
Method Article
2 en 1 : en une seule étape de Purification d’affinité pour l’analyse parallèle de protéine-protéine et protéine-métabolite Complexes
* Ces auteurs ont contribué à parts égales
Dans cet article
Résumé
Les interactions protéine-protéine et protéine-métabolite sont indispensables pour toutes les fonctions cellulaires. Ici, les auteurs décrivent un protocole qui permet une analyse parallèle de ces interactions avec une protéine de choix. Notre protocole a été optimisé pour les cultures de cellules végétales et combine purification d’affinité avec la protéine basé sur la spectrométrie de masse et de la détection de métabolite.
Résumé
Les processus cellulaires sont réglementés par des interactions entre molécules biologiques telles que des protéines, des métabolites et des acides nucléiques. Alors que l’étude des interactions protéine-protéine (PPI) n’est aucune nouveauté, approches expérimentales visant à caractériser les interactions protéine-métabolite endogène (PMI) constituent un développement assez récent. Ici, nous présentons un protocole qui permet la caractérisation simultanée de l’IPP et PMI d’une protéine de choix, dénommé comme appât. Notre protocole a été optimisé pour des Arabidopsis cell cultures et combine la purification d’affinité (AP) avec la spectrométrie de masse (MS)-fonction détection de protéines et de métabolites. En bref, les lignées transgéniques d’Arabidopsis, exprimant la protéine appât fusionnée à une balise d’affinité, sont tout d’abord lysées pour obtenir un extrait cellulaire natif. Anticorps anti-Tags servent à tirer vers le bas des partenaires protéiques et métabolite de la protéine de l’appât. Les complexes purifiés par affinité sont extraites à l’aide d’une seule étape méthyl tert-butyl éther (MTBE) / méthanol/eau méthode. Alors que les métabolites séparent dans le polaire ou la phase hydrophobe, protéines se trouvent dans le culot. Les métabolites et les protéines sont ensuite analysés par spectrométrie de masse par chromatographie liquide ultra performance (UPLC-MS ou UPLC-MS/MS). Lignes de contrôle vide-vector (EV) sont utilisés pour exclure des faux positifs. L’avantage majeur de notre protocole est qu’il permet d’identifier des partenaires protéiques et métabolite d’une protéine cible en parallèle dans des conditions physiologiques près (lysat cellulaire). La méthode présentée est simple, rapide et peut être facilement adaptée aux systèmes biologiques autres que les cultures de cellules végétales.
Introduction
La méthode décrite ici vise à l’identification des partenaires métabolite et protéine d’une protéine de choix dans des conditions de lysat cellulaire prochein vivo . Il a été spéculé que nombreux métabolites plus que caractérise aujourd'hui ont une importante fonction de réglementation1. Métabolites peuvent agir comme des auxiliaires biologiques, modification de l’activité, fonctionnalité, et/ou la localisation de leurs récepteurs protéines2,3,4. Dans la dernière décennie, plusieurs méthodes de percée, ce qui permet l’identification des PMI en vivo ou dans des conditions quasiin vivo , ont été développés5. Les approches disponibles peuvent être séparés en deux groupes. Le premier groupe comprend les techniques qui commencent avec un appât connu-métabolite afin d’emprisonner les partenaires de la nouvelle protéine. Méthodes incluent affinité chromatographie6drogues affinité cible sensible-stabilité dosage7, chimio-protéomique8et thermique proteome profilage9. Le deuxième groupe est constitué d’une seule méthode qui commence par une protéine connue afin d’identifier des ligands de petit-molécule10,11.
AP couplé avec lipidomique axée sur le MS a été utilisé pour analyser les complexes protéines-lipides dans Saccharomyces cerevisiae12. Comme point de départ, les auteurs ont utilisé des souches de levures exprimant 21 enzymes impliquées dans la biosynthèse de l’ergostérol et 103 kinases fusionnée à une purification d’affinité en tandem de tag (TAP). 70 % des enzymes et 20 % des kinases trouvées pour lier les différents ligands hydrophobes, éclairant dans le réseau d’interactions complexes protéines-lipides.
Auparavant, nous pourrions démontrer que, de la même façon aux lipides, composés polaires et semi polaires aussi restent liés à des complexes de protéine isolées de lysats cellulaires13. Se fondant sur ces constatations, nous avons décidé d’optimiser l’AP méthode publiée précédemment10,11 pour cellules végétales et des composés hydrophiles14. À cet effet, nous avons utilisé des vecteurs robinet décrites par Van lhonneux et al. 2010, utilisée avec succès dans l’usine PPI études15. Pour raccourcir le temps nécessaire pour obtenir des lignées transgéniques, nous avons décidé sur des cultures cellulaires Arabidopsis. Nous avons utilisé un one-step méthyl tert-butyl éther, (MTBE) / méthanol/eau méthode d’extraction, ce qui permet la caractérisation des protéines (pellet), lipides (phase organique) et métabolites hydrophile (phase aqueuse)16 dans une seule expérience de purification d’affinité. Les lignes de contrôle EV ont été introduites afin d’exclure des faux positifs, par exemple les protéines liant à la balise seule. Comme preuve de concept, nous avons marqué trois (sur cinq) kinase de diphosphate de nucléoside présents dans le génome d’Arabidopsis (NDPK1-NDPK3). Entre autres conclusions, nous pourrions démontrer que NDPK1 interagit avec le glutathion S-transférase et glutathion. Par conséquent, nous pourrions prouver que NDPK1 est soumis à glutathionylation14.
Pour résumer, le protocole présenté est un outil important pour caractériser les protéines et les réseaux d’interaction de protéine-petit-molécule et constitue une avancée majeure par rapport aux méthodes existantes.
Protocole
On trouvera dans17préparation des lignées transgéniques de Arabidopsis cell culture, y compris les conditions de clonage, transformation, sélection et la croissance. Notez que les lignes de contrôle EV sont recommandés pour corriger les faux positifs. Avant l’expérience, confirmer la surexpression de la protéine d’appât par analyse par western blot, par exemple en utilisant des anticorps IgG contre la partie de G-protéine de la balise d’affinité en tandem. Il est important de séparer les milieux de culture de matériel de culture de cellules végétales.
1. préparation du matériel cellulaire végétale avant l’expérience
- Cultiver une lignée de culture de cellules de PSB-L a. thaliana surexprimant la protéine d’intérêt18.
- Préparer le support MSMO, contenant 4,43 g/L que MSMO mélangé avec saccharose 30 g/L. Ajuster le pH de la mémoire tampon à 5,7 avec 1 M KOH et stériliser la solution. Avant l’expérience, compléter le milieu avec 0,5 mg/L d’acide α-naphtalène, 0,05 mg/L de kinétine et 50 kanamycine μg/mL.
- Cultiver des cultures de cellules végétales transformées dans 50 mL de milieu MSMO dans un ballon jaugé de 100 mL dans un agitateur orbital plate-forme avec agitation modérée (130 tr/min). La croissance de cellules dans une chambre de culture à 20 ° C et intensité lumineuse égale à 80 μmol m-2 s-1.
- Cellules de sous-culture dans les supports neufs tous les 7 jours, diluer 01:10.
- Prélever des cellules à la phase de croissance logarithmique à l’aide d’un entonnoir en verre combiné avec une pompe à vide, à l’aide d’une maille en nylon comme filtre. Envelopper l’infiltrat en papier d’aluminium et congeler dans l’azote liquide.
ATTENTION : N’oubliez pas que l’azote liquide est très froid. Toute manipulation incorrecte peut provoquer des brûlures. Porter un équipement de protection individuelle approprié, y compris thermiquement isolant gants, lunettes de protection et d’une blouse de laboratoire.
2. Tapez sur protocole
Remarque : L’étape suivante est adaptée de Maeda et al 201411 et Van lhonneux et coll. 201117.
- Homogénéiser les végétaux récoltés et congelés matériel de culture cellulaire à l’aide d’un mélangeur broyeur (2 min à 20 Hz) ou un mortier et pilon pour obtenir une poudre fine. Aliquote 3 g du matériel au sol (correspondant à environ 90 mg de protéine totale) par exemple. Éviter la décongélation de l’échantillon au cours de cette étape à l’aide d’azote liquide équipements préalablement réfrigérées.
Remarque : Magasin sol végétal en tube de 50 mL à – 80 ° C au début d’une procédure d’AP. - Broyer l’échantillon dans un mortier liquide-azote-prérefroidir avec 3 mL de tampon de lyse glacee (0,025 M Tris-HCl pH 7,5 ; 0,5 M de NaCl 1,5 mM MgCl20,5 mM TNT ; 1 mM NaF ; 1 mM Na3VO4; 100 x inhibiteur de la protéase commercial dilué cocktail ; 1 mM PMSF) jusqu'à ce que le matériau dégèle. Une fois l’échantillon dégèle, procéder immédiatement à l’étape suivante.
Remarque : Préparer le frais de tampon de lyse. Introduire les blancs à cette étape. Détergents ne sont pas recommandés car ils peuvent causer des problèmes chez MS détection. - Pour supprimer les débris cellulaires, diviser le matériau en tubes de microcentrifuge de 2 mL et centrifuger à 20 817 x g pendant 10 min à 4 ° C. Collecter 3 mL du lysat clair dans un tube à centrifuger conique de 15 mL.
- Au cours de l’étape de centrifugation, Equilibrer perles IgG-Sépharose. Aliquotes de 100 µL des perles par échantillon et les laver avec 1 mL de tampon de lyse. Vortex pour remettre en suspension de perles et flash-spin. Jeter le tampon de lyse et répétez l’étape deux fois. Remettre en suspension les perles dans 400 µL de tampon de lyse.
- Ajouter les perles dans la plante recueillie lysate et incuber le mélange sur une roue en rotation pendant 1 h à 4 ° C.
- Transférer le mélange dans une seringue combiné via bouchon Luer lock avec une colonne avec filtre pore taille 35 µm. Exercez une pression pour passer le lysat à travers. Perles avec des complexes ci-joint resteront sur le filtre, tandis que le lysat va passer.
Remarque : Utilisez un système collecteur d’aspiration. Assurez-vous d’appliquer une légère pression pour ne pas endommager les perles. - Laver les perles dans un premier temps avec 10 mL de tampon de lavage (0,025 M Tris-HCl pH 7,5 ; 0,5 M de NaCl) et puis avec 1 mL de tampon d’élution (pH 10 mM Tris-HCl 7,5 ; 150 mM NaCl, 0,5 mM EDTA, 1000 x dilué E64 et 1 mM PMSF). Effectuer le lavage à l’aide d’une seringue attachée à la colonne ou le collecteur d’aspiration.
Note : Quand à l’aide d’une tubulure d’aspiration s’assurer d’appliquer une légère pression pour ne pas endommager les perles. - Incuber les perles avec les 400 µL de tampon d’élution contenant 50 U d’une version améliorée du tabac etch protéase du virus (AcTEV). Utiliser le shaker table à 1 000 tr/min pendant 30 min à 16 ° C.
Remarque : N’oubliez pas d’utiliser un bouchon pour fermer la colonne en bas ajoutant de la mémoire tampon d’élution. - Ajouter une partie supplémentaire (50 U) de l’enzyme dans la colonne et incuber le mélange pendant 30 min suivantes selon les mêmes, les conditions décrites ci-dessus.
- Recueillir l’éluat dans un tube de microcentrifuge de 2 mL par collecteur sous vide ou par centrifugation (1 min, 20 817 x g). Pour supprimer les autres complexes, introduire une étape supplémentaire d’élution avec 200 µL de tampon d’élution.
Note : Conserver l’échantillon à – 20 ° C ou à – 80 ° C, ou immédiatement procéder à l’extraction de protéines et métabolite. Décongeler les échantillons congelés sur la glace.
3. immunobuvardage
- Pour confirmer la présence de l’appât protéique dans l’éluat recueillie utiliser 10 µL de l’éluat contenant des protéines – métabolite SDS-PAGE et analyse par western blot. Pour identifier la protéine d’intérêt, utilisez les anticorps primaires de souris contre la protéine de liaison streptavidine (1 : 200), une partie de la balise de robinet qui reste après clivage de protéase TEV, comme décrit dans Van lhonneux et coll. 201117. Utilisez ensuite les anticorps anti-souris de chèvre secondaire couplées à la HRP.
4. métabolite et Extraction de protéine
Remarque : Ce protocole est adapté de Giavalisco et coll. 201116.
Remarque : Cette étape a partir utiliser UPLC-MS – grade solutions.
- Ajouter 1 mL de méthyl tert-butyl éther (MTBE) / méthanol/eau solvant (3:1:1) de l’éluat collectée et mélanger l’échantillon en inversion. Assurez-vous que le solvant est refroidi à-20 ° C avant l’étape d’extraction.
ATTENTION : Le MTBE et le méthanol sont des substances nocives. Effectuez l’étape d’extraction sous la hotte aspirante et porter un équipement de protection individuelle approprié, p. ex., des gants. - Ajouter 0,4 mL de méthanol : eau solution 1:3 dans chaque échantillon et mélanger le contenu de l’échantillon par inversion.
Remarque : La supplémentation du mélange avec la solution de méthanol : eau se traduit par la séparation des phases. La phase supérieure contient des lipides, phase inférieure contient des métabolites polaires et semi polaires et les protéines se trouvent dans le culot. - Pour séparer les phases, centrifuger l’échantillon à 20 817 x g pendant 2 min à température ambiante, puis recueillir la phase supérieure pour les mesures de lipide (ne pas fait dans le présent protocole) aide d’une pipette de manutention de liquides avec une capacité de volume de 1 mL.
- Ajouter 0,2 mL de méthanol et mélanger par retournement.
- Centrifuger l’échantillon à 20 817 x g pendant 2 min à la droite, puis recueillir la phase polaire pour les mesures de métabolite (composés polaires et semi polaires). Pour éviter de déranger le culot de protéine, laissez environ 50 µL de la phase liquide au fond du tube.
- Échantillons secs pour les mesures de métabolite du jour au lendemain dans un évaporateur centrifuge. Éviter de trop sécher les pellets protéine en enlevant des échantillons de l’évaporateur après 30 à 60 min.
Remarque : Conserver les échantillons à – 20 ° C ou à – 80 ° C, ou immédiatement procéder à la préparation des protéines pour l’analyse de LC-MS/MS.
5. préparation des échantillons pour l’analyse protéomique
Remarque : Cette étape est une adaptation d’Olsen et al. 200419 et le manuel technique du Mix trypsine/Lys-C (voir Table des matières).
- Effectuer la digestion enzymatique de l’échantillon.
ATTENTION : Les solvants utilisés lors de la digestion enzymatique et dessalage de l’échantillon sont nocifs. Travailler sous la hotte aspirante et porter un équipement de protection individuelle approprié, p. ex., des gants.- Dissoudre le culot de protéine dans 30 µL de tampon de dénaturation fraîchement préparée (bicarbonate d’ammonium de 40 mM contenant de l’urée de thiourée/6 M 2 M, pH 8). Pour obtenir la meilleure solubilité de protéine, exécuter une étape de sonication 15 min. Répétez l’étape jusqu'à ce que le plomb se dissout.
- Centrifuger l’échantillon à 20 817 x g pendant 10 min à 4 ° C, puis transférer le surnageant dans un nouveau tube de microcentrifuge.
- Déterminer la concentration de protéines à l’aide de l’analyse de protéine de Bradford.
- Pour plus d’analyse, aliquote un volume équivalent à 100 µg de protéines et remplir l’échantillon jusqu'à 46 µL de tampon de dénaturation.
- Ajouter dans le µL de l’échantillon 2 de mémoire tampon de réduction fraîchement préparée (50 mM DTT dissous dans H2O) et incuber pendant 30 min à température ambiante.
- Traiter l’échantillon avec 2 µL de tampon d’alkylation fraîchement préparée (150 mM iodoacétamide dissoute dans un tampon bicarbonate d’ammonium 40 mM) et incuber le mélange dans l’obscurité pendant 20 min à température ambiante.
- Diluer l’échantillon avec 30 µL de tampon de bicarbonate d’ammonium de 40 mM et ajouter 20 µL du mélange LysC trypsine.
- Après 4 h d’incubation à 37 ° C, diluer l’échantillon avec 300 µL de tampon de bicarbonate d’ammonium de 40 mM.
- Continuer avec une nuit d’incubation à 37 ° C.
- Acidifier l’échantillon avec environ 20 µL de 10 % d’acide trifluoroacétique (TFA) obtention du pH < 2. Vérifier le pH d’échantillon à l’aide d’une bande de pH.
Note : Conserver l’échantillon à – 20 ° C ou passez à l’étape suivante.
- Dessalement des protéines digérées.
Remarque : Utilisez de préférence, une tubulure d’aspiration. Éviter de trop sécher de la colonne.- Rincer la colonne SPE C18 (voir Table des matières) avec 1 mL de 100 % MeOH et puis 1 ml de 80 % d’acétonitrile (ACN) contenant 0,1 % TFA dilué dans l’eau. Utilisation, ICIE et dans d’autres protéines-dessalage des étapes, un système de collecteur vide pour accélérer le processus. Éviter de trop sécher de la colonne.
- Equilibrer la colonne par laver deux fois avec 1 mL de 0.1 % TFA dilué dans l’eau.
- Charger l’échantillon sur la colonne. Tube de rinçage avec supplémentaires à 200 µL de 0,1 % TFA et transfert la solution sur la colonne. Exécuter les solutions par le biais de la colonne.
- Laver la colonne deux fois avec 1 mL de 0.1 % TFA.
- Éluer les peptides de morue dessalées de la colonne avec 800 µL de 60 % ACN, solution à 0,1 % TFA. Sécher la fraction recueillie dans un évaporateur centrifuge, en évitant trop de séchage de la fraction protéique en enlevant des échantillons de l’évaporateur après 30 à 60 min.
Remarque : Conserver les échantillons à – 20 ° C ou à – 80 ° C ou procéder immédiatement à l’étape suivante. Dans les étapes suivantes, conserver les échantillons sur la glace.
6. mesures a préparé des échantillons de protéine à l’aide de UPLC-MS/MS.
Remarque : Avant les mesures protéomique et métabolomique, filtrer (taille des pores de 0,2 µm) et dégazer tous les tampons à l’aide d’une pompe à vide pendant 1 h.
- Pellet de peptide remettre sec stocké dans tube de microcentrifuge de 2 mL dans 50 µL de tampon C (3 % v/v ACN, l’acide formique 0,1 % v/v) en utilisant pipette de manutention de liquides avec une capacité de volume 200 µL. Soniquer échantillons pour un 15 min dans un bain à ultrasons avec 35 kHz fréquence ultrasonique.
ATTENTION : ACN et l’acide formique sont des substances nocives. Travailler sous la hotte aspirante et porter un équipement de protection individuelle approprié, p. ex., des gants. - Centrifuger l’échantillon à 20 817 x g pendant 10 min à 4 ° C, puis transférer 20 µL du liquide surnageant dans un flacon de verre.
- Séparé peptides digérés à l’aide d’une colonne C18 à phase inverse relié à une chromatographie en phase liquide et acquièrent des spectres de masse à l’aide d’un spectromètre de masse.
- Séparer la colonne 3 µL de l’échantillon à l’aide d’un débit de 300-nl/min. Pour une phase mobile, utilisez un tampon C et D (63 % v/v ACN, l’acide formique 0,1 % v/v), formant une pente de montée en puissance de 3 % ACN à 15 % ACN pendant 20 min, puis à 30 % ACN au cours des 10 prochaine min.
Note : Conserver le reste de l’échantillon à – 20 ° C ou -80 ° C jusqu'à quelques mois. Avant la mesure de la protéomique, congeler de nouveau l’échantillon sur la glace. - Lavent les contaminants pendant 10 min à l’aide de 60 % ACN et équilibrer la colonne 5 µl de tampon C avant la mesure de l’échantillon suivant.
- Gain de spectres de masse à l’aide de la méthode MS/MS dépendant des données avec résolution fixé à 70 000, les AGC cible 3e6 ions, les temps d’injection maximal de 100 ms et un m/z allant de 300 à 1600. Acquérir un maximum de 15 MS/MS des analyses à une résolution de 17 500, les AGC cible de 1e,5, les temps d’injection maximal de 100 ms, ratio de sous-remplissage de 20 %, avec une fenêtre de l’isolement de 1,6 m/z et m/z allant de 200 à 2000. Permettent le déclencheur de l’apex (6 – 20 s), set exclusion dynamique à 15 s et excluent les frais de 1 et 5 >.
- Séparer la colonne 3 µL de l’échantillon à l’aide d’un débit de 300-nl/min. Pour une phase mobile, utilisez un tampon C et D (63 % v/v ACN, l’acide formique 0,1 % v/v), formant une pente de montée en puissance de 3 % ACN à 15 % ACN pendant 20 min, puis à 30 % ACN au cours des 10 prochaine min.
7. le traitement de données protéomiques
- Télécharger la base de données du protéome de Arabidopsis thaliana plus récent de http://www.uniprot.org/ et inclure la base de données de contaminant. Analyser des données brutes provenant de la LC-MS s’exécute à l’aide de MaxQuant avec le moteur de recherche de peptide Andromeda intégré à l’aide du paramètre par défaut avec permis LFQ normalisation20,21,22. Trouver des informations détaillées sur les paramètres utilisés dans le Tableau S1.
- Ouvrez le fichier de sortie « protéine groups.txt ». Pour une analyse ultérieure, filtrer les groupes de protéines identifiées au moins deux peptides uniques. Supprimez les groupes de protéines définies par MaxQuant comme potentielle de contaminants et de filtre pour les protéines Arabidopsis (ARATH dans la colonne d’en-têtes Fasta) présents dans la base de données.
- Pour tester l’importance de l’enrichissement en protéines entre les échantillons, utiliser des intensités de LFQ normalisé et effectuer des t-test de Student sur non appariés, bilatérale suivi de correction de comparaison multiple (par exemple Benjamini & Hochberg faux taux de découverte (FDR) correction ou correction de Bonferroni).
- Calculer valeur prédictive en comparant les intensités LFQ obtenues pour le contrôle de l’EV et NDPK1. Filtrer toutes les valeurs non déterminés. Trier les valeurs de p dans l’ordre croissant et utiliser R script ou la calculatrice en ligne (p. ex., https://www.sdmproject.com/utilities/?show=FDR), à calculer la correction des FDR. Filtrer les valeurs de FDR inférieures à 0,1.
Remarque : Considère la forme d’analyse de données approprié pour la recherche. Pour les études quantitatives (analyse de l’enrichissement en protéines entre les échantillons) utilisent la valeur « Intensité LFQ », alors que pour la recherche qualitative (présence ou absence d’une protéine particulière), choisissez la valeur « Intensité ». - Filtre pour les groupes de protéines qui sont plus abondantes dans les NDPK1 en comparant au contrôle de l’EV. Déterminer la localisation des partenaires potentiels de protéine à l’aide de la base de données SUBA23 et corriger la protéine localisée conjointement avec NDPK1.
- Calculer valeur prédictive en comparant les intensités LFQ obtenues pour le contrôle de l’EV et NDPK1. Filtrer toutes les valeurs non déterminés. Trier les valeurs de p dans l’ordre croissant et utiliser R script ou la calculatrice en ligne (p. ex., https://www.sdmproject.com/utilities/?show=FDR), à calculer la correction des FDR. Filtrer les valeurs de FDR inférieures à 0,1.
8. mesure des échantillons contenant Phase polaire à l’aide de UPLC-MS.
- Resuspendre séchée phase polaire d’étape 4.5 dans 200 µL d’eau et laisser agir l’échantillon pendant 5 min.
- Centrifuger l’échantillon à 20 817 x g pendant 10 min à 4 ° C, puis transférer le surnageant dans un flacon de verre.
Note : Conserver le reste de l’échantillon à – 20 ° C ou à – 80 ° C pendant plusieurs mois. Avant la mesure de la métabolomique, congeler de nouveau l’échantillon sur la glace. - Exécuter une étape de séparation à l’aide de UPLC couplé à une colonne C18 à phase inverse et acquisition de spectres de masse avec MS.
- Chargez à la colonne 2 de µL de l’échantillon par injection pour chaque mode d’ionisation (positif et négatif) et séparer la fraction à l’aide de 400 µL/min de débit. Pour créer la pente requise pour la mesure de métabolite, préparer la phase mobile solution comme suit : mettre en mémoire tampon un (acide formique 0,1 % H2O) et de la mémoire tampon B (0,1 % de l’acide formique chez ACN).
- Métabolites distincts à 400 µL/min et le gradient suivant : 1 min 99 % du tampon A, 11 min dégradé linéaire de 99 % des tampons A à 60 % de la mémoire tampon A, 13 min dégradé linéaire de 60 % des tampons A à 30 % de tampon A, 15 min de dégradé linéaire de 30 % de la mémoire tampon A à 1 % des tampons A, maintient la concentration de 1 % jusqu'à 16 min. à partir de 17 min, usage dégradé linéaire de 1 % des tampons A 99 % de tampon A. rééquilibrer la colonne pendant 3 min avec une concentration de 99 % des tampons A avant la mesure de l’échantillon suivant.
- Acquisition de spectres de masse couvrant la gamme de masses entre 100 et 1500 m/z avec résolution la valeur 25 000 et limité à 100 cible d’AGC mis Mme 1e6dans le temps de chargement, tension capillaire à 3kV avec une gaine gaz flux et valeur de gaz auxilliaire de 60 et 20 , respectivement. Régler température capillaire à 250 ° C et skimmer tension à 25V.
9. traitement des données de métabolomique
- Chromatogrammes de processus recueillies acquis auprès de ces deux modes d’ionisation. Utiliser un logiciel pour extraire de masse à rapport de charge (m/z), les temps de rétention (RT) et l’intensité des pics associés, par exemple, des logiciels commerciaux (voir la Table des matières) ou alternatif24.
- Démarrer le logiciel de traitement en double-cliquant sur le fichier .exe
- Créer de nouveaux flux de travail, recherchez l’activité « Load from File » et déplacer cette activité par « drag and drop » dans l’espace vide de flux de travail. Appuyez sur l’activité avec le bouton droit de la souris et ouvrir les paramètres de l’activité.
- Dans l’onglet contenant les paramètres du « Générales », définir un nom de l’expérience dans le champ « Nom » et ensuite cliquez sur « Select fichiers et dossiers » et marquez les chromatogrammes bruts.
- Dans l’onglet contenant les paramètres « Avancé », la valeur « Profil données Cutoff » intensité 0. Cliquez sur « Appliquer » et « OK ».
- Vous pouvez chercher et ajouter une activité « Données Sweep ». Appuyez sur l’activité avec le bouton droit de la souris et ouvrir les paramètres de l’activité.
- Dans l’onglet contenant les paramètres du « Générales », marque « Centroïde données » et « MS/MS ». Supprimer toutes les données de MS/MS en sélectionnant « Tout » dans le panneau de sélection.
- Vous pouvez chercher et ajouter l’activité « Chromatogramme soustraction de bruit chimique ». Appuyez sur l’activité avec le bouton droit de la souris et ouvrir les paramètres de l’activité.
- Dans l’onglet contenant les paramètres du « Générales », marque « Chromatogramme lissage » et définir nombre de scans à « 3 » et « Estimateur » à « Moyenne mobile ». La valeur « RT fenêtre » 51 scans, « Quantile » à 50 %, soustraction « Méthode » et 750 intensité « Seuil ».
- Dans l’onglet contenant les paramètres « Avancé », marque « RT Structure Removal » et affectez « Longueur minimale du RT » 5 scans.
- Dans l’onglet contenant les paramètres « Avancés », marquer « m/z Structure enlèvement » et « Minimum m/z longueur » de la valeur à 3 points.
- Vous pouvez chercher et ajouter l’activité « Chromatogramme RT alignement ». Appuyez sur l’activité avec le bouton droit de la souris et ouvrir les paramètres de l’activité.
- Dans l’onglet contenant les paramètres du « Générales », valeur « Alignement Scheme » à « Pairwise alignement arbre de Base » et « Interval de recherche RT » 0,5 min.
- Dans l’onglet contenant les paramètres « Avancé », utilisez les paramètres par défaut.
- Vous pouvez chercher et ajouter l’activité « Pics » en groupe « Chromatogramme » d’activités. Appuyez sur l’activité avec le bouton droit de la souris et ouvrir les paramètres de l’activité.
- Dans l’onglet contenant les paramètres du « Générales », la valeur « Sommation fenêtre » 0,09 min, « Taille de pointe Minimum » à 0,03 min, « Distance de fusion maximale » à 5 points et de la « Stratégie de fusionner » aux « Centres ». Dans le « pic RT Splitting » coffret « Rapport Gap/pics » à 50 %.
- Dans l’onglet contenant les paramètres « Avancé », la valeur « Lissage fenêtre » 5 points, le « Seuil de raffinement » à 80 % et le « Seuil de cohérence » à 1. Définie sur « Computation Center » « Pondérée en fonction de l’intensité » et « Seuil d’intensité » fixé à 70 %.
- Vous pouvez chercher et ajouter l’activité « Isotope Clustering » dans le groupe « Chromatogramme » des activités. Appuyez sur l’activité avec le bouton droit de la souris et ouvrir les paramètres de l’activité.
- Dans l’onglet contenant les paramètres du « Générales », définir « La tolérance RT » à 0,015 min et « m/z tolérance » à 5 ppm.
- Dans l’onglet contenant les paramètres « Montage enveloppe », définir « Méthode » comme « Aucune Restriction de forme » et « Ionisation » comme « Protonation (pour le mode positif) et « Déprotonation » (pour le mode négatif). La valeur « Minimum et Maximum de remplissage » 1 et 4, respectivement.
- Dans l’onglet contenant les paramètres « Avancé », utilisez les paramètres par défaut.
- Vous pouvez chercher et ajouter une activité « Singleton Filter ».
- Pour exporter les résultats du traitement des données, recherchez et ajoutez l’activité « Analyste » dans « Exporter » groupe d’activités.
- Dans l’onglet contenant les paramètres du « Générales », mettez « Type » comme « Grappes » et « Observables » comme « A résumé l’intensité ». Choisissez « Custom Destination » et indiquer le répertoire du fichier d’exportation.
- Dans l’onglet contenant les paramètres « Avancé », utilisez les paramètres par défaut.
- Annoter la massives caractéristiques utilisation interne composé de référence base de données.
- Analyser un ou plusieurs MS grade référence-composé UPLC-Mme utilisez la même méthode LC-MS pour l’analyse des composés de la référence et métabolites co purifiés avec la protéine d’intérêt.
Remarque : Pour cette étude, un ensemble de près de 300 dipeptides a été analysé et utilisé comme bibliothèque de référence du complexe. - Utiliser l’analyse (voir Table des matières) logiciel pour ouvrir fichier raw chromatogramme et recherche pour associées à m/z spécifique et RT mesuré composé de référence (voir le Guide de l’utilisateur).
Remarque : Les métabolites secondaires diffèrent dans types d’ionisation. Vérifier la présence des communs des adduits en recherchant des ions de masse égale à M-1.007276, de 1.007276 M + et M + 18.033823 M + 22.989218 [M-H], [M + H], [M + NH4] et [M + Na], respectivement. - Utilisation tableur pour ouvrir le fichier « Analyste » obtenu après transformation des chromatogrammes et recherche d’ion spécifique exporté massive. Comparer RT de la fonction de masse mesurée dans l’expérience et la RT de la substance de référence. Laissez un écart de 0,005 Da pour m/z et 0,1 min pour RT.
- Analyser un ou plusieurs MS grade référence-composé UPLC-Mme utilisez la même méthode LC-MS pour l’analyse des composés de la référence et métabolites co purifiés avec la protéine d’intérêt.
- Pour tester l’importance de l’enrichissement de métabolite co purifié avec une protéine particulière entre échantillons (ligne protéine surexprimée de contrôle vs EV intérêt), comparer les valeurs de crête à l’aide bilatérale non appariés de test t de Student suivi de multiples correction de comparaison (par exemple Benjamini & Hochberg découverte faux taux correction ou correction de Bonferroni).
Résultats
Dans l’étude originale, trois gènes d’Arabidopsis NDPK étaient surexprimés dans les cultures de suspension de cellules de PSB-L sous le contrôle du promoteur 35 s constitutif14 (Figure 1). Balise d’affinité en tandem a été fusionnée à une extrémité carboxy - ou amino-terminale d’une protéine de l’appât. Les complexes purifiés par affinité ont été soumis à l' extraction de MTBE/méthanol/eau16. Affinité-tiré de protéines et de petites molécules ont été identifiées à l’aide de MS (tableaux S2 et S3).
Pour corriger des faux positifs, échantillons blancs ont été utilisés pour exclure des contaminants de petites molécules des produits chimiques et consommables de laboratoire. En outre, métabolites et les protéines qui se lient à soit une balise d’affinité ou résine seul comptaient pour à l’aide de lignes de commande EV. Pour récupérer les vrais positifs, les t-test bilatéral non appariés de l’étudiant et les taux de fausse découverte Benjamini & Hochberg correction a été appliquée afin d’identifier des métabolites (Tableau S4) et protéines (Tableau S5) considérablement enrichis dans l’AP NDPK expériences (N - et C-terminale le tag NDPK) en comparaison avec les lignes de contrôle EV (FDR < 0,1). Notez que dans les travaux antérieurs, nous avons utilisé des critères absence/présence pour délimiter les Interactiens de protéines et de petites molécules.
Résultats représentatifs sont données pour NDPK1, tout en métabolite données mettant l’accent sur les dipeptides, une nouvelle classe des régulateurs petites molécules étudiées dans notre groupe. Analyse protéomique a révélé 26 partenaires de protéine putative de NDPK1. Par filtrage supplémentaire pour les protéines co localisés dans le même compartiment subcellulaire voir NDPK1 (cytosol), la liste réduite jusqu'à 13 Interactiens de protéine putative. Parmi les protéines identifiées étaient glutathion S-transférase, deux facteurs initiation, tubuline et aconitate hydratase. Métabolomique analyse a révélé quatre dipeptides Val-Leu, Ile-Glu, Leu-Ile et Ile-Phe qui spécifiquement co élué avec NDPK1 (Figure 2). Notez que tous les quatre dipeptides partagent un résidu hydrophobe dans leur extrémité N-terminale, ce qui suggère la spécificité de liaison partagée.
Chercher des complexes protéine-protéine et protéine-métabolite connus nous interrogé 13 identifiées protéines et quatre dipeptides contre le point de base de données25 (Figure 3). Plusieurs observations peuvent être faites : (i) aucun d'entre les Interactiens a été signalé auparavant pour NDPK1. (ii) APX1 orthologues a été signalé pour interagir avec l’aldéhyde déshydrogénase membre de la famille ALDH7B4, tout facteur d’initiation de traduction FBR12 avec un autre facteur d’initiation de traduction codé par le gène AT2G40290. (iii) les dipeptides identifiés ont rapporté non partenaires protéiques. Les dipeptides co éluées n’ont pas été signalés auparavant, associé à n’importe quelle protéine végétale récupérée. Cependant, ils jouent un rôle important dans d’autres organismes : Leu-Ile, par exemple, a un effet activateur neurotrophine dans une cellule humaine ligne26. Notez que l’expérience ne permet pas de déterminer la topologie exacte du système. Par exemple, un dipeptide peut-être interagir directement avec les NDPK1 mais peut bien être lié à l’une des protéines purifiées conjointement.
Pris ensemble, nos résultats montrent que la procédure établie, employant AP avec la spectrométrie de masse, facilite l’identification de protéine-protéine et protéine-petit-molécule Interactiens et contribue à générer des informations détaillées sur la interactome de la protéine cible.

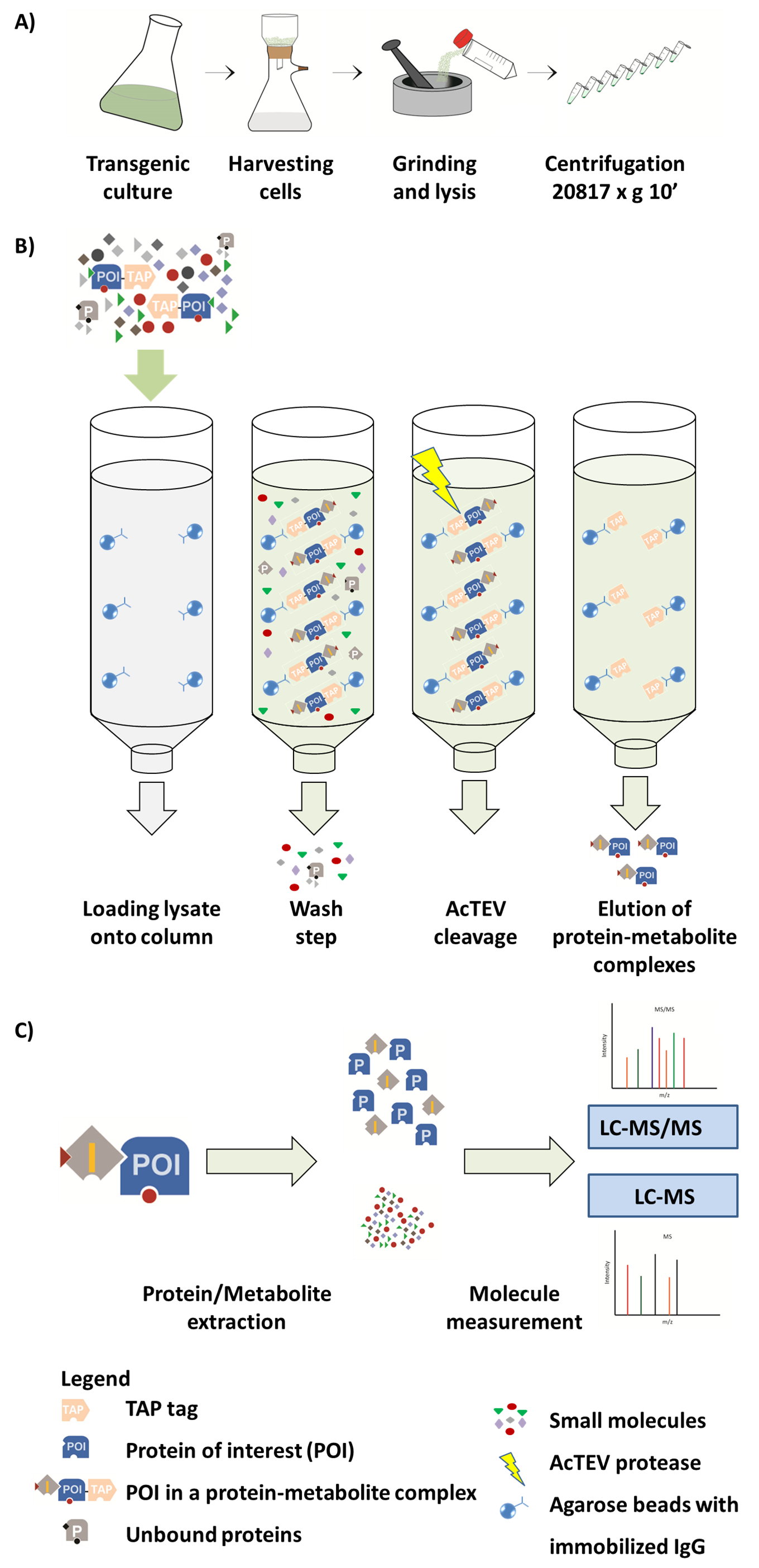
Figure 1. Schéma du flux de travail AP-MS. (A) préparation d’une fraction soluble native de culture de cellules végétales. (B) prochaines étapes dans la procédure de l’AP. Après le chargement de l’échantillon sur la colonne, la protéine d’intérêt (POI) fusionnée à une balise de robinet se lie à l’anticorps de type IgG immobilisés sur les billes de gel d’agarose. Lavage de la colonne facilite l’élimination des protéines non liées et de métabolites. Après avoir effectué un clivage AcTEV, complexes de protéine-métabolite POI sont éluées. (C) séparation des complexes en fraction de protéine et métabolite suivie d’analyse semi-quantitative de SM. Partie de cette Figure est reproduite de Luzarowski al 201714. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 2. Dipeptides spécifiquement à élution conjointement avec NDPK1. Les intensités moyennes de quatre dipeptides Val-Leu (A), Ile-Glu (B), (C)de la Leu-Ile et Ile-Phe (D) mesurée dans l’expérience de l’AP ont été tracées. Tous les quatre dipeptides montrent l’enrichissement significatif dans des échantillons de NDPK1 par rapport au témoin EV (astérisques représentent FDR < 0,1). Barres d’erreur représentent écart-type pour les 6 mesures (3 répétitions de N - et 3 de terminalement tag protéines). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 3. Réseau d’interactions de toutes les molécules à élution conjointement avec NDPK1, interrogé sur base de données de point tenant compte seulement précédent expérimentale et les preuves de base de données (confiance > 0,2). Plus de confiance envers indique des chances plus élevées de l’interaction et est calculé en fonction des données déposées. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
Tableau S1. MaxQuant sortie de table « parameters.txt ». Table inclut des valeurs de seuil pour identification et quantification, en plus d’informations sur les bases de données utilisées. S’il vous plaît cliquez ici pour télécharger ce fichier.
Tableau S2. Informations de MaxQuant sortie table « proteinGroups.txt ». Table contient une liste de tous les groupes de protéines identifiées, intensités et des informations supplémentaires telles que nombre de peptides uniques et score. S’il vous plaît cliquez ici pour télécharger ce fichier.
Tableau S3. Fichier de sortie contenant l’analyse des métabolites polaires. Table contient une liste de toutes les fonctionnalités de massives identifiées caractérisée par spécifique m/z, RT et intensité. S’il vous plaît cliquez ici pour télécharger ce fichier.
Tableau S4. Dipeptides trouvent dans des échantillons de l’AP dans les NDPK1, les NDPK2 ou les NDPK3 ont été utilisés comme appât. Dipeptides présents dans les échantillons blancs ont été exclus de la liste. Deux lignes indépendantes (taggés en soit N - ou C-terminale) pour chaque NDPK ont été exécutés en trois exemplaires. De Student t-tester et assurer la correction de la p-valeur à l’aide de Benjamini & méthode Hochberg ont servi à déterminer les partenaires interactor significativement enrichis de NDPK (FDR < 0,1). Donné est ΔRT calculé par rapport à la référence composés et Δppm en ce qui concerne le monoisotopique masse donnée dans Metlin27. S’il vous plaît cliquez ici pour télécharger ce fichier.
Table S5. Les protéines purifiées conjointement avec NDPK1. Deux lignes indépendantes (taggés en soit N - ou C-terminale) pour chaque NDPK ont été exécutés en trois exemplaires. De Student t-tester et assurer la correction de la p-valeur à l’aide de Benjamini & méthode Hochberg ont servi à déterminer les partenaires interactor significativement enrichis de NDPK (FDR < 0,1). S’il vous plaît cliquez ici pour télécharger ce fichier.
Discussion
Le protocole présenté permet une identification parallèle de PP et PM complexes d’une protéine cible. Du clonage aux résultats définitifs, l’expérience peut être complétée en aussi peu que 8 à 12 semaines. AP complète prend environ 4 à 6 h pour une série d’échantillons de 12 à 24, rendant notre protocole d’analyse de débit moyen.
Le protocole, tout en étant simple dans l’ensemble, a un certain nombre d’étapes critiques. (i) suffisamment de protéines d’entrée et des billes d’affinité est cruciale pour atteindre une plage dynamique de détection de métabolite. Lyse cellulaire efficace est donc une étape cruciale dans la procédure. Protéine pauvres rendements peuvent être une conséquence de pulvérisation insuffisante du matériel ou du ratio de lyse-tampon/matériel sous-optimale. (ii) il faut que les réactifs utilisés sont respectueux de MS. Détergents forts, glycérol ou des quantités excessives de sel doivent être évitées car ils interfèrent avec la détection de MS. (iii) l’agar-agar ne devrait pas être trop secs pendant les étapes de lavage, et lorsque vous utilisez une tubulure de vide, il est important d’appliquer un taux de débit lent afin de ne pas pour détruire les billes ou affecter la stabilité complexe.
Il y a quelques importantes modifications possibles au protocole présenté : (i) nous permet de maximiser la quantité de protéine appât le promoteur constitutif de CaMV35S. Surexpression, bien que très utile, peut avoir de graves effets sur la cellule homéostasie28 et conduisent à la formation des interactions physiologiquement non pertinentes. Expression de protéines étiquetées à l’aide de promoteurs natifs et où possible dans un contexte de perte de fonction est considéré comme supérieur afin de récupérer le vrais Interactiens biologiques. Pour les protéines normalement ne pas exprimés dans les cultures de cellules végétales, un fond de plantes peut-être s’avérer nécessaire pour identifier les Interactiens pertinentes. (ii) lorsque vous travaillez avec des protéines membranaires, le tampon de lyse doit être complété par un détergent MS-compatible. (iii) l’introduction d’une deuxième étape de purification d’affinité pourrait améliorer le nombre de faux positifs au ratio de true-positifs et éliminent la nécessité de contrôles de EV29. Une balise de roman en tandem avec deux sites de clivage protéase indépendant présente une alternative intéressante à l’étape de chromatographie liquide d’exclusion ajoutée par Maeda et al 201411, qui est laborieuse et chronophage.
Le plus grave inconvénient de l’AP est le taux élevé de faux positifs. Les raisons sont nombreuses. Surexpression constitutive a été déjà mentionnée. Une autre source d’interactions physiologiquement non pertinentes, à moins que travaillant avec des organites isolés, est préparation de mélanges de protéines et de métabolites des différents compartiments subcellulaires des lysats de cellules entières. Localisation sous-cellulaire devrait être utilisée pour filtrer les Interactiens vrais. Néanmoins, la majorité des faux positifs proviennent de liaison non-spécifique entre les protéines et les résines d’agarose. Introduction d’une deuxième étape de purification, comme décrit plus haut, offre la meilleure solution au problème, vient toutefois au détriment du temps et débit. En outre, plus faible interaction peut être perdue car le protocole s’allonge. Une autre restriction d’AP, c’est que, malgré les informations complètes qu'il fournit sur l’interactome d’une protéine cible, différencier les cibles directes et indirectes de la protéine appâtée est impossible. Les approches bimoléculaires ciblées sont nécessaires pour confirmer les interactions.
AP couplé avec métabolomique basée sur MS a permis d’étudier les protéines complexes dans S. cerevisiae12. Ce travail, ainsi que de notre précédente observation13 , que, de même aux lipides, les composés polaires et semi polaires restent liés à des complexes de protéine isolées de lysats cellulaires, a fourni les bases conceptuelles pour le protocole présenté. Notre protocole est caractérisée par trois points uniques : (i) en revanche à la levure travail12, il montre que l’AP est approprié pour récupérer non seulement les ligands protéine hydrophobe mais aussi hydrophile. (ii) en introduisant un protocole d’extraction de trois-en-un, un seul point d’accès peut servir à étudier les Interactiens de protéine et métabolite de la protéine de l’appât. (iii) nous avons adapté le protocole de cellules végétales.
Futurs efforts porteront sur la création d’une balise de roman en tandem avec deux sites de clivage protéase indépendant. Nous tenons également à explorer les qualités du protocole à la faible abondance de petites molécules telles que des hormones végétales.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous aimerions bien vouloir reconnaître Prof. Dr. Lothar Willmitzer pour son implication dans le projet, des discussions productives et grande surveillance. Nous sommes reconnaissants à m. Daniel Veyel pour aider avec les mesures de MS de protéomique. Nous remercions Mme Änne Michaelis qui nous a fourni une aide technique précieuse avec mesures LC-MS. En outre, nous tenons à remercier le Dr. Monika Kosmacz et Dr. Ewelina Sokołowska pour leur aide et leur implication dans le travail sur le manuscrit original et à Weronika Jasińska pour le support technique.
matériels
| Name | Company | Catalog Number | Comments |
| Murashige and Skoog Basal Salts with minimal organics | Sigma-Aldrich | M6899 | |
| 1-Naphthylacetic acid | Sigma-Aldrich | N1641 | |
| Kinetin solution | Sigma-Aldrich | K3253 | |
| Tris base | Sigma-Aldrich | 10708976001 | |
| NaCl | Sigma-Aldrich | S7653 | |
| MgCl2 | Carl Roth | 2189.1 | |
| EDTA | Sigma-Aldrich | 3609 | |
| NaF | Sigma-Aldrich | S6776 | |
| DTT | Sigma-Aldrich | D0632 | |
| PMSF | Sigma-Aldrich | P7626 | |
| E-64 protease inhibitor | Sigma-Aldrich | E3132 | |
| Protease Inhibitor Cocktail | Sigma-Aldrich | P9599 | |
| Na3VO4 | Sigma-Aldrich | S6508 | |
| AcTEV Protease | Thermo Fischer Scientific | 12575015 | |
| Rotiphorese Gel 30 (37,5:1) | Carl Roth | 3029.2 | |
| TEMED | Carl Roth | 2367.3 | |
| PageRuler Prestained Protein Ladder | Thermo Fischer Scientific | 26616 | |
| SBP Tag Antibody (SB19-C4) | Santa Cruz Biotechnology | sc-101595 | |
| Goat anti-mouse IgG-HRP | Santa Cruz Biotechnology | sc-2005 | |
| Bradford Reagent | Sigma-Aldrich | B6916 | |
| Trypsin/Lys-C Mix, Mass Spec Grade | Promega | V5071 | |
| Urea | Sigma-Aldrich | U5128 | |
| Thiourea | Sigma-Aldrich | T8656 | |
| Ammonium bicarbonate | Sigma-Aldrich | 9830 | |
| Iodoacetamide | Sigma-Aldrich | I1149 | |
| MTBE | Biosolve | 138906 | |
| Methanol | Biosolve | 136806 | |
| Water | Biosolve | 232106 | |
| Acetonitrile | Biosolve | 12006 | |
| Trifluoroacetic acid | Biosolve | 202341 | |
| Formic acid | Biosolve | 69141 | |
| Unimax 2010 Platform Shaker | Heidolph | 5421002000 | |
| Nylon Mesh (Wire diameter 34 µM, thickness 55 µM, open area 14%) | Prosepa | Custom order | |
| Glass Funnel, 47 mm, 300 ml | Restek | KT953751-0000 | |
| Filter Bottle Top 500 mL 0,2 µM Pes St | VWR International GmbH | 514-0340 | |
| Mixer Mill MM 400 | Retsch GmbH | 207450001 | |
| IgG Sepharose 6 Fast Flow | GE Healthcare Life Sciences | 17-0969-02 | |
| Mobicol ""Classic"" with 2 different screw caps without filters | MoBiTec GmbH | M1002 | |
| Filter (small) 35 µM pore size, for Mobicol M 1002, M1003, M1050 & M1053 | MoBiTec GmbH | M513515 | |
| Variable Speed Tube Rotator SB 3 | Carl Roth | Y550.1 | |
| Rotary dishes for rotators SB 3 | Carl Roth | Y555.1 | |
| Resprep 24-Port SPE Manifolds | Restek | 26080 | |
| Finisterre C18/17% SPE Columns 100mg / 1ml | Teknokroma | TR-F034000 | |
| Autosampler Vials | Klaus Trott Chromatographie-Zubehör | 40 11 01 740 | |
| Acclaim PepMap 100 C18 LC Column | Thermo Fischer Scientific | 164534 | |
| EASY-nLC 1000 Liquid Chromatograph | Thermo Fischer Scientific | LC120 | |
| Q Exactive Plus Hybrid Quadrupole-Orbitrap Mass Spectrometer | Thermo Fischer Scientific | IQLAAEGAAPFALGMBDK | |
| Acquity UPLC system | Waters | Custom order | |
| ACQUITY UPLC HSS C18 Column, 100A, 1.8 µM, 2.1 mM X 100 mM, 1/pkg | Waters | 186003533 | |
| High-power ultrasonic cleaning baths for aqueous cleaning solutions | Bandelin | RK 31 | |
| Genedata Expressionist | Genedata | NaN | |
| Xcalibur Software | Thermo Fischer Scientific | NaN | |
| MaxQuant | NaN | NaN |
Références
- Li, X., Snyder, M. Metabolites as global regulators: A new view of protein regulation. Bioessays. 33 (7), 485-489 (2011).
- Jacob, F., Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. Journal of Molecular Biology. 3 (3), 318-356 (1961).
- Schlattner, U., et al. Dual Function of Mitochondrial Nm23-H4 Protein in Phosphotransfer and Intermembrane Transfer a cardiolipin-dependent switch. Journal of Biological Chemistry. 288 (1), 111-121 (2013).
- Ramírez, M. B., et al. GTP binding regulates cellular localization of Parkinson's disease-associated LRRK2. Human Molecular Genetics. , ddx161 (2017).
- Jung, H. J., Kwon, H. J. Target deconvolution of bioactive small molecules: the heart of chemical biology and drug discovery. Archives of Pharmacal Research. 38 (9), 1627-1641 (2015).
- Harding, M. W., Galat, A., Uehling, D. E., Schreiber, S. L. A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature. 341 (6244), 758-760 (1989).
- Lomenick, B., et al. Target identification using drug affinity responsive target stability (DARTS). Proceedings of the National Academy of Sciences of the United States of America. 106 (51), 21984-21989 (2009).
- Manabe, Y., Mukai, M., Ito, S., Kato, N., Ueda, M. FLAG tagging by CuAAC and nanogram-scale purification of the target protein for a bioactive metabolite involved in circadian rhythmic leaf movement in Leguminosae. Chemical Communications. 46 (3), 469-471 (2010).
- Pantoliano, M. W., et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. Journal of Biomolecular Screening. 6 (6), 429-440 (2001).
- Li, X., Snyder, M. Analyzing In vivo Metabolite-Protein Interactions by Large-Scale Systematic Analyses. Current Protocols in Chemical Biology. , 181-196 (2010).
- Maeda, K., Poletto, M., Chiapparino, A., Gavin, A. -. C. A generic protocol for the purification and characterization of water-soluble complexes of affinity-tagged proteins and lipids. Nature Protocols. 9 (9), 2256-2266 (2014).
- Li, X., Gianoulis, T. A., Yip, K. Y., Gerstein, M., Snyder, M. Extensive in vivo metabolite-protein interactions revealed by large-scale systematic analyses. Cell. 143 (4), 639-650 (2010).
- Veyel, D., et al. System-wide detection of protein-small molecule complexes suggests extensive metabolite regulation in plants. Scientific Reports. 7, (2017).
- Luzarowski, M., et al. Affinity purification with metabolomic and proteomic analysis unravels diverse roles of nucleoside diphosphate kinases. Journal of Experimental Botany. , (2017).
- Van Leene, J., et al. Targeted interactomics reveals a complex core cell cycle machinery in Arabidopsis thaliana. Molecular systems biology. 6 (1), 397 (2010).
- Giavalisco, P., et al. Elemental formula annotation of polar and lipophilic metabolites using 13C, 15N and 34S isotope labelling, in combination with high-resolution mass spectrometry. The Plant Journal. 68 (2), 364-376 (2011).
- Van Leene, J., et al. Isolation of transcription factor complexes from Arabidopsis cell suspension cultures by tandem affinity purification. Plant Transcription Factors: Methods and Protocols. , 195-218 (2011).
- Van Leene, J., et al. A tandem affinity purification-based technology platform to study the cell cycle interactome in Arabidopsis thaliana. Molecular & Cellular Proteomics. 6 (7), 1226-1238 (2007).
- Olsen, J. V., Ong, S. -. E., Mann, M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Molecular & Cellular Proteomics. 3 (6), 608-614 (2004).
- Cox, J., Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology. 26 (12), 1367-1372 (2008).
- Cox, J., et al. Andromeda: A peptide search engine integrated into the MaxQuant environment. Journal of Proteome Research. 10 (4), 1794-1805 (2011).
- Tyanova, S., Temu, T., Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols. 11 (12), 2301 (2016).
- Hooper, C. M., et al. SUBAcon: a consensus algorithm for unifying the subcellular localization data of the Arabidopsis proteome. Bioinformatics. 30 (23), 3356-3364 (2014).
- Katajamaa, M., Orešič, M. Data processing for mass spectrometry-based metabolomics. Journal of Chromatography A. 1158 (1-2), 318-328 (2007).
- Szklarczyk, D., et al. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Research. 1277, (2015).
- Tanaka, K. -. i., et al. Dipeptidyl compounds ameliorate the serum-deprivation-induced reduction in cell viability via the neurotrophin-activating effect in SH-SY5Y cells. Neurological Research. 34 (6), 619-622 (2012).
- Smith, C. A., et al. METLIN: A metabolite mass spectral database. Therapeutic Drug Monitoring. 27, 747-751 (2005).
- Bhattacharyya, S., et al. Transient protein-protein interactions perturb E. coli metabolome and cause gene dosage toxicity. Elife. 5, (2016).
- Rigaut, G., et al. A generic protein purification method for protein complex characterization and proteome exploration. Nature Biotechnology. 17 (10), 1030-1032 (1999).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.