
Method Article
2 in 1: purificazione di affinità per l'analisi parallela di proteina-proteina e proteina-metabolita complessi in un unico passaggio
* Questi autori hanno contribuito in egual misura
In questo articolo
Riepilogo
Interazioni proteina-proteina e proteina-metabolita sono cruciali per tutte le funzioni cellulari. Qui, descriviamo un protocollo che permette l'analisi parallela di queste interazioni con una proteina di scelta. Il nostro protocollo è stato ottimizzato per colture di cellule vegetali e combina purificazione di affinità con proteina basati sulla spettrometria di massa e rilevamento di metabolita.
Abstract
Processi cellulari sono regolati dalle interazioni tra molecole biologiche come proteine e metaboliti acidi nucleici. Mentre l'indagine sulle interazioni proteina-proteina (PPI) è una novità, approcci sperimentali con l'obiettivo di caratterizzare le interazioni della proteina-metabolita endogeno (PMI) costituiscono uno sviluppo piuttosto recente. Qui, presentiamo un protocollo che consente la simultanea caratterizzazione del PPI e PMI di una proteina di scelta, di cui come esca. Il nostro protocollo è stato ottimizzato per Arabidopsis cell culture e combina la purificazione di affinità (AP) con spettrometria di massa (MS)-base di rilevazione della proteina e del metabolita. In breve, linee transgeniche di Arabidopsis, che esprimono la proteina esca fusa a un tag di affinità, in primo luogo vengono lisate per ottenere un estratto cellulare nativo. Anticorpi anti-tag sono usati per tirare giù partner della proteina e del metabolita della proteina esca. I complessi purificato per affinità vengono estratti mediante un One-Step metil tert-butil etere (MTBE) / metanolo/acqua metodo. Mentre metaboliti separano in polare o la fase idrofoba, proteine possono essere trovate nel pellet. Entrambi i metaboliti e proteine vengono poi analizzate tramite spettrometria della cromatografia-massa liquida ultra-prestazioni (UPLC-MS o UPLC-MS/MS). Linee di controllo di vuoto-vector (EV) vengono utilizzati per escludere falsi positivi. Il vantaggio principale del nostro protocollo è che esso consente l'identificazione dei partner della proteina e del metabolita di una proteina dell'obiettivo in parallelo in condizioni di quasi-fisiologiche (lisato cellulare). Il metodo proposto è semplice, veloce e può essere facilmente adattato a sistemi biologici diversi da colture di cellule vegetali.
Introduzione
Il metodo qui descritto mira all'identificazione di partner metabolita e proteina di una proteina di scelta in condizioni di lisato cellulare vicinoin vivo . Si è ipotizzato che molti metaboliti più che caratterizza oggi hanno un' importante funzione regolatrice1. Metaboliti possono fungere da interruttori biologici, cambiando l'attività, la funzionalità e/o la localizzazione di loro recettore proteine2,3,4. Nell'ultimo decennio diversi metodi di innovazione, che consente di identificare delle PMI in vivo o in condizioniin vivo vicino, sono stati sviluppati5. Approcci disponibili possono essere separati in due gruppi. Il primo gruppo comprende tecniche che iniziano con un noto-metabolita esca per intrappolare partner nuova proteina. Metodi includono cromatografia di affinità6, droga affinità reattivo destinazione-stabilità test7, chemio-proteomica8e proteoma termica9di profilatura. Il secondo gruppo è costituito da un singolo metodo che inizia con una proteina nota per identificare piccole molecole leganti10,11.
AP accoppiato con lipidomica basati su MS è stato utilizzato per analizzare i complessi proteina-lipide in Saccharomyces cerevisiae12. Come punto di partenza, gli autori hanno usato i ceppi di lievito che esprimono 21 enzimi coinvolti nella biosintesi dell'ergosterolo e 103 chinasi fuse con una purificazione di affinità tandem (TAP) tag. 70% degli enzimi e il 20% delle chinasi sono stati trovati per associare diversi ligandi idrofobici, mettendo in luce la rete di interazione proteina-lipide intricati.
In precedenza, potremmo dimostrare che, similmente ai lipidi, composti polari e semi-anche rimangano legati a complessi della proteina isolati dai lisati cellulari13. Basato su questi risultati, abbiamo deciso di ottimizzare l'AP metodo precedentemente pubblicato10,11 tra cellule vegetali e composti idrofili14. Per questo scopo, abbiamo utilizzato vettori TAP descritti da Van Leene et al. 2010, utilizzato con successo nella pianta PPI studi15. Per accorciare il tempo necessario per ottenere linee transgeniche, abbiamo deciso su colture cellulari di Arabidopsis. Abbiamo impiegato un One-Step metil tert-butil etere (MTBE) / metanolo/acqua estrazione metodo, permettendo la caratterizzazione delle proteine (pellet), lipidi (fase organica) e metaboliti idrofili (fase acquosa)16 in una singola esperimento di purificazione di affinità. Linee di controllo di EV sono stati introdotti per escludere falsi positivi, ad esempio proteine leganti al tag da solo. Come prova di concetto siamo taggati tre (di cinque) delle chinasi del nucleoside difosfato presentano nel genoma di Arabidopsis (NDPK1-NDPK3). Tra altri reperti, potremmo dimostrare che NDPK1 interagisce con il glutatione S-transferasi e del glutatione. Di conseguenza potremmo dimostrare che NDPK1 è sottoposto a glutationilazione14.
Per riassumere, il protocollo presentato è un importante strumento per la caratterizzazione di proteine e reti di interazione della proteina-piccolo-molecola e costituisce un importante passo in avanti rispetto ai metodi esistenti.
Protocollo
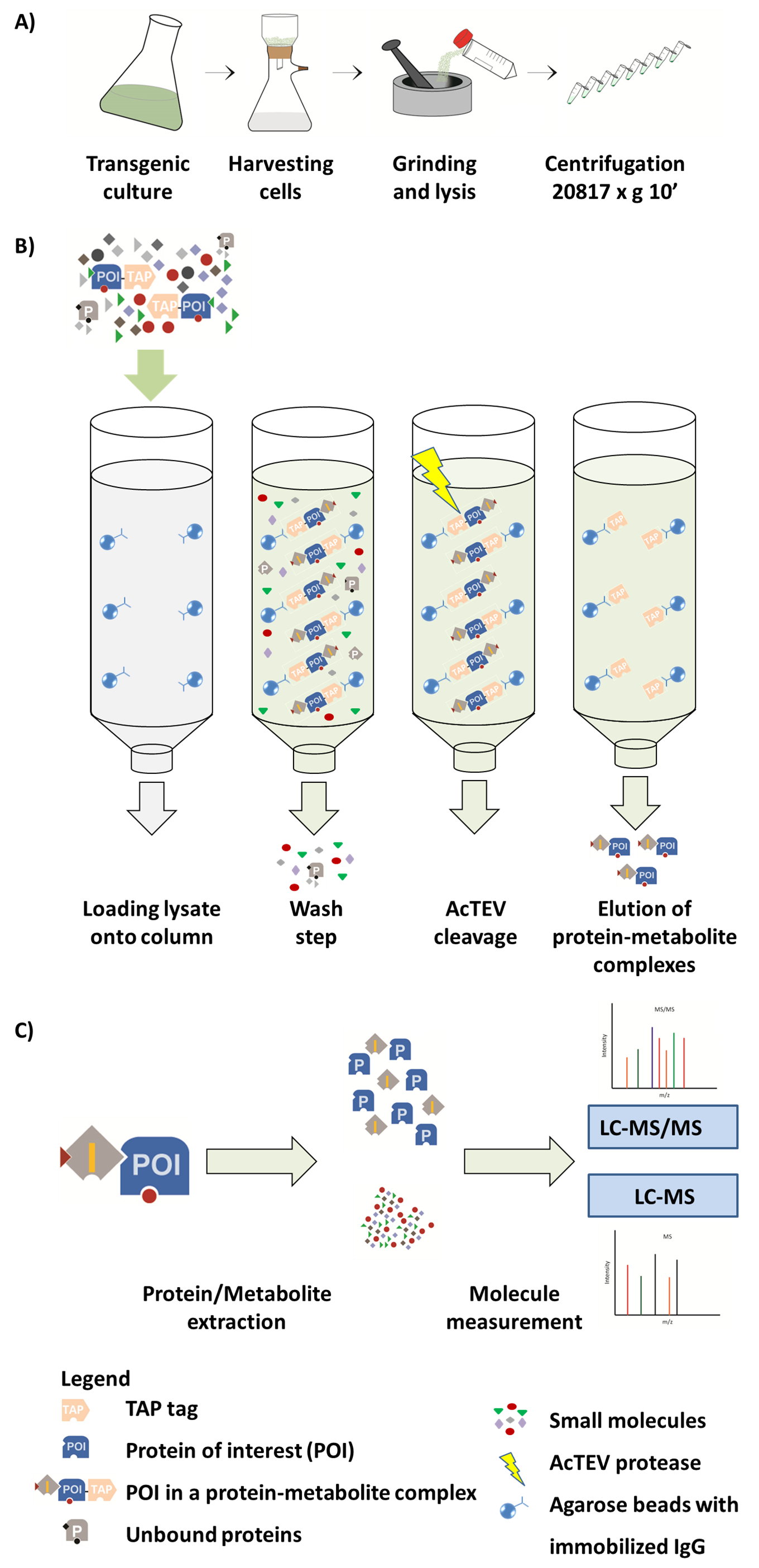
Preparazione di linee transgeniche Arabidopsis cella cultura, comprese le condizioni di clonazione, trasformazione, selezione e crescita può essere trovato in17. Si noti che le linee di controllo EV sono raccomandato per correggere per i falsi positivi. Prima dell'esperimento, è necessario confermare la sovraespressione della proteina esca da analisi western blot, per esempio usando gli anticorpi IgG contro la parte G-proteina del tag affinità tandem. È importante separare il media di crescita da materiale vegetale cella cultura.
1. preparazione di materiale cellulare vegetale prima dell'esperimento
- Crescere una linea di cultura cellulare PSB-L a. thaliana che overexpressing la proteina di interesse18.
- Preparare mezzo MSMO, che contiene 4,43 g/L che msmo mescolato con saccarosio 30 g/L. Regolare il pH del tampone a 5,7 con 1m KOH ed autoclave la soluzione. Prima dell'esperimento, integrare il mezzo con 0,5 mg/L di acido α-naftaleneacetico, chinetina 0,05 mg/L e 50 μg/mL kanamicina.
- Coltivare colture di cellule vegetali trasformati in 50 mL di terreno MSMO in un matraccio da 100 mL su agitatore orbitale piattaforma con movimentazione delicata (130 giri/min). Crescere le cellule in una camera di cultura a 20 ° C e intensità luminosa pari a 80 µmol m-2 s-1.
- Celle di sottocultura in media fresco ogni 7 giorni, diluendoli 01:10.
- Raccogliere le cellule nella fase di crescita logaritmica, utilizzando un imbuto di vetro combinato con una pompa a vuoto, utilizzando una rete di nylon come filtro. Avvolgere l'infiltrato nella carta stagnola e il congelamento in azoto liquido.
Attenzione: Tenere presente che l'azoto liquido è estremamente freddo. Gestione non corretta può causare ustioni. Indossare adeguati dispositivi di protezione personale, tra cui termicamente isolati, guanti, occhiali protettivi e un camice da laboratorio.
2. Toccare il protocollo
Nota: Il passaggio seguente è adattato da Maeda et al 201411 e Van Leene et al. 201117.
- Omogeneizzare raccolte e congelate cella cultura materiale vegetale utilizzando un Vibromulino (2 min a 20 Hz) o mortaio e pestello per ottenere polvere fine. Aliquotare 3G del materiale macinato (corrispondente a circa 90 mg di proteine totali) per campione. Evitare lo scongelamento del campione durante questo passaggio utilizzando attrezzature pre-refrigerati di azoto liquido.
Nota: Archivio terra materiale vegetale in tubo da 50 mL a-80 ° C all'inizio di una procedura di AP. - Triturare il campione in un mortaio liquido-azoto-preraffreddata con 3 mL di tampone di lisi ghiacciata (0,025 M Tris-HCl pH 7.5; 0.5 M NaCl; 1,5 mM MgCl2; 0,5 mM DTT; 1 mM NaF; 1 mM Na3VO4; 100 x cocktail l'inibitore della proteasi commerciale diluito; 1 mM PMSF) fino a quando il materiale si scioglie. Una volta che il campione si scioglie, procedere immediatamente alla fase successiva.
Nota: Preparare il fresco di tampone di lisi. Introdurre i campioni bianchi in questa fase. Detergenti non sono raccomandati come possono causare problemi nella rilevazione di MS. - Per rimuovere i detriti cellulari, dividere il materiale in provette microcentrifuga da 2 mL e centrifugare a 20.817 x g per 10 min a 4 ° C. Raccogliere 3 mL del chiaro lisato in una provetta conica per centrifuga 15 mL.
- Durante la centrifugazione, equilibrare IgG-Sepharose perline. Aliquotare 100 µ l dei branelli per campione e lavarli con 1 mL di tampone di lisi. Vortice per risospendere perline e flash-spin. Gettare il tampone di lisi e ripetere il passaggio due volte. Risospendere perline in 400 µ l di tampone di lisi.
- Aggiungere perline pianta raccolta lisata e incubare la miscela su un disco rotante per 1 h a 4 ° C.
- Trasferire il composto in una siringa combinato tramite tappo Luer-lock con una colonna di spin con filtro poro dimensioni 35 µm. applica pressione per passare il lisato attraverso. Perline con annesso complessi rimarrà sul filtro, mentre il lisato passerà attraverso.
Nota: Facoltativamente, utilizzare un impianto a collettori sottovuoto. Assicurarsi di applicare una leggera pressione per non danneggiare le perline. - Lavare le perle in un primo momento con 10 mL di tampone di lavaggio (0,025 M Tris-HCl pH 7.5; 0.5 M NaCl) e quindi con 1 mL di tampone di eluizione (10 mM Tris-HCl a pH 7.5; 150 mM NaCl, 0,5 mM EDTA; 1000 x diluito E64 e 1 mM PMSF). Eseguire il lavaggio utilizzando una siringa collegata alla colonna o al sistema collettore sottovuoto.
Nota: Quando utilizzo un impianto a collettori sottovuoto accertarsi di applicare una leggera pressione per non danneggiare le perline. - Incubare le perline con il 400 µ l di tampone di eluizione contenente 50 U di una versione migliorata del tabacco etch proteasi virus (AcTEV). Utilizzare la tabella shaker a 1.000 giri/min per 30 min a 16 ° C.
Nota: Ricordarsi di utilizzare un tappo per chiudere la colonna nella parte inferiore aggiungendo il tampone di eluizione. - Aggiungere una porzione extra (50 U) dell'enzima nella colonna e incubare la miscela per il prossimo 30 min sotto lo stesso, le condizioni descritte sopra.
- Raccogliere l'eluato in un tubo del microcentrifuge 2 mL mediante centrifugazione (1 min, 20.817 x g) o collettore sottovuoto. Per rimuovere i restanti complessi, introdurre un passaggio di eluizione aggiuntive utilizzando 200 µ l di tampone di eluizione.
Nota: Conservare il campione a-20 ° C o -80 ° C, o immediatamente procedere con la fase di estrazione della proteina e del metabolita. Scongelare i campioni congelati sul ghiaccio.
3. Western Blot analisi
- Per confermare la presenza dell'esca proteica nell'eluito raccolto utilizzare 10 µ l dell'eluato contenente proteine – metabolita per eseguire SDS-PAGE e analisi western blot. Per identificare la proteina di interesse, utilizzare contro streptavidina-legante della proteina (1: 200), una parte del tag del rubinetto restanti dopo la scissione di proteasi TEV, come descritto in Van Leene et al. 201117gli anticorpi primari del mouse. Successivamente, utilizzare anticorpi anti-topo di capra secondario accoppiati con HRP.
4. metabolita ed estrazione della proteina
Nota: Questo protocollo è adattato da Giavalisco et al. 201116.
Nota: Da questo punto in poi utilizzare soluzioni UPLC-MS – grado.
- Aggiungere 1 mL di metil tert-butil etere (MTBE) / metanolo/acqua solvente (3:1:1) per l'eluato raccolto e mescolare il campione di inversione. Assicurarsi che il solvente è raffreddato a-20 ° C prima della fase di estrazione.
Attenzione: MTBE e metanolo sono sostanze nocive. Eseguire il passaggio di estrazione sotto la cappa e indossare indumenti protettivi appropriati, ad esempio, guanti. - Aggiungere 0,4 mL di metanolo: acqua soluzione 1:3 per ogni campione e miscelare il contenuto del campione di inversione.
Nota: Il completamento della miscela con soluzione di metanolo: acqua provoca in fase di separazione. La fase superiore contiene lipidi, fase inferiore contiene metaboliti polari e semi-polari e proteine possono essere trovate nel pellet. - Per separare le fasi, centrifugare il campione a 20.817 x g per 2 min a temperatura ambiente, quindi raccogliere la fase superiore per le misure del lipido (non fatto in questo protocollo) usando una pipetta manuale dei liquidi con capacità di volume di 1 mL.
- Aggiungere 0,2 mL di metanolo e mescolare capovolgendo.
- Centrifugare il campione a 20.817 x g per 2 min a RT, quindi raccogliere la fase polare per misure di metabolita (polar e semi-composti). Per evitare di disturbare il pellet di proteina, lasciare circa 50 µ l della fase liquida nella parte inferiore del tubo.
- Campioni raccolti secchi per misurazioni di metabolita pernottamento in un evaporatore centrifugo. Evitare sovra i pellet di proteina rimuovendo campioni dall'evaporatore dopo 30-60 min.
Nota: Conservare i campioni a – 20 ° C o -80 ° C, o immediatamente procedere con la preparazione di proteine per analisi LC-MS/MS.
5. preparazione dei campioni per analisi proteomica
Nota: Questo passaggio è adattato da Olsen et al. 200419 e il manuale tecnico del Mix tripsina/Lys-C (Vedi Tabella materiali).
- Eseguire la digestione enzimatica del campione.
Attenzione: Solventi utilizzati durante la digestione enzimatica e dissalazione del campione sono nocivi. Lavorare sotto la cappa e indossare indumenti protettivi appropriati, ad esempio, guanti.- Dissolva la pallina di proteina in 30 µ l di tampone di denaturazione preparata al momento (bicarbonato d'ammonio 40 mM contenenti 2M Tiourea/6 M urea, pH 8). Per raggiungere maggiore solubilità della proteina, eseguire un passo 15-min sonicazione. Ripetere il passaggio fino a quando la pallina si dissolve.
- Centrifugare il campione a 20.817 x g per 10 min a 4 ° C, quindi trasferire il surnatante ad un nuovo tubo del microcentrifuge.
- Calcolare la concentrazione di proteina mediante l'analisi della proteina di Bradford.
- Per ulteriori analisi, aliquotare un volume equivalente a 100 µ g di proteina e riempire il campione fino a 46 µ l di tampone di denaturazione.
- Aggiungere il campione 2 µ l di tampone di riduzione preparata al momento (50 mM DTT disciolto in H2O) e incubare per 30 min a temperatura ambiente.
- Trattare il campione con 2 µ l di tampone di alchilazione preparata al momento (150mm iodoacetamide sciolto in soluzione tampone del bicarbonato di ammonio 40 mM) e incubare la miscela al buio per 20 min a temperatura ambiente.
- Diluire il campione con 30 µ l di tampone di bicarbonato di ammonio di 40 mM e aggiungere 20 µ l di LysC/tripsina Mix.
- Dopo 4 ore di incubazione a 37 ° C, è necessario diluire il campione con 300 µ l di tampone di bicarbonato di ammonio di 40 mM.
- Continuare con una notte di incubazione a 37 ° C.
- Acidificare il campione con circa 20 µ l di 10% acido trifluoroacetico (TFA) ottenere pH < 2. Controllare il pH del campione usando una striscia di pH.
Nota: Conservare il campione a-20 ° C o procedere al passaggio successivo.
- Dissalare le proteine digerite.
Nota: Utilizzare preferibilmente, un impianto a collettori sottovuoto. Evitare l'eccessivo essiccamento della colonna.- Lavare la colonna SPE C18 (Vedi Tabella materiali) con 1 mL di 100% MeOH e quindi con 1 mL di 80% acetonitrile (ACN) contenente 0,1% TFA diluito in acqua. Uso, qui e in ulteriori proteine-dissalazione passaggi, un impianto a collettori sottovuoto per accelerare il processo. Evitare l'eccessivo essiccamento della colonna.
- Equilibrare la colonna di lavaggio due volte con 1 mL di 0,1% TFA diluito in acqua.
- Caricare il campione sulla colonna. Tubo di risciacquo con ulteriori 200 µ l di 0,1% TFA e trasferire la soluzione nella colonna. Eseguire le soluzioni attraverso la colonna.
- Lavare la colonna due volte con 1 mL di 0,1% TFA.
- Eluire dissalate peptidi dalla colonna con 800 µ l di 60% ACN, soluzione 0,1% TFA. Asciugare la frazione raccolta in un evaporatore centrifugo, evitando sovra della frazione proteica rimuovendo campioni dall'evaporatore dopo 30-60 min.
Nota: Conservare i campioni a – 20 ° C o -80 ° C o immediatamente procedere al passaggio successivo. Nei passaggi successivi, mantenere i campioni su ghiaccio.
6. misura preparati campioni proteici utilizzando UPLC-MS/MS.
Nota: Prima di misurazioni di proteomica e metabolomica, filtrare (dimensione dei pori di 0,2 µm) e degassare tutti i buffer utilizzando una pompa a vuoto per 1 h.
- Pellet di peptide risospendere secchi memorizzati nel tubo del microcentrifuge 2 mL in 50 µ l di tampone C (3% v/v ACN, 0.1% v/v acido formico) utilizzando pipetta manuale dei liquidi con capacità di 200 µ l volume. Sonicare campioni per un 15 min in un bagno ad ultrasuoni con frequenza ultrasuoni 35 kHz.
Attenzione: ACN e acido formico sono sostanze nocive. Lavorare sotto la cappa e indossare indumenti protettivi appropriati, ad esempio, guanti. - Centrifugare il campione a 20.817 x g per 10 min a 4 ° C, poi il trasferimento di 20 µ l del surnatante di un flaconcino di vetro.
- Peptidi digeriti separati utilizzando una colonna a fase inversa C18 collegato ad una cromatografia liquida e acquisiscono spettri di massa utilizzando uno spettrometro di massa.
- Separare la colonna 3 µ l del campione utilizzando una portata di 300-nl/min. Per una fase mobile, utilizzare buffer C e D (63% v/v ACN, 0.1% v/v acido formico), formando una pendenza rampa dal 3% ACN al 15% ACN oltre 20 min e poi al 30% ACN sopra il prossimo 10 min.
Nota: Conservare il resto del campione a-20 ° C o -80 ° C fino a pochi mesi. Prima misurazione di proteomica, ricongelare il campione sul ghiaccio. - Lavare contaminanti per 10 min utilizzando il 60% ACN ed equilibrare la colonna con 5 µ l di tampone C prima misurazione del campione successivo.
- Acquisire spettri di massa utilizzando dati dipendente dal metodo di MS/MS con risoluzione impostata a 70.000, AGC target di 3e6 ioni, tempo di iniezione massima di 100 ms e un m/z che vanno da 300 a 1600. Acquisire il massimo di 15 MS/MS scansione a una risoluzione di 17.500, destinazione AGC di 1e5, tempo di iniezione massima di 100 ms, rapporto underfill del 20%, con una finestra di isolamento di 1,6 m/z e m/z da 200 a 2000. Abilitare il trigger di apice (6 – 20 s), esclusione set dinamico per 15 s e le tasse di esclusione di 1 e > 5.
- Separare la colonna 3 µ l del campione utilizzando una portata di 300-nl/min. Per una fase mobile, utilizzare buffer C e D (63% v/v ACN, 0.1% v/v acido formico), formando una pendenza rampa dal 3% ACN al 15% ACN oltre 20 min e poi al 30% ACN sopra il prossimo 10 min.
7. trattamento dei dati di proteomica
- Scaricare il database più recente del proteoma Arabidopsis thaliana da http://www.uniprot.org/ e includere database di contaminante. Analizzare dati grezzi ottenuti da LC-MS viene eseguita mediante MaxQuant con il motore di ricerca dei peptidi Andromeda integrato utilizzando impostazione predefinita con abilitato LFQ normalizzazione20,21,22. Informazioni dettagliate sui parametri utilizzati nella Tabella S1.
- Aprire il file di output "groups.txt della proteina". Per ulteriori analisi, filtrare per gruppi di proteine identificati con almeno due peptidi unici. Rimuovere i gruppi di proteine definiti da MaxQuant come potenziali contaminanti e filtro per proteine di a. thaliana (Gabriella nella colonna intestazioni Fasta) presenti nel database.
- Per verificare il significato di arricchimento di proteine tra campioni, utilizzare LFQ normalizzato intensità ed eseguire il test t di Student spaiati, due code seguita da correzione di confronto più (ad es. Benjamini & Hochberg false discovery rate (FDR) correzione o la correzione di Bonferroni).
- Calcolare il p-valore confrontando intensità LFQ ottenuti per il controllo di EV e NDPK1. Filtrare tutti i valori indeterminati. P-valori in ordine crescente di ordinare e utilizzare script R o calcolatore online (ad esempio, https://www.sdmproject.com/utilities/?show=FDR) per calcolare la correzione di FDR. Filtro per valori FDR sotto 0.1.
Nota: Considerare la forma di analisi dei dati adatto per la ricerca. Per gli studi quantitativi (analisi di arricchimento di proteine tra campioni) utilizzano il valore di "IFQ intensità", mentre per la ricerca qualitativa (presenza o assenza di particolare proteina) scegliere il valore di "Intensità". - Filtro per i gruppi di proteine che sono più abbondanti nel NDPK1 rispetto al controllo di EV. Determinare la localizzazione di potenziali partner di proteina utilizzando il database SUBA23 e correggere per proteina co-localizzata con NDPK1.
- Calcolare il p-valore confrontando intensità LFQ ottenuti per il controllo di EV e NDPK1. Filtrare tutti i valori indeterminati. P-valori in ordine crescente di ordinare e utilizzare script R o calcolatore online (ad esempio, https://www.sdmproject.com/utilities/?show=FDR) per calcolare la correzione di FDR. Filtro per valori FDR sotto 0.1.
8. misura di campioni contenenti fase polare utilizzando UPLC-MS.
- Risospendere secchi fase polare da passo 4.5 in 200 µ l di acqua e Sonicare il campione per 5 min.
- Centrifugare il campione a 20.817 x g per 10 min a 4 ° C, quindi trasferire il surnatante in un flaconcino di vetro.
Nota: Conservare il resto del campione a-20 ° C o -80 ° C per fino a parecchi mesi. Prima misurazione metabolomica, ricongelare il campione sul ghiaccio. - Eseguire un passaggio di separazione tramite UPLC accoppiato alla colonna in fase inversa C18 e acquisire spettri di massa con MS.
- Caricare sulla colonna 2 µ l del campione per iniezione per ogni modalità di ionizzazione (positive e negative) e separare la frazione utilizzando 400 µ l/min di portata. Per creare il gradiente necessario per la misurazione del metabolita, preparare la fase mobile soluzione come segue: un (acido formico 0.1% in H2O) nel buffer e buffer B (0,1% acido formico in ACN).
- Metaboliti separate a 400 µ l/min e la pendenza seguente: 1 min 99% del buffer A, 11-min sfumatura lineare dal 99% di tampone al 60% del buffer A, 13-min sfumatura lineare dal 60% di tampone al 30% del buffer A, 15 min sfumatura lineare dal 30% di buffer A 1% di tampone A, tenere premuto 1% di concentrazione fino a 16 min a partire da 17 min, uso sfumatura lineare dall'1% del tampone al 99% di tampone A. riequilibrare la colonna per 3 min con concentrazione di 99% di tampone A prima misurazione del campione successivo.
- Acquisire spettri di massa che copre la gamma di massa compresa tra 100 e 1500 m/z con risoluzione impostata su 25.000 e caricamento tempo limitato a 100 ms. target di AGC impostato a 1e6, tensione capillare a 3kV con una guaina gas flusso e valore di gas ausiliario di 60 e 20 , rispettivamente. Impostare temperatura capillare alla tensione di 250 ° C e skimmer a 25V.
9. trattamento dei dati di metabolomica
- Processo raccolti cromatogrammi acquisiti da entrambe le modalità di ionizzazione. Utilizzare software per estrarre messa a rapporto di carica (m/z), tempo di ritenzione (RT) e l'intensità dei picchi associati, ad esempio, software commerciale (Vedi Tabella materiali) o alternativi24.
- Avviare il software di elaborazione facendo doppio clic il file .exe
- Creare il nuovo flusso di lavoro, ricerca per attività "Carica da File" e spostare questa attività di "drag and drop" nello spazio vuoto del flusso di lavoro. Premere l'attività con il pulsante destro del mouse e aprire le impostazioni dell'attività.
- Nella scheda contenente le impostazioni di "General", impostare un nome dell'esperimento nel campo "Nome" e fare clic su Avanti "selezionare file e cartelle" e segnare cromatogrammi crudi.
- Nella scheda contenente le impostazioni "Advanced", impostare "Profilo dati Cutoff" intensità 0. Fare clic su "Applica" e "OK".
- Cercare e aggiungere attività "Sweep di dati". Premere l'attività con il pulsante destro del mouse e aprire le impostazioni dell'attività.
- Nella scheda contenente le impostazioni di "Generale", contrassegnare "Centroide dati" e "Dati di MS/MS". Rimuovere tutti i dati di MS/MS selezionando "Tutto" nel pannello di selezione.
- Cercare e aggiungere l'attività "Cromatogramma chimico rumore sottrazione". Premere l'attività con il pulsante destro del mouse e aprire le impostazioni dell'attività.
- Nella scheda contenente le impostazioni di "Generale", contrassegnare "Cromatogramma Smoothing" e impostare il numero di scansioni a "3" e "Stimatore" e "Media mobile". Impostare "RT finestra" 51 scansioni, "Quantile" al 50%, sottrazione "Metodo" e intensità 750 "Soglia".
- Nella scheda contenente le impostazioni "Advanced", contrassegnare "RT struttura rimozione" e impostare "Lunghezza minima RT" su 5 scansioni.
- Nella scheda contenente le impostazioni di "Avanzate", contrassegnare "m/z struttura rimozione" e impostare "Minimo m/z lunghezza" a 3 punti.
- Cercare e aggiungere l'attività "Cromatogramma RT allineamento". Premere l'attività con il pulsante destro del mouse e aprire le impostazioni dell'attività.
- Nella scheda contenente le impostazioni di "Generale", impostare "Schema di allineamento" a "Pairwise allineamento Base Tree" e "Intervallo di ricerca RT" a 0,5 min.
- Nella scheda contenente le impostazioni "Advanced", utilizzare i parametri predefiniti.
- Cercare e aggiungere l'attività "Rilevamento di picco" nel gruppo "Cromatogramma" delle attività. Premere l'attività con il pulsante destro del mouse e aprire le impostazioni dell'attività.
- Nella scheda contenente le impostazioni di "General", impostare "Sommatoria finestra" a 0,09 min, "Dimensione di picco minima" a 0,03 min, "Unire strategia" ai "Centri" e "Massima distanza Merge" a 5 punti. Nella "Picco RT spaccare" impostare "Gap/Peak Ratio" al 50%.
- Nella scheda contenente le impostazioni "Advanced", è possibile impostare "Smoothing finestra" a 5 punti, "Raffinatezza soglia" all'80% e "consistenza" a 1. Impostare "Centro calcolo" come "Intensità-weighted" con "Soglia di intensità" fissato al 70%.
- Cercare e aggiungere l'attività "Isotopo Clustering" in gruppo "Cromatogramma" delle attività. Premere l'attività con il pulsante destro del mouse e aprire le impostazioni dell'attività.
- Nella scheda contenente le impostazioni di "General", impostare "RT tolleranza" a min 0,015 e "m/z tolleranza" a 5 ppm.
- Nella scheda contenente le impostazioni di "raccordo busta", impostare "Metodo" come "Forma di limitazione" e "Ionizzazione" come "protonazione (per modalità positiva) e"Deprotonazione"(per modalità negativa). Impostare "minimo e massimo di carica" a 1 e 4, rispettivamente.
- Nella scheda contenente le impostazioni "Advanced", utilizzare i parametri predefiniti.
- Cercare e aggiungere attività "Singleton filtro".
- Per esportare i risultati del trattamento dei dati, cercare e aggiungere l'attività "Analista" in gruppo "Esportazione" di attività.
- Nella scheda contenente le impostazioni di "General", impostare "Tipo" come "Cluster" e "Osservabile" come "Ha riassunto intensità". Scegliere "Custom destinazione" e specificare la directory del file di esportazione.
- Nella scheda contenente le impostazioni "Advanced", utilizzare i parametri predefiniti.
- Annotare la massa caratteristiche utilizzando database interno composto di riferimento.
- Analizzare uno o più MS grado composto di riferimento utilizzando UPLC-MS. uso lo stesso metodo di LC-MS per l'analisi dei composti di riferimento e metaboliti co-purificati con la proteina di interesse.
Nota: Per questo studio, un insieme di quasi 300 dipeptidi è stato analizzato e utilizzato come composto di riferimento libreria. - Utilizzare l'analisi (Vedi Tabella materiali) software per aprire file raw cromatogramma e ricerca per specifiche m/z e RT associato misurato composto di riferimento (Vedi manuale).
Nota: Metaboliti secondari diversi tipi di ionizzazione. Verifica per la presenza del comune addotti ricercando per ion massa uguale a M-1.007276, 1.007276 M +, M + 18.033823 e M + 22.989218 [M-H], [M + H], [M + NH4] e [M + Na], rispettivamente. - Foglio di calcolo di uso per aprire file di "Analista" ottenuto dopo la trasformazione di cromatogrammi e ricerca di ioni specifici esportato massa. Confronta RT della caratteristica massa misurata nell'esperimento e RT il composto di riferimento. Consentire una deviazione di Da 0,005 per m/z e 0,1 min per RT.
- Analizzare uno o più MS grado composto di riferimento utilizzando UPLC-MS. uso lo stesso metodo di LC-MS per l'analisi dei composti di riferimento e metaboliti co-purificati con la proteina di interesse.
- Per verificare il significato di arricchimento di metabolita co-purificato con particolare proteina tra i campioni (linea con proteina overexpressed di controllo vs EV di interesse), confrontare i valori di picco usando due code non associati test t di Student-seguita da molteplici correzione di confronto (ad es. Benjamini & Hochberg false discovery rate correzione o la correzione di Bonferroni).
Risultati
Nello studio originale, tre geni di a. thaliana NDPK overexpressed in colture di sospensioni cellulari PSB-L sotto il controllo del promotore 35S costitutiva14 (Figura 1). Tag di affinità tandem è stato fuso a due estremità carbossi - o amminico-terminale di una proteina esca. I complessi purificato per affinità sono stati sottoposti a MTBE/metanolo/acqua estrazione16. Affinità-tirato proteine e piccole molecole sono stati identificati usando MS (tabelle S2 e S3).
Per correggere errori di falsi positivi, campioni bianchi sono stati utilizzati per escludere le sostanze contaminanti della piccolo-molecola da prodotti chimici e consumabili per laboratorio. Inoltre, metaboliti e proteine che legano sia un tag di affinità o resina da solo sono state contabilizzate utilizzando linee di controllo EV. Per recuperare i veri positivi, due code non associati Student t-test e tasso di falsi scoperta Benjamini & Hochberg correzione è stata applicata per identificare metaboliti (Tabella S4) e proteine (Tabella S5) arricchiti in modo significativo l'AP NDPKs esperimenti (N - e C-terminali contrassegnati NDPKs) in confronto le linee di controllo EV (FDR < 0,1). Si noti che nel lavoro precedente, abbiamo usato i criteri di presenza/assenza di delineare interattori della proteina e della piccolo-molecola.
Risultati rappresentativi sono dato per NDPK1, mentre il metabolita dati focus su dipeptidi, una nuova classe di regolatori della piccolo-molecola studiata nel nostro gruppo. Analisi proteomica ha rivelato 26 partner della proteina presunta di NDPK1. Applicando un filtro ulteriore per proteine co-localizzati nello stesso contenitore subcellulare NDPK1 (citosol), l'elenco ristretto giù per 13 interattori della proteina presunta. Tra le proteine identificate erano glutatione S-transferasi, fattori di inizio due allungamento, tubulina e aconitato idratasi. Analisi metabolomica ha rivelato quattro dipeptidi Val-Leu, Ile-Glu, Leu-Ile e Ile-Phe che specificamente co-eluiti con NDPK1 (Figura 2). Si noti che tutti i quattro dipeptidi condividono un residuo idrofobico in loro N-terminale, suggerendo la specificità di legame comune.
Per cercare noti complessi proteina-proteina e proteina-metabolita abbiamo interrogato 13 identificate proteine e quattro dipeptidi contro il Stitch database25 (Figura 3). Potrebbero essere fatto diverse osservazioni: (i) nessuna degli Interactiani precedentemente è stata segnalata per NDPK1. (ii) APX1 ortholog è stato segnalato per interagire con aldeide deidrogenasi membro della famiglia ALDH7B4, mentre il fattore di inizio di traduzione FBR12 con un altro fattore di inizio di traduzione codificato dal gene AT2G40290. (iii) i dipeptidi identificati non sono segnalati partner proteici. Co-eluiti dipeptidi non sono stati segnalati in precedenza come associati a qualsiasi proteina vegetale estratto. Tuttavia, essi svolgono i ruoli importanti in altri organismi: Leu-Ile, per esempio, ha un effetto d'attivazione neurotrophin in una linea cellulare umana26. Si noti che l'esperimento non consente di identificare l'esatta topologia del sistema. Ad esempio, un dipeptide può interagire direttamente con NDPK1 ma ben può essere collegato a qualsiasi delle proteine co-purificate.
Presi insieme, i nostri risultati indicano che la procedura stabilita, che impiegano AP insieme a spettrometria di massa, facilita l'identificazione di interattori della proteina-proteina e proteina-piccolo-molecola e aiuta a genera informazioni esaustive circa la Interactoma della proteina bersaglio.

Figura 1. Schema di flusso di lavoro AP-MS. (A) preparazione di una frazione solubile nativa da colture cellulari vegetali. (B) passaggi successivi della procedura di AP. Dopo aver caricato il campione sulla colonna, la proteina di interesse (PDI) fuso a un tag di rubinetto si lega all'anticorpo di IgG immobilizzati sulle perle di agarosio. Lavaggio della colonna facilita la rimozione delle proteine non associati e metaboliti. Dopo aver eseguito AcTEV fenditura, complessi proteina-metabolita POI vengono eluite. (C) separazione dei complessi nella frazione della proteina e del metabolita, seguita da analisi semi-quantitativa di MS. Questa figura è riprodotta da Luzarowski et al 201714. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2. Dipeptidi in particolare co-eluizione con NDPK1. Intensità media di quattro dipeptidi Val-Leu (A), Ile-Glu (B), Leu-Ile (C)e Ile-Phe (D) misurato in esperimento di AP sono state tracciate. Tutti i quattro dipeptidi Visualizza arricchimento significativo nei campioni di NDPK1 rispetto al controllo di EV (asterischi rappresentano FDR < 0,1). Barre di errore rappresentano errore standard per 6 misure (3 replicati del N - e 3 di C-terminale etichettato proteine). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3. Rete di interazione di tutte le molecole co-eluizione con NDPK1, eseguire una query su database STITCH considerando solo precedente sperimentali ed evidenze di database (fiducia > 0,2). Confidence più elevato indica una maggiore probabilità di interazione e viene calcolato in base ai dati depositati. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
Tabella S1. MaxQuant uscita tabella "parameters.txt". Tabella include i valori di soglia per identificazione e quantificazione, così come informazioni sui database utilizzati. Per favore clicca qui per scaricare questo file.
Tabella S2. Informazioni da MaxQuant uscita tabella "proteinGroups.txt". Tabella contiene un elenco di tutti i gruppi di proteine identificate, intensità e ulteriori informazioni quali numero di peptidi unici e punteggio. Per favore clicca qui per scaricare questo file.
Tabella S3. File di output contenente analisi di metaboliti polari. Tabella contiene un elenco di tutte le caratteristiche di massa identificati caratterizzata da specifiche m/z, RT e intensità. Per favore clicca qui per scaricare questo file.
Tabella S4. Dipeptidi trovano in campioni di AP in cui NDPK1, NDPK2 o NDPK3 sono stati usati come esca. Dipeptidi presenti in campioni bianchi sono stati esclusi dall'elenco. Due linee indipendenti (contrassegnati in entrambi N - o C-terminale) per ogni NDPK sono stati eseguiti in triplice copia. Dello studente t-test e ulteriore correzione del p-valore utilizzando Benjamini & Hochberg metodo sono stati usati per determinare significativamente arricchito interactor partner di NDPKs (FDR < 0,1). Dato ΔRT viene calcolato in relazione ai composti di riferimento e Δppm in relazione monoisotopic massa dato in Metlin27. Per favore clicca qui per scaricare questo file.
Tabella S5. Le proteine co-purificate con NDPK1. Due linee indipendenti (contrassegnati in entrambi N - o C-terminale) per ogni NDPK sono stati eseguiti in triplice copia. Dello studente t-test e ulteriore correzione del p-valore utilizzando Benjamini & Hochberg metodo sono stati usati per determinare significativamente arricchito interactor partner di NDPKs (FDR < 0,1). Per favore clicca qui per scaricare questo file.
Discussione
Il protocollo presentato permette l'identificazione parallela di PP e PM complessi di una proteina dell'obiettivo. Dalla clonazione ai risultati finali, l'esperimento può essere completato in appena 8-12 settimane. Completa AP prende circa 4-6 h per un set di 12 a 24 campioni, rendendo il nostro protocollo adatto per analisi di rendimento medio.
Il protocollo, pur essendo nel complesso semplice, ha un numero di passaggi critici. (i) sufficienti quantità di proteina input e branelli di affinità è cruciale per raggiungere una gamma dinamica di rilevamento del metabolita. Lisi cellulare efficiente sono quindi un passo cruciale nella procedura. Proteina poveri rendimenti possono essere una conseguenza dell'insufficiente polverizzazione del materiale o di non ottimale rapporto di lisi-buffer/materiale. (ii) dovrebbe prestare attenzione che i reagenti utilizzati sono MS-friendly. Detergenti aggressivi, glicerolo o una quantità eccessiva di sale dovrebbe essere evitata come interferiscono con rilevazione di MS. (iii) dell'agarosi branelli non dovrebbero essere eccessivamente secchi durante fasi di lavaggio, e quando si utilizza un collettore sottovuoto è importante applicare un tasso di flusso lento così come non per distruggere i branelli o influenzare la stabilità del complesso.
Ci sono alcune importanti modifiche possibili al protocollo presentato: (i) usiamo il promotore CaMV35S costitutivo per massimizzare la quantità di proteina esca. Sovraespressione, mentre molto utile, può avere gravi effetti sulla cella omeostasi28 e portano alla formazione di interazioni fisiologicamente irrilevante. Espressione di proteine etichettate utilizzando promotori nativi e dove possibile in una priorità bassa di perdita-de-funzione è considerata superiore per il recupero veri Interactiani biologici. Per le proteine non normalmente espresse in colture di cellule vegetali, una priorità bassa della pianta può rivelarsi necessaria per identificare gli interattori pertinenti. (ii) quando si lavora con proteine di membrana, il buffer di Lisi deve essere completata con un detergente compatibile con MS. (iii) l'introduzione di una seconda fase di purificazione di affinità potrebbe migliorare falsi positivi al rapporto di vero-positivi ed eliminano la necessità per EV controlli29. Un tag romanzo tandem con due siti di proteasi-taglio indipendente presenta un'alternativa attraente per il passo di cromatografia di esclusione dimensionale aggiunto da Maeda et al 201411, che è sia laboriosa e che richiede tempo.
L'inconveniente più grave dell'AP è l'alto tasso di falsi positivi. Le ragioni sono numerose. Sovraespressione costitutiva è stato già citata. Un'altra fonte di interazioni fisiologicamente irrilevante, a meno che lavorando con gli organelli isolati, è la preparazione di lisati di cellule intere contenenti miscele di proteine e metaboliti da diversi compartimenti subcellulari. Localizzazione sottocellulare dovrebbe essere utilizzata per filtrare gli Interactiani veri. Tuttavia, la maggior parte dei falsi positivi dovuti a aspecifici associazione tra proteine e resine di agarosio. Introduzione di una seconda fase di purificazione, come descritto in precedenza, offre la migliore soluzione al problema, tuttavia è disponibile al costo di tempo e velocità effettiva. Inoltre, più debole interazione può essere perso come si allunga il protocollo. Un altro avvertimento di AP è che, nonostante le informazioni complete che fornisce sui Interactoma di una proteina bersaglio, differenziando tra obiettivi diretti e indiretti della proteina innescato è impossibile. Sono necessari approcci bimolecolari mirati per confermare interazioni.
AP accoppiato con metabolomica basata su MS-è stata usata per studiare i complessi della proteina in S. cerevisiae12. Quest'opera, insieme a nostra osservazione precedente13 che, similmente ai lipidi, composti polari e semi-rimangano legati a complessi della proteina isolati da lisati cellulari, fornito basi concettuali per il protocollo presentato. Il nostro protocollo è caratterizzato da tre punti: (i) In opposizione al lievito lavorare12, dimostra che AP è adatto per il recupero non solo ligandi di proteine idrofobiche ma anche idrofili. (ii) con l'introduzione di un protocollo di estrazione di tre-in-one, un singolo punto di accesso consente di studiare la proteina ed il metabolita interattori della proteina esca. (iii) abbiamo adattato il protocollo per cellule vegetali.
Gli sforzi futuri si concentreranno sulla creazione di un tag di romanzo tandem con due siti di proteasi-taglio indipendente. Vorremmo anche esplorare l'idoneità del protocollo a basso-abbondanza di piccole molecole quali ormoni vegetali.
Divulgazioni
Gli autori non hanno nulla a rivelare.
Riconoscimenti
Vorremmo ringraziare gentilmente Dr. Lothar Willmitzer per il suo coinvolgimento nel progetto, discussioni produttive e grande vigilanza. Siamo grati al Dr. Daniel Veyel per aiutare con le misure di MS di proteomica. Apprezziamo la signora Änne Michaelis che ci ha fornito prezioso aiuto tecnico con misurazioni di LC-MS. Inoltre, vorremmo ringraziare il Dr. Monika Kosmacz e Dr. Ewelina Sokołowska per l'aiuto e il coinvolgimento nel lavoro sul manoscritto originale e di Weronika Jasińska per il supporto tecnico.
Materiali
| Name | Company | Catalog Number | Comments |
| Murashige and Skoog Basal Salts with minimal organics | Sigma-Aldrich | M6899 | |
| 1-Naphthylacetic acid | Sigma-Aldrich | N1641 | |
| Kinetin solution | Sigma-Aldrich | K3253 | |
| Tris base | Sigma-Aldrich | 10708976001 | |
| NaCl | Sigma-Aldrich | S7653 | |
| MgCl2 | Carl Roth | 2189.1 | |
| EDTA | Sigma-Aldrich | 3609 | |
| NaF | Sigma-Aldrich | S6776 | |
| DTT | Sigma-Aldrich | D0632 | |
| PMSF | Sigma-Aldrich | P7626 | |
| E-64 protease inhibitor | Sigma-Aldrich | E3132 | |
| Protease Inhibitor Cocktail | Sigma-Aldrich | P9599 | |
| Na3VO4 | Sigma-Aldrich | S6508 | |
| AcTEV Protease | Thermo Fischer Scientific | 12575015 | |
| Rotiphorese Gel 30 (37,5:1) | Carl Roth | 3029.2 | |
| TEMED | Carl Roth | 2367.3 | |
| PageRuler Prestained Protein Ladder | Thermo Fischer Scientific | 26616 | |
| SBP Tag Antibody (SB19-C4) | Santa Cruz Biotechnology | sc-101595 | |
| Goat anti-mouse IgG-HRP | Santa Cruz Biotechnology | sc-2005 | |
| Bradford Reagent | Sigma-Aldrich | B6916 | |
| Trypsin/Lys-C Mix, Mass Spec Grade | Promega | V5071 | |
| Urea | Sigma-Aldrich | U5128 | |
| Thiourea | Sigma-Aldrich | T8656 | |
| Ammonium bicarbonate | Sigma-Aldrich | 9830 | |
| Iodoacetamide | Sigma-Aldrich | I1149 | |
| MTBE | Biosolve | 138906 | |
| Methanol | Biosolve | 136806 | |
| Water | Biosolve | 232106 | |
| Acetonitrile | Biosolve | 12006 | |
| Trifluoroacetic acid | Biosolve | 202341 | |
| Formic acid | Biosolve | 69141 | |
| Unimax 2010 Platform Shaker | Heidolph | 5421002000 | |
| Nylon Mesh (Wire diameter 34 µM, thickness 55 µM, open area 14%) | Prosepa | Custom order | |
| Glass Funnel, 47 mm, 300 ml | Restek | KT953751-0000 | |
| Filter Bottle Top 500 mL 0,2 µM Pes St | VWR International GmbH | 514-0340 | |
| Mixer Mill MM 400 | Retsch GmbH | 207450001 | |
| IgG Sepharose 6 Fast Flow | GE Healthcare Life Sciences | 17-0969-02 | |
| Mobicol ""Classic"" with 2 different screw caps without filters | MoBiTec GmbH | M1002 | |
| Filter (small) 35 µM pore size, for Mobicol M 1002, M1003, M1050 & M1053 | MoBiTec GmbH | M513515 | |
| Variable Speed Tube Rotator SB 3 | Carl Roth | Y550.1 | |
| Rotary dishes for rotators SB 3 | Carl Roth | Y555.1 | |
| Resprep 24-Port SPE Manifolds | Restek | 26080 | |
| Finisterre C18/17% SPE Columns 100mg / 1ml | Teknokroma | TR-F034000 | |
| Autosampler Vials | Klaus Trott Chromatographie-Zubehör | 40 11 01 740 | |
| Acclaim PepMap 100 C18 LC Column | Thermo Fischer Scientific | 164534 | |
| EASY-nLC 1000 Liquid Chromatograph | Thermo Fischer Scientific | LC120 | |
| Q Exactive Plus Hybrid Quadrupole-Orbitrap Mass Spectrometer | Thermo Fischer Scientific | IQLAAEGAAPFALGMBDK | |
| Acquity UPLC system | Waters | Custom order | |
| ACQUITY UPLC HSS C18 Column, 100A, 1.8 µM, 2.1 mM X 100 mM, 1/pkg | Waters | 186003533 | |
| High-power ultrasonic cleaning baths for aqueous cleaning solutions | Bandelin | RK 31 | |
| Genedata Expressionist | Genedata | NaN | |
| Xcalibur Software | Thermo Fischer Scientific | NaN | |
| MaxQuant | NaN | NaN |
Riferimenti
- Li, X., Snyder, M. Metabolites as global regulators: A new view of protein regulation. Bioessays. 33 (7), 485-489 (2011).
- Jacob, F., Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. Journal of Molecular Biology. 3 (3), 318-356 (1961).
- Schlattner, U., et al. Dual Function of Mitochondrial Nm23-H4 Protein in Phosphotransfer and Intermembrane Transfer a cardiolipin-dependent switch. Journal of Biological Chemistry. 288 (1), 111-121 (2013).
- Ramírez, M. B., et al. GTP binding regulates cellular localization of Parkinson's disease-associated LRRK2. Human Molecular Genetics. , ddx161 (2017).
- Jung, H. J., Kwon, H. J. Target deconvolution of bioactive small molecules: the heart of chemical biology and drug discovery. Archives of Pharmacal Research. 38 (9), 1627-1641 (2015).
- Harding, M. W., Galat, A., Uehling, D. E., Schreiber, S. L. A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature. 341 (6244), 758-760 (1989).
- Lomenick, B., et al. Target identification using drug affinity responsive target stability (DARTS). Proceedings of the National Academy of Sciences of the United States of America. 106 (51), 21984-21989 (2009).
- Manabe, Y., Mukai, M., Ito, S., Kato, N., Ueda, M. FLAG tagging by CuAAC and nanogram-scale purification of the target protein for a bioactive metabolite involved in circadian rhythmic leaf movement in Leguminosae. Chemical Communications. 46 (3), 469-471 (2010).
- Pantoliano, M. W., et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. Journal of Biomolecular Screening. 6 (6), 429-440 (2001).
- Li, X., Snyder, M. Analyzing In vivo Metabolite-Protein Interactions by Large-Scale Systematic Analyses. Current Protocols in Chemical Biology. , 181-196 (2010).
- Maeda, K., Poletto, M., Chiapparino, A., Gavin, A. -. C. A generic protocol for the purification and characterization of water-soluble complexes of affinity-tagged proteins and lipids. Nature Protocols. 9 (9), 2256-2266 (2014).
- Li, X., Gianoulis, T. A., Yip, K. Y., Gerstein, M., Snyder, M. Extensive in vivo metabolite-protein interactions revealed by large-scale systematic analyses. Cell. 143 (4), 639-650 (2010).
- Veyel, D., et al. System-wide detection of protein-small molecule complexes suggests extensive metabolite regulation in plants. Scientific Reports. 7, (2017).
- Luzarowski, M., et al. Affinity purification with metabolomic and proteomic analysis unravels diverse roles of nucleoside diphosphate kinases. Journal of Experimental Botany. , (2017).
- Van Leene, J., et al. Targeted interactomics reveals a complex core cell cycle machinery in Arabidopsis thaliana. Molecular systems biology. 6 (1), 397 (2010).
- Giavalisco, P., et al. Elemental formula annotation of polar and lipophilic metabolites using 13C, 15N and 34S isotope labelling, in combination with high-resolution mass spectrometry. The Plant Journal. 68 (2), 364-376 (2011).
- Van Leene, J., et al. Isolation of transcription factor complexes from Arabidopsis cell suspension cultures by tandem affinity purification. Plant Transcription Factors: Methods and Protocols. , 195-218 (2011).
- Van Leene, J., et al. A tandem affinity purification-based technology platform to study the cell cycle interactome in Arabidopsis thaliana. Molecular & Cellular Proteomics. 6 (7), 1226-1238 (2007).
- Olsen, J. V., Ong, S. -. E., Mann, M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Molecular & Cellular Proteomics. 3 (6), 608-614 (2004).
- Cox, J., Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology. 26 (12), 1367-1372 (2008).
- Cox, J., et al. Andromeda: A peptide search engine integrated into the MaxQuant environment. Journal of Proteome Research. 10 (4), 1794-1805 (2011).
- Tyanova, S., Temu, T., Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols. 11 (12), 2301 (2016).
- Hooper, C. M., et al. SUBAcon: a consensus algorithm for unifying the subcellular localization data of the Arabidopsis proteome. Bioinformatics. 30 (23), 3356-3364 (2014).
- Katajamaa, M., Orešič, M. Data processing for mass spectrometry-based metabolomics. Journal of Chromatography A. 1158 (1-2), 318-328 (2007).
- Szklarczyk, D., et al. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Research. 1277, (2015).
- Tanaka, K. -. i., et al. Dipeptidyl compounds ameliorate the serum-deprivation-induced reduction in cell viability via the neurotrophin-activating effect in SH-SY5Y cells. Neurological Research. 34 (6), 619-622 (2012).
- Smith, C. A., et al. METLIN: A metabolite mass spectral database. Therapeutic Drug Monitoring. 27, 747-751 (2005).
- Bhattacharyya, S., et al. Transient protein-protein interactions perturb E. coli metabolome and cause gene dosage toxicity. Elife. 5, (2016).
- Rigaut, G., et al. A generic protein purification method for protein complex characterization and proteome exploration. Nature Biotechnology. 17 (10), 1030-1032 (1999).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati