo manoscritto sono offerti clinicamente, la più importante considerazione pratica è il controllo della qualità. In particolare, stretta considerazione deve essere prestata alla qualità e quantità di DNA estratto. Ciò è particolarmente importante per campioni FFPE che sono spesso altamente degradato con resa DNA variabile. Un metodo isopropanolo precipitazione è stata sviluppata al fine di massimizzare il rendimento del DNA da campioni FFPE come metodi basati su colonne sono stati trovati talvolta portare alla tranciatura DNA con volumi di eluizione limitati. Pertanto, la maggior parte del tempo in cui un esemplare produce concentrazione troppo bassa o troppo degradato per il test, è molto probabilmente a causa delle dimensioni del tessuto, tipo o fissaggio, e non il processo di estrazione. Per i campioni di sangue / midollo osseo, se vi è un guasto di estrazione, di solito è dovuto ad un campione di hemodilute essere (cioè, non avendo il numero sufficiente di sangue o di tumore bianchi cellule in quel pareggio) o chemio ablazione.
. Nt "> Durante la convalida, tagli di accettabilità della qualità del DNA e la quantità dovrebbe essere istituito L'ingresso raccomandata di 100-250 ng è spesso usato nel saggio, tuttavia, se la qualità del DNA è buona, allora quantità di input più bassi possono avere successo. Inoltre, se la qualità del DNA è scarsa (cioè, la quantità di DNA amplificabile è inferiore a 100 - 250 ng) allora importi più elevati dei fattori possono migliorare la qualità dei risultati di sequenziamento (poiché la quantità di DNA amplificabile raggiungerà l'ingresso raccomandato) . Metriche per la qualità del DNA e la quantità dovrebbe essere applicato a ogni campione prima di passare il DNA in preparazione biblioteca. I campioni in una "zona grigia" (vedi figura 2) dovrebbe essere eseguito a discrezione del direttore del laboratorio o designato. Attualmente il migliore senso predire se il DNA non eseguirà bene durante sequenziamento è quello di eseguire un saggio qPCR-based che permette di valutare la quantificazione e la qualità del DNA di ingresso. Questo approccio risolve il bioavailabilità di frammenti di dimensioni diverse nel campione, attraverso l'amplificazione di frammenti di dimensioni diverse (ad esempio, 100 bp, 150 bp, 200 bp e 300 bp) e confronto resa.Attualmente, la preparazione biblioteca coinvolge un gran numero di operazioni manuali, dove un passo falso in uno dei numerosi frangenti può causare la libreria sia sicuro o essere di scarsa qualità. L'analisi di gel microfluidica è l'unico passo QC per controllare un problema di biblioteca di preparazione prima di sequenziamento. Di conseguenza, ci sono diversi passaggi critici in cui la consapevolezza extra può aumentare la probabilità di una reazione di successo. È indispensabile garantire il campione corretto e piscina oligonucleotide sono utilizzati per ciascun campione. Garantire e registrare correttamente che ogni campione contiene una delle 96 combinazioni uniche di coppie dual-indicizzato di primer PCR riduce la possibilità per un campione confusione. Inoltre, è importante garantire la piastra filtrante (FPU) drena correttamente; se non scarica adeguatamente questo può causare la extepasso nsion-legatura della preparazione libreria per eseguire subottimale e portare a dati di sequenziamento scarsa qualità. Dopo libreria QC, è fondamentale garantire che le perline LNB1 siano completamente risospese e che la soluzione LNB1 / LNA1 è ben miscelato prima di aggiungere ai campioni come la concentrazione di questa miscela è utilizzato per determinare la molarità della libreria. Infine, se il passo tallone eluizione porta ad una quantità ottimale di biblioteca eluendo fuori le perline diminuirà la densità di clustering e causare la libreria di non ottenere un'adeguata copertura medio. Al contrario, un eccesso di libreria comporterà qualità inferiore legge. Pertanto, è importante essere coerenti nella fase di normalizzazione tallone basata per garantire il pool ottimale e raggruppamento delle librerie del sequencer.
Oltre alla preparazione biblioteca, è fondamentale per convalidare un oleodotto bioinformatica che produrrà le chiamate mutazione accurati dai, i file RAW de-multiplexing FASTQ. la scelta di unsoluzione personalizzata può essere in termini di tempo in quanto vi sono molti allineatori open source e disponibili in commercio, i chiamanti variante, e pacchetti software NGS che uno avrebbe dovuto passare al setaccio. dovranno algoritmi personalizzati per essere progettato per estrarre statistiche essenziali delle prestazioni, identificare le mutazioni ricorrenti uniche che sfuggivano strumenti più open source, e di determinare lo stato del numero di copie su ciascuno dei loci. Durante il processo di convalida di una conduttura bioinformatica, è importante determinare i tagli di informativa per le varianti che soddisfano o superano entrambi una profondità minima di copertura dopo filtrazione qualità (per esempio, un minimo di 250 letture) e una frequenza minima allele (ad esempio, 4 %). Poiché questo un saggio amplicone basato multiplato, è importante determinare la media minima profondità di copertura (ad esempio, 1,000x) che la biblioteca deve raggiungere per poter ottenere l'amplicone eseguendo basso alla profondità minima di letture. Inoltre, la natura multiplato del test fa cause off effetti bersaglio e questi 'artefatti' dovrà essere scoperto e completamente controllati prima del lancio. Un'altra importante limitazione al test descritto è la necessità di campioni di contenere più del 10% del tumore al fine di conseguire l'allele frequenza minima convalidato.
Il rilevamento di bassa frequenza, 1%, inserzioni FLT3 è prova che revisione manuale è ancora desiderabile in questo processo. Anche con una frequenza allele cut-off del 5%, alcune mutazioni importanti forse senza risposta e revisione in tal modo manuale saranno essenziali per accertare queste varianti. Per FLT3-DTI, ispezione visiva dell'esone 14 viene eseguita per tutti i pazienti con LMA per garantire un livello basso o grande inserimento / duplicazione non passare inosservato. Inoltre, HER2 esone 20 inserzioni che sono comunemente accanto alla sequenza primer devono intervento manuale. Pur avendo una robusta pipeline di bioinformatica, alcune varianti potrebbero andare trascurati, che è solo la natura di avere un taglio durooff per la maggior parte le statistiche di cui sopra. bioinformatica Meglio sarebbero necessari per contribuire ad alleviare questo problema, così come una migliore biblioteca preparazione e / o sequenziamento metodologie, perché è più vantaggioso disporre di dati di buona qualità a tagli ancora più bassi che contengono un minor numero di artefatti e falsi positivi.
Il rilevamento e l'interpretazione di frequenze alleliche possono essere difficile a causa della difficoltà di determinazione percentuale tumore e polarizzazione amplificazione alcune regioni del genoma. Inoltre, le frequenze alleliche oltre 50% possono essere rilevate, come osservato nel caso 2. Questo viene interpretato come una perdita di eterozigosi (LOH) evento, sia a causa di perdita dell'allele normale, portando alla apparente aumento del mutante legge, un guadagno del allele mutante (ad esempio, 2 mutante e una copia normale) o altri meccanismi. Questi meccanismi possono essere chiariti utilizzando array di ibridazione genomica comparativa (CGH 19) e / o un array di genotipizzazione SNP. 20.
Le attuali metodologie di arricchimento obiettivo si basano su procedure di un'intera giornata di entrambi cattura ibrido inefficiente o tecniche di PCR multiplex che si traducono nella necessità di una maggiore copertura sequenziamento di un singolo campione e più fuori sequenza bersaglio legge. Ulteriori applicazioni per NGS oncologia molecolare ci si attende in un prossimo futuro includeranno più semplici metodi di preparazione libreria che può essere completamente automatizzabili e in grado di elaborare i campioni con quantità molto basse di DNA di ingresso (ad esempio, meno di 1 ng), così come i campioni con altamente degradati DNA. Per affrontare queste sfide, la maggior parte dei metodi saranno presumibilmente basati sulla PCR, sia essere un approccio a più fasi PCR o un approccio massicciamente parallelo singleplex PCR. Inoltre, codici a barre molecolare dei singoli ampliconi ha dimostrato di ridurre drasticamente il rumore di fondo di sequenziamento e consentirà analisi di campioni con proporzioni minori di cellule tumorali per ottenere basse frequenze alleliche e muoversi verso l'acquisizione circulating cellule tumorali.
Rilevazione delle mutazioni associate alla malattia nei campioni tumorali è stata standard di cura per decenni. Storicamente, i geni sono stati spesso testati in sequenza, un gene / esone alla volta, con l'identificazione di una mutazione che porta alla fine della sequenza di test. L'avvento di NGS ha consentito un approccio meno polarizzate ad sequenziare più geni associati con molti tumori in parallelo conducono alla identificazione di mutazioni multiple che sono associati con neoplasia. L'utilità clinica della NGS per la rivelazione di mutazioni somatiche nel cancro è sempre più evidente. Infatti, l'analisi NGS basata su campioni di tumore rappresenta un nuovo paradigma che sfida tradizionale, singolo test genetici, ma l'utilità clinica è molto chiaro. laboratori clinici di oggi hanno l'eccitante opportunità di coniugare un'attenta validazione del metodo e interpretazione del test con l'applicazione di questa potente tecnologia.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Gli autori vorrebbero riconoscere l'assistenza di Daniel Wild per la lettura del manoscritto e l'assistenza nella produzione.
Materiali
| Name | Company | Catalog Number | Comments |
| Genomic DNA ScreenTape | Agilent Technology | 5067-5365 | |
| Genomic DNA Reagents | Agilent Technology | 5067-5366 | |
| High Sensitivity D1000 ScreenTape | Agilent Technology | 5067-5584 | |
| High Sensitivity D1000 Reagents | Agilent Technology | 5067-5585 | |
| TapeStation 2200 | Agilent Technology | G2965A | |
| TapeStation Analysis Software | Agilent Technology | A.01.04 or higher | |
| 96-well Tube Storage Racks | Any Vendor | ||
| 15/50 ml Tube Rack | Any Vendor | ||
| 96-well Plate Rack | Any Vendor | ||
| Pipette, single-channel, 0.5–2.5 μl | Any Vendor | ||
| Pipette, single-channel, 1–10 μl | Any Vendor | ||
| Pipette, single-channel, 2–20 μl | Any Vendor | ||
| Pipette, single-channel, 10–100 μl | Any Vendor | ||
| Pipette, single-channel, 20–200 μl | Any Vendor | ||
| Pipette, single-channel, 100–1,000 μl | Any Vendor | ||
| Serological Pipettor | Any Vendor | ||
| Vortexer | Any Vendor | ||
| Ice bucket | Any Vendor | ||
| Microcentrifuge (for tubes and strip tubes) | Any Vendor | ||
| Freezer, -20 °C | Any Vendor | ||
| 4 °C Refrigerator | Any Vendor | ||
| Water or Bead Bath | Any Vendor | ||
| Incubator (37 °C) | Any Vendor | ||
| Serological Pipettes, 1 ml | Any Vendor | ||
| Serological Pipettes, 5 ml | Any Vendor | ||
| Serological Pipettes, 10 ml | Any Vendor | ||
| Serological Pipettes, 25 ml | Any Vendor | ||
| Gloves | Any Vendor | ||
| Razor Blades/Scaples | Any Vendor | ||
| KimWipes | Any Vendor | ||
| 15 ml Conical Tube | Any Vendor | ||
| 50 ml Conical Tube | Any Vendor | ||
| Paper Towels | Any Vendor | ||
| 200 proof Ethanol | Any Vendor | Store in Flammable Cabinet | |
| 2-Propanol (Isopropanol) | Any Vendor | Store in Flammable Cabinet | |
| 25 ml Reservoirs | Any Vendor | ||
| 10 N NaOH | Any Vendor | ||
| Pipette, 8-channel, 1 – 10 μl | Any Vendor | ||
| Pipette, 8-channel, 10 – 100 μl | Any Vendor | ||
| Pipette, 8-channel, 20 – 300 μl | Any Vendor | ||
| Ice Bucket | Any Vendor | ||
| Water Squirt Bottle | Any Vendor | ||
| Alcohol Squirt Bottle | Any Vendor | ||
| Lens Cleaning Paper | Any Vendor | ||
| Plates, 96-well PCR, Semi-Skirted | Any Vendor | ||
| Tube strips, 8-well, 0.2 ml | Any Vendor | ||
| Agencourt AMPure XP Beads | Beckman Coulter | A63881 | |
| BioShake IQ or 3000-T elm | Bulldog Bio/Q.Instruments | 1808-0506/ 1808-0517 | |
| DropPlate96 S - LabChipDS | Caliper | 128876 | |
| DropPlate96 D - LabChipDS | Caliper | 132848 | |
| DropSense96 | Caliper (Trinean) | ||
| DropQuant Software | Caliper (Trinean) | ||
| Plate Sealing Film | Denville | B1212-5S | |
| Aluminum Seal Foil | Denville | B1212-6S | |
| Nuclease-Free, Pure Water System | EMD Millipore | ||
| 5424 centrifuge | Eppendorf | 22621408 | |
| 5804R centrifuge | Eppendorf | 22623508 | Both 15 ml tube and plate rotators, preferably a centrifuge that can go up to 2,500 x g. |
| Safe-Lock Tube 1.5 ml, Natural | Eppendorf | 22431021 | |
| 5 ml Tube, DNA LoBind Tube | Eppendorf | 30108310 | |
| 5430R Centrifuge | Eppendorf | 022620645 | Any plate rotator centrifuge will work |
| Hybex Microsample Incubator | Fisher Scientific | 1057-30-0 | |
| Hybex 0.2 ml Tube Block | Fisher Scientific | 1057-31-0 | |
| TruSeq Amplicon – Cancer Panel | Illumina | FC-130-1008 | 96 reactions |
| TruSeq Custom Amplicon | Illumina | PE-940-1011 | 96 reactions |
| TruSeq Custom Amplicon Index Kit | Illumina | FC-130-1003 | 96 Indices, 384 Samples |
| MiSeq Reagent Kit v3, 500 Cycles | Illumina | MS-102-3003 | |
| MiSeq Reagent Kit v2, 300 Cycles | Illumina | MS-102-2002 | |
| MiSeq Reagent Kit v2, 500 Cycles | Illumina | MS-102-2003 | |
| Experiment Manager | Illumina | 1.3 or higher | |
| MiSeq Reporter | Illumina | 2.0 or higher | |
| Sequencing Analysis Viewer | Illumina | 1.8 or higher | |
| TruSeq Index Plate Fixture and Collar Kit | Illumina | FC-130-1007 | |
| MiSeq v2 | Illumina | SY-410-1003 | |
| TruSeq Custom Amplicon Filter Plate | Illumina | FC-130-1006 | |
| Index Adapter Replacement Caps | Illumina | 11294657 | |
| Qubit 2.0 | Invitrogen | Q32866 | |
| Qubit 0.5 ml Tubes | Invitrogen | Q32856 | |
| Qubit dsDNA Broad Range Assay Kit | Invitrogen | Q32853 | |
| DynaMa6-96 Magnetic Stand, Side Skirted | Invitrogen | 120.27 | |
| GeneAmp PCR System 9700 (gold/silver block) | Life Technologies | N8050200 | |
| Gentra Puregene Blood Kit | Qiagen | 158489 | |
| Deparaffinization Solution (16 ml) | Qiagen | 19093 | |
| Buffer ATL (4 x 50ml) | Qiagen | 939011 | |
| Protein Precipitation Solution (50 ml) | Qiagen | 158910 | |
| DNA Hydration Solution (100 ml) | Qiagen | 158914 | |
| Glycogen Solution (500 μl) | Qiagen | 158930 | |
| Qiagen Proteinase K | Qiagen | 19133 | |
| Rnase (5 ml) | Qiagen | 158924 | |
| Nuclease-Free Water (10 x 50 ml) | Qiagen | 129114 | |
| Pestles | USA Scientific | 1415-5390 | |
| TipOne RPT 10 µl elongated filter pipet tips in sterilized racks, 10 racks of 96 tips (960 tips). | USA Scientific | 1180-3810 | |
| TipOne RPT 100 µl natural, beveled filter pipet tips in sterilized racks, 10 racks of 96 tips (960 tips) | USA Scientific | 1180-1840 | |
| TipOne RPT 200 μl natural, beveled filter pipet tips in racks, sterilized racks, 10 racks of 96 tips (960 tips) | USA Scientific | 1180-8810 | |
| TipOne RPT 20 μl natural, beveled filter pipet tips in racks, sterilized racks, 10 racks of 96 tips (960 tips) | USA Scientific | 1180-1810 | |
| TipOne RPT 1,000 μl natural, graduated XL filter pipet tips in | USA Scientific | 1182-1830 |
Riferimenti
- Gerlinger, M., et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 366 (10), 883-892 (2012).
- Campbell, P. J., et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 467 (7319), 1109-1113 (2010).
- Ding, L., et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 481 (7382), 506-509 (2012).
- Frampton, G. M., et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nature Biotechnol. 31 (11), 1023-1031 (2013).
- Patel, J. P., et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 366 (12), 1079-1089 (2012).
- Forbes, S. A., et al. COSMIC (the Catalogue of Somatic Mutations in Cancer ): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 38 (Database Issue), 652-657 (2010).
- Shih, A. H., Abdel-wahab, O., Patel, J. P., Levine, R. L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 12 (9), 599-612 (2012).
- Liersch, R., Müller-Tidow, C., Berdel, W. E., Krug, U. Prognostic factors for acute myeloid leukaemia in adults - biological significance and clinical use. Br J Haematol. 165 (1), 17-38 (2014).
- Bacher, U., Schnittger, S., Haferlach, T. Molecular genetics in acute myeloid leukemia. Curr Opin Oncol. 22 (6), 646-655 (2010).
- Subramanian, J., Govindan, R. Lung cancer in "Never-smokers": a unique entity. Oncology (Williston Park). 24 (1), 29-35 (2010).
- Sakashita, S., Sakashita, M., Tsao, M. S. Genes and pathology of non-small cell lung carcinoma. Semin Oncol. 41 (1), 28-39 (2014).
- Arcila, M. E., Chaft, J. E., Nafa, K. Prevalence clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin Cancer Res. 18 (18), (2012).
- Gandhi, L., et al. Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2-dependent and other solid tumors. J Clin Oncol. 32 (2), 68-75 (2014).
- Sheikhha, M. H., Awan, A., Tobal, K., Liu Yin, J. A. Prognostic significance of FLT3 ITD and D835 mutations in AML patients. Hematol J. 4 (1), 41-46 (2003).
- Mazières, J., et al. Lung cancer that harbors an HER2 mutation epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 31 (16), 1-8 (2014).
- Robinson, J. T., et al. Integrative Genomics Viewer. Nat Biotechnol. 29 (1), 495-500 (2011).
- Forbes, S. A., et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 43 (Database issue), D805-D811 (2014).
- Daber, R., Sukhadia, S., Morrissette, J. J. Understanding the limitations of next generation sequencing informatics, an approach to clinical pipeline validation using artificial data sets. Cancer Genetics. 206 (12), 441-448 (2013).
- Haraksingh, R. R., et al. Genome-Wide Mapping of Copy Number Variation in Humans: Comparative Analysis of High Resolution Array Platforms. PLoS ONE. 6 (11), e27859(2011).
- de Leeuw, N., et al. SNP Array Analysis in Constitutional and Cancer Genome Diagnostics - Copy Number Variants, Genotyping and Quality Control. Cytogenet Genome Res. 135 (3-4), 212-221 (2011).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati