
Method Article
2 en 1: purificación de la afinidad para el análisis paralelo de proteínas y complejos de proteína-metabolito de un solo paso
* Estos autores han contribuido por igual
En este artículo
Resumen
Las interacciones proteína-proteína y proteína-metabolito son cruciales para todas las funciones celulares. Adjunto, describimos un protocolo que permite el análisis paralelo de estas interacciones con una proteína de elección. Nuestro protocolo fue optimizado para cultivos de células vegetales y combina la purificación de la afinidad con la proteína basada en espectrometría de masa y detección de metabolitos.
Resumen
Procesos celulares están regulados por interacciones entre moléculas biológicas como proteínas, metabolitos, ácidos nucleicos. Mientras que la investigación de las interacciones proteína-proteína (PPI) no es ninguna novedad, aproximaciones experimentales con el objetivo de caracterizar las interacciones proteína-metabolito endógeno (PMI) constituyen un desarrollo bastante reciente. Adjunto, presentamos un protocolo que permite la caracterización simultánea de los PPI y PMI de una proteína de elección, que se refiere como cebo. Nuestro protocolo fue optimizado para celulares de Arabidopsis culturas y combina la purificación de la afinidad (AP) con espectrometría de masas (MS)-basado en la detección de proteínas y metabolitos. En definitiva, líneas transgénicas de Arabidopsis, expresan la proteína cebo fusionada a una etiqueta de afinidad, primero son lisis para obtener un extracto celular nativo. Se utilizan anticuerpos contra etiquetas bajar asociados proteína y metabolito de la proteína cebo. Los complejos purificados por afinidad son extraídos utilizando un solo metil tert-butil éter (MTBE) / metanol/agua método. Mientras que los metabolitos se separan en la polar o la fase hidrofóbica, las proteínas pueden encontrarse en el sedimento. Metabolitos y proteínas son luego analizadas por ultra performance líquida cromatografía-espectrometría de masas (UPLC-MS o UPLC-MS/MS). Líneas de control de vacío-vector (EV) se utilizan para excluir falsos positivos. La gran ventaja de nuestro protocolo es que permite la identificación de proteínas y metabolitos asociados de una proteína diana en paralelo en condiciones fisiológicas cerca (lisado celular). El método presentado es sencillo, rápido y puede ser adaptado fácilmente a los sistemas biológicos que no sean de cultivos de células vegetales.
Introducción
El método aquí había descrito tiene como objetivo la identificación de los socios de metabolitos y proteínas de una proteína de elección en condiciones de lisado celular cercain vivo . Se ha especulado que muchos metabolitos más que caracteriza hoy en día tienen una importante función reguladora1. Metabolitos pueden actuar como interruptores biológicos, cambiar la actividad, función o localización de su receptor proteínas2,3,4. En la última década varios métodos de avance, que permitan identificar de PMI en vivo o en condiciones de cercain vivo , han sido desarrollados5. Enfoques disponibles pueden dividirse en dos grupos. El primer grupo comprende técnicas que comienzan con un metabolito conocido cebo para atrapar a socios de la nueva proteína. Métodos incluyen cromatografía de afinidad6, drogas afinidad sensible objetivo estabilidad ensayo7, quimio-proteómica8y proteoma térmico perfilado9. El segundo grupo consiste en un método único que comienza con una proteína conocida con el fin de identificar ligandos moléculas pequeñas10,11.
AP juntada con anti-lípidos basada en MS se utilizó para analizar los complejos proteína-lípido en Saccharomyces cerevisiae12. Como punto de partida, los autores utilizaron levaduras expresando 21 enzimas implicadas en la biosíntesis de ergosterol y 103 quinasas fundieron a una purificación de la afinidad de tándem etiqueta (TAP). se encontraron 70% de las enzimas y el 20% de las quinasas para enlazar diferentes ligandos hidrofóbicos, arrojando luz a la red de interacción de complejos proteína-lípido.
Anteriormente, podríamos demostrar que, semejantemente a los lípidos, compuestos polares y los polares también permanecen enlazados a complejos de la proteína aislados de la de Lisados celulares13. Basado en estos resultados, decidimos optimizar la AP método anteriormente publicado de10,11 células de la planta y compuestos hidrófilos14. Para ello, utilizamos vectores de corriente descritos por Van Leene et al. 2010, utilizado con éxito en planta PPI estudios15. Para acortar el tiempo necesario para obtener líneas transgénicas, decidimos en cultivos celulares de Arabidopsis. Se empleó un solo metil tert-butil éter, (MTBE) / metanol/agua método de extracción, lo que permite la caracterización de las proteínas (pellet), lípidos (fase orgánica) y metabolitos hidrofílicos (fase acuosa)16 en un solo experimento de la purificación de la afinidad. Líneas de control de EV se introdujeron para excluir falsos positivos, por ejemplo proteínas vinculantes a la etiqueta solamente. Como prueba de concepto que etiqueta tres (de cinco) kinases del difosfato de nucleósido presentan en el genoma de Arabidopsis (NDPK1 NDPK3). Entre otros resultados, podríamos demostrar que NDPK1 interactúa con glutatión S-transferasa y el glutatión. Por lo tanto podríamos demostrar que NDPK1 está sometido a glutathionylation14.
En definitiva, el protocolo presentado es una herramienta importante para la caracterización de proteínas y redes de interacción de la molécula de proteína pequeña y constituye un gran avance sobre los métodos existentes.
Protocolo
Preparación de Arabidopsis de la célula cultura líneas transgénicas, incluyendo las condiciones de reproducción, transformación, selección y crecimiento puede encontrarse en17. Tenga en cuenta que las líneas de control de EV se recomiendan para corregir de falsos positivos. Antes del experimento, confirmar la sobreexpresión de la proteína cebo por análisis de western blot, por ejemplo usando los anticuerpos IgG contra la parte de la proteína G de la etiqueta de afinidad de tándem. Es importante separar los medios de crecimiento material de la cultura de célula vegetal.
1. preparación Material de la célula de la planta antes del experimento
- Crecer una línea de cultivo celular de PSB-L a. thaliana overexpressing la proteína de interés18.
- Preparar medio del MSMO, que contiene 4,43 g/L que MSMO mezclado con sacarosa 30 g/L. Ajustar el pH del buffer a 5.7 con 1 M de KOH y autoclave la solución. Antes del experimento, complementar el medio con 0,5 mg/L de ácido α-naftalenacético, cinetina 0,05 mg/L y 50 de μg/mL kanamicina.
- Cultivar cultivos de células de la planta transformada en 50 mL de medio MSMO en un matraz de 100 mL en un agitador orbital plataforma con agitación suave (130 rpm). Crecen las células en una sala de la cultura en 20 ° C y la intensidad de la luz igual a 80 μmol m-2 s-1.
- Células del subcultivo en medios frescos cada 7 días, ellos diluir 1:10.
- Recoger las células en la fase de crecimiento logarítmico con un embudo de vidrio combinado con una bomba de vacío, usando una malla de nylon como filtro. Envuelva el infiltrado en papel aluminio y congelar en nitrógeno líquido.
PRECAUCIÓN: Recuerda que el nitrógeno líquido es extremadamente frío. Una manipulación incorrecta puede causar quemaduras. Usar equipo de protección personal, incluyendo térmicamente aislado, guantes, gafas protectoras y una bata de laboratorio.
2. selecciona el protocolo
Nota: El siguiente paso es adaptado de Maeda et al 201411 y Leene Van et al. 201117.
- Homogeneizar el material de cultura celular planta cosechada y congelados utilizando un molino mezclador (a 2 min a 20 Hz) o mortero y una maja para obtener polvo fino. Alícuota 3 g del material molido (correspondiente a unos 90 mg de proteína total) por muestra. Evite la descongelación de la muestra durante este paso utilizando equipo previamente refrigerado de nitrógeno líquido.
Nota: Tienda planta de triturado en tubo de 50 mL a –80 ° C al inicio de un procedimiento de AP. - Triturate la muestra en un mortero preenfriado de nitrógeno líquido con 3 mL de tampón de lisis helada (0.025 M Tris-HCl, pH 7,5; 0,5 M NaCl 1.5 mM MgCl20,5 mM DTT; 1 mM NaF; 1 mM Na3VO4; 100 x inhibidor de la proteasa comercial diluido cóctel; 1 mM PMSF) hasta que el material se descongela. Una vez que se descongela la muestra, proceder inmediatamente al siguiente paso.
Nota: Preparar el dulce de buffer de lisis. Introducir las muestras en blanco en este paso. Detergentes no se recomiendan ya que pueden causar problemas en la detección de MS. - Para quitar la ruina celular, dividir el material en tubos de microcentrífuga de 2 mL y centrifugar a 20.817 x g durante 10 min a 4 ° C. Recoger 3 mL del lisado claro en un tubo de centrífuga cónico de 15 mL.
- Durante el paso de centrifugación, equilibrar cuentas IgG-Sepharose. Alícuota de 100 μl de los granos por muestra y lavarlas con 1 mL de tampón de lisis. Vortex para suspender cuentas y flash-spin. Desechar el tampón de lisis y repita el paso dos veces. Resuspender los granos en 400 μL de tampón de lisis.
- Agregar granos a la planta de recogida lisada e incubar la mezcla sobre una rueda giratoria de 1 h a 4 ° C.
- Transferencia de la mezcla en una jeringa combinado vía tapa Luer-lock con una columna de vuelta con filtro poro tamaño 35 μm. ejerza presión para pasar el lisado a través. Granos con complejos conectados permanecerá en el filtro, mientras que el lisado irá a través.
Nota: Opcionalmente, utilice un sistema colector de vacío. Asegúrese de aplicar una presión suave para no dañar los granos. - Lavar los granos al principio con 10 mL de tampón de lavado (0.025 M Tris-HCl, pH 7,5; 0,5 M NaCl) y luego con 1 mL de tampón de elución (0,5 mM EDTA, 10 mM Tris-HCl de pH 7.5; 150 mM NaCl; 1000 x diluido E64 y 1 mM PMSF). Realizar el lavado mediante una jeringa conectada a la columna o sistema colector de vacío.
Nota: Cuando utilizando un sistema colector de vacío Asegúrese de aplicar una presión suave para no dañar los granos. - Incubar los granos con el 400 μL de tampón de elución que contiene 50 U de una versión mejorada del tabaco etch proteasa del virus (AcTEV). Usar coctelera de tabla a 1.000 rpm durante 30 min a 16 ° C.
Nota: No olvide utilizar un tapón para cerrar la columna en la parte inferior añadir el tampón de elución. - Agregar una porción adicional (50 U) de la enzima en la columna e incubar la mezcla para los próximo 30 min bajo el mismo, las condiciones descritas anteriormente.
- Recoger el eluido en un tubo de microcentrífuga de 2 mL por centrifugación (1 min, 20.817 x g) o múltiple de vacío. Para quitar los restantes complejos, introducir un paso adicional de elución con 200 μL de tampón de elución.
Nota: Almacene la muestra a – 20 ° C o –80 ° C, o proceder inmediatamente con el paso de extracción de proteínas y metabolitos. Descongelar las muestras congeladas en el hielo.
3. Western Blot análisis
- Para confirmar la presencia del cebo proteína en el efluente recogido utilizar 10 μl del efluente que contiene proteína – metabolito para realizar SDS-PAGE y western blot análisis. Para identificar la proteína de interés, utilizar anticuerpos primarios de ratón contra la proteína de unión a estreptavidina (1: 200), una parte de la etiqueta de grifo que queda después de la hendidura de la proteasa TEV, como se describe en Leene Van et al. 201117. A continuación, utilizar anticuerpos anti-ratón de cabra secundaria juntados con HRP.
4. metabolito y extracción de proteínas
Nota: Este protocolo se ha adaptado de Giavalisco et al. 201116.
Nota: De este paso adelante use soluciones UPLC-MS – grado.
- Añadir 1 mL de metil tert-butil éter (MTBE) / metanol/agua solvente (3:1:1) al efluente recogido y mezclar la muestra por inversión. Asegúrese de que el solvente se enfría a – 20 ° C antes de la etapa de extracción.
PRECAUCIÓN: MTBE y metanol son sustancias nocivas. Realizar el paso de extracción bajo la campana y usar equipos de protección personal apropiados, por ejemplo, guantes. - Agregar 0,4 mL de metanol: agua 1:3 solución a cada muestra y mezclar el contenido de la muestra por inversión.
Nota: Resultados de la suplementación de la mezcla con solución de metanol: agua en separación de fases. La fase superior contiene lípidos, fase inferior contiene metabolitos polares y los polares, y las proteínas pueden encontrarse en el sedimento. - Para separar las fases, centrifugar la muestra a 20.817 x g durante 2 min a temperatura ambiente, luego recolectar la fase superior para la medición de lípidos (no hecho en este protocolo) usando una pipeta manual de manejo de líquidos con capacidad de volumen de 1 mL.
- Añadir 0,2 mL de metanol y mezclar por inversión.
- Centrifugar la muestra a 20.817 x g durante 2 min a temperatura ambiente, luego recolectar la fase polar para la medición de metabolitos (compuestos polares y los polares). Para evitar perturbar el sedimento de la proteína, deja alrededor de 50 μl de la fase líquida en la parte inferior del tubo.
- Muestras colectadas seco para la medición de metabolitos durante la noche en un evaporador centrífugo. Evitar que se sequen demasiado los gránulos de proteína mediante la eliminación de las muestras del evaporador después de 30 – 60 min.
Nota: Almacenar las muestras a – 20 ° C o –80 ° C, o inmediatamente proceder con la preparación de proteínas para análisis por LC-MS/MS.
5. preparación de muestras para análisis proteómico
Nota: Este paso es una adaptación de Olsen et al. 200419 y el manual técnico de la mezcla de tripsina/Lys-C (véase Tabla de materiales).
- Realizar la digestión enzimática de la muestra.
PRECAUCIÓN: Solventes usados durante la digestión enzimática y la desalación de la muestra son perjudiciales. Trabajar bajo campana extractora y usar equipos de protección personal apropiados, por ejemplo, guantes.- Disolver los pellets de proteína en 30 μl de tampón de desnaturalización recién preparada (bicarbonato de amonio 40 mM que contiene urea de tiourea/6 M de 2 M, pH 8). Para lograr la mejor solubilidad de la proteína, realizar un paso de sonicación de 15 minutos. Repita el paso hasta que el precipitado se disuelva.
- Centrifugar la muestra a 20.817 x g por 10 min a 4 ° C, y luego transferir el sobrenadante a un tubo nuevo de microcentrífuga.
- Determinar la concentración de proteína usando el análisis de la proteína de Bradford.
- Para más análisis, alícuotas de un volumen equivalente a 100 μg de proteína y llenan la muestra hasta 46 μl de tampón de desnaturalización.
- Añadir a la muestra 2 μl de buffer de reducción recién preparada (50 mM TDT disuelto en H2O) e incubar durante 30 min a temperatura ambiente.
- Tratar la muestra con 2 μl de tampón de alquilación recién preparada (Yodoacetamida 150 mM disuelto en buffer de bicarbonato de amonio 40 mM) e incubar la mezcla en la oscuridad por 20 min a temperatura ambiente.
- Diluir la muestra con 30 μl de tampón de bicarbonato de amonio 40 mM y añadir 20 μl de mezcla de LysC/tripsina.
- Después de 4 h de incubación a 37 ° C, diluir la muestra con 300 μL de tampón de bicarbonato de amonio 40 mM.
- Continuar con la incubación a 37 ° C.
- Acidificar la muestra con aproximadamente 20 μl de 10% de ácido trifluoroacético (TFA) para obtener pH < 2. Compruebe el pH de la muestra usando una tira de pH.
Nota: Almacene la muestra a – 20 ° C o proceder al siguiente paso.
- Desalar las proteínas digeridas.
Nota: Preferiblemente, utilizar un sistema de colector de vacío. Evitar el exceso de secado de la columna.- Enjuagar la columna SPE C18 (véase Tabla de materiales) con 1 mL de 100% MeOH y luego con 1 mL de 80% de acetonitrilo (ACN) conteniendo 0.1% TFA diluido en agua. Pasos de uso, aquí y en otras proteínas-la desalación, un sistema múltiple de vacío para acelerar el proceso. Evitar el exceso de secado de la columna.
- Equilibrar la columna lavando dos veces con 1 mL de 0.1% TFA diluido en agua.
- Cargar la muestra en la columna. Enjuague el tubo con adicional 200 μL de 0,1% TFA y la transferencia de la solución en la columna. Ejecutar las soluciones a través de la columna.
- Lavar la columna dos veces con 1 mL de 0.1% TFA.
- Eluir péptidos desaladas de la columna con 800 μl de 60% ACN, solución al 0.1% TFA. Seque la fracción recogida en un evaporador centrífugo, evitando exceso de secado de la fracción de proteína quitando muestras del evaporador después de 30-60 min.
Nota: Almacenar las muestras a – 20 ° C o –80 ° C o proceder inmediatamente al siguiente paso. Realizar los siguientes pasos, mantener las muestras en hielo.
6. la medida había preparado muestras de proteína mediante UPLC-MS/MS.
Nota: Antes de las mediciones de proteómicos y metabolómicos, filtro (tamaño de poro de 0.2 μm) y degas todas las memorias intermedias utilizando una bomba de vacío de 1 h.
- Pellets de péptido Resuspenda cuidadosamente secado almacenado en el tubo de microcentrífuga de 2 mL en 50 μl de tampón C (3% v/v ACN, el ácido fórmico 0.1% v/v) usando pipeta manual de manejo de líquidos con capacidad de volumen de 200 μl. Someter a ultrasonidos muestras de 15 minutos en un baño ultrasónico con frecuencia ultrasónica de 35 kHz.
PRECAUCIÓN: ACN y el ácido fórmico son sustancias nocivas. Trabajar bajo campana extractora y usar equipos de protección personal apropiados, por ejemplo, guantes. - Centrifugar la muestra a 20.817 x g por 10 min a 4 ° C, y luego transferir 20 μl del sobrenadante a un frasco de vidrio.
- Péptidos digeridos separados utilizando una columna de fase inversa C18 conectado a una cromatografía en fase líquida y adquieran espectros de masas utilizando un espectrómetro de masas.
- Separar en la columna 3 μl de la muestra utilizando un caudal de 300 nl/min. Para una fase móvil, utilice tampón C y D (63% v/v ACN, el ácido fórmico 0.1% v/v), formando un gradiente de reducción del 3% ACN 15% ACN durante 20 min y luego al 30% ACN durante los próximos 10 minutos.
Nota: Guarde el resto de la muestra a – 20 ° C o –80 ° C hasta unos meses. Antes de la medición de la proteómica, vuelva a congelar la muestra en hielo. - Eliminar contaminantes durante 10 min con 60% ACN y equilibrar la columna con 5 μl de tampón de C antes de la medición de la muestra siguiente.
- Obtener espectros de masas utilizando método de MS/MS dependientes datos con resolución conjunto en 70.000, objetivo AGC de 3e6 iones, tiempo de inyección máxima de 100 ms y un m/z que van desde 300 a 1600. Adquirir el máximo de 15 MS/MS explora en una resolución de 17.500, objetivo AGC de 1e5, tiempo de inyección máxima de 100 ms, ratio de subutilización del 20%, con una ventana de aislamiento de 1,6 m/z y m/z desde 200 hasta 2000. Activar el gatillo de apex (6 – 20 s), exclusión dinámico set a 15 s y excluir los cargos de 1 y > 5.
- Separar en la columna 3 μl de la muestra utilizando un caudal de 300 nl/min. Para una fase móvil, utilice tampón C y D (63% v/v ACN, el ácido fórmico 0.1% v/v), formando un gradiente de reducción del 3% ACN 15% ACN durante 20 min y luego al 30% ACN durante los próximos 10 minutos.
7. procesamiento de datos proteómicos
- Descargar la más reciente base de datos de proteoma de thaliana de Arabidopsis de http://www.uniprot.org/ e incluyen base de datos de contaminantes. Analizar datos obtenidos de la LC-MS se ejecuta usando MaxQuant con el motor de búsqueda de péptido de Andrómeda integrado utilizando la configuración predeterminada con habilitado LFQ normalización20,21,22. Encuentre información detallada acerca de los parámetros utilizados en la Tabla S1.
- Abra el archivo de salida "groups.txt de la proteína". Para su posterior análisis, filtro de grupos de proteínas identificados con al menos dos péptidos únicos. Quitar grupos de proteína definidos por MaxQuant como potenciales contaminantes y filtro para proteínas de a. thaliana (ARATH en columna de cabeceras de Fasta) presentes en la base de datos.
- Para probar la importancia del enriquecimiento de proteínas entre muestras, utilizar intensidades LFQ normalizado y realizar prueba-t de Student no pareado, dos colas seguida de corrección de comparación múltiple (por ejemplo Benjamini & Hochberg false discovery rate (FDR) rectificación o corrección de Bonferroni).
- Calcular el valor p comparando intensidades LFQ obtenidos para el control de EV y NDPK1. Filtrar todos los valores de indeterminado. Ordenar en orden ascendente los valores de p y usar escritura de R o Calculadora en línea (por ejemplo, https://www.sdmproject.com/utilities/?show=FDR) para calcular la corrección del FDR. Filtro para valores FDR por debajo de 0.1.
Nota: Utilizar el formulario de análisis de datos adecuado para la investigación. Para estudios cuantitativos (análisis de enriquecimiento de proteínas entre muestras) usan el valor de "Intensidad de LFQ", mientras que para la investigación cualitativa (presencia o ausencia de determinada proteína), elija el valor de "Intensidad". - Filtro para los grupos de proteínas que son más abundantes en las NDPK1 en comparación con el control de EV. Determinar la localización de posibles socios de proteína utilizando la base de datos SUBA23 y correcto para la proteína Co localizada con NDPK1.
- Calcular el valor p comparando intensidades LFQ obtenidos para el control de EV y NDPK1. Filtrar todos los valores de indeterminado. Ordenar en orden ascendente los valores de p y usar escritura de R o Calculadora en línea (por ejemplo, https://www.sdmproject.com/utilities/?show=FDR) para calcular la corrección del FDR. Filtro para valores FDR por debajo de 0.1.
8. medición de las muestras que contienen Polar fase utilizando UPLC-MS.
- Secado fase polar de paso 4.5 en 200 μL de agua de suspender y someter a ultrasonidos la muestra durante 5 minutos.
- Centrifugar la muestra a 20.817 x g por 10 min a 4 ° C, y luego transferir el sobrenadante a un frasco de vidrio.
Nota: Guarde el resto de la muestra a – 20 ° C o –80 ° C de hasta varios meses. Antes de la medición de la metabolómica, vuelva a congelar la muestra en hielo. - Realizar un paso de separación mediante UPLC acoplado a la columna de fase inversa C18 y adquirir espectros de masas con MS.
- Carga en la columna 2 μl de la muestra por inyección para cada modo de ionización (positiva y negativa) y separar la fracción usando 400 μL/min de caudal. Para crear el gradiente necesario para la medición del metabolito, preparar la solución de la fase móvil como sigue: un (ácido fórmico 0.1% en H2O) de tampón y tampón B (0,1% de ácido fórmico en ACN).
- Diferentes metabolitos en 400 μL/min y el siguiente gradiente: 1 min 99% de tampón A, degradado lineal de 11 min del 99% de tampón A 60% de tampón A, degradado lineal de 13 min entre el 60% de tampón A 30% de tampón A, 15 min de degradado lineal del 30% de tampón A 1% de tampón A, mantener 1% concentración hasta 16 minutos a partir de 17 min, uso degradado lineal del 1% de tampón A 99% de tampón A. volver a equilibrar la columna por 3 min con 99% de concentración de tampón A antes de la medición de la muestra siguiente.
- Adquisición de espectros de masas abarcan masa entre 100 y 1500 m/z con resolución establece en 25.000 y carga limitada a 100 objetivo ajustar AGC Sra. 1e6, capilar tensión 3kV con una envoltura de gas flujo y gas auxiliar valor de 60 y 20 , respectivamente. Establezca la temperatura capilar a 250 ° C y skimmer voltaje de 25V.
9. tratamiento de los datos de la metabolómica
- Proceso de recogida cromatogramas de ambos modos de ionización. Utilizar software para extraer la masa a la proporción de carga (m/z), tiempo de retención (RT) y la intensidad de los picos asociados, por ejemplo, software comercial (véase Tabla de materiales) o alternativos24.
- Iniciar proceso de software haciendo doble clic en el archivo .exe
- Crear nuevo flujo de trabajo, buscar actividad "Carga de archivos" y mover esta actividad por "arrastrar y soltar" en el espacio de flujo de trabajo en blanco. Presione la actividad con el botón derecho del ratón y abrir opciones de la actividad.
- En la ficha que contiene la configuración "General", establece un nombre de la experiencia en el campo "Nombre" y a continuación, haga clic en "seleccionar archivos y carpetas" y marcar raws cromatogramas.
- En la ficha que contiene la configuración de "Avanzado", establece "Perfil de datos de corte" en intensidad 0. Haga clic en "Aplicar" y "OK".
- Buscar y agregar actividad "Barrido de datos". Presione la actividad con el botón derecho del ratón y abrir opciones de la actividad.
- En la ficha que contiene la configuración "General", marca "Centroide datos" y "MS/MS". Quitar todos los datos de MS/MS por seleccionar "Todo" en el panel de selección.
- Busca y añade la actividad «Cromatograma química ruido resta». Presione la actividad con el botón derecho del ratón y abrir opciones de la actividad.
- En la ficha que contiene la configuración "General", marca "Atenuación del cromatograma" y número de análisis a "3" y "Estimador" a "Promedio móvil". Conjunto "RT ventana" análisis 51, "Quantile" al 50%, intensidad 750 «Umbral» y resta «Método».
- En la ficha que contiene la configuración de "Avanzado", marca "RT eliminación de estructura" y "Longitud mínima de RT" análisis 5.
- En la ficha que contiene configuraciones "Avanzadas", marcar "m/z eliminación de estructura" y "Mínimo m/z longitud" a 3 puntos.
- Busca y añade la actividad «Cromatograma RT alineación». Presione la actividad con el botón derecho del ratón y abrir opciones de la actividad.
- En la ficha que contiene la configuración "General", establece "Esquema de alineación" a "Pairwise alineación Base de árbol" y "RT intervalo de búsqueda" en 0,5 minutos.
- En la ficha que contiene la configuración de "Avanzado", utilizar parámetros por defecto.
- Buscar y agregar la actividad "Detección de pico" en grupo "Cromatograma" de actividades. Presione la actividad con el botón derecho del ratón y abrir opciones de la actividad.
- En la ficha que contiene la configuración "General", establece "Suma ventana" en min 0,09, "Tamaño de pico mínimo" min 0,03, "Combinar la distancia máxima" a 5 puntos y "Combinar estrategia" a los "Centros". En el "pico RT partir" caja establece "Cociente de brecha/Peak" en 50%.
- En la ficha que contiene la configuración de "Avanzado", establece "Ventana de suavizado" en 5 puntos, "Umbral de refinamiento" en un 80% y "Umbral de coherencia" a 1. Conjunto de "Centro de cómputo" como "Cargado de intensidad" con "Intensidad umbral" al 70%.
- Buscar y agregar la actividad "Isótopo Clustering" en grupo "Cromatograma" de actividades. Presione la actividad con el botón derecho del ratón y abrir opciones de la actividad.
- En la ficha que contiene la configuración "General", establece "Tolerancia RT" minuto 0,015 y "m/z tolerancia" a 5 ppm.
- En la ficha que contiene configuración de "Ajuste envolvente", sistema "Método" como "Ionización" y "Restricción de forma de No" como "protonación (para el modo positivo) y"Deprotonation"(para el modo negativo). Configurar "mínimo y máximo de carga" en 1 y 4, respectivamente.
- En la ficha que contiene la configuración de "Avanzado", utilizar parámetros por defecto.
- Buscar y agregar actividad "Filtro de Singleton".
- Para exportar los resultados de procesamiento de datos, buscar y agregar la actividad "Analista" en grupo "Exportación" de actividades.
- En la ficha que contiene la configuración "General", sistema "Tipo" como "Clusters" y "Observables" como "Suma intensidad". Elija "Custom Destination" y especificar el directorio de archivo de exportación.
- En la ficha que contiene la configuración de "Avanzado", utilizar parámetros por defecto.
- Anotar características masa con base de datos compuesto de referencia.
- Analizar MS de uno o varios grado compuesto de referencia mediante UPLC-Sra. uso el mismo método de LC-MS para análisis de compuestos de referencia y metabolitos Co purificados con la proteína de interés.
Nota: Para este estudio, un conjunto de casi 300 dipéptidos fue analizado y utilizado como compuesto de referencia biblioteca. - Análisis (véase Tabla de materiales) software para abrir archivo raw cromatograma y buscar para específico de m/z y RT asociaron a medida compuesto de referencia (consulte la guía del usuario).
Nota: Metabolitos secundarios difieren en tipos de ionización. Verificar la presencia del común aductos buscando para iones de masa igual a M-1.007276, 1.007276 M + y M + 18.033823 M + 22.989218 [M-H], [M + H], [M + NH4] y [M +], respectivamente. - Hoja de cálculo de uso para abrir exporta archivo "Analista" obtenido después del procesamiento de los cromatogramas y búsqueda de ion específico masa. Comparar el RT de la función de masa medida en el experimento y RT del compuesto de referencia. Permite una desviación de 0.005 Da de m/z y 0,1 min para RT.
- Analizar MS de uno o varios grado compuesto de referencia mediante UPLC-Sra. uso el mismo método de LC-MS para análisis de compuestos de referencia y metabolitos Co purificados con la proteína de interés.
- Para probar la significación de enriquecimiento metabolito Co purificado con proteína particular entre muestras (línea con Proteína sobreexpresada de control de interés vs EV), comparar los valores de pico con dos colas no apareado prueba t de Student-seguido por múltiples corrección de comparación (por ejemplo Benjamini & Hochberg false discovery tipo corrección o corrección de Bonferroni).
Resultados
En el estudio original, tres genes a. thaliana NDPK se sobreexpresa en cultivos de suspensión de células de PSB-L bajo el control de la de promotor constitutivo 35S14 (figura 1). Etiqueta de afinidad tándem fue fundido a ambos extremos de la terminal carboxilo o amino de una proteína de cebo. Los complejos purificados por afinidad fueron sometidos a extracción de MTBE y metanol/agua16. Tiró de afinidad de las proteínas y moléculas pequeñas fueron identificadas utilizando MS (tablas de S2 y S3).
Para corregir para falsos positivos, las muestras en blanco fueron utilizadas para excluir contaminantes de molécula pequeña de los productos químicos y consumibles de laboratorio. Además, metabolitos y proteínas que se unen a cualquiera una etiqueta de afinidad o la resina solo se representaron mediante el uso de líneas de control de EV. Para recuperar los verdaderos positivos, prueba t de dos colas no apareado del estudiante y Benjamini & Hochberg tarifa falsa del descubrimiento se aplicó corrección para identificar metabolitos (Tabla S4) y proteínas (Tabla S5) considerablemente enriquecidas en el AP NDPKs experimentos (N - y C-terminales etiquetados NDPKs) en comparación con las líneas de control de EV (FDR < 0.1). Note que en el trabajo anterior, hemos utilizado criterios de ausencia/presencia para delinear interactianos proteínas y moléculas pequeñas.
Resultados representativos se dan para NDPK1, mientras que el enfoque de datos de metabolitos en dipéptidos, una nueva clase de los reguladores de molécula pequeña estudiado en nuestro grupo. Análisis proteómico revelaron a 26 socios putativos proteínas NDPK1. Al filtrar más proteínas localizadas en el mismo compartimento subcelular como NDPK1 Co (citosol), la lista se redujo a 13 interactianos de proteína supuesta. Entre las proteínas identificadas fueron glutatión S-transferasa, factores de iniciación elongación dos, tubulina y aconitate hydratase. Análisis metabolómicos reveló cuatro dipéptidos Val-Leu, Glu Ile, Leu-Ile y Ile Phe que específicamente eluyen conjuntamente con NDPK1 (figura 2). Observe que los cuatro dipéptidos comparten un residuo hidrofóbico en su N-terminal, sugiriendo la especificidad de unión común.
Buscar complejos proteína-proteína y proteína-metabolito conocidos hacemos consultado 13 identificado proteínas y cuatro dipéptidos contra la base de datos de punto25 (figura 3). Podrían hacerse varias observaciones: (i) ninguno de los interactianos fue divulgado previamente para NDPK1. (ii) APX1 ortholog informó a interactuar con aldehído deshidrogenasa familiar ALDH7B4, mientras que el factor de la iniciación de la traducción FBR12 con otro factor de iniciación de traducción codificada por el gen AT2G40290. (iii) los dipéptidos identificadas no han reportado a socios de proteína. Dipéptidos Co eluídas no se informaron más temprano asociado a cualquier proteína vegetal obtenido. Sin embargo, desempeñan importantes funciones en otros organismos: Leu-Ile, por ejemplo, tiene un efecto activador de neurotrophin en una línea de células humanas26. Tenga en cuenta que el experimento no permite identificar la topología exacta del sistema. Por ejemplo, un dipéptido puede interactuar directamente con NDPK1 pero también puede deberse a cualquiera de las proteínas Co purificadas.
Tomados en conjunto, nuestros resultados muestran que el procedimiento establecido, empleando AP junto con espectrometría de masas, facilita la identificación de proteínas y moléculas de proteína pequeñas interactianos y ayuda a genera información extensa sobre el interactoma de la proteína diana.

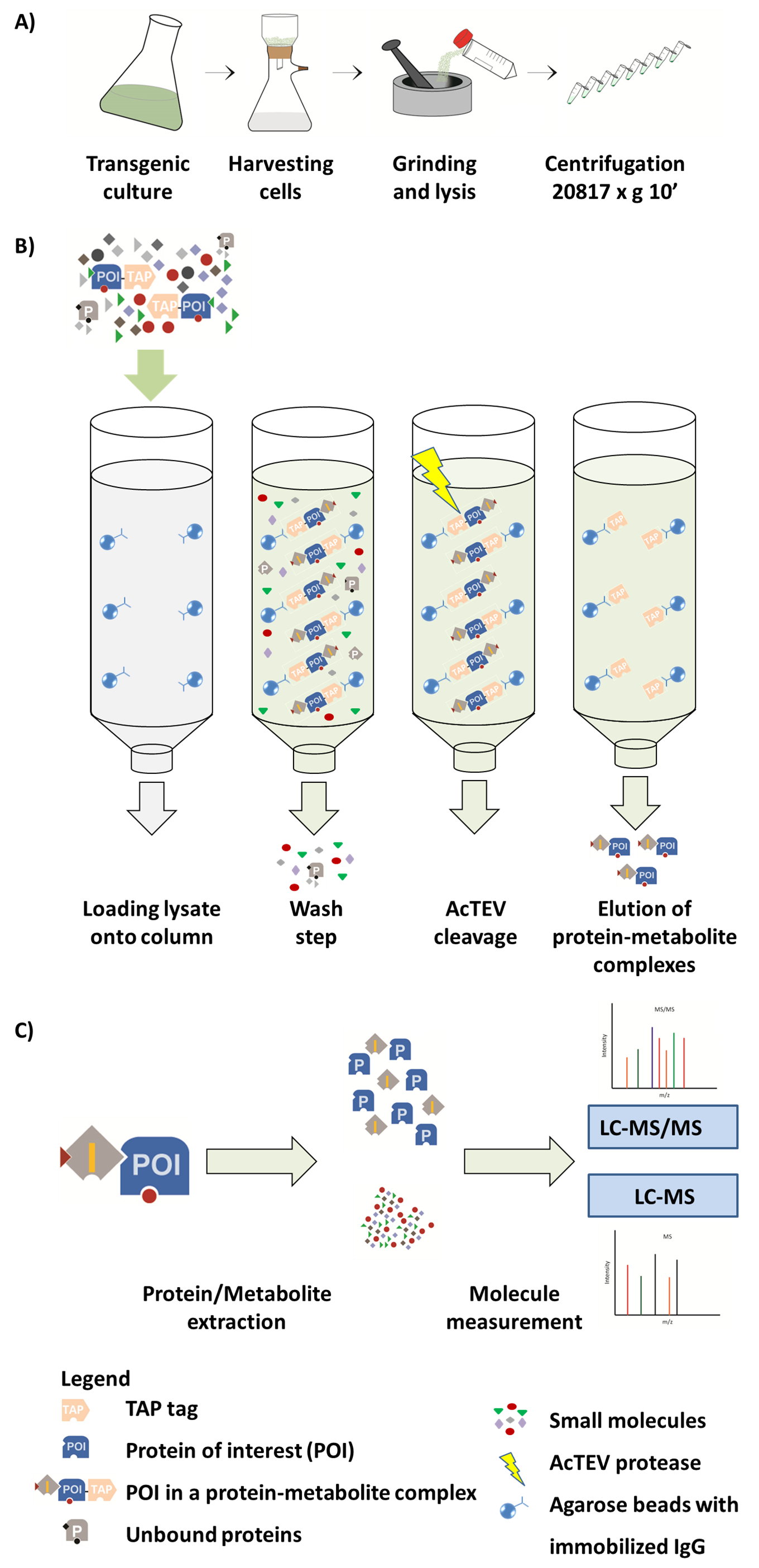
Figura 1. Esquema del flujo de trabajo AP-MS. (A) preparación de una fracción soluble nativa de cultivo de células vegetales. (B) pasos en el procedimiento de AP. Después de cargar la muestra en la columna, la proteína de interés (POI) fusionado a una etiqueta TAP se une al anticuerpo IgG inmovilizado en las perlas de agarosa. Lavado de la columna facilita la eliminación de las proteínas no Unidas y metabolitos. Después de realizar el escote AcTEV, complejos de proteína-metabolito POI se eluyen. (C) separación de complejos en fracción de proteínas y metabolitos seguida de análisis semicuantitativo de MS. Parte de esta figura es reproducida de Luzarowski et al. 201714. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2. Dipéptidos específicamente Co liberador con NDPK1. Intensidad promedio de cuatro dipéptidos Val-Leu (A), Glu Ile (B), Leu-Ile (C)y Ile Phe (D) medidos en experimento de AP se trazaron. Los cuatro dipéptidos muestran significativo enriquecimiento en NDPK1 muestras en comparación con el control de EV (asteriscos representan FDR < 0.1). Barras de error representan el error estándar de 6 medidas (3 repeticiones de n y 3 de C-terminal de proteínas etiquetadas). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3. Red de interacción de las moléculas co liberador con NDPK1, consultar base de datos de puntada sólo considerando anterior experimentales y evidencias de la base de datos (confianza > 0,2). Confianza superior indica mayor posibilidad de interacción y se calcula en base a los datos depositados. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Tabla S1. MaxQuant salida de mesa "parameters.txt". Tabla incluye valores umbral para la identificación y cuantificación, así como información sobre las bases de datos utilizado. Haga clic aquí para descargar este archivo.
Tabla S2. Información de MaxQuant salida de mesa "proteinGroups.txt". La tabla contiene una lista de todos los grupos de proteína identificada, intensidades e información adicional como número de péptidos únicos y puntuación. Haga clic aquí para descargar este archivo.
Tabla S3. Archivo de salida conteniendo análisis de metabolitos polares. Tabla contiene una lista de todas las características identificadas masa caracterizado por específica m/z, la RT y la intensidad. Haga clic aquí para descargar este archivo.
Tabla S4. Dipéptidos encontraron en muestras de AP NDPK1, NDPK2 o NDPK3 eran utilizados como cebo. Dipéptidos presentes en muestras en blanco fueron excluidos de la lista. Dos líneas independientes (con la etiqueta en cualquier terminal N o C) para cada NDPK fueron funcionadas por triplicado. De Student t-test y más corrección de p-valor usando Benjamini & Hochberg método fueron utilizados para determinar significativamente enriquecidos socios interactor de NDPKs (FDR < 0.1). Dado ΔRT calcula en relación con los compuestos de referencia y Δppm en relación con el monoisotopic masa dada en Metlin27. Haga clic aquí para descargar este archivo.
Tabla S5. Proteínas NDPK1 Co depuradas. Dos líneas independientes (con la etiqueta en cualquier terminal N o C) para cada NDPK fueron funcionadas por triplicado. De Student t-test y más corrección de p-valor usando Benjamini & Hochberg método fueron utilizados para determinar significativamente enriquecidos socios interactor de NDPKs (FDR < 0.1). Haga clic aquí para descargar este archivo.
Discusión
El protocolo presentado permite paralelo identificación de complejos PP y PM de una proteína diana. De la clonación a resultados finales, el experimento puede realizarse en tan sólo 8-12 semanas. AP completa toma aproximadamente 4-6 horas para un grupo de 12 a 24 muestras, haciendo nuestro protocolo de análisis de rendimiento medio.
El protocolo, a pesar de ser sencillo en general, tiene una serie de pasos críticos. (i) suficiente cantidad de cuentas de afinidad y entrada de la proteína es crucial para alcanzar un rango dinámico de detección de metabolitos. Lisis celular eficiente por lo tanto es un paso crucial en el procedimiento. Rendimientos pobres de proteína pueden ser una consecuencia de pulverización insuficiente del material o del cociente de tampón de lisis/material subóptima. (ii) debe tener cuidado que los reactivos utilizados son MS respetuoso. Detergentes fuertes, glicerol o cantidades excesivas de sal se deben evitar que interfieran con la detección de MS. (iii) perlas de agarosa no deben ser secados demasiado durante el lavado los pasos, y cuando se usa un colector de vacío es importante aplicar una tasa de flujo lento para no destruir los granos o afectan la estabilidad compleja.
Hay algunas posibles modificaciones importantes en el protocolo presentado: (i) utilizamos el promotor CaMV35S constitutivo para maximizar la cantidad de proteína cebo. Sobreexpresión, aunque muy útil, puede tener efectos graves sobre la homeostasis de la célula28 y llevar a la formación de interacciones fisiológicamente irrelevantes. Expresión de proteínas etiquetadas usando promotores nativos y donde sea posible en un fondo de pérdida de función se considera superior para recuperar los interactianos biológicos verdadero. Las proteínas que normalmente no se expresa en cultivos de células de la planta, un fondo de planta puede resultar necesario identificar relevantes interactianos. (ii) cuando se trabaja con proteínas de la membrana, el tampón de lisis debe complementarse con un detergente compatible con MS. (iii) la introducción de un segundo paso de purificación de la afinidad podría mejorar la proporción de verdaderos positivos falsos positivos y eliminar la necesidad de controles de EV29. Una etiqueta de novela tandem con dos sitios independientes proteasa-escote presenta una alternativa atractiva a la cromatografía por exclusión de tamaño paso Añadida por Maeda et al 201411, que es laborioso y consume tiempo.
El inconveniente más grave de la AP es la alta tasa de falsos positivos. Las razones son numerosas. Ya se ha mencionado la sobre-expresión constitutiva. Otra fuente de interacciones fisiológicamente irrelevantes, a menos que se ocupan de los organelos aislados, es preparación de lisados de células completas que contienen mezclas de proteínas y metabolitos de diferentes compartimentos subcelulares. Localización subcelular puede usarse para filtrar ciertos interactianos. Sin embargo, la mayoría de los falsos positivos resultado de uniones inespecíficas entre proteínas y resinas de agarosa. Introducción de un segundo paso de purificación, como se describe anteriormente, ofrece la mejor solución al problema, sin embargo viene a costa de tiempo y rendimiento. Por otra parte, la interacción más débil puede perder como alarga el protocolo. Otra advertencia de AP es que a pesar de la amplia información que ofrece sobre el interactoma de una proteína objetivo, distinguiendo entre objetivos directos e indirectos de la proteína con cebo es imposible. Enfoques bimoleculares específicos son necesarios para confirmar las interacciones.
AP juntada con metabolómica basada en MS se utilizó para el estudio de complejos de la proteína en S. cerevisiae12. Este trabajo, junto con nuestra anterior observación13 que, de manera similar a los lípidos, compuestos polares y los polares siendo enlazados a complejos de la proteína aislados de Lisados celulares, proporciona bases conceptuales para el protocolo presentado. Nuestro protocolo se caracteriza por tres únicos puntos: () en contraste a la levadura trabajar12, demuestra que el AP es conveniente para recuperar no sólo ligandos de la proteína hidrofóbica, pero también hidrofílicas. (ii) mediante la introducción de un protocolo de extracción de tres-en-uno, un solo AP puede utilizarse para estudiar proteínas y metabolitos interactianos de la proteína cebo. (iii) adaptar el protocolo para las células de la planta.
Esfuerzos futuros se centrarán en crear un tag nuevo tándem con dos sitios independientes proteasa-escote. También le gustaría explorar la idoneidad del Protocolo de baja abundancia de pequeñas moléculas como hormonas vegetales.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Nos gustaría reconocer amablemente Prof. Dr. Lothar Willmitzer por su participación en el proyecto, discusiones productivas y gran control. Estamos agradecidos con el Dr. Daniel Veyel para ayudar con las mediciones de MS proteómica. Agradecemos a la Sra. Änne Michaelis que nos proporcionó una inestimable ayuda técnica LC-MS medidas. Además, nos gustaría agradecer al Dr. Monika Kosmacz y Dr. Ewelina Sokołowska por su ayuda y participación en el trabajo en el manuscrito original y a Weronika Jasińska soporte técnico.
Materiales
| Name | Company | Catalog Number | Comments |
| Murashige and Skoog Basal Salts with minimal organics | Sigma-Aldrich | M6899 | |
| 1-Naphthylacetic acid | Sigma-Aldrich | N1641 | |
| Kinetin solution | Sigma-Aldrich | K3253 | |
| Tris base | Sigma-Aldrich | 10708976001 | |
| NaCl | Sigma-Aldrich | S7653 | |
| MgCl2 | Carl Roth | 2189.1 | |
| EDTA | Sigma-Aldrich | 3609 | |
| NaF | Sigma-Aldrich | S6776 | |
| DTT | Sigma-Aldrich | D0632 | |
| PMSF | Sigma-Aldrich | P7626 | |
| E-64 protease inhibitor | Sigma-Aldrich | E3132 | |
| Protease Inhibitor Cocktail | Sigma-Aldrich | P9599 | |
| Na3VO4 | Sigma-Aldrich | S6508 | |
| AcTEV Protease | Thermo Fischer Scientific | 12575015 | |
| Rotiphorese Gel 30 (37,5:1) | Carl Roth | 3029.2 | |
| TEMED | Carl Roth | 2367.3 | |
| PageRuler Prestained Protein Ladder | Thermo Fischer Scientific | 26616 | |
| SBP Tag Antibody (SB19-C4) | Santa Cruz Biotechnology | sc-101595 | |
| Goat anti-mouse IgG-HRP | Santa Cruz Biotechnology | sc-2005 | |
| Bradford Reagent | Sigma-Aldrich | B6916 | |
| Trypsin/Lys-C Mix, Mass Spec Grade | Promega | V5071 | |
| Urea | Sigma-Aldrich | U5128 | |
| Thiourea | Sigma-Aldrich | T8656 | |
| Ammonium bicarbonate | Sigma-Aldrich | 9830 | |
| Iodoacetamide | Sigma-Aldrich | I1149 | |
| MTBE | Biosolve | 138906 | |
| Methanol | Biosolve | 136806 | |
| Water | Biosolve | 232106 | |
| Acetonitrile | Biosolve | 12006 | |
| Trifluoroacetic acid | Biosolve | 202341 | |
| Formic acid | Biosolve | 69141 | |
| Unimax 2010 Platform Shaker | Heidolph | 5421002000 | |
| Nylon Mesh (Wire diameter 34 µM, thickness 55 µM, open area 14%) | Prosepa | Custom order | |
| Glass Funnel, 47 mm, 300 ml | Restek | KT953751-0000 | |
| Filter Bottle Top 500 mL 0,2 µM Pes St | VWR International GmbH | 514-0340 | |
| Mixer Mill MM 400 | Retsch GmbH | 207450001 | |
| IgG Sepharose 6 Fast Flow | GE Healthcare Life Sciences | 17-0969-02 | |
| Mobicol ""Classic"" with 2 different screw caps without filters | MoBiTec GmbH | M1002 | |
| Filter (small) 35 µM pore size, for Mobicol M 1002, M1003, M1050 & M1053 | MoBiTec GmbH | M513515 | |
| Variable Speed Tube Rotator SB 3 | Carl Roth | Y550.1 | |
| Rotary dishes for rotators SB 3 | Carl Roth | Y555.1 | |
| Resprep 24-Port SPE Manifolds | Restek | 26080 | |
| Finisterre C18/17% SPE Columns 100mg / 1ml | Teknokroma | TR-F034000 | |
| Autosampler Vials | Klaus Trott Chromatographie-Zubehör | 40 11 01 740 | |
| Acclaim PepMap 100 C18 LC Column | Thermo Fischer Scientific | 164534 | |
| EASY-nLC 1000 Liquid Chromatograph | Thermo Fischer Scientific | LC120 | |
| Q Exactive Plus Hybrid Quadrupole-Orbitrap Mass Spectrometer | Thermo Fischer Scientific | IQLAAEGAAPFALGMBDK | |
| Acquity UPLC system | Waters | Custom order | |
| ACQUITY UPLC HSS C18 Column, 100A, 1.8 µM, 2.1 mM X 100 mM, 1/pkg | Waters | 186003533 | |
| High-power ultrasonic cleaning baths for aqueous cleaning solutions | Bandelin | RK 31 | |
| Genedata Expressionist | Genedata | NaN | |
| Xcalibur Software | Thermo Fischer Scientific | NaN | |
| MaxQuant | NaN | NaN |
Referencias
- Li, X., Snyder, M. Metabolites as global regulators: A new view of protein regulation. Bioessays. 33 (7), 485-489 (2011).
- Jacob, F., Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. Journal of Molecular Biology. 3 (3), 318-356 (1961).
- Schlattner, U., et al. Dual Function of Mitochondrial Nm23-H4 Protein in Phosphotransfer and Intermembrane Transfer a cardiolipin-dependent switch. Journal of Biological Chemistry. 288 (1), 111-121 (2013).
- Ramírez, M. B., et al. GTP binding regulates cellular localization of Parkinson's disease-associated LRRK2. Human Molecular Genetics. , ddx161 (2017).
- Jung, H. J., Kwon, H. J. Target deconvolution of bioactive small molecules: the heart of chemical biology and drug discovery. Archives of Pharmacal Research. 38 (9), 1627-1641 (2015).
- Harding, M. W., Galat, A., Uehling, D. E., Schreiber, S. L. A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature. 341 (6244), 758-760 (1989).
- Lomenick, B., et al. Target identification using drug affinity responsive target stability (DARTS). Proceedings of the National Academy of Sciences of the United States of America. 106 (51), 21984-21989 (2009).
- Manabe, Y., Mukai, M., Ito, S., Kato, N., Ueda, M. FLAG tagging by CuAAC and nanogram-scale purification of the target protein for a bioactive metabolite involved in circadian rhythmic leaf movement in Leguminosae. Chemical Communications. 46 (3), 469-471 (2010).
- Pantoliano, M. W., et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. Journal of Biomolecular Screening. 6 (6), 429-440 (2001).
- Li, X., Snyder, M. Analyzing In vivo Metabolite-Protein Interactions by Large-Scale Systematic Analyses. Current Protocols in Chemical Biology. , 181-196 (2010).
- Maeda, K., Poletto, M., Chiapparino, A., Gavin, A. -. C. A generic protocol for the purification and characterization of water-soluble complexes of affinity-tagged proteins and lipids. Nature Protocols. 9 (9), 2256-2266 (2014).
- Li, X., Gianoulis, T. A., Yip, K. Y., Gerstein, M., Snyder, M. Extensive in vivo metabolite-protein interactions revealed by large-scale systematic analyses. Cell. 143 (4), 639-650 (2010).
- Veyel, D., et al. System-wide detection of protein-small molecule complexes suggests extensive metabolite regulation in plants. Scientific Reports. 7, (2017).
- Luzarowski, M., et al. Affinity purification with metabolomic and proteomic analysis unravels diverse roles of nucleoside diphosphate kinases. Journal of Experimental Botany. , (2017).
- Van Leene, J., et al. Targeted interactomics reveals a complex core cell cycle machinery in Arabidopsis thaliana. Molecular systems biology. 6 (1), 397 (2010).
- Giavalisco, P., et al. Elemental formula annotation of polar and lipophilic metabolites using 13C, 15N and 34S isotope labelling, in combination with high-resolution mass spectrometry. The Plant Journal. 68 (2), 364-376 (2011).
- Van Leene, J., et al. Isolation of transcription factor complexes from Arabidopsis cell suspension cultures by tandem affinity purification. Plant Transcription Factors: Methods and Protocols. , 195-218 (2011).
- Van Leene, J., et al. A tandem affinity purification-based technology platform to study the cell cycle interactome in Arabidopsis thaliana. Molecular & Cellular Proteomics. 6 (7), 1226-1238 (2007).
- Olsen, J. V., Ong, S. -. E., Mann, M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Molecular & Cellular Proteomics. 3 (6), 608-614 (2004).
- Cox, J., Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology. 26 (12), 1367-1372 (2008).
- Cox, J., et al. Andromeda: A peptide search engine integrated into the MaxQuant environment. Journal of Proteome Research. 10 (4), 1794-1805 (2011).
- Tyanova, S., Temu, T., Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols. 11 (12), 2301 (2016).
- Hooper, C. M., et al. SUBAcon: a consensus algorithm for unifying the subcellular localization data of the Arabidopsis proteome. Bioinformatics. 30 (23), 3356-3364 (2014).
- Katajamaa, M., Orešič, M. Data processing for mass spectrometry-based metabolomics. Journal of Chromatography A. 1158 (1-2), 318-328 (2007).
- Szklarczyk, D., et al. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Research. 1277, (2015).
- Tanaka, K. -. i., et al. Dipeptidyl compounds ameliorate the serum-deprivation-induced reduction in cell viability via the neurotrophin-activating effect in SH-SY5Y cells. Neurological Research. 34 (6), 619-622 (2012).
- Smith, C. A., et al. METLIN: A metabolite mass spectral database. Therapeutic Drug Monitoring. 27, 747-751 (2005).
- Bhattacharyya, S., et al. Transient protein-protein interactions perturb E. coli metabolome and cause gene dosage toxicity. Elife. 5, (2016).
- Rigaut, G., et al. A generic protein purification method for protein complex characterization and proteome exploration. Nature Biotechnology. 17 (10), 1030-1032 (1999).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados