Method Article
Next Generation Sequencing für den Nachweis von Mutationen umsetzbare in fester und flüssiger Tumoren
In diesem Artikel
Zusammenfassung
This manuscript describes clinical protocols for two next-generation sequencing panels. One panel interrogates hematologic malignancies while the other panel targets genes commonly mutated in solid tumors. Molecular classification of driver mutations in human malignancies offers valuable prognostic and predictive information.
Zusammenfassung
As our understanding of the driver mutations necessary for initiation and progression of cancers improves, we gain critical information on how specific molecular profiles of a tumor may predict responsiveness to therapeutic agents or provide knowledge about prognosis. At our institution a tumor genotyping program was established as part of routine clinical care, screening both hematologic and solid tumors for a wide spectrum of mutations using two next-generation sequencing (NGS) panels: a custom, 33 gene hematological malignancies panel for use with peripheral blood and bone marrow, and a commercially produced solid tumor panel for use with formalin-fixed paraffin-embedded tissue that targets 47 genes commonly mutated in cancer. Our workflow includes a pathologist review of the biopsy to ensure there is adequate amount of tumor for the assay followed by customized DNA extraction is performed on the specimen. Quality control of the specimen includes steps for quantity, quality and integrity and only after the extracted DNA passes these metrics an amplicon library is generated and sequenced. The resulting data is analyzed through an in-house bioinformatics pipeline and the variants are reviewed and interpreted for pathogenicity. Here we provide a snapshot of the utility of each panel using two clinical cases to provide insight into how a well-designed NGS workflow can contribute to optimizing clinical outcomes.
Einleitung
Next-Generation-Sequencing (NGS) der klinischen Onkologie Proben wurde weiter verbreitet mehrere Jahre in den letzten werden als wissenschaftliche Literatur weist auf die Bedeutung der Identifizierung anzielbare genetischen Veränderungen und prädiktive / prognostische molekulare Marker wächst. Multi-Gen - Panel analysiert und ganze Exoms Sequenzierungsuntersuchungen in beiden epithelialen 1,2 und hämatologischen malignen Erkrankungen 3 haben das Konzept der Tumorheterogenität und klonalen Evolution als Krankheit fortschreitet und Schübe verfestigt. Zusätzlich, im Gegensatz zu konkurrierenden Technologien wie beispielsweise Polymerasekettenreaktion (PCR) oder Sanger - Sequenzierung, kann NGS meisten genomischen Veränderungen in allen klinisch relevanten Krebsgene in einem einzigen Assay - 4 ermitteln.
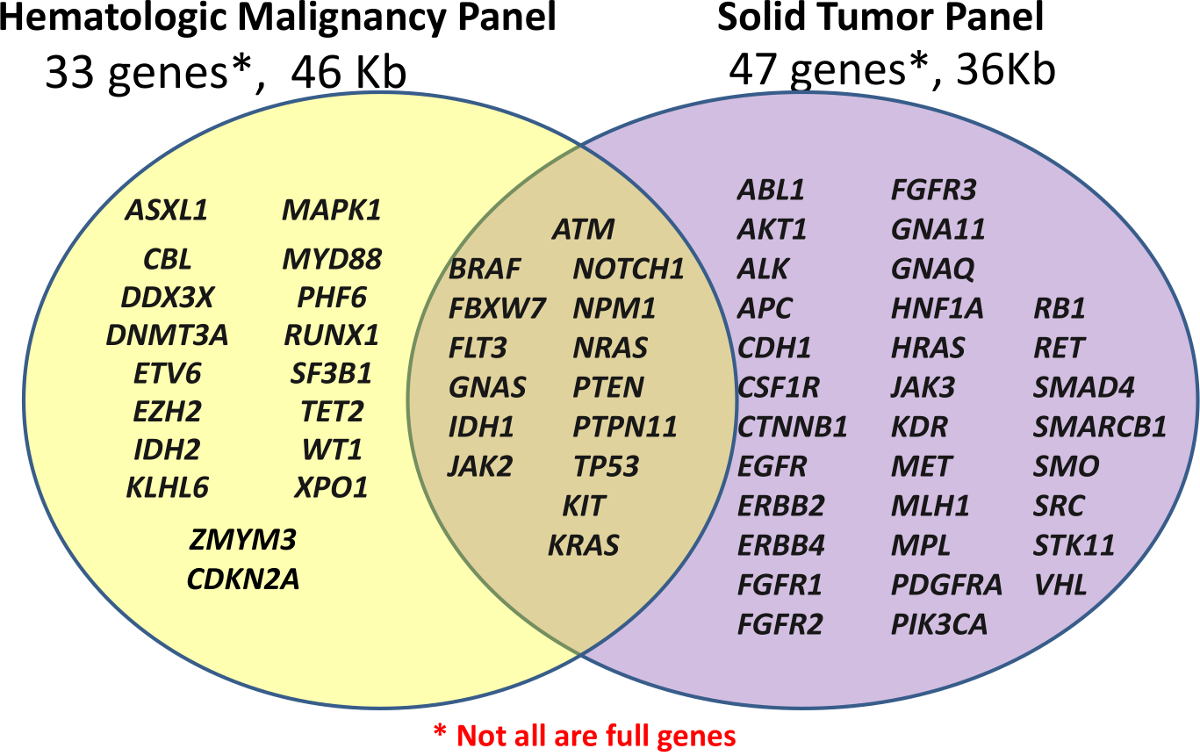
Das Zentrum für personalisierte Diagnose zunächst mit zwei klinischen NGS-Panels, eine benutzerdefinierte Hämatologische Panel (Häm-NGS Panel) und einem Off-the-shelf-Krebs-Panel für FFPE Proben (Solid-NGS Panel) ins Leben gerufen (siehe 1). Diese Platten bedecken klinisch relevant oder hohe Interesse Regionen der ausgewählten Gene; nicht alle Gene oder Exons vollständig abgedeckt sind. Amplicons werden durch Sondenhybridisierung, die durch Verlängerung und Ligation. Die Zielregionen werden weiter verstärkt für die Sequenzierung unter Verwendung gepoolten PCR mit universellem Dual-indexiert Primern, so dass bis zu 96 Proben werden.

Abb . 1: Liste der in den Panels Covered Gene Bibliothek Vorbereitung wird durchgeführt , und zwar mit dem benutzerdefinierten Hämatologische Panel (Häm-NGS Panel) von 33 Genen oder der Off-the-Shelf - Amplicon Krebs - Panel (Solid-NGS) von 47 Genen. Nicht alle Gene oder Exons sind in voller Höhe gedeckt, da einige Amplikons nur bestimmte Hotspots abdecken. Bitte hier klicken , um eine größere Version dieser Figur zu sehen. </ A>
{kind=link}
Der Inhalt für die Häm-NGS - Panel wurde von verschiedenen Quellen abgeleitet, sondern dreht sich um 16 Gene bei akuter myeloischer Leukämie mutiert (AML) , die zuvor als Beweis ein hohes Maß an klinischen Nutzen 5 beschrieben. Das Solid-NGS Das Bedienfeld ist im Handel mit den Zielregionen basieren auf allgemein mutierte Gene in der Krebs produziert , wie im Katalog der somatische Mutationen in Krebs (COSMIC) Datenbank 6 gemeldet.
Mehrere wichtige Schritte charakterisieren den gesamten Workflow für die klinische NGS. Nachdem der Arzt den Test bestellt, bestimmt ein Pathologe Angemessenheit der Probe für die Tumor Prozentsatz und Probenvolumen folgende Analyse. In unserer Einrichtung, benötigen wir mindestens 10% Tumor aufgrund des Hintergrundsequenzierungsfehlerrate ( "Rauschen") der Technologie und die Effizienz der gezielten Ansatz. Wenn das Gewebe für den Test ausreichend ist, wird genomische DNA extrahiert. Diese DNA wird dann auf mehrere Qualitätskontrolle unterzogen (QC) Schritte. Wenn der DNA QC passiert, wird eine Amplicon-Bibliothek generiert und sequenziert. Die resultierenden Daten werden durch eine in-house Bioinformatik Pipeline analysiert. Nach bioinformatische Analyse werden Varianten manuell überprüft und für die Pathogenität vor dem Einbau in einer klinischen Bericht interpretiert. Im Folgenden zwei Fälle beschreiben, die durch diesen strengen Workflow ging und führte schließlich in klinische Behandlung zu Veränderungen.
Fall 1 - Akute myeloische Leukämie
Eine Knochenmark-Biopsie von Patient A war Diagnose für AML, ohne Reifung. Zytogenetische Untersuchungen wurden an der Knochenmarksprobe und zeigten eine normale weibliche Karyotyp gesendet. Es gab 95% zirkulierenden Blasten vorhanden, so dass eine periphere Blutprobe wurde für personalisierte Diagnosetests auf der Häm-NGS-Panel gesendet.
Akute myeloische Leukämie (AML) ist eine hämatologische Malignität der myeloischen Linie von weißen Blutkörperchen. die Detektionsvon Gen - Mutationen bei AML hat für Prognose und Therapie zunehmend an Bedeutung gewonnen, mit rezidivierenden Genmutationen 7 als wichtig in der Pathogenese und Prognose anerkannt. Mutationen in NPM1 und CEBPA wurden mit einer günstigen Prognose Risiko verbunden, während interne Tandemverdopplungen (ITD) in FLT3 haben mit einem weniger günstigen Ergebnis 8 in Verbindung gebracht worden. Eine wachsende Zahl von Beweisen unterstützt eine pathogene Rolle für diese und andere Mutationen bei AML 9.
Fall 2 - Lungenadenokarzinom
Eine Biopsie eines linken supraklavikulären Masse von Patient B zeigte Lungenadenokarzinom. Biopsy Material aus der Formalin-fixierten und in Paraffin eingebetteten (FFPE) Lymphknoten Masse wurde für genomische Tests (Solid-NGS Panel) gesendet, wie Rollen / Locken mit mehr als 50% Tumor, zu erkennen, ob eine Mutation für gezielte therapeutische Intervention anwesend war.
Lung cancer ist die häufigste Ursache von Krebs bedingte Sterblichkeit in den Vereinigten Staaten und gliedert sich in zwei Haupttypen, nicht-kleinzelligem Lungenkrebs (NSCLC) und kleinzelligen Lungenkarzinom (SCLC). NSCLC kann weiter als entweder Adenokarzinom oder Plattenepithelkarzinom definiert werden, bezogen auf die Histologie der Läsion. Lung Adenokarzinom ist die häufigste Subtyp von Lungenkrebs, beide gesehen bei Rauchern und Nichtrauchern, und ist die häufigste Form von Lungenkrebs für Nichtraucher 10. Molekulare Untersuchungen der Lunge Adenokarzinome haben Mutation in mehrere Onkogene 11 identifiziert. Die am häufigsten verwendeten Treiber - Mutationen bei Rauchern identifiziert sind Mutationen im KRAS und BRAF. Die häufigsten Mutationen bei Nichtrauchern sind Mutationen in EGFR und Umlagerungen die Gene ALK, RET und ROS1 beteiligt sind . Lungentumore wurden mit einem in-frame Exon 20 Insertion in dem Gen , ERBB2 (HER2 / neu) beschrieben. Die häufigste Anomalie in HER2 / neu ist eine Verstärkung dieses Locus in Brustkrebs , für die eine gezielte Therapie verfügbar ist (Trastuzumab: ein humanisierter monoklonaler Antikörper gegen HER2 / neu). Die HER2 / neu - Exon 20 Insertion , die in 2 beobachtet wird - 4% der Lunge adenocarcimomas 12 Teilantwort auf die Kombinationstherapie mit HER2 / neu und mTOR - Inhibitoren (Neratinib und Temsirolimus, jeweils) 13 gezeigt hat.
Protokoll
Dieses Protokoll umfasst die wesentlichen Schritte der zwei validierten Labor entwickelten Tests für die genomische Profilierung von fester und flüssiger Tumoren, respectively. Die Tests im Labor durchgeführt wird, in Übereinstimmung mit den Anforderungen der Clinical Laboratory Improvement Amendments (CLIA) von 1988 getan.
1. DNA-Extraktion aus dem peripheren Blut oder Knochenmark
- Ermitteln Sie, wie viel Blut oder Knochenmark nimmt , basierend auf Tabelle 1.
| Sample / WBC | Menge wie 1 ml Blut zu behandelnden |
| Knochenmark | 250 ul |
| Blut WBC 12.000 - 50.000 | 1 ml |
| Blut WBC 50.000 - 100.000 | 500 ul |
| Blut WBC 100.000 - 200.000 | 200 & mgr; l |
| Blut WBC> 200.000 | 100 ul |
| * For Blood WBC <12.000, nehmen Sie 2 ml Blut | |
Tabelle 1:. Blut / Knochenmark - Volume - Diagramm zu verwenden , da die Anzahl der weißen Blutkörperchen von der Probe variieren zu probieren, ist es schwierig , ein spezifisches Volumen von Blut zu spezifizieren zu verwenden. Daher, indem man die Anzahl der weißen Blutkörperchen (WBC) vor dem Start des Assays bestimmt werden, die Menge an Blut für den Test zu verwenden. Obwohl weniger Blut verwendet wird, sollte sie immer noch als behandelt werden, wenn ihre 1 ml, da das Volumen von Blut verwendet wird verringert, da die Anzahl der vorhandenen Zellen größer als normal ist.
- Folgen Sie den im Handel Protokoll erhältlichen Kits der genomischen DNA zu isolieren.
2. DNA-Extraktion aus Formalin-fixierten und in Paraffin eingebetteten (FFPE) Gewebe
- Beyogen aufder Tumorregion der Pathologe auf der H & E Dia eingekreist, säumen die ungefärbten Folien mit der Führung H & E Folie nach oben und einen ähnlichen Bereich für die Extraktion zu skizzieren. Für Makro-Dissektion Prozess nur eine Probe / Patienten Reihe von Dias auf einmal.
- Erhitzen Sie die Dias auf einer 45 ° C Wärmeblock leicht das Paraffin zu schmelzen. Sorgfältig schaben das Gewebe innerhalb der Linien, die auf der Folie markiert sind, ein neues Skalpell für jede Probe unter Verwendung extrahiert werden. Legen Sie die Wachs Scharren in den entsprechend markierten 1,5-ml-Röhrchen. Seien Sie vorsichtig, da die geschabt Wachs sehr elektrostatische und kann aus der Röhre springen.
- In 320 ul Entparaffinierung Lösung für alle fünf vor sechs 5 um Abschnitte (25 bis 30 & mgr; m insgesamt). Wenn beispielsweise ein Rohr 3 Teilen einer 10 & mgr; m Roll- / curl enthaltenden wird verarbeitet werden, verwenden dann 320 & mgr; l, aber wenn 5 Abschnitte mit der gleichen Dicke dann 640 ul verwenden erhalten wurden.
- Vortex kräftig aq mindestens 10 sec und ausführenuick Spin in einer Mikro das Gewebe / Wachs von den Seiten und der Kappe und in die Lösung zu entfernen. Inkubieren bei 56 ° C für 3 min und dann bei RT inkubieren. 5 - 10 min.
- Im Anschluss an die RT Inkubation geben 180 & mgr; l ATL-Puffer für jede 320 ul Entparaffinierung Lösung gegeben. Mince das Gewebe zehnmal mit einem sterilen Mini-Stößel einen neuen Stößel für jede Probe verwenden. Stellen Sie sicher, da kein Gewebe mit dem Stößel stecken, da es sehr klebrig sein kann. Vortex die Suspension kräftig für 3 Sekunden, dann bei maximaler Geschwindigkeit Zentrifuge für 1 min.
- In 10 ul Proteinase K in die untere klare Phase. Vorsichtig mischen durch Pipettieren nach oben und unten das Gewebe, um sicherzustellen, erneut suspendiert. Nicht mit dem Vortex. bei 56 ° C Inkubieren über Nacht bei 400 unter Schütteln - 500 Umdrehungen pro Minute.
- Am nächsten Morgen, prüfen Sie, ob das Gewebe vollständig aufgelöst ist (dies geschehen ist, wenn die untere Lösung klar ist). Wenn die untere Lösung nicht klar ist, dann kräftig vortexen für 3 Sekunden und Zentrifuge bei maximaler Geschwindigkeit for 1 min. Fügen Sie ein zusätzliches von 5 bis 10 & mgr; l Proteinase K und Inkubation bei 56 ° C für weitere 30 - 60 min.
- 1 h bei 90 ° C inkubieren Formaldehyd Vernetzung zu helfen, umzukehren. Werden die Proben für 5 auf Raumtemperatur abkühlen - 10 min und dann kurz jedes Röhrchen Zentrifuge, die Flüssigkeit zu konsolidieren.
- Übertragen Sie die untere klare Phase in ein beschriftetes 1,5-ml-Röhrchen. Wenn mehrere Rohre aus den gleichen Patientenprobe (wie zum Beispiel der Fall, wenn mehrere Walzen verwendet werden) sind, rekombinieren die unteren Phasen in einem Rohr an dieser Stelle. Hinweis: Übertragen von kleinen Mengen der Entparaffinierung Lösung sollte nicht mit dem Reinigungsverfahren stören, aber es besteht die Gefahr, wenn eine große Menge übertragen wird.
- Hinzufügen, 2 ul RNase A-Lösung. Vortex sanft oder Invertzucker 25-fache und schnelle Drehung in einer Mikro. 5 min bei RT inkubiert.
- In 200 ul Protein Fällungslösung. Wenn eine oder zwei Rollen tun, 200 & mgr; l verwendet werden. Wenn dabei drei Rolleauf einmal s, dann 400 & mgr; l verwenden. Vortex kräftig bei hoher Geschwindigkeit für 30 Sekunden, um gleichmäßig die Lysepuffern mischen. Inkubieren auf Eis für 5 min oder die Proben können bis zu einer Stunde auf Eis bleiben.
- Zentrifuge bei 5.000 xg für 5 min. Das gefällte Protein sollte ein dichtes, weißes Pellet bilden. Gießen Sie den Überstand in ein beschriftetes 1,5-ml-Röhrchen, und brüten dann die Proben auf Eis für mindestens 3 min. Zentrifuge bei 5000 × g für 3 min.
- In 200 ul 2-Propanol (Isopropanol) pro 180 ul Puffer ATL hinzugefügt früher zu einem markierten 1,5-ml-Röhrchen (es könnte notwendig sein, eine 2-ml-Röhrchen zu verwenden). Zum Beispiel, wenn dabei drei Rollen mit 600 & mgr; l Isopropanol. In 1 ul Glykogen für jede 180 ul Puffer ATL hinzugefügt früher Isopropanol und wird das Röhrchen mehrmals zu mischen.
- Vorsichtig den Überstand von dem Schritt Proteinfällung in das Isopropanolgemisch. Mischen Sie die Röhrchen vorsichtig mindestens 50-mal umgekehrt wird.
- Zentrifuge bei maximaler speed 3 min. Die DNA wird als kleine weiße Pellet am Boden des Röhrchens sichtbar.
- Gießen oder den Überstand in den entsprechenden Abfallrohr absaugen. Halten Sie einen separaten 1,5 ml Abfallrohr für jede Probe, falls das Pellet verdrängt kommt, damit es nicht mit dem Abfall von anderen Proben verloren gehen oder gemischt. Lassen Sie das Rohr auf einem Papiertuch und sorgen für die Mehrheit des Isopropanols entfernt wird.
- In 300 ul frisch 70% Ethanol hergestellt. Drehen Sie das Röhrchen mehrmals vorsichtig das Pellet zu waschen. Versuchen Sie, das Pellet, um sicherzustellen, kommt abgelösten eine gründlichere Reinigung zu gewährleisten.
- Zentrifuge bei maximaler Geschwindigkeit für 5 min. und entfernen Sie dann vorsichtig das Ethanol. Das Pellet kann locker sein, so gießen oder absaugen langsam und das Pellet beobachten. Das überschüssige Ethanol aus dem Inneren des Rohres, ohne das Pellet zu berühren. Lassen Sie die Proben an der Luft trocknen für 5 bis 15 min, wobei darauf geachtet, nicht über die Probe trocknen.
- Fügen Sie zwischen 25 bis 100 & mgr; l DNA Hydratationslösung bis eACH Probe basierend auf der Größe des DNA-Pellets und die Ausgangsmenge von Gewebe. Vortex die Röhrchen kräftig und kurz in einer Mikro drehen. bei 65 ° C für 1 h inkubieren vollständig die DNA rehydrieren.
3. Genomic DNA Qualitätskontrolle
Hinweis: Es gibt drei unabhängige Schritte für die DNA-Qualitätskontrolle (QC). Siehe Tabelle 2 für eine weitere Erklärung, warum jeder QC Schritt durchgeführt wird.
| Instrument | Ergebnis | Indikation | Ideal Reichweite |
| DropSense96 | A260 / A230 | Identifizierung von chemischen Verunreinigungen (zB Ethanol) | 1,50-2,2 |
| DropSense96 | A260 / A280 | Identifizierung von Proteinverunreinigungen | 1,60-2,2 |
| DropSense96 | Konzentration | DNA-Quantifizierung | > 1 ng / ul |
| Tape | DNA-Abstrich | Bestimmung der DNA - Integrität (zB Abbau / Fragmentierung der extrahierten DNA) | 50%> 1000 bp |
| Qubit 2.0 | Konzentration | Genauere DNA Quantifizierung | > 1 ng / ul |
Tabelle 2:. DNA QC Erwartete Ergebnisse Alle diese Werte werden berücksichtigt vor dem Ausführen einer Probe ermöglicht der Bibliothek Vorbereitungsphase , um fortzufahren.
- Nach dem Protokoll des Herstellers laufen 2 ul der extrahierten DNA auf einem Fluorometer die Arbeitskonzentration (ng / ul) der Probe zu erhalten.
- Führen Sie 1 bis 2 ul jeder Probe auf einem UV / VIS-Spektrophotometer die Qualität der Probe (A260 / A230 und A260 / A280 Ratte zu überprüfenios) gemäß den Anweisungen des Herstellers.
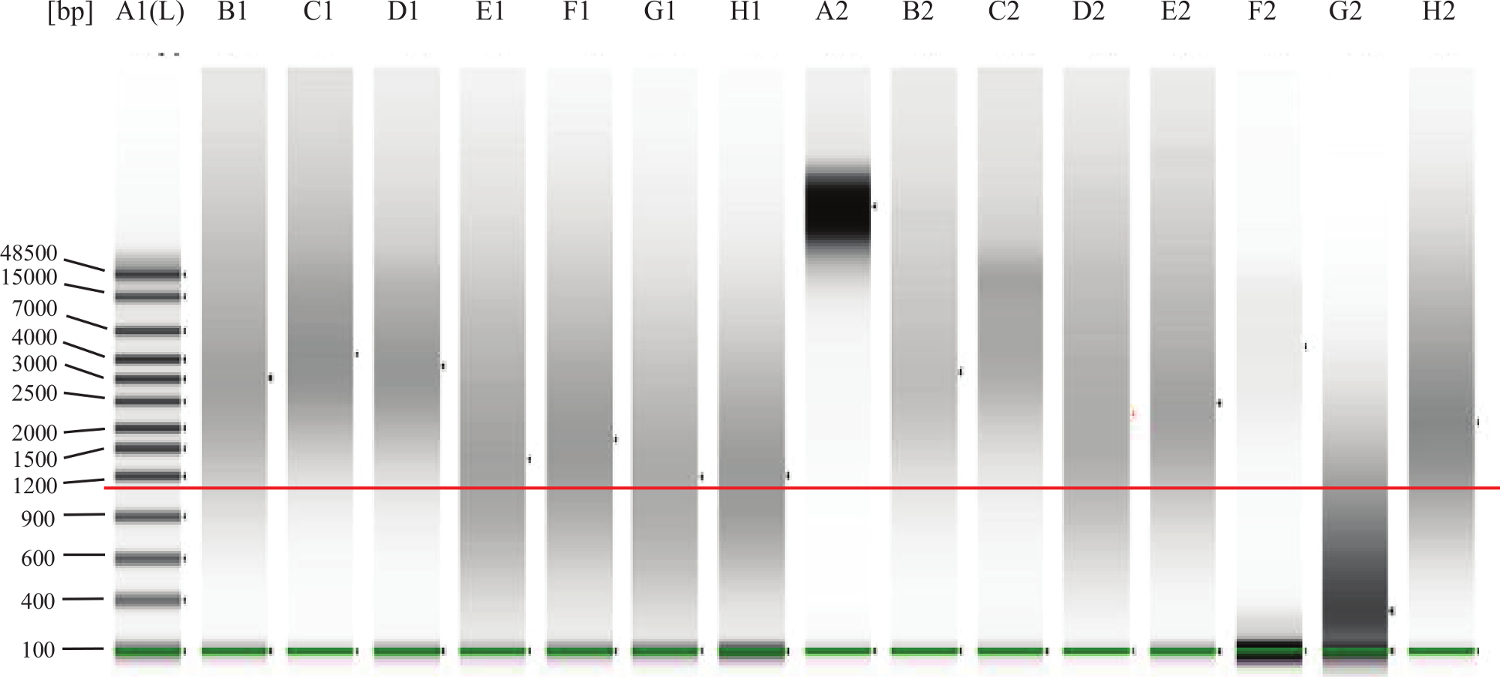
- Für FFPE Proben: Nach den Anweisungen des Herstellers, führen 1 ul jeder Probe auf einem Mikrofluidik-Gel-Elektrophorese-System Abbau / Fragmentierung der DNA zu bewerten. Siehe Abbildung 2 für Beispiele.

Abbildung 2:. Genomic DNA QC Gel Beispiel Die grüne Linie über die unteren Bänder ist die untere Markierung , um anzuzeigen. Der Mehr in rote Linie ist etwa 1000 bp anzuzeigen. Lane A2 stellt völlig intakte DNA, wie man aus frischen tis sue (zB dem peripheren Blut oder Knochenmark) erwarten würde. Die Bahnen B1, C1, D1, B2, C2, E2 sind Beispiele für gute, intakte FFPE DNA. Die DNA in der Spur F2 erscheint intakt, aber bei einer zu niedrigen Konzentration zu sein. Die DNA in Bahn G2 verschlechtert oder fragmentiert und werden in dem Test nicht. Lanes E1, F1, G1, H1, D2,H2 repräsentieren DNA , die in der "Grauzone" fällt was bedeutet , dass der Test gut funktionieren könnte, aber einige der DNA zu stark beschädigt werden könnten oder vernetzt und so wird es nicht gut in dem Test durchführen. Bitte klicken Sie hier um ein , um zu vergrößern Version dieser Figur.
{kind=link}
4. Amplicon Bibliothek Vorbereitung
- Die Hybridisierung von Oligo Pool und Erweiterungs-Ligatur von Bound Oligos
- In der Pre-PCR - Raum, fügen Sie das notwendige Volumen von Low EDTA TE, 5 ul des Oligos Röhre Panel (zB Fest NGS - Panel oder Häm-NGS Panel), und 100 bis 250 ng genomischer DNA in jede Vertiefung entspricht , die in ein semi-skirted 96-Well-Platte als Hybridisierung (HYB) Platte markiert.
- In 40 ul Oligo Hybridisierung für Sequencing Reagenz 1 (OHS1) zu jeder Probe in der HYB Platte. Pipette vorsichtig nach oben und unten mindestens 5 - 6 mal zu mischen. Ändern Spitzen nach jeder Spalte zu vermeidenKreuzkontamination.
- Verschließen Sie die HYB Platte mit Kleber Aluminiumfolie und Zentrifuge bei 1000 × g für 30 Sekunden.
- Inkubiere die HYB Platte in der vorgewärmten Hybridisierungs Inkubator bei 95 ° C für 1 min. Eingestellt, die Temperatur des Hybridisierungs Inkubator auf 40 ° C. So lange fortgesetzt, wie es das Inkubieren des Inkubators nimmt von 95 ° C bis 40 ° C (~ 90 min) zu verringern. Hinweis: Diese allmähliche Abkühlung für die richtige Hybridisierung kritisch ist.
- Während der letzten 15 bis 20 min der Hybridisierungs Inkubation vorwaschen der Filterplatte Unit (FPU). Bereiten Sie nur die Vertiefungen im aktuellen Test verwendet werden, das heißt, nur verwenden frische / nicht verwendeten Wells einer zuvor geöffneten Filterplatte, aber nie wiederverwenden Brunnen , die verwendet wurden. Hinweis: Dies sollte über die Platte verwendet, basierend auf dem Kit-Nummer auf der Filterplatte, die Markierungen auf der Filterplatte und dem Dichtmittel klar sein.
- Mit Hilfe einer Mehrkanalpipette, fügen Sie 45 ul Strikte Wash 1 (SW1) in jede Vertiefung.
ACHTUNG: Enthält Formamid. Deckel und Zentrifuge die FPU bei 2.250 × g für 3 min bei 20 ° C. Überprüfen Sie jede einzelne gut für Restflüssigkeit (> 15 & mgr; l / Well). - Wenn Restflüssigkeit vorhanden ist, drehen Sie den FPU 180 ° und wiederholen Sie die Zentrifuge Schritt für weitere 3 min. Wenn die Restflüssigkeit durch die zweite Zeit dreht, dann mit dem nächsten Schritt fort, wenn nicht, dann gibt es einen Defekt in der Filterplatte sein kann, und die aktuelle Platte muss ersetzt werden.
- Mit Hilfe einer Mehrkanalpipette, fügen Sie 45 ul Strikte Wash 1 (SW1) in jede Vertiefung.
- Nachdem die Hybridisierungs Inkubator auf 40 ° C abgekühlt ist, um die Platte zentrifugieren bei 1.000 xg für mindestens 30 sec bei 20 ° C Kondensation zu sammeln. Übertragen, das gesamte Volumen jeder Probe aus der HYB Platte auf die Mitte der entsprechenden vorgewaschenen Vertiefungen der FPU. Ändern Sie Spitzen nach jeder Spalte eine Kreuzkontamination zu vermeiden. Decken Sie die FPU und Zentrifuge bei 2.250 × g für 3 min bei 20 ° C.
- Hinzuzufügen 45 ul SW1 und zentrifugieren bei 2.250 × g für 3 min bei 20 ° C. Wiederholen für insgesamt zwei Waschungen. Drehen Sie den FPU 180 ° und Zentrifuge wieder bei 2.250 × g für 3 min vollständig alle SW1 entfernen.
- Heucheln die FPU. Entsorgen Sie alle Durchfluss (mit Formamid) in den richtigen Abfallbehälter. Bauen Sie die FPU eine andere MIDI-Abfallsammelplatte verwenden. Hinzuzufügen 45 ul Universalpuffer 1 (UB1) in jede Probenvertiefung und zentrifugieren bei 2.250 × g für 3 min bei 20 ° C. VORSICHT: Enthält Formamid.
- In 45 ul Erweiterung Ligationsmix 3 (ELM3) zu jeder Probe auf dem besten FPU Platte und Pipette nach oben und unten 3 Mal zu mischen. Hinweis: Die Erweiterung-Ligatur Reaktion auf der Filterplatte Membran stattfindet.
- Verschließen Sie die FPU Platte mit Kleber Aluminiumfolie und inkubieren die gesamte FPU Montage in einem vorgewärmten 37 ° C Inkubator für 45 min.
- Indizierung und PCR - Amplifikation
- Aliquote wobei die Indizes zu den entsprechenden Vertiefungen in der indizierte Amplification Pl verwendethorizontal i5 Primer Rohre (weiße Kappen, klare Lösung) vertikal, mit Reihen A bis H ausgerichtet, i7 Primer Rohre (orange Kappen, gelbe Lösung): ate (IAP) durch die Primer in der Indexplattenbefestigung Anordnung, in der folgenden Weise angeordnet ausgerichtet, 12. mit den Spalten 1 bis Verwendung eines p10 Mehrkanalpipette, fügen Sie 4 ul i7-Primer (gelbe Lösung) zu jeder Zeile des IAP und fügen 4 ul i5-Primer (klare Lösung) zu jeder Spalte des IAP.
- Auf Eis oder einem Kühlblock, bereiten Sie die PCR Master Mix durch Zugabe von 0,5 ul DNA-Polymerase 1 (TDP1) bis 25 & mgr; l PCR Master Mix 2 (PMM2) pro Probe. Invert, schnell Wirbel, und kurz zentrifugiert werden die PCR Master Mix zu mischen.
- In 22 ul des PCR Master Mix zu jeder Vertiefung der IAP und Pipette nach oben und unten 3 Mal zu mischen. Ändern Sie Spitzen zwischen den Wells. Halten Sie das IAP bei 4 ° C.
- Nach der 45 min Verlängerung-Ligatur-Reaktion (Schritt 4.1.10), die FPU aus dem Inkubator entfernen und entfernen Sie vorsichtig die aluminum Foliendichtung. Deckel mit dem Deckel und Zentrifuge bei 2.250 × g für 3 min bei 20 ° C.
- In 25 ul 50 mM NaOH zu jeder Probe auf dem besten FPU. Pipette auf und ab mindestens 6 mal; die Pipettenspitzen kommen in Kontakt mit der Membran zu gewährleisten. Ändern Spitzen nach jeder Spalte. Inkubieren bei Raumtemperatur für 5 min.
- Transfer Proben aus der FPU an die IAP eluiert, wie folgt:
- Mit einer p100 Pipette Mehrkanal bis 20 & mgr; l eingestellt, Pipette die NaOH auf der FPU Platte nach oben und unten mindestens 6 mal. Etwas die FPU Platte kippen ein vollständiges Absaugen von der Platte zu gewährleisten.
- Transfer 20 ul von der FPU an die entsprechende Spalte des IAP. Pipette vorsichtig auf und ab mindestens 6 mal gründlich, um die DNA mit der PCR Master Mix kombinieren. Verschließen Sie den IAP mit Klebefolie und Zentrifuge bei 1000 × g für 1 min bei 20 ° C.
- Bringen Sie die PCR-Platte in die Post-PCR-Raum und laden Sie die Platte auf einem Thermocycler. Führen Sie die PCR-program eines Wärme aus Schritt 3 min bei 95 ° C Denaturierung; gefolgt von 25 Zyklen von 95 ° C für 30 sec, 62 ° C für 30 sec, 72 ° C für 60 sec; durch eine abschließende Extension von 72 ° C für 5 min, gefolgt; bei 10 ° C mit einer Halte Schlichten. Wenn nicht zur nächsten Stufe ausgehend nach Abschluss der PCR kann die Platte auf dem Thermocycler bleiben über Nacht, oder es kann bis zu zwei Tage bei 2 bis 8 ° C gelagert werden.
- PCR - Aufreinigung und Bead-basierte Normalisierungs
- Entfernen Sie die magnetischen Reinigungsperlen, Elutionspuffer (EBT) und Gel-Elektrophorese Reagenzien aus dem 4 ° C Kühlschrank und Platz bei RT mindestens 20 min vor der nächsten Stufe.
- Sobald die PCR abgeschlossen ist, Zentrifuge bei 1000 × g für 1 min bei 20 ° C Kondensation zu sammeln. Übertragung 1 ul jeder PCR-Reaktionsröhrchen / Platte Vertiefungen mit 4 ul Wasser abzustreifen die Proben 1/5 zu verdünnen. Pipette nach oben und unten zu mischen.
- In 2 ul derPCRed Proben 2 ul mikrofluidischen-Gel-Puffer verdünnt. Verschließen Sie die Streifen / Platte. Schütteln bei 1.800 Umdrehungen pro Minute für mindestens 30 sec und Zentrifuge bei 1000 × g für 30 Sekunden. Nach Herstellerprotokoll, führen Sie die Mischung auf einem Mikrofluidik - Gel zu beurteilen , ob die Bibliothek Vorbereitung eine akzeptable Bibliothek ergab (siehe Abbildung 3).
- Vortex, um die magnetische Reinigung Perlen, bis sie gut suspendiert und die Farbe erscheint homogen. Hinzufügen, 45 & mgr; l der Kügelchen zu jeder Probe.
- Verschließen Sie die Platte mit klaren Klebefilm und schütteln Sie die Platte bei 1.800 Umdrehungen pro Minute für 2 min. Inkubieren bei Raumtemperatur für 10 min ohne Schütteln.
- Legen Sie die Platte auf einen magnetischen Ständer. Nachdem der Überstand gelöscht hat, sorgfältig entfernen und den Überstand verwerfen. Wenn irgendwelche Perlen versehentlich in die Spitzen angesaugt werden, verzichten die Perlen auf die Platte zurück und lassen Sie die Platte ruhen auf dem Magneten für 2 min und bestätigen, dass der Überstand gelöscht hat.
- Mit der Platte auf ter Magnetfuß, 200 & mgr; l frisch zubereiteter 80% Ethanol zu jeder Vertiefung Probe. Bewegen Sie die Platte hin und her ein paar Mal. Die Platte auf dem Magnetständer für 30 Sekunden. Entfernen Sie vorsichtig und den Überstand verwerfen. Wiederholen für insgesamt zwei Waschungen.
- Entfernen Sie das überschüssige Ethanol durch kurzes Drücken der Platte bei 1000 × g zentrifugiert jede Ethanol auf den Rohrseiten zu senken, setzen die Platte wieder auf die magnetische Halterung, und unter Verwendung eines p10 Mehrkanalpipette auf 10 ul eingestellt um das Ethanol zu entfernen. Lassen Sie die Perlen an der Luft trocknen für 5 bis 8 min.
- Mit einer p100 Mehrkanalpipette, fügen Sie 30 ul EBT in jede Vertiefung. Pipette nach oben und unten ein paar Mal, um sicherzustellen, die Perlen an der Seite des Rohres abspringen. Verschließen Sie die Platte mit klaren Klebefilm und schütteln Sie die Platte bei 1.800 Umdrehungen pro Minute für 2 min.
- Inkubieren bei Raumtemperatur für 3 Minuten ohne Schütteln. Wenn es Proben, in denen die Kügelchen nicht vollständig suspendiert, sanft Pipette nach oben und unten um die Perlen zu resuspendieren.
- Legen Sie die Platte auf einem magnetischen Ständer. Mit einer p100 Pipette Mehrkanal-Übertragung 20 & mgr; l des Überstandes auf eine ganz neue Platte namens Bibliothek Normalisierungs Platte (LNP). Übertragen Sie die restlichen ~ 10 & mgr; l der einzelnen Sequenzierungsbibliotheken zu einer separaten Platte genannt Rest aufgeräumte Bibliothek Platte (RCLP). Bewahren Sie diese Platte zusammen mit der endgültigen Bibliothek prep, da sie als Reserve für eine zweite Sequenzierungslauf verwendet werden kann, falls erforderlich. Wenn nicht in die nächste Stufe fortfahren kann die LNP und RCLP bei -15 bis -25 ° C gelagert werden.
- Energisch verwirbeln und die Bibliothek Normalisierungs Beads 1 (LNB1) resuspendieren. Es ist entscheidend, die LNB1 bead Pellet am Boden des Röhrchens vollständig zu resuspendieren. Bereiten Sie die Normalisierungs Mischung, durch Mischen von 8 ul LNB1 mit 44 & mgr; l-Bibliothek Normalisierungs Additives 1 (LNA1) pro Probe. Energisch den Normalisierungs Mix verwirbeln für 10 - 20 sec.
ACHTUNG: LNA1 enthält Formamid. - Mit intermittierenden Inversion einnd Verwirbelung des Normalisierungs Mix, fügen Sie 45 ul jeder Probe des LNP. Verschließen Sie die Platte mit klaren Klebefilm und schütteln Sie die Platte bei 1.800 Umdrehungen pro Minute für 30 Minuten. Diese 30 min Inkubation ist entscheidend für die richtige Bibliothek Normalisierung als Inkubationszeiten von mehr oder weniger als 30 min kann Bibliothek Darstellung und Clusterdichte beeinflussen.
- Während der 30-minütigen Inkubation, bereiten die Reagenzien für die Sequenzierung durch die Reagenzienkartusche und Hyb Puffer Auftauen (HT1) [ACHTUNG: Beide enthalten Formamid]. Darüber hinaus Eis für einen späteren Schritt erhalten und einen Wärmeblock für 1,5 ml Zentrifugenröhrchen geeignet sicherzustellen, wird auf 96 ° C.
- Wenn die 30 Minuten Mischschritt abgeschlossen ist, auf einem Magnetfuß die LNP platzieren. Sobald der Überstand gelöscht hat, mit einem Pipetten mehrkanaligen sorgfältig zu entfernen, und der Überstand in dem entsprechenden Abfallbehälter verwerfen.
- Entfernen Sie die LNP vom magnetischen stehen und waschen Sie die Perlen mit Bibliothek Normalisierungs Wash 1 (LNW1) wie folgt:
- In 45ul LNW1 gut zu jeder Probe.
VORSICHT: Enthält Formamid. Verschließen Sie die Platte mit klaren Klebefilm und schütteln Sie die Platte bei 1.800 Umdrehungen pro Minute für 5 min. Wiederholen für insgesamt zwei Waschungen. Stellen Sie sicher, alle LNW1 nach der zweiten Wäsche zu entfernen.
- In 45ul LNW1 gut zu jeder Probe.
- Entfernen Sie die LNP vom magnetischen stehen und eine Mehrkanalpipette verwenden, fügen Sie 30 ul 0,1 N NaOH (weniger als eine Woche alt) in jede Vertiefung, die Probe zu eluieren. Verschließen Sie die Platte mit klaren Klebefilm und schütteln Sie die Platte bei 1.800 Umdrehungen pro Minute für 5 min.
- Während der 5 min Elution, fügen Sie 30 ul-Bibliothek Normalisierungsspeicherpuffer 1 (LNS1) in jede Vertiefung in einer neuen Platte der Ablageplatte (SGP) mit dem Namen verwendet werden.
- Legen Sie die LNP auf dem Magnetständer. Sobald der Überstand gelöscht hat, übertragen Sie die 30 ul Elution auf die LNS1 im SWP. Ändern Sie Spitzen zwischen den Proben eine Kreuzkontamination zu vermeiden.
- Verschließen Sie die Platte mit Klebefolie und Zentrifuge bei 1000 × g für mindestens 30 sec.
- In 51; l jeder Probe mit einer markierten Pooled Amplicon Library (PAL) 1,5-ml-Röhrchen sequenziert werden. Mischen Sie den PAL zu mischen und kurz zentrifugieren in einer Mikro.
- Je nachdem, welche Sequenzierungschemie verwendet werden (V2 oder V3) in 4 bis 10 & mgr; l des PAL 590-596 ul HT1. Im Allgemeinen, fügen 5,8 ul PAL bis 595 & mgr; l von HT1 für V2 Chemie und 8,5 ul PAL bis 592 & mgr; l von HT1 für V3 Chemie. Beschriften Sie diese Röhre als das verwässerte Amplicon Library (DAL) Röhre.
- Vortex die DAL und kurz drehen, um in einer Mikro. Inkubieren Sie die DAL bei 96 ° C für 2 min. Kehren Sie die DAL Röhre 3 Mal und legen Sie die DAL auf Eis für mindestens 5 min, während der Sequenzer für die Sequenzierung vorbereitet. Nach 5 min ist die DAL fertig geladen werden.
- Wenn mit dem SWP abgeschlossen, versiegeln Sie die Platte mit Klebstoff Aluminiumfolie Film und beschriften Sie es mit dem Datum und der Platte ID. Lagern Sie den versiegelten SGP und PAL bei -15 bis -25 ° C.

Abbildung 3:. Bibliothek Prep QC Gel Beispiel Die grüne Linie über die unteren Bänder ist die untere Markierung und die violette Linie über den oberen Bänder , um anzuzeigen , ist der höhere Marker anzuzeigen. Alles funktionierte gut für Bahnen H1, A2, B2, C2, E2 und G2. Die Bibliothek prep nicht optimal arbeiten, für Bahnen D2, F2 und H2, aber die Ergebnisse werden noch könnten sie erhalten nicht nur eine angemessene Deckung aufweisen. Für A3, kaum die Bibliothek prep gearbeitet und die meisten wahrscheinlich, dass diese DNA-Probe war für den Test nicht ausreichend. Die unteren Bänder oberhalb der unteren Markierung sind die nicht verwendeten Primer, weil die aliquoten gut direkt aus dem PCRed genommen wird. Der NTC - Probe sollte nur den nicht verwendeten Primer - Band haben, und sonst nichts. Bitte klicken Sie hier , um eine größere Version dieser Figur zu sehen.
{kind=link}
5. Sequenzierung
- Stellen Sie sicher, eine korrekte SampleSheet.csv hat für den Lauf gemacht worden. Siehe Ergänzende Abbildung 1 zeigt ein Beispiel.
- Spülen und trocknen die Durchflusszelle und fügen Sie 600 ul der DAL zu dem aufgetauten Reagenzpatrone.
- Bereiten Sie den Sequenzer für die Sequenzierung, indem Sie den Anweisungen auf dem Bildschirm.
6. Datenanalyse
- Führen Sie die Bioinformatik Pipeline. Nutzen Sie die in-house, kundenspezifische bioinformatischen Pipeline zu identifizieren Mutationen, Insertionen, Deletionen und Amplifikationen 18.
- Nachdem die Pipeline abgeschlossen hat, überprüfen Sie die Protokolldateien auf Fehler / Warnung, wie jeder erhebliche Fehler / Warnungen Hilfe bei der QC der Pipeline-Verarbeitung.
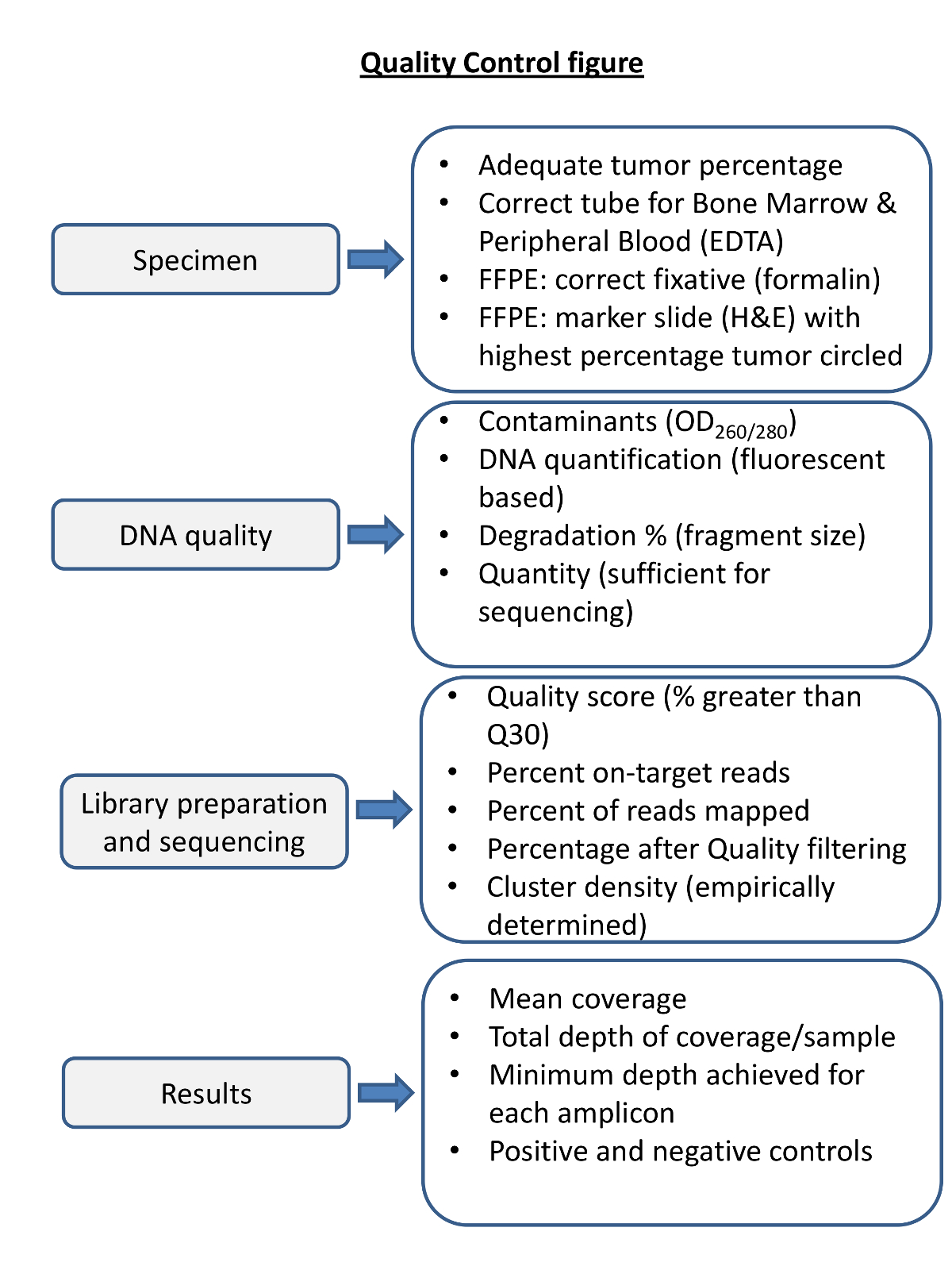
- Analysieren Sie die Laufstatistiken (Tabelle 3) , um die Bibliothek sequenziert , um sicherzustellen , ist vergangen das Labor bestimmt QC - Metriken (Abbildung 4). Überprüfen Sie manuell jede Variante durch die .bam Dateien in einer genomischen Daten - Viewer angezeigt werden (zB die Integrative Genomics ViEwer 16 (IGV)).
ANMERKUNG: Nur die Varianten innerhalb der validierten Allel-Frequenzbereich und über der minimalen Tiefe der Berichterstattung nach Qualität Filterung berichtet (Human Genome Variation Society (Lkw) Nomenklatur verwendet wird). Für die endgültige Berichterstattung wurde jede exonische Variante in kategorisiert: pathogen, wahrscheinlich Krankheit assoziiert sind, Variante unbekannter Signifikanz (VUS), wahrscheinlich gutartig und gutartig. Alle Varianten über den Berichtskriterien von 5% Allelfrequenz, mit Ausnahme der als gutartiger Varianten und auch Änderungen, berichtet.

Abbildung 4. Überblick über die Qualitätskontrolle Schritte für NGS. Die Qualitätskontrolle von jedem Schritt in dem Verfahren ist notwendig , um die Sequenzierung , um sicherzustellen , werden die Ergebnisse liefern , so dass vor und nach der Sequenzierung Metriken betrachtet werden. Geeignete Probenbehandlung ist für qualitativ hochwertige DNA. Blut undKnochenmark in ungeeigneten Fixierungen können niedrige Qualität DNA ergeben. Unangemessen Fixierung von festen Tumorproben können degradieren DNA (zB Fixierung in B5). DNA-Qualität sollte für Protein- und RNA-Kontamination durch spektrophotometrische Mittel, und genau beurteilt für die Menge und die Integrität der DNA beurteilt werden. Die Sequenzierungsmetriken müssen empirisch in der Sequenzierung Labor und anschließend für jede Sequenzierungsreaktion, und jede Probe bestimmt werden. Vor der Sequenzierung Ergebnisse für jede Probe zu berichten sollte für die Berichterstattung, Tiefe und eine angemessene Leistung von positiven und negativen Kontrollen. Beurteilen Bitte klicken Sie hier um eine größere Version dieser Figur zu sehen.
{kind=link}
Ergebnisse
Fall 1 - Häm-NGS - Panel
Die DNA aus leukämischen peripheren Blut extrahiert war von ausreichender Qualität und Menge (176 ng / ul) für die Häm-NGS-Panel. Die gesamte mittlere Tiefe der Berichterstattung war 4,933x (über der minimalen mittleren Tiefe der Berichterstattung von 1,000x). Zusätzliche Laufstatistiken sind in Tabelle 3 dar . Von den 8 Regionen unterhalb 250x Abdeckung 3 durch unsachgemäße Beschneiden der Primer waren (dh, wurde die Primersequenz nicht richtig durch Sequenzierungsfehler entfernt), 1 war ein bekannter Artefakt des Assays, und die anderen vier waren Teilbereiche der Exons verschiedener Gene ohne berichtspflichtigen Varianten. Obwohl unsere einzige klinische Protokollvarianten von mindestens 250 liest abgedeckt Berichterstattung umfasst, werden alle Daten mit mindestens 100 liest in die Datenbank für die Überprüfung importiert.
Datenverarbeitung from der Bioinformatik Pipeline entdeckt drei berichts, krankheitsassoziierte Mutationen; eine Missense - Mutation in FLT3, eine Missense - Mutation in IDH2 und eine Frameshift - Mutation in NPM1. Die exonische Varianten mit ihren Allelfrequenzen sind in Tabelle 4 beschrieben ist . Eine gemeinsame Mutation in FLT3 ist die FLT3 -Interne Tandemverdopplungs (ITD) , die nicht automatisch von der Pipeline und erfordert eine Sichtprüfung von Exon 14. Sichtprüfung von Exon 14 aufgerufen von FLT3 zeigte in der eingereichten Probe keine Insertion oder Vervielfältigung. Die FLT3 I836del war bei 1% Allelfrequenz und wurde nicht in den Abschlussbericht aufgenommen , weil sie unter dem validierten Mindest Allelfrequenz von 5%. Diese Mutation ist nicht auf dem gleichen DNA - Molekül als FLT3 D835Y Veränderung (dh in der gleichen Amplikon Region beobachtet, aber nicht in "cis" in einem der Sequenzierung liest) , und es wurde nur durch eine manuelle Überprüfung der .bam fi beobachtetles bei der Überprüfung der p.D835Y ändern. Die unteren Allelfrequenzen der beiden FLT3 Mutationen schlägt diese Mutationen Heterogenität und / oder klonalen Evolution darstellen können; jedoch könnte der Unterschied zu Testleistung für das Amplikon oder ein single-nucleotide polymorphism (SNP) liegen, in der Nähe von oder überlappend die Primersequenz, die die Amplifikation dieses Allels beeinflusst.
Die Häm-NGS - Panel - Ergebnisse für Fall 1 mit AML identifizierten Mutationen in FLT3, IDH2 und NPM1, drei häufigsten mutierten Gene in AML. Mutationen im FLT3 in ~ 25% der erwachsenen Patienten mit AML (COSMIC Datenbank 17) und sind entweder beobachtet interne Tandemverdopplungen (ITD) oder Missense-Mutationen in der Tyrosinkinase-Domäne. Die FLT3-ITD sind die häufigsten Mutation und sind mit einer schlechten Reaktion auf Standard - Chemotherapie, während die prognostische Bedeutung der FLT3 - Kinase - domain Punktmutationen, wie sie in dieser AML Patienten gesehen, hat eine unklare Auswirkungen auf die Prognose 14. Isozitratdehydrogenase 2 (NADP +), mitochondriale (IDH2) ist ein Gen , das eine epigenetische Modifier kodiert , das häufig bei AML mutiert ist. Mutationen in der epigenetischen Modifikatoren sind relativ häufig bei AML, mit Mutationen in IDH1 und Dnmt3a repräsentieren andere Mutationen in dieser Klasse von Genen , die zu Gen Dysregulation führen. Mutationen in dem Gen für Nucleophosmin (NPM1) sind eine der häufigsten erworbene Mutationen in AML und werden in der Regel ein guter prognostischer Faktor sein (in Abwesenheit eines FLT3 - ITD) betrachtet.
Co-Mutationen von NPM1 und IDH2 wurden in der Literatur als günstige prognostischer Indikator 5, mit einem 3-Jahres - Überlebensrate von 89% beschrieben. Dies stellt einen signifikanten Vorteil Überleben im Vergleich zur Gesamt 3-Jahres-ÜberlebenWildtyp NPM1 und IDH2 von 31%. Zum Beispiel enthält Mutation Standard-Pflege Praxis - Analyse für NPM 1 und FLT3-ITD - Mutationen. In diesem Szenario würde der Nachweis von nur einer NPM1 - Mutation nicht in geeigneter Weise stratify Patienten für Risiko, da die sekundären Mutationen günstig sein können (zB IDH2) oder ungünstig (zB TET2), das Vertrauen der Reduktion einer Knochenmarktransplantation zur Linderung.
Fall 2 - Fest NGS - Panel
DNA aus dem Gewebe extrahiert FFPE war von ausreichender Qualität und Quantität für die Fest NGS-Panel, mit einer Konzentration von 252 ng / ul und nur 4% der DNA unter 1000 Basenpaare (bp). Nach der Datenanalyse war die mittlere Tiefe der Berichterstattung 9167 liest (weit über unsere Mindesttiefe Cutoff von 1000 liest) ohne Regionen unter 250 Lesetiefe. Zusätzliche QC-Metriken sind shown in Tabelle 3.
Daten werden verarbeitet durch die Bioinformatik Pipeline entdeckt zwei krankheitsassoziierte Mutationen: eine In-Frame Insertion in Exon 20 von ErbB2 (HER2 / neu) und eine Missense - Mutation in TP53. Alle exonische Varianten mit ihren Allelfrequenzen sind in Tabelle 5 dargestellt. Die Insertion in ERBB2 stellt tatsächlich einen tandem dupliziert (Her2 / neu) -Sequenz, wie in der Nomenklatur reflektiert. Identifizierung und Bestätigung der in-Rahmeneinfügeschaltung erforderlichen manuellen Überprüfung der Sequenzierung liest durch IGV. Die Detektion von Mutation Frequenzen von mehr als 50%, wie sie sowohl für die TP53 und der Her2 / neu - Mutationen ist von einem Verlust von Heterozygotie (LOH) Ereignis suggestive gesehen (siehe Diskussion).
Die Fest NGS-Panel-Ergebnisse für Fall 2 mit Adenokarzinom der Lunge entdeckt Mutationen in ErbB2 (HER2 / neu) und TP53, zwei Gene nicht häufig getestet als Teil des aktuellen Standard-of-Care für Patienten mit Lungenkrebs. HER2 / neu kodiert für ein Tyrosinkinase-Rezeptor ähnlich zu einem anderen häufig mutierte Gen bei Lungenkrebs, EGFR . Aktivieren HER2 / neu Exon 20 Einfügungen in 2 beobachtet werden , - 4% der Lungenadenokarzinomen, entfallen für die Mehrheit der HER2 / neu - Mutationen in Lungenkrebs beobachtet, und sie werden typischerweise in Tumoren ohne Mutationen in anderen Treiber Gene wie EGFR und ALK 12 gesehen . Es gibt mehrere Beweislinien zeigen Potential für verschiedene Behandlungsmöglichkeiten für Patienten mit aktivierenden HER2 / neu Einfügungen, einschließlich Teilantwort auf die Kombinationstherapie mit HER2 / neu und mTOR - Inhibitoren 13 und signifikante Krankheitsbekämpfung mit dem monoklonalen Antikörper Trastuzumab in Verbindung mit einer Chemotherapie 15. Eine TP53 Veränderung zu entdecken , ist nicht uncommon in Krebs, aber zu diesem Zeitpunkt gibt es keine umsetzbaren Therapien.
| Fall 1 | Fall 2 | |
| Insgesamt starten Liest | 2215926 | 2129110 |
| Prozentsatz der Liest Mapping | 98.42% | 98.29% |
| Prozentsatz der Liest auf Ziel | 99.01% | 97.29% |
| Prozentsatz der Liest auf Ziel Nach Filter | 97.60% | 95.45% |
| Prozentsatz der verwendbaren Liest | 94.87% | 91.79% |
| Prozentsatz der Basen über 250x Coverage | 98.40% | 100% |
| Prozentsatz der Basen above 1000x Coverage | 95.90% | 99.70% |
| Coverage Unter 250x - Amplicon Nummer | 8 | 0 |
Tabelle 3:. Sequencing Run QC Metrics Dies ist eine Zusammenfassung der wichtigsten Lauf Statistiken, ohne die mittlere Abdeckung, die für die Datenüberprüfung verwendet werden , um festzustellen , ob eine Bibliothek prep Probe QC bestanden hat. Der gesamte Prozess ist erfolgreich, wenn alle Prozentsätze über 90% sind, aber es ist möglich, mit Verschleppung von SW1 oder UB1 in der FPU Waschschritt oder Primer Übersprechen, haben niedrigere "Prozent auf Target 'im Bereich von 80 - 90%. Wenn der "Percent Mapped" zu niedrig ist, das wäre eine Kontamination von Bakterien oder anderen DNA zeigen, wie alle Proben für die menschliche ausrichten sollte. Wenn eine dieser Spezifikationen unter 80% sind, wird die Probe zur weiteren Überprüfung markiert festzustellen, wie proceed und den Prozess zu verbessern.

Tabelle 4:.. Fall 1 Ergebnisse Erkannte pathogen, Krankheit assoziiert sind , Variante unbekannter Signifikanz (VUS) und wahrscheinlich gutartig exonische Varianten über den Berichtskriterien von 5% Allelfrequenz aufgelistet Bitte hier klicken , um diese Tabelle zu laden.

Tabelle 5:.. Fall 2 Ergebnisse Erkannte pathogen, Krankheit assoziiert sind , Variante unbekannter Signifikanz (VUS) und wahrscheinlich gutartig exonische Varianten über den Berichtskriterien von 5% Allelfrequenz aufgelistet Bitte hier klicken , um diese Tabelle zu downloaden.
Supplemental Figur 1: Ein Beispiel für ein Amplikon-basierten SampleSheet.csv. Dieses Blatt vermittelt dem Sequenzer , welche Chemie (in diesem Fall Amplicon) zu laufen, was für Workflow (zB GenerateFastq), welche Anwendung und Assay (zB FastqOnly), wie viele Basen (oder liest) zu Folge (in diesem Fall 186 bp x 186 bp), und schließlich, was Proben mit bestimmten Indizes (in diesem Fall Dual Indizierung verbunden sind). Die Teile , die gelb markiert werden können, was auch immer geändert werden , der Experimentator will, aber in diesem Fall ist das Labor diese Parameter verwendet. Bitte klicken Sie hier , um diese Figur zum Download bereit .
Diskussion
Da die beiden in diesem Manuskript beschrieben NGS Tests klinisch angeboten werden, ist die wichtigste praktische Überlegung Qualitätskontrolle. Insbesondere müssen daraufhin geprüft werden, um die Qualität und die Menge der extrahierten DNA bezahlt werden. Dies ist besonders wichtig für FFPE-Proben, die häufig mit variablen DNA-Ausbeute stark abgebaut werden. Eine Isopropanolpräzipitation Methode wurde zu maximieren, um DNA-Ausbeute von FFPE-Proben als spaltenbasierte Methoden manchmal mit begrenzten Elutionsvolumina zu DNA Scher gefunden wurden entwickelt, zu führen. Daher die meiste Zeit, wenn eine Probe zu niedrige Konzentration ergibt oder ist zu abgebauten für den Assay, ist es sehr wahrscheinlich aufgrund der Gewebegröße, Typ oder Fixierung und nicht das Extraktionsverfahren. Für Blut / Knochenmark - Proben, wenn ein Extraktionsversagen ist, ist es normalerweise wegen einer Probe ist hemodilute (dh nicht ausreichende Anzahl von weißen Blut oder Tumorzellen in diesem DRAW) oder ablatiert chemo.
. Nt "> Bei der Validierung, Cutoffs für die Akzeptanz von DNA-Qualität und Menge festgelegt werden Die empfohlene Eingabe von 100-250 ng oft wird in dem Test verwendet, aber wenn die DNA-Qualität ist gut, dann niedrigere Eingangsmengen erfolgreich sein können. Darüber hinaus (ist also die Menge an amplifizierbaren DNA weniger als 100 bis 250 ng) , wenn die DNA - Qualität schlecht ist , dann höhere Eingangs Mengen , die Qualität der Sequenzierungsergebnisse verbessern kann (da die Menge an amplifizierbaren DNA erreichen die empfohlene Eingang) . Metriken für die DNA - Qualität und Quantität sollte vor dem Vorrücken der DNA in die Bibliothek Vorbereitung auf jede Probe angewendet werden. Diese Proben in einer "Grauzone" (siehe Abbildung 2) sollte im Ermessen des Laborleiters oder designee betrieben werden. Derzeit ist die beste Möglichkeit, vorherzusagen, wenn die DNA nicht gut durchführen während der Sequenzierung ist eine qPCR-basierte Assay durchzuführen, die für die Quantifizierung und Qualitätsbeurteilung von Input-DNA ermöglicht. Dieser Ansatz befasst sich mit der bioavailability von unterschiedlich großen Fragmente in der Probe, durch die Amplifikation von unterschiedlich großen Fragmente (beispielsweise 100 bp, 150 bp, 200 bp und 300 bp) und Vergleich Ausbeute.Derzeit beinhaltet Bibliothek Vorbereitung eine große Anzahl von manuellen Schritten, wo ein Fehltritt an einem von mehreren Verbindungsstellen der Bibliothek entweder scheitern lassen kann oder von schlechter Qualität sein. Die Mikrofluidik-Gel-Analyse ist die einzige QC Schritt eine Bibliothek prep Problem vor Sequenzierung zu überprüfen. Dementsprechend gibt es mehrere kritische Schritte, bei denen zusätzliche Achtsamkeit die Wahrscheinlichkeit einer erfolgreichen Reaktion erhöhen kann. Es ist zwingend notwendig, die richtige Probe und Oligonukleotidpool zu gewährleisten werden für jede Probe verwendet. Die Sicherstellung und richtig Aufnahme, dass jede Probe eine von 96 einzigartige Kombinationen von Dual-indiziert PCR Primerpaare enthält reduziert Chance für eine Probe mischen. Auch ist es wichtig, die Filterplatte (FPU) entwässert richtig zu gewährleisten; wenn es nicht richtig abfließt dies kann die exte verursachennsion-Ligatur Schritt der Bibliothek Vorbereitung suboptimal durchzuführen und zu schlechter Qualität Sequenzierungsdaten führen. Nach Bibliothek QC, ist es entscheidend, sicherzustellen, dass die LNB1 Kügelchen vollständig resuspendiert werden und dass die LNB1 / LNA1 Lösung wird gut gemischt, bevor es auf die Proben als die Konzentration dieser Mischung Zugabe verwendet wird, um die Molarität der Bibliothek zu bestimmen. Schließlich, wenn der Wulst Elutionsschritt zu einer suboptimalen Menge der Bibliothek führt die Perlen eluierenden off wird es clustering Dichte verringern und möglicherweise die Bibliothek verursachen nicht ausreichende mittlere Abdeckung erhalten. Im Gegensatz dazu wird ein Überschuss an Bibliothek führen zu einer schlechteren Qualität liest. Daher ist es wichtig, an der Sicke basierten Normalisierungsschritt konsistent sein optimale Bündelung zu gewährleisten und Clustering der Bibliotheken auf dem Sequenzer.
Neben der Bibliothek Vorbereitung ist es entscheidend, eine Bioinformatik Pipeline zu validieren, die genaue Mutation Anrufe aus den rohen, demultiplexten fastq Dateien erzeugen. Die Wahl eineskundenspezifische Lösung kann sehr zeitaufwändig sein, da es viele Open-Source und kommerziell verfügbare Aligner, Variante Anrufer und NGS Software-Pakete sind, die man haben würde zu sichten. Benutzerdefinierte Algorithmen werden müssen, mit denen wesentliche Performance-Statistiken zu extrahieren, zu identifizieren einzigartige wiederkehrende Mutationen, die die meisten Open-Source-Tools ausgewichen, und Kopienzahl Status über jeden der Loci bestimmen. Während der Validierung einer Bioinformatik - Pipeline, ist es wichtig , die berichtspflichtigen Cutoffs für Varianten zu bestimmen , die den Anforderungen sowohl eine minimale Tiefe der Berichterstattung nach Qualität Filterung überschreiten (zB ein Minimum von 250 mal gelesen) und eine minimale Allelfrequenz (zB 4 %). Da dies ein Multiplex - Amplikon-basierten Assays, ist es wichtig , die minimale Tiefe der Berichterstattung bedeuten , um zu bestimmen (zB 1,000x) , dass die Bibliothek muss erreichen zu können , die leistungsschwächsten Amplicon auf die minimale Tiefe zu bekommen von liest. Darüber hinaus bedeutet das gemultiplexte Art des Assays cAUSE Tor Effekte und diese "Artefakte" müssen vor dem Start entdeckt und sind vollständig überprüft werden. Eine weitere wichtige Einschränkung für die beschriebenen Test besteht ein Bedarf für Proben enthalten mehr als 10% Tumor um die validierten Mindest Allelfrequenz zu erzielen.
Die Detektion von niedriger Frequenz, 1%, FLT3 Einfügungen ist Beweis dafür , dass eine manuelle Überprüfung in diesem Prozess noch wünschenswert ist. Selbst bei einer Allelfrequenz Cutoff-Gehalt von 5%, einige wichtige Mutationen vielleicht verpasst und somit manuelle Überprüfung ist unerlässlich, diese Varianten zu ermitteln. Für FLT3-ITD, 14 Sichtprüfung von Exon wird für alle AML - Patienten durchgeführt , bleibt nicht unbemerkt eine geringe oder große Ein- / Doppelarbeit zu gewährleisten. HER2 Exon 20 Einfügungen zusätzlich die üblicherweise neben der Primersequenz sind, müssen manuelle Eingriffe. Trotz einer robusten Bioinformatik Pipeline hat, könnte noch einige Varianten zu übersehen, die nur die Natur ist ein harter Schnitt zu habenoff für die meisten die Statistiken oben erwähnt. Bessere Bioinformatik wäre notwendig, um dieses Problem zu lindern, ebenso wie eine bessere Bibliothek Vorbereitung und / oder Sequenzierung Methoden, weil es vorteilhafter ist, Daten von guter Qualität haben bei noch niedrigeren Cutoffs, die weniger Artefakte und False Positives enthalten.
Die Detektion und Interpretation von Allelfrequenzen kann aufgrund der Schwierigkeit bei der Tumorprozentbestimmung und Amplifizierung Vorspannung von einigen Regionen des Genoms schwierig sein. Zusätzlich kann Allelfrequenzen über 50% festgestellt werden, wie im Fall beobachtet 2. Diese als Verlust der Heterozygotie interpretiert wird (LOH) -Ereignis, entweder durch den Verlust des normalen Allels, auf die scheinbare Zunahme der Mutante führt liest ein Verstärkung des mutanten Allels (zB 2 - Mutante und eine normale Kopie) oder andere Mechanismen. Diese Mechanismen können durch die Verwendung von Array komparative genomische Hybridisierung (aCGH 19) und / oder eine SNP - Genotypisierung Array erläutert. 20.
Die aktuellen Ziel Anreicherung Methoden stützen sich auf ganztägige Verfahren entweder ineffizient oder Hybrid-Capture-Multiplex-PCR-Techniken, die für mehr Sequenzierung Abdeckung einer einzigen Probe und mehr vom Ziel-Sequenzierung liest in der Notwendigkeit, zur Folge haben. Weitere Anwendungen für NGS Molekulare Onkologie in naher Zukunft erwartet wird einfacher Bibliothek Herstellungsverfahren umfassen , die vollständig automatisierbar und in der Lage sein können Proben mit sehr geringen Mengen an Ausgangs - DNA zu verarbeiten (dh weniger als 1 ng) sowie Proben mit stark abgebaut DNA. Um diesen Herausforderungen zu begegnen, wird die meisten Methoden vermutlich sein PCR-Basis, entweder ein mehrstufiges PCR Ansatz zu sein oder eine massiv-parallele Singleplex PCR-Ansatz. Darüber hinaus hat Molekular Strichcode- einzelner Amplicons gezeigt worden, um dramatisch Hintergrund Sequenzierungs Rauschen zu reduzieren und Untersuchung von Proben mit geringeren Anteilen an Tumorzellen ermöglichen unteren Allelfrequenzen erreichen und bewegen circulat zur Erfassunging Tumorzellen.
Nachweis von krankheitsassoziierten Mutationen bei Krebsproben seit Jahrzehnten Sorgfalt Standard. Historisch wurden oft sequentiell getestet, Gene ein Gen / Exon zu einer Zeit, mit der Identifizierung eines an das Ende der Testsequenz führende Mutation. Das Aufkommen von NGS hat mehrere Gene zu sequenzieren, die mit vielen Krebsarten in parallel, was zu der Identifizierung von Mehrfachmutationen für eine weniger vorgespannten Ansatz erlaubt, die mit Neoplasie assoziiert sind. Die klinische Brauchbarkeit von NGS zum Nachweis von somatischen Mutationen in Krebs ist immer deutlicher. Tatsächlich stellt NGS-basierte Analyse von Tumorproben ein neues Paradigma, das traditionelle, einzelnes Gen-Tests in Frage stellt, aber die klinischen Nutzen ist sehr klar. Klinische Laboratorien haben heute die spannende Möglichkeit, sorgfältige Methodenvalidierung und Testinterpretation mit der Anwendung dieser leistungsfähigen Technologie vermählen.
Offenlegungen
Die Autoren haben nichts zu offenbaren.
Danksagungen
Die Autoren möchten die Hilfe von Daniel Wild anerkennen für bei der Herstellung des Manuskripts und Hilfe zu lesen.
Materialien
| Name | Company | Catalog Number | Comments |
| Genomic DNA ScreenTape | Agilent Technology | 5067-5365 | |
| Genomic DNA Reagents | Agilent Technology | 5067-5366 | |
| High Sensitivity D1000 ScreenTape | Agilent Technology | 5067-5584 | |
| High Sensitivity D1000 Reagents | Agilent Technology | 5067-5585 | |
| TapeStation 2200 | Agilent Technology | G2965A | |
| TapeStation Analysis Software | Agilent Technology | A.01.04 or higher | |
| 96-well Tube Storage Racks | Any Vendor | ||
| 15/50 ml Tube Rack | Any Vendor | ||
| 96-well Plate Rack | Any Vendor | ||
| Pipette, single-channel, 0.5–2.5 μL | Any Vendor | ||
| Pipette, single-channel, 1–10 μL | Any Vendor | ||
| Pipette, single-channel, 2–20 μL | Any Vendor | ||
| Pipette, single-channel, 10–100 μL | Any Vendor | ||
| Pipette, single-channel, 20–200 μL | Any Vendor | ||
| Pipette, single-channel, 100–1000 μL | Any Vendor | ||
| Serological Pipettor | Any Vendor | ||
| Vortexer | Any Vendor | ||
| Ice bucket | Any Vendor | ||
| Microcentrifuge (for tubes and strip tubes) | Any Vendor | ||
| Freezer, -20 °C | Any Vendor | ||
| 4 °C Refrigerator | Any Vendor | ||
| Water or Bead Bath | Any Vendor | ||
| Incubator (37 oC) | Any Vendor | ||
| Serological Pipettes, 1 mL | Any Vendor | ||
| Serological Pipettes, 5 mL | Any Vendor | ||
| Serological Pipettes, 10 mL | Any Vendor | ||
| Serological Pipettes, 25 mL | Any Vendor | ||
| Gloves | Any Vendor | ||
| Razor Blades/Scaples | Any Vendor | ||
| KimWipes | Any Vendor | ||
| 15 mL Conical Tube | Any Vendor | ||
| 50 mL Conical Tube | Any Vendor | ||
| Paper Towels | Any Vendor | ||
| 200 proof Ethanol | Any Vendor | Store in Flammable Cabinet | |
| 2-Propanol (Isopropanol) | Any Vendor | Store in Flammable Cabinet | |
| 25ml Reservoirs | Any Vendor | ||
| 10N NaOH | Any Vendor | ||
| Pipette, 8-channel, 1–10 μL | Any Vendor | ||
| Pipette, 8-channel, 10–100 μL | Any Vendor | ||
| Pipette, 8-channel, 20–300 μL | Any Vendor | ||
| Ice Bucket | Any Vendor | ||
| Water Squirt Bottle | Any Vendor | ||
| Alcohol Squirt Bottle | Any Vendor | ||
| Lens Cleaning Paper | Any Vendor | ||
| Plates, 96-well PCR, Semi-Skirted | Any Vendor | ||
| Tube strips, 8-well, 0.2 mL | Any Vendor | ||
| Agencourt AMPure XP Beads | Beckman Coulter | A63881 | |
| BioShake IQ or 3000-T elm | Bulldog Bio/Q.Instruments | 1808-0506/ 1808-0517 | |
| DropPlate96 S - LabChipDS | Caliper | 128876 | |
| DropPlate96 D - LabChipDS | Caliper | 132848 | |
| DropSense96 | Caliper (Trinean) | ||
| DropQuant Software | Caliper (Trinean) | ||
| Plate Sealing Film | Denville | B1212-5S | |
| Aluminum Seal Foil | Denville | B1212-6S | |
| Nuclease-Free, Pure Water System | EMD Millipore | ||
| 5424 centrifuge | Eppendorf | 22621408 | |
| 5804R centrifuge | Eppendorf | 22623508 | Both 15 ml tube and plate rotators, preferably a centrifuge that can go up to 2,500 x g. |
| Safe-Lock Tube 1.5 mL, Natural | Eppendorf | 22431021 | |
| 5 mL Tube, DNA LoBind Tube | Eppendorf | 30108310 | |
| 5430R Centrifuge | Eppendorf | 022620645 | Any plate rotator centrifuge will work |
| Hybex Microsample Incubator | Fisher Scientific | 1057-30-0 | |
| Hybex 0.2 mL Tube Block | Fisher Scientific | 1057-31-0 | |
| TruSeq Amplicon – Cancer Panel | Illumina | FC-130-1008 | 96 reactions |
| TruSeq Custom Amplicon | Illumina | PE-940-1011 | 96 reactions |
| TruSeq Custom Amplicon Index Kit | Illumina | FC-130-1003 | 96 Indices, 384 Samples |
| MiSeq Reagent Kit v3, 500 Cycles | Illumina | MS-102-3003 | |
| MiSeq Reagent Kit v2, 300 Cycles | Illumina | MS-102-2002 | |
| MiSeq Reagent Kit v2, 500 Cycles | Illumina | MS-102-2003 | |
| Experiment Manager | Illumina | 1.3 or higher | |
| MiSeq Reporter | Illumina | 2.0 or higher | |
| Sequencing Analysis Viewer | Illumina | 1.8 or higher | |
| TruSeq Index Plate Fixture and Collar Kit | Illumina | FC-130-1007 | |
| MiSeq v2 | Illumina | SY-410-1003 | |
| TruSeq Custom Amplicon Filter Plate | Illumina | FC-130-1006 | |
| Index Adapter Replacement Caps | Illumina | 11294657 | |
| Qubit 2.0 | Invitrogen | Q32866 | |
| Qubit 0.5 ml Tubes | Invitrogen | Q32856 | |
| Qubit dsDNA Broad Range Assay Kit | Invitrogen | Q32853 | |
| DynaMa6-96 Magnetic Stand, Side Skirted | Invitrogen | 120.27 | |
| GeneAmp PCR System 9700 (gold/silver block) | Life Technologies | N8050200 | |
| Gentra Puregene Blood Kit | Qiagen | 158489 | |
| Deparaffinization Solution (16ml) | Qiagen | 19093 | |
| Buffer ATL (4x50ml) | Qiagen | 939011 | |
| Protein Precipitation Solution (50 ml) | Qiagen | 158910 | |
| DNA Hydration Solution (100ml) | Qiagen | 158914 | |
| Glycogen Solution (500 μl) | Qiagen | 158930 | |
| Qiagen Proteinase K | Qiagen | 19133 | |
| Rnase (5ml) | Qiagen | 158924 | |
| Nuclease-Free Water (10 x 50 ml) | Qiagen | 129114 | |
| Pestles | USA Scientific | 1415-5390 | |
| TipOne RPT 10 ul elongated filter pipet tips in sterilized racks, 10 racks of 96 tips (960 tips). | USA Scientific | 1180-3810 | |
| TipOne RPT 100 ul natural, beveled filter pipet tips in sterilized racks, 10 racks of 96 tips (960 tips) | USA Scientific | 1180-1840 | |
| TipOne RPT 200 μl natural, beveled filter pipet tips in racks, sterilized racks, 10 racks of 96 tips (960 tips) | USA Scientific | 1180-8810 | |
| TipOne RPT 20 μl natural, beveled filter pipet tips in racks, sterilized racks, 10 racks of 96 tips (960 tips) | USA Scientific | 1180-1810 | |
| TipOne RPT 1000 μl natural, graduated XL filter pipet tips in | USA Scientific | 1182-1830 |
Referenzen
- Gerlinger, M., et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 366 (10), 883-892 (2012).
- Campbell, P. J., et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 467 (7319), 1109-1113 (2010).
- Ding, L., et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 481 (7382), 506-509 (2012).
- Frampton, G. M., et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nature Biotechnol. 31 (11), 1023-1031 (2013).
- Patel, J. P., et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 366 (12), 1079-1089 (2012).
- Forbes, S. A., et al. COSMIC (the Catalogue of Somatic Mutations in Cancer ): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 38 (Database Issue), 652-657 (2010).
- Shih, A. H., Abdel-wahab, O., Patel, J. P., Levine, R. L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 12 (9), 599-612 (2012).
- Liersch, R., Müller-Tidow, C., Berdel, W. E., Krug, U. Prognostic factors for acute myeloid leukaemia in adults - biological significance and clinical use. Br J Haematol. 165 (1), 17-38 (2014).
- Bacher, U., Schnittger, S., Haferlach, T. Molecular genetics in acute myeloid leukemia. Curr Opin Oncol. 22 (6), 646-655 (2010).
- Subramanian, J., Govindan, R. Lung cancer in "Never-smokers": a unique entity. Oncology (Williston Park). 24 (1), 29-35 (2010).
- Sakashita, S., Sakashita, M., Tsao, M. S. Genes and pathology of non-small cell lung carcinoma. Semin Oncol. 41 (1), 28-39 (2014).
- Arcila, M. E., Chaft, J. E., Nafa, K. Prevalence clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin Cancer Res. 18 (18), (2012).
- Gandhi, L., et al. Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2-dependent and other solid tumors. J Clin Oncol. 32 (2), 68-75 (2014).
- Sheikhha, M. H., Awan, A., Tobal, K., Liu Yin, J. A. Prognostic significance of FLT3 ITD and D835 mutations in AML patients. Hematol J. 4 (1), 41-46 (2003).
- Mazières, J., et al. Lung cancer that harbors an HER2 mutation epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 31 (16), 1-8 (2014).
- Robinson, J. T., et al. Integrative Genomics Viewer. Nat Biotechnol. 29 (1), 495-500 (2011).
- Forbes, S. A., et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 43 (Database issue), D805-D811 (2014).
- Daber, R., Sukhadia, S., Morrissette, J. J. Understanding the limitations of next generation sequencing informatics, an approach to clinical pipeline validation using artificial data sets. Cancer Genetics. 206 (12), 441-448 (2013).
- Haraksingh, R. R., et al. Genome-Wide Mapping of Copy Number Variation in Humans: Comparative Analysis of High Resolution Array Platforms. PLoS ONE. 6 (11), e27859 (2011).
- de Leeuw, N., et al. SNP Array Analysis in Constitutional and Cancer Genome Diagnostics - Copy Number Variants, Genotyping and Quality Control. Cytogenet Genome Res. 135 (3-4), 212-221 (2011).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten