
Method Article
Cálculo de las concentraciones atmosféricas de los cúmulos moleculares a partir de la termoquímica ab initio
En este artículo
Resumen
Las concentraciones atmosféricas de cúmulos moleculares débilmente unidos se pueden calcular a partir de las propiedades termoquímicas de las estructuras de baja energía que se encuentran a través de una metodología de muestreo configurational de varios pasos utilizando un algoritmo genético y una química cuántica semiempírica y ab initio.
Resumen
El estudio computacional de la formación y crecimiento de aerosoles atmosféricos requiere una superficie de energía libre de Gibbs precisa, que se puede obtener de la estructura electrónica de fase de gas y los cálculos de frecuencia vibratoria. Estas cantidades son válidas para aquellos cúmulos atmosféricos cuyas geometrías corresponden a un mínimo en sus superficies de energía potenciales. La energía libre de Gibbs de la estructura de energía mínima se puede utilizar para predecir las concentraciones atmosféricas del racimo en una variedad de condiciones tales como temperatura y presión. Presentamos un procedimiento computacionalmente barato basado en un muestreo de configuración basado en algoritmos genéticos seguido de una serie de cálculos de cribado cada vez más precisos. El procedimiento comienza generando y evolucionando las geometrías de un gran conjunto de configuraciones utilizando modelos semiempíricos y luego refina las estructuras únicas resultantes en una serie de niveles de teoría ab initio de alto nivel. Por último, las correcciones termodinámicas se calculan para el conjunto resultante de estructuras de energía mínima y se utilizan para calcular las energías libres de Formación, constantes de equilibrio y concentraciones atmosféricas de Gibbs. Presentamos la aplicación de este procedimiento al estudio de racimos de glicina hidratada en condiciones ambientales.
Introducción
El parámetro más incierto en los estudios atmosféricos del cambio climático es la medida exacta en que las partículas de nubes reflejan la radiación solar entrante. Los aerosoles, que son partículas suspendidas en un gas, forman partículas de nubes llamadas núcleos de condensación de nubes (CCN) que dispersan la radiación entrante, evitando así su absorción y el posterior calentamiento de la atmósfera1. Una comprensión detallada de este efecto de enfriamiento neto requiere una comprensión del crecimiento de aerosoles en CCN, lo que a su vez requiere una comprensión del crecimiento de pequeños racimos moleculares en partículas de aerosol. Trabajos recientes han sugerido que la formación de aerosoles se inicia mediante racimos moleculares de 3 nm de diámetro o menos2; sin embargo, este régimen de tamaño es difícil de acceder utilizando técnicas experimentales3,4. Por lo tanto, se desea un enfoque de modelado computacional para superar esta limitación experimental.
Utilizando nuestro enfoque de modelado que se describe a continuación, podemos analizar el crecimiento de cualquier racimo hidratado. Debido a que estamos interesados en el papel del agua en la formación de grandes moléculas biológicas a partir de componentes más pequeños en entornos prebióticos, ilustramos nuestro enfoque con glicina. Los retos encontrados y las herramientas necesarias para abordar esas cuestiones de investigación son muy similares a los que intervienen en el estudio de aerosoles atmosféricos y grupos de prenucleación5,6,7,8,9,10,11,12,13,14,15. Aquí, examinamos los racimos de glicina hidratados a partir de una molécula de glicina aislada seguida de una serie de adiciones escalonadas de hasta cinco moléculas de agua. El objetivo final es calcular las concentraciones de equilibrio de gly(H2O)n-0-5 clusters en la atmósfera a temperatura ambiente a nivel del mar y una humedad relativa (RH) del 100 %.
Un pequeño número de estos cúmulos moleculares subnanómetros se convierten en un cúmulo crítico metastable (1-3 nm de diámetro) ya sea mediante la adición de otras moléculas de vapor o la coagulación en los racimos existentes. Estos cúmulos críticos tienen un perfil de crecimiento favorable que conduce a la formación de núcleos de condensación de nubes mucho más grandes (hasta 50-100 nm) (CCN), que afectan directamente a la eficiencia de precipitación de las nubes, así como a su capacidad para reflejar la luz incidente. Por lo tanto, tener una buena comprensión de la termodinámica de los cúmulos moleculares y sus distribuciones de equilibrio debería conducir a predicciones más precisas del impacto de los aerosoles en el clima global.
Un modelo descriptivo de formación de aerosoles requiere una termodinámica precisa de la formación de racimos moleculares. El cómputo de la termodinámica precisa de la formación de racimos moleculares requiere la identificación de las configuraciones más estables, lo que implica encontrar el mínimo global y local en la superficie de energía potencial del clúster (PES)16. Este proceso se denomina muestreo de configuración y se puede lograr a través de una variedad de técnicas, incluidas las basadas en la dinámica molecular (MD)17,,18,19,20,Monte Carlo (MC)21,,22,y algoritmos genéticos (GA)23,,24,25.
A lo largo de los años se han desarrollado diferentes protocolos para obtener la estructura y la termodinámica de los hidratos atmosféricos a un alto nivel de teoría. Estos protocolos diferían en la elección de (i) método de muestreo de configuración, (ii) naturaleza del método de bajo nivel utilizado en el muestreo de configuración, y (iii) la jerarquía de métodos de nivel superior utilizados para refinar los resultados en los pasos posteriores.
Los métodos de muestreo de configuración incluyeron intuición química26,muestreo aleatorio27,28,dinámica molecular (MD)29,30,salto de cuenca (BH)31,y algoritmo genético (GA)24,,25,32. Los métodos de bajo nivel más comunes empleados con estos métodos de muestreo son campos de fuerza o modelos semiempíricos como PM6, PM7 y SCC-DFTB. Estos son seguidos a menudo por cálculos DFT con conjuntos de base cada vez más grandes y funciones más confiables de los peldaños más altos de la escalera33de Jacob. En algunos casos, estos son seguidos por métodos de función de onda de nivel superior como MP2, CCSD(T), y el rentable DLPNO-CCSD(T)34,35.
Kildgaard y otros36 desarrollaron un método sistemático en el que las moléculas de agua se añaden en puntos en las esferas de Fibonacci37 alrededor de racimos más pequeños hidratados o no hidratados para generar candidatos para racimos más grandes. Los candidatos no físicos y redundantes se eliminan en función de los umbrales de contacto cercanos y la distancia entre diferentes conformadores. Las optimizaciones posteriores utilizando el método semiempírico PM6 y una jerarquía de métodos DFT y wavefunction se utilizan para obtener un conjunto de conformadores de baja energía en un alto nivel de teoría.
El algoritmo38 de la colonia de abejas artificiales (ABC) es un nuevo enfoque de muestreo configurational que ha sido implementado recientemente por Zhang y otros para estudiar los cúmulos moleculares en un programa llamado ABCluster39. 40 utilizaron ABCluster para el muestreo de configuración seguido de reoptimizaciones de bajo nivel utilizando el método semiempírico GFN-xTB41de unión ajustada. Refinaron aún más las estructuras y energías utilizando métodos DFT seguidos de energías finales usando DLPNO-CCSD(T).
Independientemente del método, el muestreo de configuración comienza con una distribución aleatoria o no aleatoria de puntos en el PES. Cada punto corresponde a una geometría específica del cúmulo molecular en cuestión y es generado por el método de muestreo. A continuación, se encuentra el mínimo local más cercano para cada punto siguiendo la dirección "descenso" en el PES. El conjunto de mínimos así encontrados corresponden a aquellas geometrías del cúmulo molecular que son estables, al menos durante algún tiempo. Aquí, la forma del PES y la evaluación de la energía en cada punto de la superficie serán sensibles a la descripción física del sistema donde una descripción física más precisa resulta en un cálculo de energía más costoso computacionalmente. Utilizaremos específicamente el método GA implementado en el programa OGOLEM25, que se ha aplicado con éxito a una variedad de problemas de optimización global y muestreo de configuración42,,43,,44,,45,para generar el conjunto inicial de puntos de muestreo. El PES será descrito por el PM7 modelo46 implementado en el programa MOPAC201647. Esta combinación se emplea porque genera una mayor variedad de puntos en comparación con los métodos MD y MC y encuentra el mínimo local más rápido que las descripciones más detalladas del PES.
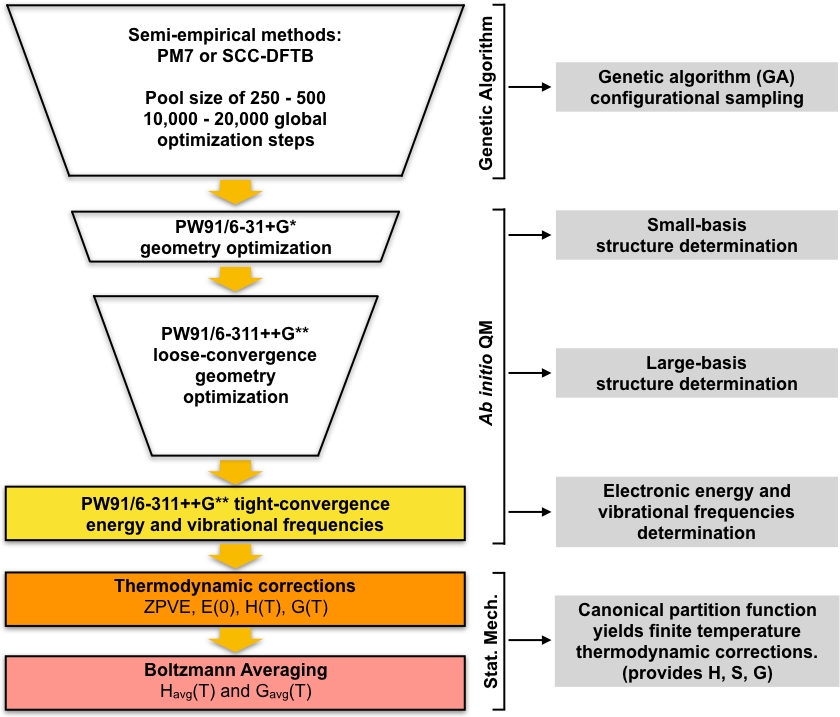
El conjunto de mínimos locales optimizados para GA se toman como geometrías iniciales para una serie de pasos de cribado, que conducen a un conjunto de energía mínima de baja altitud. Esta parte del protocolo comienza optimizando el conjunto de estructuras únicas optimizadas para GA utilizando la teoría de la densidad-funcional (DFT) con un pequeño conjunto de bases. Este conjunto de optimizaciones generalmente dará un conjunto más pequeño de estructuras mínimas locales únicas que se modelan con más detalle en comparación con las estructuras semiempíricas optimizadas para GA. A continuación, se realiza otra ronda de optimizaciones DFT en este conjunto más pequeño de estructuras utilizando un conjunto de base más grande. Una vez más, este paso generalmente dará un conjunto más pequeño de estructuras únicas que se modelan con más detalle en comparación con el paso DFT de base pequeña. El conjunto final de estructuras únicas se optimizan a una convergencia más estricta y se calculan las frecuencias vibratorias armónicas. Después de este paso tenemos todo lo que necesitamos para calcular las concentraciones de equilibrio de los cúmulos en la atmósfera. El enfoque general se resume diagramamáticamente en la Figura 1. Utilizaremos la correlación de intercambio PW9148 de aproximación de gradiente generalizado (GGA) funcional en la implementación Gaussian0949 de DFT junto con dos variaciones del conjunto de base Pople50 (6-31+G* para el paso de base pequeño y 6-311++G** para el paso de base grande). Esta combinación particular de conjunto funcional y base de correlación de intercambio fue elegida debido a su éxito anterior en la computación de energías libres Gibbs precisas de formación para los clusters atmosféricos51,52.
Este protocolo supone que el usuario tiene acceso a un clúster de computación de alto rendimiento (HPC) con el sistema por lotes portátil53 (PBS), MOPAC2016 (http://openmopac.net/MOPAC2016.html)47, OGOLEM (https://www.ogolem.org)25, Gaussian 09 (https://gaussian.com)49y OpenBabel54 (http://openbabel.org/wiki/Main_Page) software instalado siguiendo sus instrucciones de instalación específicas. Cada paso de este protocolo también utiliza un conjunto de scripts internos de shell y Python 2.7 que deben guardarse en un directorio que se incluye en la variable de entorno $PATH del usuario. Todos los módulos ambientales necesarios y permisos de ejecución para ejecutar todos los programas anteriores también deben cargarse en la sesión del usuario. El uso de disco y memoria por el código GA (OGOLEM) y los códigos semiempíricos (MOPAC) son muy pequeños según los estándares modernos de recursos informáticos. El uso general de memoria y disco para OGOLEM/MOPAC depende de cuántos subprocesos se quieran utilizar, e incluso entonces, el uso de recursos será pequeño en comparación con las capacidades de la mayoría de los sistemas HPC. Las necesidades de recursos de los métodos QM dependen del tamaño de los clústeres y del nivel de teoría utilizado. La ventaja de utilizar este protocolo es que se puede variar el nivel de teoría para poder calcular el conjunto final de estructuras de baja energía, teniendo en cuenta que por lo general los cálculos más rápidos conducen a una mayor incertidumbre en la precisión de los resultados.
En aras de la claridad, el equipo local del usuario se denominará"equipo local",mientras que el clúster de HPC al que tienen acceso se denominará"clúster remoto".
Protocolo
1. Encontrar la estructura energética mínima de glicina y agua aisladas
NOTA: El objetivo aquí es doble: (i) obtener estructuras energéticas mínimas de moléculas aisladas de agua y glicina para su uso en el muestreo configurational del algoritmo genético, (ii) y calcular las correcciones termodinámicas a las energías de fase gaseosa de estas moléculas para su uso en el cálculo de las concentraciones atmosféricas.
- En el equipo local, abra una nueva sesión de Avogadro.
- Haga clic en Construir > Insertar > Péptido y seleccione Gly en la ventana Insertar péptido para generar un monómero de glicina en la ventana de visualización.
- Haga clic en Extensiones > Gaussian y edite la primera línea en el cuadro de texto para leer ' á pw91pw91/6-311++G** int(Acc2E-12,UltraFine) scf(conver-12) opt(tight,maxcyc-300) freq'. Haga clic en Generar y guarde el archivo de entrada como glycine.com.
- Tenga en cuenta que si la molécula tiene una flexibilidad conformacional significativa, como la glicina hace55, es fundamental realizar análisis de conformación para identificar la estructura mínima global y otros conformadores de baja altitud. OpenBabel54 proporciona sólidas herramientas de búsqueda conformacional utilizando diferentes algoritmos y campos de fuerza rápida. Mientras que a los conformadores se les permite relajarse e interconvertir durante GA y cálculos posteriores, a veces es necesario ejecutar varios cálculos de GA, cada uno comenzando con un conformador diferente.
- En el equipo local, abra una nueva sesión en Avogadro.
- Haga clic en Construir > Insertar > Fragmento y busque "agua" en la ventana Insertar fragmento para obtener las coordenadas del agua.
- Haga clic en Extensiones > Gaussian y edite la primera línea en el cuadro de texto para leer ' á pw91pw91/6-311++G** int(Acc2E-12,UltraFine) scf(conver-12) opt(tight,maxcyc-300) freq'. Haga clic en Generar y guarde el archivo de entrada como water.com.
- Transfiera los dos archivos .com al clúster remoto. Una vez que inicie sesión en el clúster remoto, llame a Gaussian 09 en un script de envío por lotes para iniciar el cálculo. Cuando terminen los cálculos, extraiga las coordenadas cartesianas (archivos.xyz) de las estructuras de energía mínima llamando a OpenBabel. Para glycine, el comando que se ejecuta es:
obabel -ig09 glycine.log -oxyz > glycine.xyz
Estos dos archivos .xyz serán utilizados por el muestreo de configuración de GA en el siguiente paso.
2. Muestreo de configuración basado en algoritmos genéticos de gly(H2O)n-1-5 clusters
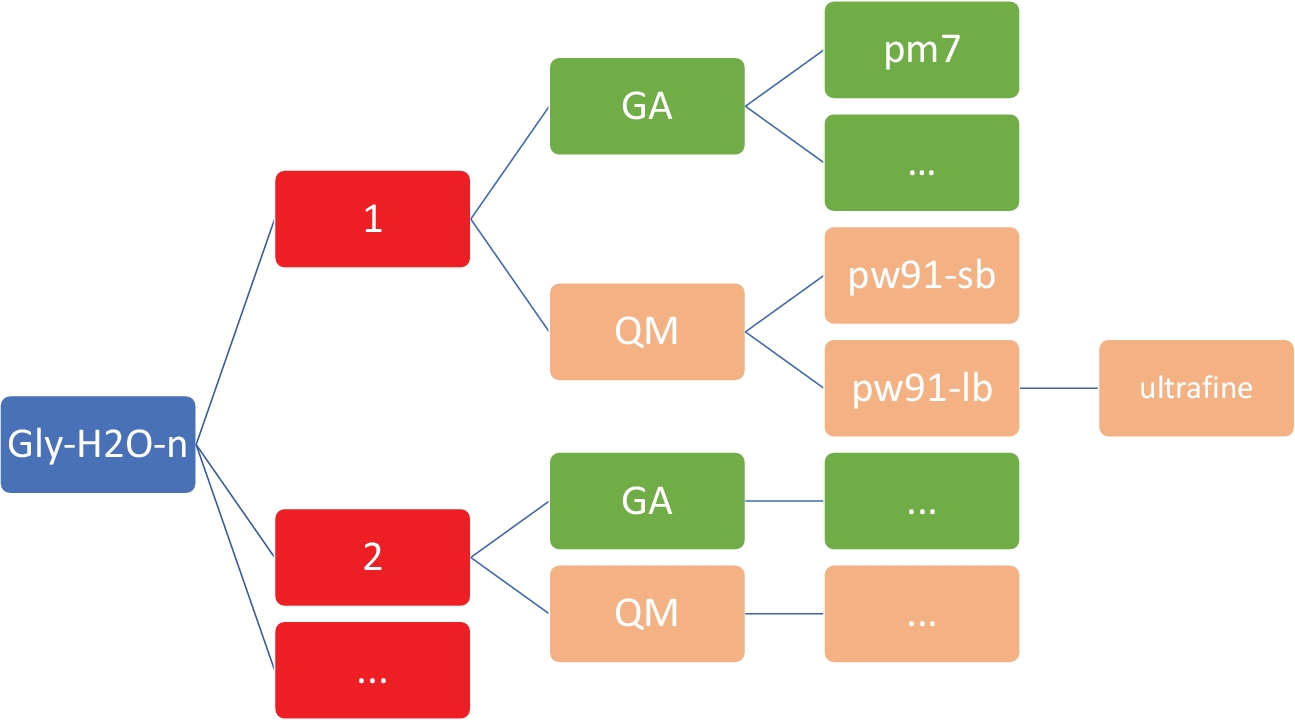
NOTA: El objetivo aquí es obtener un conjunto de estructuras de baja energía para Gly(H2O)n-1-5 en el nivel semiempírico de bajo costo de la teoría, utilizando el modelo PM746 implementado en MOPAC47. Es imperativo que el directorio de trabajo tenga la organización y la estructura exactas como se muestra en la Figura 2. Esto es para asegurarse de que el shell personalizado y los scripts de Python funcionan sin errores.
- Copie todos los scripts necesarios en el clúster remoto y agregue su ubicación a $PATH
- Coloque todos los scripts y archivos de plantilla en una carpeta (por ejemplo, scripts) y cópielo en el clúster remoto
- Asegúrese de que todos los scripts son ejecutables
- Agregue la ubicación del directorio de scripts a la variable de $PATH de medio ambiente introduciendo los siguientes comandos en un terminal. La ubicación predeterminada de los scripts se establece en $HOME/JoVE-demo/scripts, sin embargo, se puede definir una variable de medio ambiente llamada $SCRIPTS_HOME apuntando al directorio que contiene los scripts y agregar $SCRIPTS_HOME a la ruta de acceso de uno
- Concha Bash:
exportación SCRIPTS_HOME/ruta/a/scripts
exportar PATH-$-SCRIPTS_HOME-:$-PATH- - Carcasa Tcsh/Csh:
setenv SCRIPTS_HOME /path/to/scripts
setenv PATH $-SCRIPTS_HOME-:$-PATH-
- Concha Bash:
- En el clúster remoto, configure y ejecute un cálculo de GA:
- Cree un directorio llamado gly-h2o-n donde n es el número de moléculas de agua.
- Cree un subdirectorio llamado GA en el directorio gly-h2o-n para ejecutar cálculos de algoritmos genéticos.
- Copie los archivos de entrada de OGOLEM (por ejemplo, pm7.ogo), las coordenadas cartesianas de monómeros (por ejemplo, glycine.xyz, water.xyz) y el script de envío por lotes PBS (por ejemplo, run.pbs) en el directorio GA.
- Realice los cambios necesarios en el archivo de entrada OGOLEM y en el archivo de envío por lotes.
- Envíe el cálculo. Cuando se inicia el cálculo, OGOLEM creará un nuevo directorio denominado como prefijo del archivo de entrada OGOLEM (Eg. pm7) en el directorio GA y almacenará allí las coordenadas recién generadas.
- Una vez completado el cálculo, compile las energías y las constantes de rotación, y utilice esa información para determinar cuáles son las estructuras únicas de baja energía:
- Cambie el directorio a gly-h2o-n/GA/pm7 y
- Extraiga las energías y calcule las constantes de rotación de los clústeres optimizados para GA con el comando:
getRotConsts-GA.csh N 0 99
donde N es el número de átomos en el cúmulo molecular y '0 99' indica que el tamaño de la agrupación GA es 100, con índices que oscilan entre 0 y 99. Esto generará un archivo llamado rotConstsData_C que contiene una lista ordenada de todas las configuraciones de clúster optimizadas para GA, sus energías y sus constantes de rotación. - Ejecute el comando:
similarityAnalysis.py rotConstsData_C pm7
donde pm7 se utilizará como una etiqueta de nombre de archivo, para buscar y guardar los clústeres únicos optimizados para GA. Esto generará un archivo llamado uniqueStructures-pm7.data que contiene una lista ordenada de las configuraciones únicas optimizadas para GA. Esta es una lista de estructuras mínimas locales únicas para el clúster Gly(H2O)n optimizado en el nivel de teoría PM7, y estas estructuras ahora están listas para ser refinadas usando DFT.
- Vaya al directorio gly-h2o-n/GA y combine los resultados de varias ejecuciones de GA comparables utilizando el script combine-GA.csh. La sintaxis es:
combine-GA.csh
3. Refinamiento utilizando el método QM con una base pequeña establecida
NOTA: El objetivo aquí es refinar el muestreo de configuración de los clústeres Gly(H2O)n-1-5 utilizando una mejor descripción cuántica-mecánica para obtener un conjunto más pequeño pero más preciso de estructuras de clúster Gly(H2O)n-1-5. Las estructuras iniciales para este paso son las salidas del paso 2.
- Preparar y ejecutar el pequeño conjunto de bases de cálculo DFT:
- Cree un subdirectorio llamado QM en el directorio gly-h2o-n. En el directorio QM, cree otro subdirectorio denominado pw91-sb.
- Copie la lista de estructuras únicas (uniqueStructures-pm7.data) del directorio gly-h2o-n/GA al directorio QM/pw91-sb.
- Cambie el directorio a ese gly-h2o-n/QM/pw91-sb.
- Ejecute el script de muestreo de configuración DFT del conjunto de bases pequeñas utilizando el comando:
run-pw91-sb.csh uniqueStructures-pm7.data sb QUEUE 10
donde sb es una etiqueta para este conjunto de cálculos, QUEUE es la cola preferida en el clúster informático y 10 indica que 10 cálculos se deben agrupar en un trabajo por lotes. Este script generará automáticamente las entradas para Gaussian 09 y enviará todos los cálculos. Introduzca 'test' para que 'QUEUE' realice una ejecución en seco.
- Una vez completados los cálculos enviados, extraiga y analice los resultados.
- Extraiga las energías y calcule las constantes de rotación de los clústeres optimizados para bases pequeñas mediante el comando:
getRotConsts-dft-sb.csh pw91 N
donde pw91 indica que se utilizó la densidad PW91 funcional, y N es el número de átomos en el clúster. Esto creará un archivo denominado rotConstsData_C. - Ahora identifique las estructuras únicas con el comando:
similarityAnalysis.py sb rotConstsData_C
donde sb se utiliza como una etiqueta de nombre de archivo. Ahora habrá una lista de configuraciones únicas optimizadas en el nivel de teoría PW91/6-31+G* guardada en el archivo uniqueStructures-sb.data.
- Extraiga las energías y calcule las constantes de rotación de los clústeres optimizados para bases pequeñas mediante el comando:
- Vaya al directorio gly-h2o-n/QM y combine los resultados de varias ejecuciones de QM comparables utilizando el script combine-QM.csh. La sintaxis es:
combine-QM.csh
4. Mayor refinamiento utilizando el método QM con una gran base establecida
NOTA: El objetivo aquí es refinar aún más el muestreo de configuración de los clústeres Gly(H2O)n-1-5 utilizando una mejor descripción cuántica-mecánica. Las estructuras iniciales para este paso son las salidas del paso 3.
- Envíe cálculos más fiables utilizando un conjunto de base más grande.
- Cree un subdirectorio llamado pw91-lb en el directorio QM.
- Copie la lista de estructuras únicas (uniqueStructures-sb.data) del directorio gly-h2o-n/QM al directorio gly-h2o-n/QM/pw91-lb y cambie a ese directorio.
- Ejecute el script de muestreo de configuración DFT de base grande con el comando:
run-pw91-lb.csh uniqueStructures-sb.data lb QUEUE 10
donde lb es una etiqueta para este conjunto de cálculos, QUEUE es la cola preferida en el clúster informático y 10 indica que 10 cálculos se van a agrupar en un trabajo por lotes. Este script generará automáticamente las entradas para Gaussian 09 y enviará todos los cálculos. Escriba 'test' para que 'QUEUE' realice una prueba de funcionamiento en seco.
- Una vez completados los cálculos presentados, extraiga y analice los datos
- Calcular las constantes de rotación de los clústeres optimizados para bases grandes con el comando:
getRotConsts-dft-lb.csh pw91 N
donde pw91 indica que se utilizó la densidad PW91 funcional, y N es el número de átomos en el clúster. - Ahora identifique las estructuras únicas con el comando:
similarityAnalysis.py rotConstsData_C lb
donde lb se utiliza como una etiqueta de nombre de archivo. Ahora tiene una lista de configuraciones únicas optimizadas en el nivel de teoría PW91/6-311++G** guardada en el archivo uniqueStructures-lb.data.
- Calcular las constantes de rotación de los clústeres optimizados para bases grandes con el comando:
5. Cálculos finales de energía y corrección termodinámica
NOTA: El objetivo aquí es obtener la estructura vibratoria y las energías de los clústeres Gly(H2O)n-1-5 utilizando un conjunto de base grande y una cuadrícula de integración ultrafina para calcular las correcciones termoquímicas deseadas.
- A partir de los resultados del paso anterior, envíe cálculos más fiables.
- Cree un subdirectorio denominado ultrafine en el directorio QM/pw91-lb. A continuación, copie la lista de estructuras únicas (uniqueStructures-lb.data) del directorio QM/pw91-lb al directorio QM/pw91-lb/ultrafine y cambie a ese directorio.
- Envíe el script DFT ultrafino de gran base con el comando:
run-pw91-lb-ultrafine.csh uniqueStructures-lb.data uf QUEUE 10
donde uf es una etiqueta para este conjunto de cálculos, QUEUE es la cola preferida en el clúster informático y 10 indica que 10 cálculos se van a agrupar en un trabajo por lotes. Este script generará automáticamente las entradas para Gaussian 09 y enviará todos los cálculos. Escriba 'test' para que 'QUEUE' realice una prueba de funcionamiento en seco.
- Una vez completados los cálculos presentados, extraiga y analice los datos
- Extraiga las energías y calcule las constantes de rotación de los clústeres optimizados para bases grandes con el comando:
getRotConsts-dft-lb-ultrafine.csh pw91 N
donde pw91 indica que se utilizó la densidad PW91 funcional, y N es el número de átomos en el clúster. - Ahora identifique las estructuras únicas con el comando:
similarityAnalysis.py uf rotConstsData_C
donde uf se utiliza como una etiqueta de nombre de archivo. Ahora tiene una lista de configuraciones únicas optimizadas en el nivel de teoría PW91/6-311++G** guardada en el archivo uniqueStructures-uf.data.
- Extraiga las energías y calcule las constantes de rotación de los clústeres optimizados para bases grandes con el comando:
- Realice una extracción final de la información necesaria para calcular las correcciones termodinámicas. Utilice esa información para calcular las correcciones termodinámicas.
- Extraiga las energías electrónicas finales, las constantes de rotación y las frecuencias vibratorias, y utilícelas para calcular las correcciones termodinámicas mediante el comando:
run-thermo-pw91.csh uniqueStructures-uf.data - Copie/pegue la salida de la línea de comandos en la hoja 'Raw_Energies' de la hoja de cálculo de Excel denominada 'gly-h2o-n.xlsx'. Usted tendría que hacer esto para los monómeros (glicina y agua) así como el miembro de energía más baja de cada hidrato (gly-h2o-n, donde n-1,2, ...).
- A medida que las energías bruta se añaden a la primera hoja de la hoja de cálculo 'gly-h2o-n.xlsx', las hojas posteriores 'Binding_Energies' y 'Hydrate_Distribution' se actualizan automáticamente. En particular, la lámina 'Hydrate_Distribution' produce la concentración de equilibrio de hidratos a diferentes temperaturas (Ej. 298.15K), humedad relativa (20%, 50%, 100%) y concentraciones iniciales de agua ([H2O]) y glicina ([Glicina]). La teoría detrás de estos cálculos se describe en el siguiente paso.
- Extraiga las energías electrónicas finales, las constantes de rotación y las frecuencias vibratorias, y utilícelas para calcular las correcciones termodinámicas mediante el comando:
6. Calcular las concentraciones atmosféricas de Gly(H2O)n.o 0-5 racimos a temperatura ambiente a nivel del mar
NOTA: Esto se logra copiando primero los datos termodinámicos generados en el paso anterior en una hoja de cálculo y calculando las energías libres de Gibbs de hidratación secuencial. Luego, las energías libres de Gibbs se utilizan para calcular las constantes de equilibrio para cada hidratación secuencial. Finalmente, se resuelve un conjunto de ecuaciones lineales para obtener la concentración de equilibrio de los hidratos para una concentración dada de monómeros, temperatura y presión.
- Comience por establecer un sistema de equilibrios químicos para la hidratación secuencial de la glicina como se muestra a continuación:

- Calcular las constantes de equilibrio Kn usando Kn á e-- Gn/(kBT), donde n es el nivel de hidratación, Gn es el cambio de energía libre de Gibbs de la reacción de hidratación nth, kB es constante de Boltzmann, y T es la temperatura.

- Configurar la ecuación para la conservación de la masa, utilizando la suposición de que la suma de las concentraciones de equilibrio de los racimos de glicina hidratado y no hidratado equivale a la concentración inicial de glicina aislada [Gly]0. Reescribir este sistema de seis ecuaciones simultáneas, utilizando una reorganización algebraica de las expresiones constantes de equilibrio, como

- Resolver el sistema de ecuaciones mostrado anteriormente para obtener las concentraciones de equilibrio de Gly(H2O)n á 0-5 utilizando un valor experimental56,57,58 para la concentración de glicina en la atmósfera, [Gly]0 x 2,9 x 106 cm-3, y la concentración de agua en la atmósfera a 100% de humedad relativa y una temperatura de 298,15 K59, [H2O] a 7,7 x 1017 cm-3.
Resultados
El primer conjunto de resultados de este protocolo debe ser un conjunto de estructuras de baja energía de Gly(H2O)n-1-5 que se encuentran a través del procedimiento de muestreo de configuración. Estas estructuras se han optimizado en el nivel de teoría PW91/6-311+G** y se supone que son precisas para el propósito de este artículo. No hay evidencia que sugiera que PW91/6-311++G** subestime o sobreestime constantemente la energía de unión de estos clústeres. Su capacidad para predecir energías de unión en relación con MP2/CBS32 y [DLPNO-]CCSD(T)/CBS60,61 estimaciones y experimento52 muestra una gran cantidad de fluctuaciones. Lo mismo ocurre con la mayoría de las otras funciones de densidad. Por lo general, cada valor de n - 1 – 5 debe producir un puñado de estructuras de baja energía dentro de alrededor de 5 kcal mol-1 de la estructura de menor energía. Aquí, nos centramos en la primera estructura producida por el script run-thermo-pw91.csh para la brevedad. La Figura 3 muestra los isómeros de energía electrónica más bajos de los clústeres de Gly(H2O)n-0-5. Se puede ver que la red de enlaces de hidrógeno crece en complejidad a medida que aumenta el número de moléculas de agua, e incluso pasa de una red mayoritariamente plana a una estructura tridimensional similar a una jaula en n.o 5. El resto de este texto utiliza las energías y cantidades termodinámicas correspondientes a estos cinco clusters específicos.
La Tabla 1 contiene las cantidades termodinámicas necesarias para llevar a cabo el protocolo. La Tabla 2 muestra un ejemplo de la salida del script run-thermo-pw91.csh donde se imprimen las energías electrónicas, las correcciones vibratorias de punto cero y las correcciones termodinámicas a tres temperaturas diferentes. Para cada clúster (fila), E[PW91/6-311+G**] corresponde a las energías electrónicas de fase gaseosa en el nivel de teoría PW91/6-311++G** calculado en redes de integración ultrafinas en unidades de Hartree, así como la energía vibratoria de punto cero (ZPVE) en unidades de kcal mol-1. A cada temperatura, 216.65 K, 273.15 K, y 298.15 K, se enumeran las correcciones termodinámicas, H la entalpía de formación en unidades de kcal mol-1, S la entropía de formación en unidades de molcal -1, y G la energía libre de formación Gibbs en unidades de molkcal -1. La Tabla 3 muestra un ejemplo de cálculo del cambio total de energía libre de Gibbs de hidratación, así como para la hidratación secuencial. Un ejemplo de cálculo del cambio total de energía libre de Gibbs de hidratación para la reacción

comienza con el cómputo de la energía electrónica EPW91 como

donde EPW91[Gly(H2O)] se toma de la Columna 2 De la columna C, y EPW91[Gly] y EPW91[H2O] se toman de la Columna 1 de la columna B. A continuación, calculamos el cambio total de energía de la fase gaseosaE(0) incluyendo el cambio en la energía vibratoria de punto cero de la reacción como

para obtener la columna D. En este punto, el CuadroPW91/6-311+G** se toma de la Columna 3 de la columna C, EZPVE[Gly á (H2O)] de la Tabla 2 de la columna D, y EZPVE[Gly] y EZPVE[H2O] de la Columna 1 C. En aras de la brevedad, pasaremos a los clústeres de temperatura ambiente, por lo que nos saltamos los datos de 216,65 K y 273,15 K. A temperatura ambiente, calculamos el cambio de entalpía de la reacciónH corrigiendo el cambio de energía de la fase gaseosa como

donde se tomaE(0) de la columna D de la Tabla 3, setoman de la Tabla 1 [Gly-(H2O)] y se toman los puntosdela columna K de la Tabla 2, y los siguientes los datos de la tabla H [Gly] y el valor De [Gly] y el valor H [Gly] y el valor deH[H2O] se toman de la columna J de la Tabla 1. Por último, calculamos el cambio de energía libre de Gibbs de la reacciónG como

En los casos en que se tomaH de la columna 3 I, S[Gly-(H2O)] se toma de la tabla 2 de la columna L, y S[Gly] y S[H2O] se toman de la tabla 1 de la columna K. Tenga en cuenta aquí que los valores de entropía deben convertirse en unidades de mol-1 K -1 k-1 durante este paso.
Ahora tenemos las cantidades necesarias para calcular las concentraciones atmosféricas de glicina hidratada como se muestra en el paso 6. Los resultados deben ser similares a los datos que se muestran en la Tabla 4,pero cabe esperar pequeñas diferencias numéricas. La Tabla 4 muestra las concentraciones de hidratos de equilibrio encontradas a partir de la formulación del sistema de seis ecuaciones en el Paso 6.2 en una ecuación de matriz y su solución posterior. Comenzamos reconociendo el hecho de que el sistema de ecuaciones se puede escribir como

donde Kn es la constante de equilibrio para lana hidratación secuencial de glicina, w es la concentración de agua en la atmósfera, g es la concentración inicial de glicina aislada en la atmósfera, y gn es la concentración de equilibrio de Gly(H2O)n. Si reescribimos la ecuación anterior como Ax a b, obtenemos x a A-1b donde A-1 es la inversa de la matriz A. Esto inverso se puede calcular fácilmente utilizando funciones de hoja de cálculo integradas como se muestra en la Tabla 4 para obtener los resultados finales.
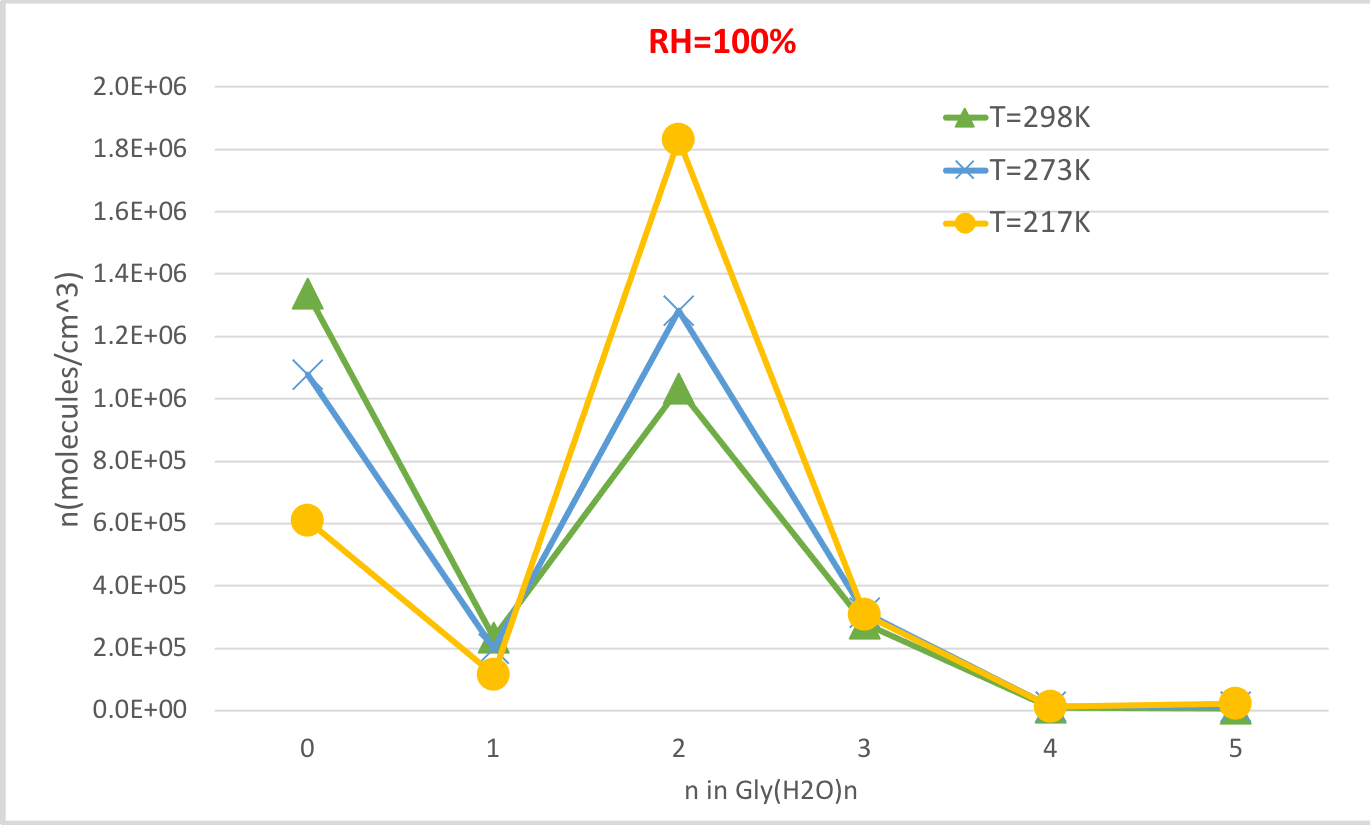
La Figura 4 muestra la concentración de equilibrio de glicina hidratada calculada en la Tabla 4 en función de la temperatura a 100% de humedad relativa y 1 presión de la atmósfera. Muestra que, a medida que la temperatura disminuye de 298.15K a 216.65K, la concentración de glicina no hidratada (n-0) disminuye y aumenta la de la glicina hidratada. La glicina dihidrato (n-2) en particular aumenta drásticamente con la disminución de la temperatura, mientras que el cambio en la concentración de otros hidratos es menos notable. Esta correlación inversa entre la temperatura y la concentración de hidratos es consistente con la expectativa de que las energías libres de Gibbs más bajas de hidratación a temperaturas más bajas favorecen la formación de hidratos.
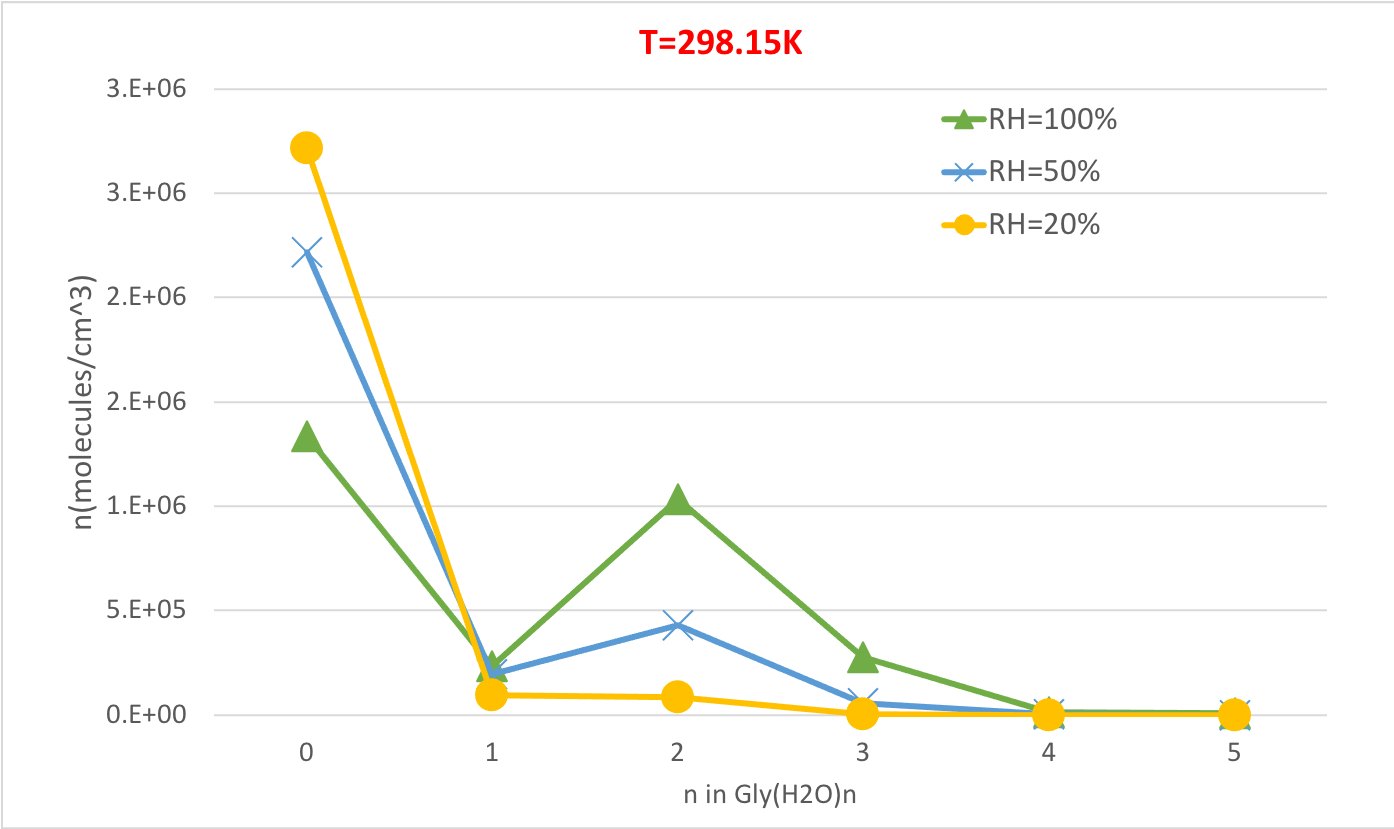
La Figura 5 ilustra la dependencia relativa de humedad de la concentración de equilibrio de hidratos de glicina a 298.15K y 1 presión de atmósfera. Demuestra claramente que a medida que la humedad relativa aumenta del 20% al 100%, la concentración de hidratos (n>0) aumenta a expensas de la glicina no hidratada (n-0). Una vez más la correlación directa entre la humedad relativa y la concentración de hidratos es consistente con la idea de que la presencia de más moléculas de agua en RH más alta promueve la formación de hidratos.
Como se presenta, este protocolo proporciona una comprensión cualitativa de las poblaciones de glicina hidratada en la atmósfera. Suponiendo una concentración inicial de glicina aislada de 2,9 millones de moléculas por centímetro cúbico, vemos que la glicina no hidratada (n-0) es la especie más abundante en la mayoría de las condiciones excepto T-216.65K y RH-100%. El dihidrato (n-2), que tiene la menor energía de hidratación libre de Gibbs secuencial a las tres temperaturas, es el hidrato más abundante en las condiciones consideradas aquí. Se prevé que los hidratos monohidratos (n-1) y los hidratos más grandes (n-3) se encuentren en cantidades insignificantes. Tras la inspección de la Figura 3,la abundancia de los racimos n-1-4 puede estar relacionada con la estabilidad y la tensión en la red de enlaces de hidrógeno de los racimos. Estos cúmulos tienen las moléculas de agua de hidrógeno unidas a la mitad del ácido carboxílico de la glicina en una geometría que se asemeja mucho a las de varias estructuras de anillo sacadas de hidrógeno, haciéndolos especialmente estables.

Figura 1: Descripción esquemática del procedimiento actual. Un gran conjunto de estructuras de conjeturas generadas por el algoritmo genético (GA) se refina mediante una serie de optimizaciones de geometría PW91 hasta que se obtiene un conjunto de estructuras convergentes. Las frecuencias vibratorias de estas estructuras se calculan y se utilizan para calcular la energía libre de formación de Gibbs, que a su vez se utiliza para calcular las concentraciones de equilibrio de los clústeres en condiciones ambientales. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Estructura de directorios representativa para cada clúster. Los scripts internos incluidos en este protocolo requieren la estructura de directorios mostrada anteriormente, donde n es el número de moléculas de agua. Para cada n en gly-h2o-n, hay los siguientes subdirectorios: GA para algoritmo genético con un directorio GA/pm7, QM para mecánica cuántica con QM/pw91-sb para PW91/6-31+G*, QM/pw91-lb para PW91/6-311++G** y QM/pw91-lb/ultrafine para optimizaciones y cálculos vibratorios finales en la integración ultra. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Estructuras representativas de baja energía de Gly(H2O)n-0-5. Estos cúmulos fueron el mínimo global de energía electrónica optimizado en el nivel de teoría PW91/6-311++G**. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Dependencia de la temperatura de Gly(H2O)n-0-5 como 100% de humedad relativa y 1 atm de presión. La concentración de los hidratos se da en unidades de moléculas cm-3. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Dependencia relativa de la humedad de Gly(H2O)n-0-5 como 298.15 K y 1 atm de presión. La concentración de los hidratos se da en unidades de moléculas cm-3. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| E[PW91/6-311++G**] | 216.65 K | 273.15 K | 298.15 K | ||||||||
| LB-UF | ZPVE | H -H | S | G | H -H | S | G | H -H | S | G | |
| Agua | -76.430500 | 13.04 | 1.72 | 42.59 | 5.54 | 2.17 | 44.44 | 3.08 | 2.37 | 45.14 | 1.96 |
| Glicina | -284.434838 | 48.55 | 2.65 | 69.53 | 36.14 | 3.70 | 73.81 | 32.09 | 4.22 | 75.61 | 30.22 |
Tabla 1: Energías monómeros. Las energías electrónicas están en unidades de Hartree mientras que todas las demás cantidades están en unidades de kcal mol-1. El agua y la glicina se optimizaron en el nivel PW91/6-311++G** de teoría y se calcularon las frecuencias vibratorias. Las correcciones termodinámicas para una presión de 1 atm y una temperatura de 298,15 K se calcularon utilizando el script thermo.pl.
| E[PW91/6-311++G**] | 0 K | 216.65 K | 273.15 K | 298.15 K | ||||||||
| N | Nombre | LB-UF | ZPVE | H -H | S | G | H -H | S | G | H -H | S | G |
| 1 | gly-h2o-1 | -360.88481 | 63.96 | 3.61 | 80.12 | 50.22 | 5.12 | 86.27 | 45.52 | 5.85 | 88.83 | 43.33 |
| 2 | gly-h2o-2 | -437.33763 | 79.33 | 4.53 | 90.86 | 64.17 | 6.46 | 98.78 | 58.81 | 7.40 | 102.06 | 56.30 |
| 3 | gly-h2o-3 | -513.78620 | 94.52 | 5.67 | 105.08 | 77.42 | 8.08 | 114.94 | 71.19 | 9.23 | 119.00 | 68.27 |
| 4 | glu-h2o-4 | -590.23667 | 109.80 | 6.03 | 104.98 | 91.30 | 8.78 | 116.21 | 84.40 | 10.11 | 120.87 | 81.14 |
| 5 | gly-h2o-5 | -666.68845 | 125.80 | 7.26 | 121.70 | 106.69 | 10.47 | 134.83 | 99.44 | 12.01 | 140.24 | 96.00 |
Tabla 2: Energías de racimo. Las energías de las estructuras Gly(H2O)n-1-5 de menor energía encontradas utilizando nuestro procedimiento descrito en la Figura 1. Las energías electrónicas están en unidades de Hartree mientras que todas las demás cantidades están en unidades de kcal mol-1.
| Hidratación total: Gly + nH2O <-> Gly(H2O)n | Hidratación secuencial: Gly(H2O)n-1 + H2O <-> Gly(H2O)n | ||||||||||||||||
| E[PW91/6-311++G**] | 216.65 | 273.15 | 298.15 | 216.65 | 273.15 | 298.15 | |||||||||||
| N | nombre del sistema | LB-UF | E(0) | H (T) | G (T) | H (T) | G (T) | H (T) | G (T) | LB-UF | E(0) | H (T) | G (T) | H(T) | G (T) | H (T) | G (T) |
| 1 | gly-h2o-1 | -12.22 | -9.85 | -10.61 | -3.68 | -10.61 | -1.87 | -10.59 | -1.07 | -12.22 | -9.85 | -10.61 | -3.68 | -10.61 | -1.87 | -10.59 | -1.07 |
| 2 | gly-h2o-2 | -26.22 | -21.53 | -23.10 | -9.27 | -23.11 | -5.66 | -23.09 | -4.06 | -14.00 | -11.68 | -12.49 | -5.59 | -12.50 | -3.79 | -12.50 | -2.99 |
| 3 | gly-h2o-3 | -37.56 | -30.72 | -32.88 | -12.90 | -32.87 | -7.69 | -32.82 | -5.38 | -11.34 | -9.19 | -9.78 | -3.63 | -9.76 | -2.03 | -9.73 | -1.32 |
| 4 | glu-h2o-4 | -50.10 | -40.34 | -43.48 | -15.87 | -43.54 | -8.71 | -43.51 | -5.55 | -12.54 | -9.62 | -10.60 | -2.97 | -10.67 | -1.02 | -10.69 | -0.17 |
| 5 | gly-h2o-5 | -63.45 | -51.41 | -55.42 | -20.58 | -55.51 | -11.48 | -55.48 | -7.45 | -13.35 | -11.07 | -11.94 | -4.71 | -11.97 | -2.77 | -11.97 | -1.90 |
Tabla 3: Energías hidratantes. La energía total de hidratación y energía de hidratación secuencial para Gly(H2O)n-1-5 en unidades de kcal mol-1. Aquí, E[PW91/6-311++G**] es el cambio en la energía electrónica, e(0) es el cambio de energía vibratoria de punto cero (ZPVE) corregido en la energía, H (T) es el cambio de entalpía a la temperatura T, y g(T) es el cambio de energía libre de Gibbs de hidratación de cada gly(H2O)n-1-5 cluster.
| Distribución de hidratos de equilibrio en función de la temperatura y la humedad relativa | |||||||||
| T-298.15K | T-273.15K | T-216.65K | |||||||
| Gly(H2O)n | RH-100% | RH-50% | RH-20% | RH-100% | RH-50% | RH-20% | RH-100% | RH-50% | RH-20% |
| 0 | 1.3E+06 | 2.2E+06 | 2.7E+06 | 1.1E+06 | 2.0E+06 | 2.7E+06 | 6.1E+05 | 1.5E+06 | 2.5E+06 |
| 1 | 2.3E+05 | 1.9E+05 | 9.5E+04 | 2.0E+05 | 1.9E+05 | 9.9E+04 | 1.2E+05 | 1.5E+05 | 9.5E+04 |
| 2 | 1.0E+06 | 4.3E+05 | 8.4E+04 | 1.3E+06 | 6.1E+05 | 1.3E+05 | 1.8E+06 | 1.1E+06 | 3.0E+05 |
| 3 | 2.8E+05 | 5.8E+04 | 4.5E+03 | 3.2E+05 | 7.4E+04 | 6.3E+03 | 3.1E+05 | 9.6E+04 | 1.0E+04 |
| 4 | 1.1E+04 | 1.1E+03 | 3.4E+01 | 1.3E+04 | 1.5E+03 | 5.0E+01 | 1.1E+04 | 1.8E+03 | 7.5E+01 |
| 5 | 7.5E+03 | 3.9E+02 | 4.9E+00 | 1.2E+04 | 7.2E+02 | 9.7E+00 | 2.4E+04 | 1.9E+03 | 3.1E+01 |
Tabla 4: Concentraciones de hidratos de equilibrio de Gly(H2O)n-0-5 como temperatura de funcionamiento (T-298.15K, 273.15K, 216.65K) y humedad relativa (RH-100%, 50%, 20%). La concentración de los hidratos se da en unidades de moléculas cm-3 asumiendo valores experimentales56,57,58, de [Gly]0 a 2,9 x 106 cm-3 y [H2O] a 7,7 x 1017 cm-3, 1,6 x 1017 cm-3 y 9,9 x 1014 cm-3 a 100% de humedad relativa y T a 298,15 K, 273.15 K y 216.65 K, respectivamente59.
Archivos suplementarios. Haga clic aquí para descargar estos archivos.
Discusión
La exactitud de los datos generados por este protocolo depende principalmente de tres cosas: (i) la variedad de configuraciones muestreadas por el Paso 2, (ii) la exactitud de la estructura electrónica del sistema, (iii) y la exactitud de las correcciones termodinámicas. Cada uno de estos factores se puede abordar modificando el método editando los scripts incluidos. El primer factor se supera fácilmente con el uso de un grupo inicial más grande de estructuras generadas aleatoriamente, más numerosas iteraciones de la GA y una definición más flexible de los criterios involucrados en el GA. Además, se puede utilizar un método semiempírico diferente, como el modelo autoconsistente de enlace estrecho funcional de densidad de carga (SCC-DFTB)y el modelo eficaz de potencial de fragmentos (EFP)63 para explorar los efectos de diferentes descripciones físicas. La principal limitación aquí es la incapacidad del método para formar o romper enlaces covalentes, lo que significa que los monómeros están congelados. El procedimiento GA sólo encuentra las posiciones relativas más estables de estos monómeros congelados de acuerdo con la descripción semiempírica.
La precisión de la estructura electrónica del sistema se puede mejorar de diversas maneras, cada una con su costo computacional. Uno puede elegir una mejor densidad funcional, como M06-2X64 y wB97X-V65,o el método mecánico cuántico (QM), como el método de perturbación de m-ller-Plesset66,67,68 (MPn) y los métodos acoplados69 (CC) para mejorar la descripción física del sistema. En la jerarquía de las funciones funcionales, el rendimiento generalmente mejora al pasar de las funciones de aproximación de gradiente generalizado (GGA) como PW91 a las funciones híbridas separadas por rango como las funciones híbridas wB97X-D y meta-GGA como M06-2X.
La desventaja de los métodos DFT es que no es posible una convergencia sistemática hacia un valor preciso; sin embargo, los métodos DFT son computacionalmente baratos y hay una amplia variedad de funciones para una amplia variedad de aplicaciones.
Energías calculadas utilizando métodos de función de onda como MP2 y CCSD(T) junto con conjuntos de base consistentes de correlación de número cardinal creciente ([aug-]cc-pV[D,T,Q,...] Z) convergen hacia su límite de conjunto de base completo sistemáticamente, pero el costo computacional de cada cálculo se vuelve prohibitivo a medida que crece el tamaño del sistema. El perfeccionamiento adicional de la estructura electrónica se puede lograr mediante el uso de conjuntos de base explícitamente correlacionados70 y extrapolando al límite71 del conjunto de bases completos (CBS). Nuestro trabajo reciente sugiere que un enfoque perturbador de segundo orden de segundo orden (DF-MP2-F12) explícitamente correlacionado con la densidad produce energías que se acercan a la de los cálculos MP2/CBS32. La modificación del protocolo actual para utilizar diferentes métodos de estructura electrónica implica dos pasos: (i) preparar un archivo de entrada de plantilla siguiendo la sintaxis dada por el software, (ii) y editar los scripts run-pw91-sb.csh, run-pw91-lb.cshy run-pw91-lb-ultrafine.csh para generar la sintaxis correcta del archivo de entrada, así como el script de envío correcto para el software.
Por último, la precisión de las correcciones termodinámicas depende del método de estructura electrónica, así como de la descripción del PES en torno al mínimo global. Una descripción precisa del PES requiere el cálculo de derivados de tercer y mayor orden del PES con respecto a los desplazamientos en los grados nucleares de libertad, como el campo de fuerza cuartica72,73 (QFF), que es una tarea excepcionalmente costosa. El protocolo actual utiliza la aproximación del oscilador armónico a las frecuencias vibratorias, lo que resulta en la necesidad de calcular sólo hasta un segundo derivados del PES. Este enfoque se vuelve problemático en sistemas con alta anarmónica, tales como moléculas muy flácidas y potenciales simétricos de doble pocación debido a la gran diferencia en el verdadero PES y el PES armónico. Además, el costo de tener un PES de alta calidad a partir de un método de estructura electrónica computacionalmente exigente sólo agrava el problema del costo para los cálculos de frecuencia de vibración. Un enfoque para superar esto es utilizar las energías electrónicas de un cálculo de estructura electrónica de alta calidad junto con frecuencias vibratorias calculadas en un PES de menor calidad, lo que resulta en un equilibrio entre costo y precisión. El protocolo actual se puede modificar para utilizar diferentes descripciones PES como se describe en el párrafo anterior; sin embargo, también se pueden editar las palabras clave de frecuencia vibratoria en los scripts y plantillas para calcular las frecuencias vibratorias anarmáticas.
Dos problemas cruciales para cualquier protocolo de muestreo de configuración son el método inicial para muestrear la superficie de energía potencial y los criterios utilizados para identificar cada clúster. Hemos hecho un uso extensivo de una variedad de métodos en nuestro trabajo anterior. Para el primer número, el método inicial para el muestreo de la superficie de energía potencial, hemos tomado la decisión de utilizar GA con métodos semiempíricos basados en estos factores. El muestreo configurational utilizando la intuición química26, muestreo aleatorio, y dinámica molecular (MD)29,30, no encuentra minima global putativo regularmente para racimos de más de 10 monómeros, como observamos en nuestros estudios de cúmulos de agua18. Hemos utilizado con éxito el salto de cuenca (BH) para estudiar el complejo PES de (H2O)1174, pero requería la inclusión manual de algunos isómeros potenciales de baja energía que el algoritmo BH no encontró. Una comparación del desempeño de BH y GA en la búsqueda del mínimo global de los cúmulos de agua, (H2O)n-10-20 demostró que GA constantemente encontró el mínimo global más rápido que BH75. GA como se implementa en OGOLEM y CLUSTER es muy versátil porque se puede aplicar a cualquier clúster molecular y puede interactuar con un gran número de paquetes con campo de fuerza clásico, semiempírica, densidad funcional y capacidades ab initio. La elección de PM7 es impulsada por su velocidad y precisión razonable. Prácticamente cualquier otro método semiempírico tendría un costo computacional significativamente mayor.
En cuanto al segundo número, hemos explorado el uso de diferentes criterios para identificar estructuras únicas que van desde energías electrónicas, momentos de dipolo, RMSD superpuestas y constantes de rotación. El uso de momentos dipolo resultó difícil porque ambos componentes del momento del dipolo dependían de la orientación de la molécula y el momento total del dipolo era muy sensible a las diferencias de geometría de tal manera que era difícil establecer umbrales determinando que las estructuras son las mismas o únicas. Una combinación de energías electrónicas y constantes de rotación resultó ser más útil.
Los criterios actuales para considerar dos estructuras únicas se basan en un umbral de diferencia de energía de 0,10 kcal mol-1 y una diferencia constante rotacional del 1%. Por lo tanto, dos estructuras se consideran diferentes si sus energías difieren en más de 0,10 kcal mol-1 (0,00015 a.u.) Y cualquiera de sus tres constantes de rotación (A, B, C) difieren en más de 1%. Los puntos de referencia internos sustanciales a lo largo de los años constataron que estos umbrales eran opciones razonables. Nuestro enfoque de muestreo de configuración y metodología de cribado se ha aplicado a racimos muy débilmente unidos, como hidrocarburos poliaromáticos complejos con agua76,,77, así como hidratos de sulfato ternario fuertemente unidos que contienen amoníaco y aminas32. Para los cúmulos donde hay diferentes estados de protonación a tener en cuenta, el mejor enfoque es ejecutar varios cálculos de GA, cada uno comenzando con monómeros en diferentes estados de protonación. Esto garantiza que las estructuras con diferentes estados de protonación se consideran cuidadosamente. Sin embargo, los cálculos DFT de bajo nivel a menudo permiten que los estados de protonación cambien durante el transcurso de la optimización de la geometría, lo que produce el estado de protonación más estable independientemente de la geometría inicial.
Nuestros métodos de muestreo de configuración de GA deben funcionar bien incluso para moléculas de disquete, siempre y cuando los códigos GA estén intercomunicados con métodos generales no parametrizados que permitan a los monómeros adoptar diferentes configuraciones durante el transcurso de la ejecución de GA. Por ejemplo, la interconexión de GA con PM7 permitiría que las estructuras de los monómeros cambien, pero si sus lazos se rompen como sucedería cuando cambian los estados de protonación, las estructuras pueden descartarse como candidatos inaceptables.
Hemos considerado diferentes formas de corregir las deficiencias de la aproximación armónica, especialmente las derivadas de frecuencias vibratorias bajas. Incorporar la aproximación cuasi-armónica en la metodología actual no es difícil. Sin embargo, todavía hay preguntas sobre el método cuasi armónico, especialmente cuando se trata de la frecuencia de corte por debajo de la cual se aplicará. Además, no hay trabajos de benchmarking rigurosos que examinen la fiabilidad de la aproximación cuasi-RRHO a pesar de que la sabiduría convencional sugiere que debería ser una mejora con respecto a la aproximación de RRHO.
El protocolo así presentado puede generalizarse en cualquier sistema de cúmulos moleculares de fase de gas no ligados a la cocovalente. También se puede generalizar para utilizar cualquier método semiempírico, método de estructura electrónica y software, y método de análisis de vibración y software mediante la edición de los scripts y plantillas. Esto supone que el usuario se siente cómodo con la interfaz de línea de comandos de Linux, secuencias de comandos de Python y la informática de alto rendimiento. La sintaxis desconocida y el aspecto del sistema operativo Linux y la falta de experiencia en scripting es el mayor escollo en este protocolo y es donde los nuevos estudiantes más luchan. Este protocolo se ha utilizado con éxito en una variedad de implementaciones durante años en nuestro grupo, centrándose principalmente en los efectos del ácido sulfúrico y el amoníaco en la formación de aerosoles. Otras mejoras en este protocolo implicarán una interfaz más robusta para un software de estructura más electrónica, implementaciones alternativas del algoritmo genético y posiblemente el uso de métodos más nuevos para cálculos más rápidos de energías electrónicas y vibratorias. Nuestras aplicaciones actuales de este protocolo están explorando la importancia de los aminoácidos en las primeras etapas de la formación de aerosoles en la atmósfera actual y en la formación de moléculas biológicas más grandes en entornos prebióticos.
Divulgaciones
Ninguno.
Agradecimientos
Este proyecto fue apoyado por las subvenciones CHE-1229354, CHE-1662030, CHE-1721511 y CHE-1903871 de la National Science Foundation (GCS), el Arnold and Mabel Beckman Foundation Beckman Scholar Award (AGG) y la Barry M. Goldwater Scholarship (AGG). Se utilizaron recursos informáticos de alto rendimiento del Consorcio MERCURY (http://www.mercuryconsortium.org).
Materiales
| Name | Company | Catalog Number | Comments |
| Avogadro | https://avogadro.cc | Open-source molecular visualization program | |
| Gaussian [09/16] Software | http://www.gaussian.com/ | Commercial ab initio electronic structure program | |
| MOPAC 2016 | http://openmopac.net/MOPAC2016.html | Open-source semi-empirical program | |
| OGOLEM Software | https://www.ogolem.org | Genetic algorithm-based global optimization program | |
| OpenBabel | http://openbabel.org/wiki/Main_Page | Open-source cheminformatics library | |
| calcRotConsts.py | Shields Group, Department of Chemistry, Furman University | Python script to compute rotational constants | |
| calcSymmetry.csh | Shields Group, Department of Chemistry, Furman University | Shell script to calculate symmetry number of a molecule given Cartesian coordinates | |
| combine-GA.csh | Shields Group, Department of Chemistry, Furman University | Shell script to combine energy and rotational constants from different GA directories | |
| combine-QM.csh | Shields Group, Department of Chemistry, Furman University | Shell script to combine energy and rotational constants from different QM directories | |
| gaussianE.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract Gaussian 09 energies | |
| gaussianFreqs.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract Gaussian 09 vibrational frequencies | |
| getrotconsts | Shields Group, Department of Chemistry, Furman University | Executable to calculate rotational constants given a molecule's Cartesian coordinates | |
| getRotConsts-dft-lb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of large basis DFT optimized structures | |
| getRotConsts-dft-lb-ultrafine.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of ultrafine DFT optimized structures | |
| getRotConsts-dft-sb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of small basis DFT optimized structures | |
| getRotConsts-GA.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of genetic algorithm optimized structures | |
| global-minimum-coords.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of global minimum structures of gly-(h2o)n, where n=0-5 | |
| make-thermo-gaussian.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract data from Gaussian output files and make input files for the thermo.pl script | |
| ogolem-input-file.ogo | Shields Group, Department of Chemistry, Furman University | Ogolem sample input file | |
| ogolem-submit-script.pbs | Shields Group, Department of Chemistry, Furman University | PBS batch submission file for Ogolem calculations | |
| README.docx | Shields Group, Department of Chemistry, Furman University | Clarifications to help readers use the scripts effectively | |
| runogolem.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run OGOLEM | |
| run-pw91-lb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of large basis DFT optimization calculations | |
| run-pw91-lb-ultrafine.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of ultrafine DFT optimization calculations | |
| run-pw91-sb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of small basis DFT optimization calculations | |
| run-thermo-pw91.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute the thermodynamic corrections for a batch of DFT optimized structures | |
| similarityAnalysis.py | Shields Group, Department of Chemistry, Furman University | Python script to determine unique structures based on rotational constants and energies | |
| symmetry | Shields Group, Department of Chemistry, Furman University | Executable to calculate molecular symmetry given Cartesian coordinates | |
| symmetry.c | (C) 1996, 2003 S. Patchkovskii, Serguei.Patchkovskii@sympatico.ca | C code to determine the molecular symmstry of a molecule given Cartesian coordinates | |
| template-marcy.pbs | Shields Group, Department of Chemistry, Furman University | Template for a PBS submit script which uses OGOLEM | |
| template-pw91.com | Shields Group, Department of Chemistry, Furman University | Template Gaussian 09 input | |
| template-pw91-HL.com | Shields Group, Department of Chemistry, Furman University | Template Gaussian 09 input for ultrafine DFT optimization | |
| thermo.pl | https://www.nist.gov/mml/csd/chemical-informatics-research-group/products-and-services/program-computing-ideal-gas | Perl open-source script to compute ideal gas thermodynamic corrections | |
| gly-h2o-n.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet for the complete protocol | |
| table-1.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-2.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-3.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-4.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| water.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of water | |
| glycine.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of glycine |
Referencias
- Foster, P., Ramaswamy, V., Solomon, S., Qin, D., Manning, M., Chen, Z., Marquis, M., Averyt, K. B., Tignor, M., Miller, H. L. . Climate Change 2007 The Scientific Basis. , (2007).
- Kulmala, M., et al. Toward direct measurement of atmospheric nucleation. Science. 318 (5847), 89-92 (2007).
- Sipila, M., et al. The role of sulfuric acid in atmospheric nucleation. Science. 327 (5970), 1243-1246 (2010).
- Jiang, J., et al. First measurement of neutral atmospheric cluster and 1 - 2 nm particle number size distributions during nucleation events. Aerosol Science and Technology. 45 (4), (2011).
- Dunn, M. E., Pokon, E. K., Shields, G. C. Thermodynamics of forming water clusters at various Temperatures and Pressures by Gaussian-2, Gaussian-3, Complete Basis Set-QB3, and Complete Basis Set-APNO model chemistries; implications for atmospheric chemistry. Journal of the American Chemical Society. 126 (8), 2647-2653 (2004).
- Pickard, F. C., Pokon, E. K., Liptak, M. D., Shields, G. C. Comparison of CBSQB3, CBSAPNO, G2, and G3 thermochemical predictions with experiment for formation of ionic clusters of hydronium and hydroxide ions complexed with water. Journal of Chemical Physics. 122, 024302 (2005).
- Pickard, F. C., Dunn, M. E., Shields, G. C. Comparison of Model Chemistry and Density Functional Theory Thermochemical Predictions with Experiment for Formation of Ionic Clusters of the Ammonium Cation Complexed with Water and Ammonia; Atmospheric Implications. Journal of Physical Chemistry A. 109 (22), 4905-4910 (2005).
- Alongi, K. S., Dibble, T. S., Shields, G. C., Kirschner, K. N. Exploration of the Potential Energy Surfaces, Prediction of Atmospheric Concentrations, and Vibrational Spectra of the HO2•••(H2O)n (n=1-2) Hydrogen Bonded Complexes. Journal of Physical Chemistry A. 110 (10), 3686-3691 (2006).
- Allodi, M. A., Dunn, M. E., Livada, J., Kirschner, K. N. Do Hydroxyl Radical-Water Clusters, OH(H2O)n, n=1-5, Exist in the Atmosphere. Journal of Physical Chemistry A. 110 (49), 13283-13289 (2006).
- Kirschner, K. N., Hartt, G. M., Evans, T. M., Shields, G. C. In Search of CS2(H2O)n=1-4 Clusters. Journal of Chemical Physics. 126, 154320 (2007).
- Hartt, G. M., Kirschner, K. N., Shields, G. C. Hydration of OCS with One to Four Water Molecules in Atmospheric and Laboratory Conditions. Journal of Physical Chemistry A. 112 (19), 4490-4495 (2008).
- Morrell, T. E., Shields, G. C. Atmospheric Implications for Formation of Clusters of Ammonium and 110 Water Molecules. Journal of Physical Chemistry A. 114 (12), 4266-4271 (2010).
- Temelso, B., et al. Quantum Mechanical Study of Sulfuric Acid Hydration: Atmospheric Implications. Journal of Physical Chemistry A. 116 (9), 2209 (2012).
- Husar, D. E., Temelso, B., Ashworth, A. L., Shields, G. C. Hydration of the Bisulfate Ion: Atmospheric Implications. Journal of Physical Chemistry A. 116 (21), 5151-5163 (2012).
- Bustos, D. J., Temelso, B., Shields, G. C. Hydration of the Sulfuric Acid – Methylamine Complex and Implications for Aerosol Formation. Journal of Physical Chemistry A. 118 (35), 7430-7441 (2014).
- Wales, D. J., Scheraga, H. A. Global optimization of clusters, crystals, and biomolecules. Science. 27 (5432), 1368-1372 (1999).
- Day, M. B., Kirschner, K. N., Shields, G. C. Global search for minimum energy (H2O)n clusters, n = 3 - 5. The Journal of Physical Chemistry A. 109 (30), 6773-6778 (2005).
- Shields, R. M., Temelso, B., Archer, K. A., Morrell, T. E., Shields, G. C. Accurate predictions of water cluster formation, (H2O)n=2-10. The Journal of Physical Chemistry A. 114 (43), 11725-11737 (2010).
- Temelso, B., Archer, K. A., Shields, G. C. Benchmark structures and binding energies of small water clusters with anharmonicity corrections. The Journal of Physical Chemistry A. 115 (43), 12034-12046 (2011).
- Temelso, B., Shields, G. C. The role of anharmonicity in hydrogen-bonded systems: The case of water clusters. The Journal of Chemical Theory and Computation. 7 (9), 2804-2817 (2011).
- Von Freyberg, B., Braun, W. Efficient search for all low energy conformations of polypeptides by Monte Carlo methods. The Journal of Computational Chemistry. 12 (9), 1065-1076 (1991).
- Rakshit, A., Yamaguchi, T., Asada, T., Bandyopadhyay, P. Understanding the structure and hydrogen bonding network of (H2O)32 and (H2O)33: An improved Monte Carlo temperature basin paving (MCTBP) method of quantum theory of atoms in molecules (QTAIM) analysis. RSC Advances. 7 (30), 18401-18417 (2017).
- Deaven, D. M., Ho, K. M. Molecular geometry optimization with a genetic algorithm. Physical Review Letters. 75, 288-291 (1995).
- Hartke, B. Application of evolutionary algorithms to global cluster geometry optimization. Applications of Evolutionary Computation in Chemistry. , (2004).
- Dieterich, J. M., Hartke, B. OGOLEM: Global cluster structure optimization for arbitrary mixtures of flexible molecules. A multiscaling, object-oriented approach. Molecular Physics. 108 (3-4), 279-291 (2010).
- Herb, J., Nadykto, A. B., Yu, F. Large ternary hydrogen-bonded pre-nucleation clusters in the Earth's atmosphere. Chemical Physics Letters. 518, 7-14 (2011).
- Ortega, I. K., et al. From quantum chemical formation free energies to evaporation rates. Atmospheric Chemistry and Physics. 12 (1), 225-235 (2012).
- Elm, J., Bilde, M., Mikkelsen, K. V. Influence of Nucleation Precursors on the Reaction Kinetics of Methanol with the OH Radical. Journal of Physical Chemistry A. 117 (30), 6695-6701 (2013).
- Loukonen, V., et al. Enhancing effect of dimethylamine in sulfuric acid nucleation in the presence of water - a computational study. Atmospheric Chemistry and Physics. 10 (10), 4961-4974 (2010).
- Temelso, B., Phan, T. N., Shields, G. C. Computational study of the hydration of sulfuric acid dimers: implications for acid dissociation and aerosol formation. Journal of Physical Chemistry A. 116 (39), 9745-9758 (2012).
- Jiang, S., et al. Study of Cl-(H2O)n (n = 1-4) using basin-hopping method coupled with density functional theory. Journal of Computational Chemistry. 35 (2), 159-165 (2014).
- Temelso, B., et al. Effect of mixing ammonia and alkylamines on sulfate aerosol formation. Journal of Physical Chemistry A. 122 (6), 1612-1622 (2018).
- Perdew, J. P., Ruzsinszky, A., Tao, J. Prescription for the design and selection of density functional approximations: More constraint satisfaction with fewer fits. Journal of Chemical Physics. 123, 062201 (2005).
- Riplinger, C., Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. Journal of Chemical Physics. 138, 034106 (2013).
- Riplinger, C., Pinski, P., Becker, U., Valeev, E. F., Neese, F. Sparse maps--A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory. Journal of Chemical Physics. 144 (2), 024109 (2016).
- Kildgaard, J. V., Mikkelsen, K. V., Bilde, M., Elm, J. Hydration of atmospheric molecular clusters: a new method for systematic configurational sampling. Journal of Physical Chemistry A. 122 (22), 5026-5036 (2018).
- González, &. #. 1. 9. 3. ;. Measurement of areas on a sphere Using Fibonacci and latitude-longitude lattices. Mathematical Geosciences. 42, 49-64 (2010).
- Karaboga, D., Basturk, B. On the performance of artificial bee colony (ABC) algorithm. Applied Soft Computing. 8 (1), 687-697 (2008).
- Zhang, J., Doig, M. Global optimization of rigid molecules using the artificial bee colony algorithm. Physical Chemistry Chemical Physics. 18 (4), 3003-3010 (2016).
- Kubecka, J., Besel, V., Kurten, T., Myllys, N., Vehkamaki, H. Configurational sampling of noncovalent (atmospheric) molecular clusters: sulfuric acid and guanidine. Journal of Physical Chemistry A. 123 (28), 6022-6033 (2019).
- Grimme, S., Bannwarth, C., Shushkov, P. A Robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent Interactions of large molecular systems parametrized for all spd-block elements (Z = 1-86). Journal of Chemical Theory and Computation. 13 (5), 1989-2009 (2017).
- Buck, U., Pradzynski, C. C., Zeuch, T., Dieterich, J. M., Hartke, B. A size resolved investigation of large water clusters. Physical Chemistry Chemical Physics. 16 (15), 6859 (2014).
- Forck, R. M., et al. Structural diversity in sodium doped water trimers. Physical Chemistry Chemical Physics. 14 (25), 9054-9057 (2012).
- Witt, C., Dieterich, J. M., Hartke, B. Cluster structures influenced by interaction with a surface. Physical Chemistry Chemical Physics. 20 (23), 15661-15670 (2018).
- Freitbert, A., Dieterich, J. M., Hartke, B. Exploring self-organization of molecular tether molecules on a gold surface by global structure optimization. The Journal of Computational Chemistry. 40 (22), 1978-1989 (2019).
- Stewart, J. J. P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. The Journal of Molecular Modeling. 19 (1), 1-32 (2013).
- Burke, K., Perdew, J. P., Wang, Y. Derivation of a generalized gradient approximation: The PW91 density functional. Electronic Density Functional Theory. , 81-111 (1998).
- Frisch, M. J., et al. . Gaussian 09, Revision A.02. , (2016).
- Ditchfield, R., Hehre, W. J., Pople, J. A. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. The Journal of Chemical Physics. 54 (2), 724 (1971).
- Elm, J., Bilde, M., Mikkelsen, K. V. Assessment of density functional theory in predicting structures and free energies of reaction of atmospheric prenucleation clusters. The Journal of Chemical Theory and Computation. 8 (6), 2071-2077 (2012).
- Elm, J., Mikkelsen, K. V. Computational approaches for efficiently modelling of small atmospheric clusters. Chemical Physics Letters. 615, 26-29 (2014).
- Bayucan, A., et al. . PBS Portable Batch System. , (1999).
- O'Boyle, N. M., et al. Open Babel: An open chemical toolbox. Journal of Cheminformatics. 3, 33 (2011).
- Csaszar, A. G. Conformers of gaseous glycine. Journal of the American Chemical Society. 114 (24), 9568-9575 (1992).
- Zhang, Q., Anastasio, C. Free and combined amino compounds in atmospheric fine particles (PM2.5) and fog waters from Northern California. Atmospheric Environment. 37 (16), 2247-2258 (2003).
- Matsumoto, K., Uematsu, M. Free amino acids in marine aerosols over the western North Pacific Ocean. Atmospheric Environment. 39 (11), 2163-2170 (2005).
- Mandalakis, M., Apostolaki, M., Stephanou, E. G. Trace analysis of free and combined amino acids in atmospheric aerosols by gas chromatography-mass spectrometry. Journal of Chromatography A. 1217 (1), 143-150 (2010).
- Seinfeld, J. H., Pandis, S. N. . Atmospheric Chemistry and Physics, 3rd Ed. , (2016).
- Myllys, N., Elm, J., Halonen, R., Kurten, T., Vehkamaki, H. Coupled cluster evaluation of atmospheric acid-base clusters with up to 10 molecules. The Journal of Physical Chemistry A. 120 (4), 621-630 (2016).
- Elm, J., Bilde, M., Mikkelsen, K. V. Assessment of binding energies of atmospherically relevant clusters. Physical Chemistry Chemical Physics. 15 (39), (2013).
- Elstner, M. The SCC-DFTB method and its application to biological systems. Theoretical Chemistry Accounts. 116 (1-3), 316-325 (2006).
- Kaliman, I. A., Slipchenko, L. V. LIBEFP: A new parallel implementation of the effective fragment potential method as a portable software library. The Journal of Computational Chemistry. 34 (26), 2284-2292 (2013).
- Zhao, Y., Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and trasition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theoretical Chemistry Accounts. 120 (1-3), 215-241 (2008).
- Mardirossian, N., Head-Gordon, M. wB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy. Physical Chemistry Chemical Physics. 16 (21), 9904-9924 (2014).
- Head-Gordon, M., Pople, J. A., Frisch, M. J. MP2 energy evaluation by direct methods. Chemical Physics Letters. 153 (6), 503-506 (1988).
- Pople, J. A., Seeger, R., Krishnan, R. Variational configuration interaction methods and comparison with perturbation theory. The International Journal of Quantum Chemistry. 12, 149-163 (1977).
- Pople, J. A., Binkley, J. S., Seeger, R. Theoretical models incorporating electron correlation. The International Journal of Quantum Chemistry. 10 (10), 1-19 (1976).
- Monkhorst, H. J. Calculation of properties with the coupled-cluster method. The International Journal of Quantum Chemistry. 12 (11), 421-432 (1977).
- Klopper, W., Manby, F. R., Ten-No, S., Valeev, E. F. R12 methods in explicitly correlated molecular electronic structure theory. International Reviews in Physical Chemistry. 25, 427-468 (2006).
- Hattig, C. Optimization of auxiliary basis sets for RI-MP2 and RI-CC2 calculations: Core-valence and quintuple-z basis sets for H to Ar and QZVPP basis sets for Li to Kr. Physical Chemistry Chemical Physics. 7 (1), 59-66 (2005).
- Barone, V. Anharmonic vibrational properties by a fully automated second-order perturbative approach. The Journal of Chemical Physics. 122, 014108 (2005).
- Barone, V. Vibrational zero-point energies and thermodynamic functions beyond the harmonic approximation. The Journal of Chemical Physics. 120 (7), 3059-3065 (2004).
- Temelso, B., et al. Exploring the Rich Potential Energy Surface of (H2O)11 and Its Physical Implications. Journal of Chemical Theory and Computation. 14 (2), 1141-1153 (2018).
- Kabrede, H., Hentschke, R. Global minima of water clusters (H2O)N, N≤25, described by three empirical potentials. Journal of Physical Chemistry B. 107 (16), (2003).
- Steber, A. L., et al. Capturing the Elusive Water Trimer from the Stepwise Growth of Water on the Surface of a Polycyclic Aromatic Hydrocarbon Acenaphthene. Journal of Physical Chemistry Letters. 8 (23), 5744-5750 (2017).
- Perez, C., et al. Corrannulene and its complex with water: A tiny cup of water. Physical Chemistry Chemical Physics. 19 (22), 14214-14223 (2017).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados