
Method Article
Computation of Atmospheric Concentrations of Molecular Clusters from ab initio Thermochemistry
In This Article
Summary
The atmospheric concentrations of weakly bound molecular clusters can be computed from the thermochemical properties of low energy structures found through a multi-step configurational sampling methodology utilizing a genetic algorithm and semi-empirical and ab initio quantum chemistry.
Abstract
The computational study of the formation and growth of atmospheric aerosols requires an accurate Gibbs free energy surface, which can be obtained from gas phase electronic structure and vibrational frequency calculations. These quantities are valid for those atmospheric clusters whose geometries correspond to a minimum on their potential energy surfaces. The Gibbs free energy of the minimum energy structure can be used to predict atmospheric concentrations of the cluster under a variety of conditions such as temperature and pressure. We present a computationally inexpensive procedure built on a genetic algorithm-based configurational sampling followed by a series of increasingly accurate screening calculations. The procedure starts by generating and evolving the geometries of a large set of configurations using semi-empirical models then refines the resulting unique structures at a series of high-level ab initio levels of theory. Finally, thermodynamic corrections are computed for the resulting set of minimum-energy structures and used to compute the Gibbs free energies of formation, equilibrium constants, and atmospheric concentrations. We present the application of this procedure to the study of hydrated glycine clusters under ambient conditions.
Introduction
The most uncertain parameter in atmospheric studies of climate change is the exact extent to which cloud particles reflect incoming solar radiation. Aerosols, which are particulate matter suspended in a gas, form cloud particles called cloud condensation nuclei (CCN) that scatter incoming radiation, thus preventing its absorption and the subsequent heating of the atmosphere1. A detailed understanding of this net cooling effect requires an understanding of the growth of aerosols into CCNs, which in turn requires an understanding of the growth of small molecular clusters into aerosol particles. Recent work has suggested that aerosol formation is initiated by molecular clusters of 3 nm in diameter or less2; however, this size regime is difficult to access using experimental techniques3,4. Therefore, a computational modeling approach is desired in order to overcome this experimental limitation.
Using our modeling approach described below, we can analyze the growth of any hydrated cluster. Because we are interested in the role of water in the formation of large biological molecules from smaller constituents in pre-biotic environments, we illustrate our approach with glycine. The challenges encountered and tools needed to address those research questions are very similar to those involved in the study of atmospheric aerosols and prenucleation clusters5,6,7,8,9,10,11,12,13,14,15. Here, we examine hydrated glycine clusters starting from an isolated glycine molecule followed by a series of stepwise additions of up to five water molecules. The final goal is to calculate the equilibrium concentrations of Gly(H2O)n=0-5 clusters in the atmosphere at room temperature at sea-level and a relative humidity (RH) of 100 %.
A small number of these sub-nanometer molecular clusters grow into a metastable critical cluster (1-3 nm in diameter) either by adding other vapor molecules or coagulating on existing clusters. These critical clusters have a favorable growth profile leading to the formation of much larger (up to 50-100 nm) cloud condensation nuclei (CCN), which directly affect the precipitation efficiency of clouds as well their ability to reflect incident light. Therefore, having a good understanding of the thermodynamics of molecular clusters and their equilibrium distributions should lead to more accurate predictions of the impact of aerosols on the global climate.
A descriptive model of aerosol formation requires accurate thermodynamics of molecular cluster formation. The computation of accurate thermodynamics of molecular cluster formation requires the identification of the most stable configurations, which involves finding the global and local minima on the cluster's potential energy surface (PES)16. This process is called configurational sampling and can be achieved through a variety of techniques, including those based on molecular dynamics (MD)17,18,19,20, Monte Carlo (MC)21,22, and genetic algorithms (GA)23,24,25.
Different protocols have been developed over the years to obtain the structure and thermodynamics of atmospheric hydrates at a high level of theory. These protocols differed in the choice of (i) configurational sampling method, (ii) nature of low-level method used in the configurational sampling, and (iii) the hierarchy of higher-level methods used to refine the results in the subsequent steps.
The configurational sampling methods included chemical intuition26, random sampling27,28, molecular dynamics (MD)29,30, basin hopping (BH)31, and genetic algorithm (GA)24,25,32. The most common low-level methods employed with these sampling methods are force fields or semi-empirical models such as PM6, PM7 and SCC-DFTB. These are often followed by DFT calculations with increasingly larger basis sets and more reliable functionals from the higher rungs of Jacob's ladder33. In some cases, these are followed by higher level wavefunction methods such as MP2, CCSD(T), and the cost efficient DLPNO-CCSD(T)34,35.
Kildgaard et al.36 developed a systematic method where water molecules are added at points on the Fibonacci spheres37 around smaller hydrated or unhydrated clusters to generate candidates for larger clusters. Unphysical and redundant candidates are removed based on close contact thresholds and root-mean-square distance between different conformers. Subsequent optimizations using the PM6 semi-empirical method and a hierarchy of DFT and wavefunction methods are used to get a set of low energy conformers at a high level of theory.
The artificial bee colony (ABC) algorithm38 is a new configurational sampling approach that has recently been implemented by Zhang et al. to study molecular clusters in a program called ABCluster39. Kubecka et al.40 used ABCluster for configurational sampling followed by low-level reoptimizations using the tight-binding GFN-xTB semi-empirical method41. They further refined the structures and energies using DFT methods followed by final energies using DLPNO-CCSD(T).
Regardless of the method, configurational sampling starts with a randomly- or nonrandomly-generated distribution of points on the PES. Each point corresponds to a specific geometry of the molecular cluster in question and is generated by the sampling method. Then the closest local minimum is found for each point by following the "downhill" direction on the PES. The set of minima thus found correspond to those geometries of the molecular cluster that are stable, at least for some time. Here, the shape of the PES and the evaluation of the energy at each point on the surface will be sensitive to the physical description of the system where a more accurate physical description results in a more computationally expensive energy calculation. We will specifically use the GA method implemented in the OGOLEM25 program, which has been successfully applied to a variety of global optimization and configurational sampling problems42,43,44,45, to generate the initial set of sampling points. The PES will be described by the PM7 model46 implemented in the MOPAC2016 program47. This combination is employed because it generates a larger variety of points compared to the MD and MC methods and finds the local minima faster than more-detailed descriptions of the PES.
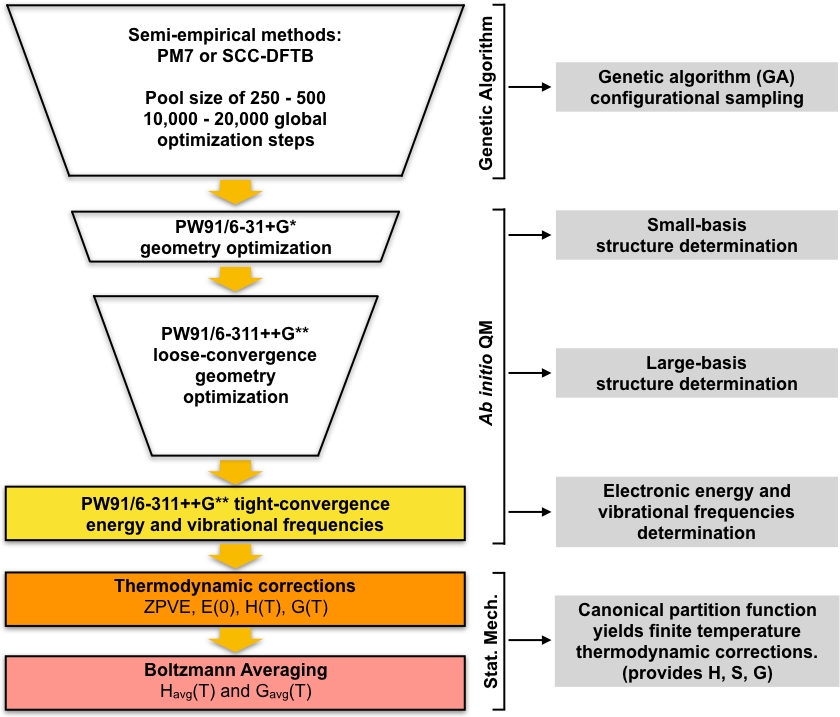
The set of GA-optimized local minima are taken as the starting geometries for a series of screening steps, which lead to a set of low-lying minimum energy. This part of the protocol begins by optimizing the set of unique GA-optimized structures using density-functional theory (DFT) with a small basis set. This set of optimizations will generally give a smaller set of unique local minimum structures which are modeled in more detail compared to the GA-optimized semi-empirical structures. Then another round of DFT optimizations are performed on this smaller set of structures using a larger basis set. Again, this step will generally give a smaller set of unique structures which are modeled in more detail compared to the small basis DFT step. The final set of unique structures are then optimized to a tighter convergence and the harmonic vibrational frequencies are calculated. After this step we have everything we need to compute the equilibrium concentrations of the clusters in the atmosphere. The overall approach is summarized diagrammatically in Figure 1. We will use the PW9148 generalized-gradient approximation (GGA) exchange-correlation functional in the Gaussian0949 implementation of DFT along with two variations of the Pople50 basis set (6-31+G* for the small basis step and 6-311++G** for the large basis step). This particular combination of exchange-correlation functional and basis set was chosen due to its previous success in computing accurate Gibbs free energies of formation for atmospheric clusters51,52.
This protocol assumes that the user has access to a high-performance computing (HPC) cluster with the portable batch system53 (PBS), MOPAC2016 (http://openmopac.net/MOPAC2016.html)47, OGOLEM (https://www.ogolem.org)25, Gaussian 09 (https://gaussian.com)49, and OpenBabel54 (http://openbabel.org/wiki/Main_Page) software installed following their specific installation instructions. Each step in this protocol also uses a set of in-house shell and Python 2.7 scripts which must be saved to a directory that is included in the user's $PATH environmental variable. All necessary environmental modules and execution permissions to run all of the above programs must also be loaded into the user's session. The disk and memory usage by the GA code (OGOLEM) and semi-empirical codes (MOPAC) are very small by modern computer resource standards. The overall memory and disk usage for OGOLEM/MOPAC depends on how many threads one wants to use, and even then, the resource usage will be small compared to the capabilities of most HPC systems. The resource needs of the QM methods depend on the size of the clusters and the level of theory used. The advantage of using this protocol is that one can vary the level of theory to be able to calculate the final set of low energy structures, keeping in mind that usually faster calculations lead to more uncertainty in accuracy of the results.
For the sake of clarity, the user's local computer will be referred to as "local computer" while the HPC cluster they have access to will be referred to as "remote cluster".
Protocol
1. Finding the minimum energy structure of isolated glycine and water
NOTE: The goal here is two-fold: (i) to obtain minimum energy structures of isolated water and glycine molecules for use in the genetic algorithm configurational sampling, (ii) and to compute the thermodynamic corrections to the gas phase energies of these molecules for use in the calculation of atmospheric concentrations.
- On your local computer, open a new session of Avogadro.
- Click Build > Insert > Peptide and select Gly from the Insert Peptide window to generate a glycine monomer in the visualization window.
- Click Extensions > Gaussian and edit the first line in the text box to read '# pw91pw91/6-311++G** int(Acc2E=12,UltraFine) scf(conver=12) opt(tight,maxcyc=300) freq'. Click Generate and save the input file as glycine.com.
- Please note that if the molecule has significant conformational flexibility, as glycine does55, it is critical to perform conformational analysis to identify the global minimum structure and other low-lying conformers. OpenBabel54 provides robust conformational search tools utilizing different algorithms and quick force fields. While conformers are allowed to relax and interconvert during GA and subsequent calculations, it is sometimes necessary to run multiple GA calculations, each starting with a different conformer.
- On your local computer, open a new session in Avogadro.
- Click Build > Insert > Fragment and search for "water" from the Insert Fragment window to get the coordinates of water.
- Click Extensions > Gaussian and edit the first line in the text box to read '# pw91pw91/6-311++G** int(Acc2E=12,UltraFine) scf(conver=12) opt(tight,maxcyc=300) freq'. Click Generate and save the input file as water.com.
- Transfer the two .com files to the remote cluster. Once you log into the remote cluster, call Gaussian 09 in a batch submission script to start the calculation. When the calculations finish, extract the Cartesian coordinates (.xyz files) of the minimum energy structures by calling OpenBabel. For glycine, the command to execute is:
obabel -ig09 glycine.log -oxyz > glycine.xyz
These two .xyz files will be used by the GA configurational sampling in the next step.
2. Genetic-algorithm-based configurational sampling of Gly(H2O)n=1-5 clusters
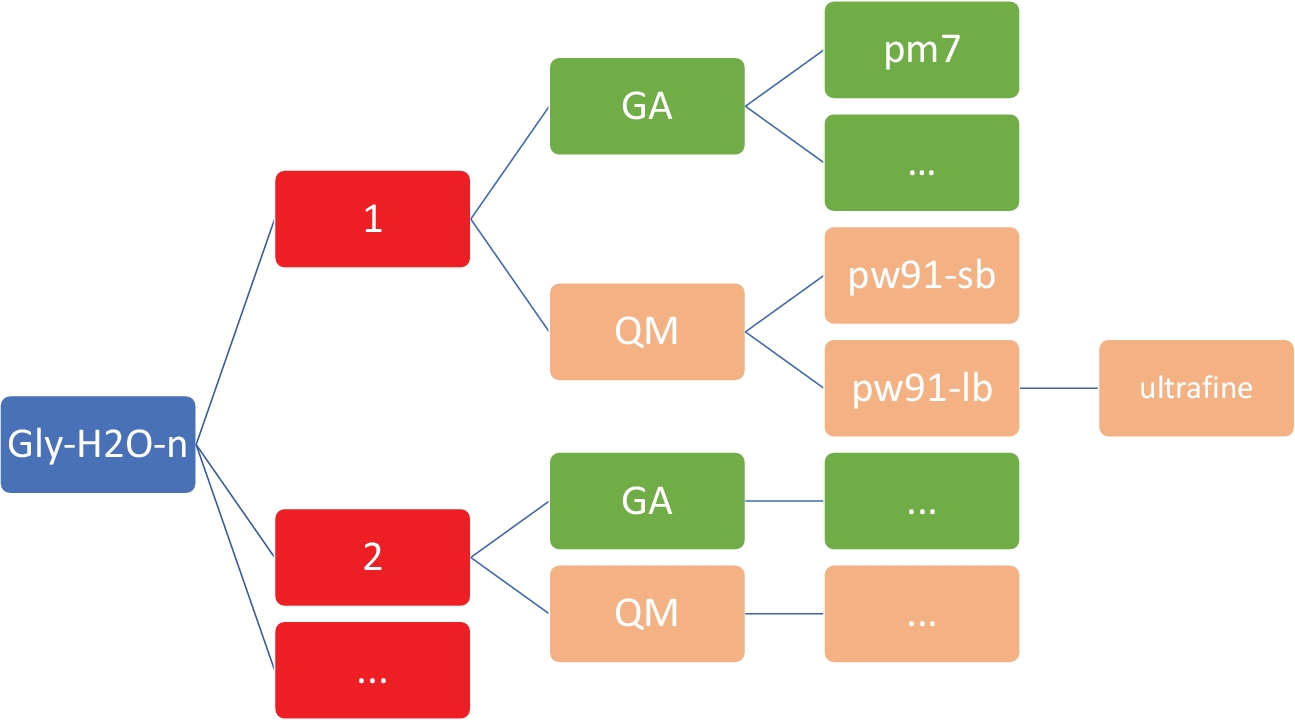
NOTE: The goal here is to obtain a set of low-energy structures for Gly(H2O)n=1-5 at the inexpensive semi-empirical level of theory, using the PM746 model implemented in MOPAC47. It is imperative that the working directory has the exact organization and structure as shown in Figure 2. This is to ensure that the custom shell and Python scripts work without failures.
- Copy all necessary scripts to the remote cluster and add their location to $PATH
- Put all scripts and template files to a folder (Eg. scripts) and copy it to the remote cluster
- Make sure all the scripts are executable
- Add the location of the scripts directory to the $PATH environmental variable by entering the following commands in a terminal. The default location of the scripts is set to $HOME/JoVE-demo/scripts, however, one can define an environmental variable called $SCRIPTS_HOME pointing to the directory containing the scripts and add $SCRIPTS_HOME to one's path
- Bash shell:
export SCRIPTS_HOME=/path/to/scripts
export PATH=${SCRIPTS_HOME}:${PATH} - Tcsh/Csh shell:
setenv SCRIPTS_HOME /path/to/scripts
setenv PATH ${SCRIPTS_HOME}:${PATH}
- Bash shell:
- On the remote cluster, set up and run a GA calculation:
- Create a directory called gly-h2o-n where n is the number of water molecules.
- Create a subdirectory called GA under the gly-h2o-n directory to run genetic algorithm calculations.
- Copy the OGOLEM input files (Eg. pm7.ogo), monomers Cartesian coordinates (Eg. glycine.xyz, water.xyz) and PBS batch submission script (Eg. run.pbs) into the GA directory.
- Make the necessary changes to the OGOLEM input file and batch submission file.
- Submit the calculation. When the calculation starts, OGOLEM will create a new directory named as the prefix of the OGOLEM input file (Eg. pm7) in the GA directory and store newly generated coordinates there.
- Once the calculation is complete, compile the energies and rotational constants, and use that information to determine which are the unique low-energy structures:
- Change directory to gly-h2o-n/GA/pm7 and
- Extract the energies and compute the rotational constants of the GA-optimized clusters with the command:
getRotConsts-GA.csh N 0 99
where N is the number of atoms in the molecular cluster and '0 99' indicates that the GA pool size is 100, with indices running from 0 through 99. This will generate a file called rotConstsData_C which contains a sorted list of all the GA-optimized cluster configurations, their energies, and their rotational constants. - Execute the command:
similarityAnalysis.py pm7 rotConstsData_C
where pm7 will be used as a file-naming label, to find and save the unique GA-optimized clusters. This will generate a file called uniqueStructures-pm7.data which contains a sorted list of the unique GA-optimized configurations. This is a list of unique local minimum structures for the Gly(H2O)n cluster optimized at the PM7 level of theory, and these structures are now ready to be refined using DFT.
- Go up to the gly-h2o-n/GA directory and combine the results from multiple comparable GA runs using the combine-GA.csh script. The syntax is:
combine-GA.csh <label> <list of directories with GA runs>
In this particular case, the command:
combine-GA.csh pm7 pm7
will generate a new unique structures list named 'uniqueStructures-pm7.data' in the gly-h2o-n/GA directory.
3. Refinement using QM method with a small basis set
NOTE: The goal here is to refine the configurational sampling of the Gly(H2O)n=1-5 clusters using a better quantum-mechanical description to obtain a smaller but more accurate set of Gly(H2O)n=1-5 cluster structures. The starting structures for this step are the outputs of Step 2.
- Prepare and run the small basis set DFT calculation:
- Create a subdirectory called QM under the gly-h2o-n directory. Under the QM directory, create another subdirectory named pw91-sb.
- Copy the unique structures list (uniqueStructures-pm7.data) from the gly-h2o-n/GA directory to the QM/pw91-sb directory.
- Change directory to that gly-h2o-n/QM/pw91-sb.
- Run the small basis set DFT configurational sampling script using the command:
run-pw91-sb.csh uniqueStructures-pm7.data sb QUEUE 10
where sb is a label for this set of calculations, QUEUE is the preferred queue on the computing cluster, and 10 indicates that 10 calculations are to be grouped into one batch job. This script will automatically generate the inputs for Gaussian 09 and submit all the calculations. Enter 'test' for the 'QUEUE' to do a dry run.
- Once the submitted calculations are complete, extract and analyze the results.
- Extract the energies and compute the rotational constants of the small-basis-optimized clusters using the command:
getRotConsts-dft-sb.csh pw91 N
where pw91 indicates that the PW91 density functional was used, and N is the number of atoms in the cluster. That will create a file named rotConstsData_C. - Now identify the unique structures with the command:
similarityAnalysis.py sb rotConstsData_C
where sb is used as a file-naming label. There will now be a list of unique configurations optimized at the PW91/6-31+G* level of theory saved in the file uniqueStructures-sb.data.
- Extract the energies and compute the rotational constants of the small-basis-optimized clusters using the command:
- Go up to the gly-h2o-n/QM directory and combine the results from multiple comparable QM runs using the combine-QM.csh script. The syntax is:
combine-QM.csh <label> <list of directories with QM calcs>
In this particular case, the command:
combine-QM.csh sb pw91-sb
will generate a new unique structures list named 'uniqueStructures-sb.data' in the gly-h2o-n/QM directory.
4. Further refinement using QM method with a large basis set
NOTE: The goal here is to further refine the configurational sampling of the Gly(H2O)n=1-5 clusters using a better quantum-mechanical description. The starting structures for this step are the outputs of Step 3.
- Submit more reliable calculations using a larger basis set.
- Create a subdirectory called pw91-lb under the QM directory.
- Copy the unique structures list (uniqueStructures-sb.data) from the gly-h2o-n/QM directory to the gly-h2o-n/QM/pw91-lb directory and change to that directory.
- Run the large-basis DFT configurational sampling script with the command:
run-pw91-lb.csh uniqueStructures-sb.data lb QUEUE 10
where lb is a label for this set of calculations, QUEUE is the preferred queue on the computing cluster, and 10 indicates that 10 calculations are to be grouped into one batch job. This script will automatically generate the inputs for Gaussian 09 and submit all the calculations. Enter 'test' for the 'QUEUE' to do a dry run testing.
- Once the submitted calculations are complete, extract and analyze the data
- Compute the rotational constants of the large-basis-optimized clusters with the command:
getRotConsts-dft-lb.csh pw91 N
where pw91 indicates that the PW91 density functional was used, and N is the number of atoms in the cluster. - Now identify the unique structures with the command:
similarityAnalysis.py lb rotConstsData_C
where lb is used as a file-naming label. You now have a list of unique configurations optimized at the PW91/6-311++G** level of theory saved in the file uniqueStructures-lb.data.
- Compute the rotational constants of the large-basis-optimized clusters with the command:
5. Final Energy and Thermodynamic Correction Calculations
NOTE: The goal here is to obtain the vibrational structure and energies of the Gly(H2O)n=1-5 clusters using a large basis set and an ultrafine integration grid in order to compute the desired thermochemical corrections.
- Starting with results from the previous step, submit more reliable calculations.
- Create a subdirectory called ultrafine under the QM/pw91-lb directory. Then copy the unique structures list (uniqueStructures-lb.data) from the QM/pw91-lb directory to the QM/pw91-lb/ultrafine directory and change to that directory.
- Submit the ultrafine large-basis DFT script with the command:
run-pw91-lb-ultrafine.csh uniqueStructures-lb.data uf QUEUE 10
where uf is a label for this set of calculations, QUEUE is the preferred queue on the computing cluster, and 10 indicates that 10 calculations are to be grouped into one batch job. This script will automatically generate the inputs for Gaussian 09 and submit all the calculations. Enter 'test' for the 'QUEUE' to do a dry run testing.
- Once the submitted calculations are complete, extract and analyze the data
- Extract the energies and compute the rotational constants of the large-basis-optimized clusters with the command:
getRotConsts-dft-lb-ultrafine.csh pw91 N
where pw91 indicates that the PW91 density functional was used, and N is the number of atoms in the cluster. - Now identify the unique structures with the command:
similarityAnalysis.py uf rotConstsData_C
where uf is used as a file-naming label. You now have a list of unique configurations optimized at the PW91/6-311++G** level of theory saved in the file uniqueStructures-uf.data.
- Extract the energies and compute the rotational constants of the large-basis-optimized clusters with the command:
- Perform a final extraction of information needed to calculate thermodynamic corrections. Use that information to compute the thermodynamic corrections.
- Extract the final electronic energies, rotational constants and vibrational frequencies, and use them to calculate thermodynamic corrections using the command:
run-thermo-pw91.csh uniqueStructures-uf.data - Copy/paste the command-line output to the 'Raw_Energies' sheet of the Excel spreadsheet named 'gly-h2o-n.xlsx'. You would need to do this for the monomers (glycine and water) as well as the lowest energy member of each hydrate (gly-h2o-n, where n=1,2, …).
- As the raw energies are added to the first sheet of the 'gly-h2o-n.xlsx' spreadsheet, the subsequent 'Binding_Energies' and 'Hydrate_Distribution' sheets are automatically updated. In particular, the 'Hydrate_Distribution' sheet yields the equilibrium concentration of hydrates at different temperatures (Eg. 298.15K), relative humidity (20%, 50%, 100%) and initial concentrations of water ([H2O]) and glycine ([Glycine]). The theory behind these calculations is described in the next step.
- Extract the final electronic energies, rotational constants and vibrational frequencies, and use them to calculate thermodynamic corrections using the command:
6. Computing atmospheric concentrations of Gly(H2O)n=0-5 clusters at room temperature at sea-level
NOTE: This is accomplished by first copying the thermodynamic data generated in the previous step into a spreadsheet and calculating the Gibbs free energies of sequential hydration. Then, the Gibbs free energies are used to calculate equilibrium constants for each sequential hydration. Finally, a set of linear equations are solved to get the equilibrium concentration of the hydrates for a given concentration of monomers, temperature and pressure.
- Start by setting up a system of chemical equilibria for the sequential hydration of glycine as shown below:

- Compute the equilibrium constants Kn using Kn = e-ΔGn/(kBT), where n is the level of hydration, ΔGn is the Gibbs free energy change of the nth hydration reaction, kB is Boltzmann's constant, and T is temperature.

- Set up the equation for the conservation of mass, using the assumption that the sum of the equilibrium concentrations of the hydrated and un-hydrated glycine clusters equals the initial concentration of isolated glycine [Gly]0. Rewrite this system of six simultaneous equations, using some algebraic rearrangement of the equilibrium constant expressions, as

- Solve the system of equations shown above to obtain the equilibrium concentrations of Gly(H2O)n = 0-5 using an experimental value56,57,58 for the concentration of glycine in the atmosphere, [Gly]0 = 2.9 x 106 cm-3, and the concentration of water in the atmosphere at 100% relative humidity and a temperature of 298.15 K59, [H2O] = 7.7 x 1017 cm-3.
Results
The first set of results from this protocol should be a set of low-energy structures of Gly(H2O)n=1-5 found through the configurational sampling procedure. These structures have been optimized at the PW91/6-311++G** level of theory and are assumed to be accurate for the purpose of this paper. There is no evidence to suggest that PW91/6-311++G** consistently underestimates or overestimates the binding energy of these clusters. Its ability to predict binding energies relative to MP2/CBS32 and [DLPNO-]CCSD(T)/CBS60,61 estimates and experiment52 shows a lot of fluctuations. The same is true of most other density functionals. Generally, each value of n = 1 – 5 should yield a handful of low-energy structures within around 5 kcal mol-1 of the lowest-energy structure. Here, we focus on the first structure produced by the run-thermo-pw91.csh script for brevity. Figure 3 shows the lowest electronic energy isomers of Gly(H2O)n=0-5 clusters. One can see that the hydrogen bond network grows in complexity as the number of water molecules increases, and even goes from a mostly planar network to a three-dimensional cage-like structure at n = 5. The rest of this text uses the energies and thermodynamic quantities corresponding to these five specific clusters.
Table 1 contains the thermodynamic quantities necessary to carry out the protocol. Table 2 shows an example of the output of the run-thermo-pw91.csh script where the electronic energies, vibrational zero-point corrections, and the thermodynamic corrections at three different temperatures are printed. For each cluster (row), E[PW91/6-311++G**] corresponds to the gas phase electronic energies at the PW91/6-311++G** level of theory calculated on ultrafine integration grids in units of Hartree, as well as the zero-point vibrational energy (ZPVE) in units of kcal mol-1. At each temperature, 216.65 K, 273.15 K, and 298.15 K, the thermodynamic corrections are listed, ∆H the enthalpy of formation in units of kcal mol-1, S the entropy of formation in units of cal mol-1, and ∆G the Gibbs free energy of formation in units of kcal mol-1. Table 3 shows an example computation of the total Gibbs free energy change of hydration, as well as for sequential hydration. An example computation of the total Gibbs free energy change of hydration for the reaction

starts with the computation of the electronic energy EPW91 as

where EPW91[Gly∙(H2O)] is taken from Table 2 column C, and EPW91[Gly] and EPW91[H2O] are taken from Table 1 column B. Next we calculate the total gas phase energy change ΔE(0) by including the change in the zero-point vibrational energy of the reaction as

to obtain column D. Here, ΔEPW91/6−311++G** is taken from Table 3 column C, EZPVE[Gly ∙ (H2O)] from Table 2 column D, and EZPVE[Gly] and EZPVE[H2O] from Table 1 column C. For the sake of brevity, we will move on to room temperature clusters, so we skip over the 216.65 K and 273.15 K data. At room temperature, we then calculate the enthalpy change of the reaction ΔH by correcting the gas phase energy change as

where ΔE(0) is taken from Table 3 column D, ΔH[Gly∙(H2O)] is taken from Table 2 column K, and ΔH[Gly] and ΔH[H2O] are taken from Table 1 column J. Finally, we calculate the Gibbs free energy change of the reaction ΔG as

where ΔH is taken from Table 3 column I, S[Gly∙(H2O)] is taken from Table 2 column L, and S[Gly] and S[H2O] are taken from Table 1 column K. Note here that the entropy values must be converted to units of kcal mol-1 K-1 during this step.
We now have the necessary quantities to compute the atmospheric concentrations of hydrated glycine as shown in Step 6. The results should resemble the data shown in Table 4, but small numerical differences are to be expected. Table 4 shows the equilibrium hydrate concentrations found from the formulation of the system of six equations in Step 6.2 into one matrix equation and its subsequent solution. We start by acknowledging the fact that the system of equations can be written as

where Kn is the equilibrium constant for the nth sequential hydration of glycine, w is the concentration of water in the atmosphere, g is the initial concentration of isolated glycine in the atmosphere, and gn is the equilibrium concentration of Gly(H2O)n. If we rewrite the above equation as Ax = b, we get x = A−1b where A−1 is the inverse of matrix A. This inverse can be easily computed using built-in spreadsheet functions as shown in Table 4 to obtain the final results.
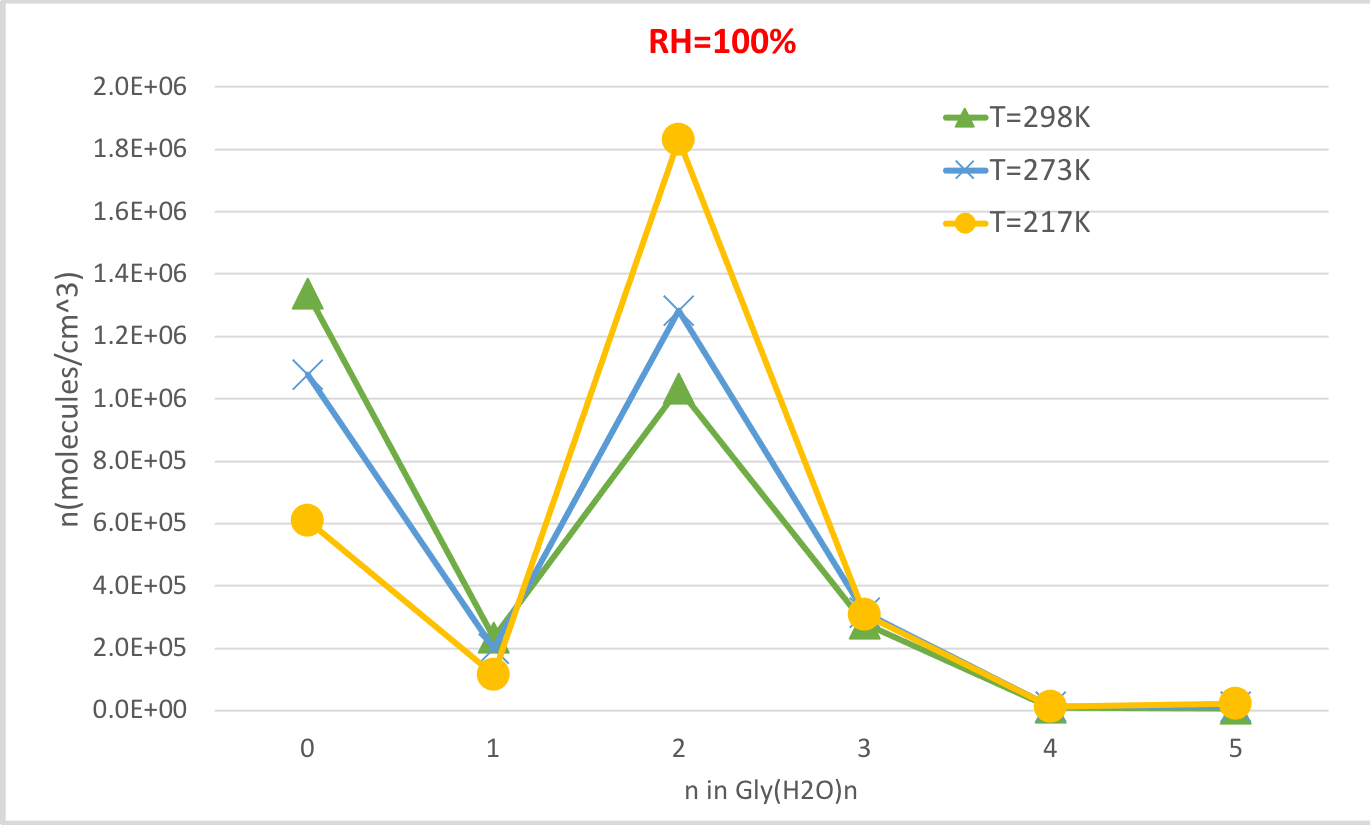
Figure 4 shows the equilibrium concentration of hydrated glycine calculated in Table 4 as a function of temperature at 100% relative humidity and 1 atmosphere pressure. It shows that, as temperature decreases from 298.15K to 216.65K, the concentration of unhydrated glycine (n=0) decreases and those of hydrated glycine increases. The glycine dihydrate (n=2) in particular increases dramatically with decreasing temperature while the change in the concentration of other hydrates is less noticeable. These inverse correlation between temperature and hydrate concentration is consistent with the expectation that lower Gibbs free energies of hydrations at lower temperatures favor the formation of hydrates.
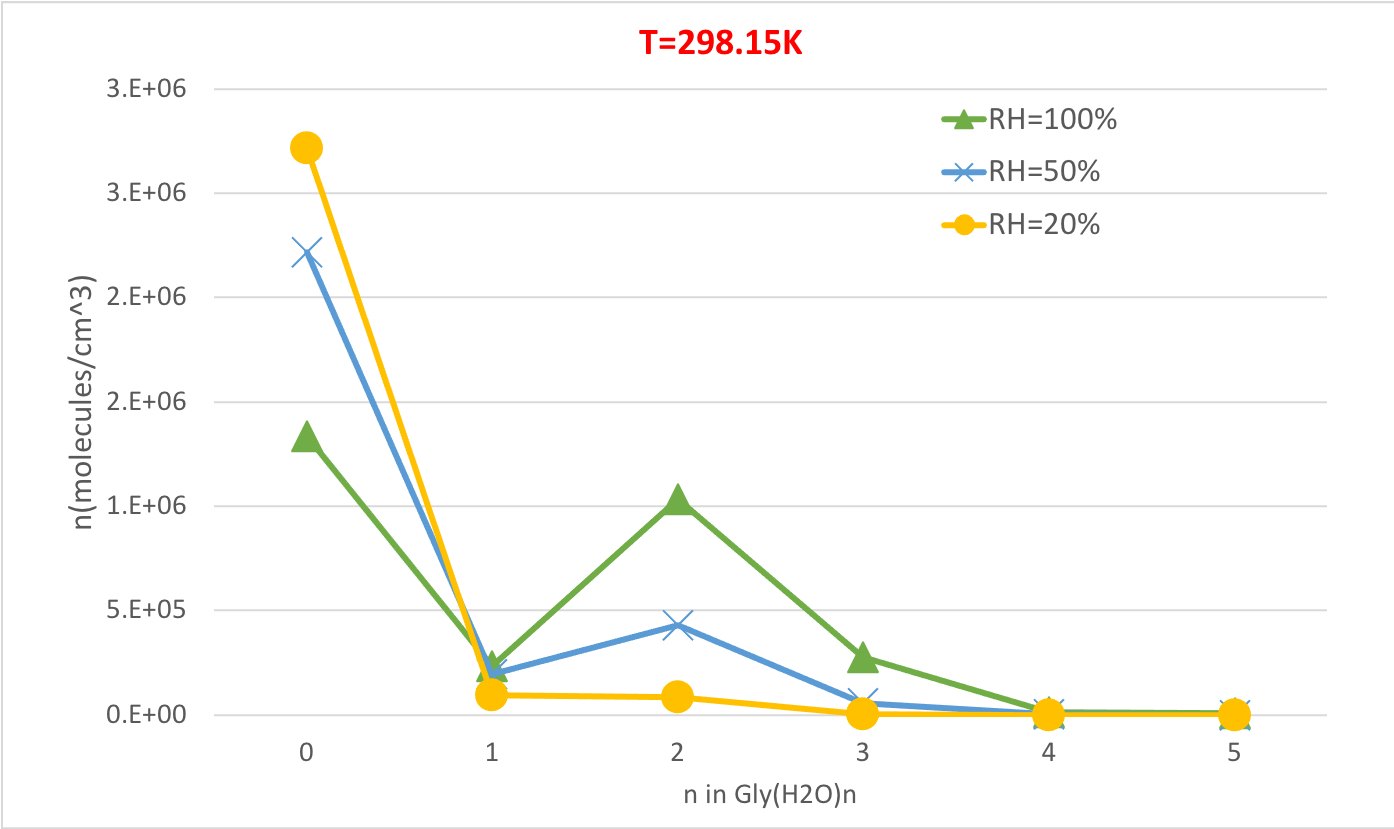
Figure 5 illustrates the relative humidity dependence of equilibrium concentration of glycine hydrates at 298.15K and 1 atmosphere pressure. It clearly demonstrates that as RH increases from 20% to 100%, the concentration of hydrates (n>0) increase at the expense of unhydrated glycine (n=0). Once again the direct correlation between the relative humidity and concentration of hydrates is consistent with the idea that the presence of more water molecules at higher RH promotes the formation of hydrates.
As presented, this protocol gives a qualitative understanding of the hydrated glycine populations in the atmosphere. Assuming an initial concentration of isolated glycine of 2.9 million molecules per cubic centimeter, we see that the unhydrated glycine (n=0) is the most abundant species under most conditions except T=216.65K and RH=100%. The dihydrate (n=2), which has the lowest sequential Gibbs free energy of hydration at all three temperatures, is the most abundant hydrate at the conditions considered here. The monohydrate (n=1) and larger hydrates (n≥3) are predicted to be found in negligible amounts. Upon inspection of Figure 3, the abundance of the n = 1–4 clusters can be related to the stability and strain in the hydrogen bond network of the clusters. These clusters have the water molecules hydrogen bonded to the carboxylic acid moiety of glycine in a geometry closely resembling those of various hydrogen-bonded ring structures, making them especially stable.

Figure 1: Schematic description of the current procedure. A large pool of guess structures generated by the genetic algorithm (GA) is refined by a series of PW91 geometry optimizations until a set of converged structures are obtained. The vibrational frequencies of these structures are computed and used to compute the Gibbs free energy of formation, which is in turn used to compute the equilibrium concentrations of the clusters under ambient conditions. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Representative directory structure for each cluster. The in-house scripts included in this protocol require the directory structure shown above, where n is the number of water molecules. For each n in gly-h2o-n, there are the following subdirectories: GA for genetic algorithm with a GA/pm7 directory, QM for quantum mechanics with QM/pw91-sb for PW91/6-31+G*, QM/pw91-lb for PW91/6-311++G**, and QM/pw91-lb/ultrafine for optimizations and final vibrational calculations on ultrafine integration grids. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Representative low energy structures of Gly(H2O)n=0-5. These clusters were the electronic energy global minima optimized at the PW91/6-311++G** level of theory. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Temperature dependence of Gly(H2O)n=0-5 as 100% relative humidity and 1 atm pressure. The concentration of the hydrates is given in units of molecules cm-3. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Relative humidity dependence of Gly(H2O)n=0-5 as 298.15 K and 1 atm pressure. The concentration of the hydrates is given in units of molecules cm-3. Please click here to view a larger version of this figure.
{kind=link}
| E[PW91/6-311++G**] | 216.65 K | 273.15 K | 298.15 K | ||||||||
| LB-UF | ZPVE | ∆H | S | ∆G | ∆H | S | ∆G | ∆H | S | ∆G | |
| water | -76.430500 | 13.04 | 1.72 | 42.59 | 5.54 | 2.17 | 44.44 | 3.08 | 2.37 | 45.14 | 1.96 |
| glycine | -284.434838 | 48.55 | 2.65 | 69.53 | 36.14 | 3.70 | 73.81 | 32.09 | 4.22 | 75.61 | 30.22 |
Table 1: Monomer energies. Electronic energies are in units of Hartree while all other quantities are in units of kcal mol-1. Water and glycine were optimized at the PW91/6-311++G** level of theory and vibrational frequencies were computed. The thermodynamic corrections for a pressure of 1 atm and temperature of 298.15 K were computed using the thermo.pl script.
| E[PW91/6-311++G**] | 0 K | 216.65 K | 273.15 K | 298.15 K | ||||||||
| n | name | LB-UF | ZPVE | ∆H | S | ∆G | ∆H | S | ∆G | ∆H | S | ∆G |
| 1 | gly-h2o-1 | -360.88481 | 63.96 | 3.61 | 80.12 | 50.22 | 5.12 | 86.27 | 45.52 | 5.85 | 88.83 | 43.33 |
| 2 | gly-h2o-2 | -437.33763 | 79.33 | 4.53 | 90.86 | 64.17 | 6.46 | 98.78 | 58.81 | 7.40 | 102.06 | 56.30 |
| 3 | gly-h2o-3 | -513.78620 | 94.52 | 5.67 | 105.08 | 77.42 | 8.08 | 114.94 | 71.19 | 9.23 | 119.00 | 68.27 |
| 4 | gly-h2o-4 | -590.23667 | 109.80 | 6.03 | 104.98 | 91.30 | 8.78 | 116.21 | 84.40 | 10.11 | 120.87 | 81.14 |
| 5 | gly-h2o-5 | -666.68845 | 125.80 | 7.26 | 121.70 | 106.69 | 10.47 | 134.83 | 99.44 | 12.01 | 140.24 | 96.00 |
Table 2: Cluster energies. The energies of the lowest-energy Gly(H2O)n=1-5 structures found using our procedure outlined in Figure 1. Electronic energies are in units of Hartree while all other quantities are in units of kcal mol-1.
| Total Hydration: Gly + nH2O <-> Gly(H2O)n | Sequential Hydration: Gly(H2O)n-1 + H2O <-> Gly(H2O)n | ||||||||||||||||
| E[PW91/6-311++G**] | 216.65 | 273.15 | 298.15 | 216.65 | 273.15 | 298.15 | |||||||||||
| n | system name | LB-UF | ∆E(0) | ∆H(T) | ∆G(T) | ∆H(T) | ∆G(T) | ∆H(T) | ∆G(T) | LB-UF | ∆E(0) | ∆H(T) | ∆G(T) | H(T) | ∆G(T) | ∆H(T) | ∆G(T) |
| 1 | gly-h2o-1 | -12.22 | -9.85 | -10.61 | -3.68 | -10.61 | -1.87 | -10.59 | -1.07 | -12.22 | -9.85 | -10.61 | -3.68 | -10.61 | -1.87 | -10.59 | -1.07 |
| 2 | gly-h2o-2 | -26.22 | -21.53 | -23.10 | -9.27 | -23.11 | -5.66 | -23.09 | -4.06 | -14.00 | -11.68 | -12.49 | -5.59 | -12.50 | -3.79 | -12.50 | -2.99 |
| 3 | gly-h2o-3 | -37.56 | -30.72 | -32.88 | -12.90 | -32.87 | -7.69 | -32.82 | -5.38 | -11.34 | -9.19 | -9.78 | -3.63 | -9.76 | -2.03 | -9.73 | -1.32 |
| 4 | gly-h2o-4 | -50.10 | -40.34 | -43.48 | -15.87 | -43.54 | -8.71 | -43.51 | -5.55 | -12.54 | -9.62 | -10.60 | -2.97 | -10.67 | -1.02 | -10.69 | -0.17 |
| 5 | gly-h2o-5 | -63.45 | -51.41 | -55.42 | -20.58 | -55.51 | -11.48 | -55.48 | -7.45 | -13.35 | -11.07 | -11.94 | -4.71 | -11.97 | -2.77 | -11.97 | -1.90 |
Table 3: Hydration energies. The total energy of hydration and energy of sequential hydration for Gly(H2O)n=1-5 in units of kcal mol-1. Here, E[PW91/6-311++G**] is the change in the electronic energy, ∆E(0) is the zero-point vibrational energy (ZPVE) corrected change in energy, ∆H(T) is the enthalpy change at temperature T, and ∆G(T) is the Gibbs free energy change of hydration of each Gly(H2O)n=1-5 cluster.
| Equilibrium Hydrate Distribution as a function of temperature and relative humidity | |||||||||
| T=298.15K | T=273.15K | T=216.65K | |||||||
| Gly(H2O)n | RH=100% | RH=50% | RH=20% | RH=100% | RH=50% | RH=20% | RH=100% | RH=50% | RH=20% |
| 0 | 1.3E+06 | 2.2E+06 | 2.7E+06 | 1.1E+06 | 2.0E+06 | 2.7E+06 | 6.1E+05 | 1.5E+06 | 2.5E+06 |
| 1 | 2.3E+05 | 1.9E+05 | 9.5E+04 | 2.0E+05 | 1.9E+05 | 9.9E+04 | 1.2E+05 | 1.5E+05 | 9.5E+04 |
| 2 | 1.0E+06 | 4.3E+05 | 8.4E+04 | 1.3E+06 | 6.1E+05 | 1.3E+05 | 1.8E+06 | 1.1E+06 | 3.0E+05 |
| 3 | 2.8E+05 | 5.8E+04 | 4.5E+03 | 3.2E+05 | 7.4E+04 | 6.3E+03 | 3.1E+05 | 9.6E+04 | 1.0E+04 |
| 4 | 1.1E+04 | 1.1E+03 | 3.4E+01 | 1.3E+04 | 1.5E+03 | 5.0E+01 | 1.1E+04 | 1.8E+03 | 7.5E+01 |
| 5 | 7.5E+03 | 3.9E+02 | 4.9E+00 | 1.2E+04 | 7.2E+02 | 9.7E+00 | 2.4E+04 | 1.9E+03 | 3.1E+01 |
Table 4: Equilibrium hydrate concentrations of Gly(H2O)n=0-5 as a function temperature (T=298.15K, 273.15K, 216.65K) and relative humidity (RH=100%, 50%, 20%). The concentration of the hydrates is given in units of molecules cm-3 assuming experimental values56,57,58, of [Gly]0 = 2.9 x 106 cm-3 and [H2O] = 7.7 x 1017 cm-3, 1.6 x 1017 cm-3 and 9.9 x 1014 cm-3 at 100% relative humidity and T = 298.15 K, 273.15 K, and 216.65 K, respectively59.
Supplemental Files. Please click here to download these files.
Discussion
The accuracy of the data generated by this protocol depends mainly on three things: (i) the variety of configurations sampled by Step 2, (ii) the accuracy of the electronic structure of the system, (iii) and the accuracy of the thermodynamic corrections. Each of these factors can be addressed by modifying the method by editing the included scripts. The first factor is easily overcome with the use of a larger initial pool of randomly generated structures, more numerous iterations of the GA, and a looser definition of the criteria involved in the GA. In addition, one may use a different semi-empirical method such as the self-consistent charge density-functional tight-binding (SCC-DFTB)62 model and the effective fragment potential (EFP)63 model in order to explore the effects of different physical descriptions. The main limitation here is the inability of the method to form or break covalent bonds, meaning that the monomers are frozen. The GA procedure only finds the most stable relative positions of these frozen monomers according to the semi-empirical description.
The accuracy of the electronic structure of the system can be improved in a variety of ways, each with its computational cost. One may choose a better density functional, such as M06-2X64 and wB97X-V65, or quantum mechanical (QM) method such as the Møller-Plesset66,67,68 (MPn) perturbation theories and coupled-cluster69 (CC) methods in order to improve the physical description of the system. In the hierarchy of functionals, the performance generally improves upon going from generalized-gradient approximation (GGA) functionals like PW91 to range-separated hybrid functionals like wB97X-D and meta-GGA hybrid functionals like M06-2X.
The disadvantage of DFT methods is that a systematic convergence towards an accurate value is not possible; however, DFT methods are computationally inexpensive and there is a wide variety of functionals for a wide variety of applications.
Energies calculated using wavefunction methods like MP2 and CCSD(T) in conjunction with correlation consistent basis sets of increasing cardinal number ([aug-]cc-pV[D,T,Q,...]Z) converge towards their complete basis set limit systematically, but the computational cost of each calculation becomes prohibitive as the system size grows. Further refinement of the electronic structure can be accomplished by using explicitly correlated basis sets70 and by extrapolating to the complete basis set (CBS)71 limit. Our recent work suggests that a density-fitted explicitly correlated second-order Møller-Plesset (DF-MP2-F12) perturbative approach yields energies approaching that of MP2/CBS computations32. Modification of the current protocol to use different electronic structure methods involves two steps: (i) prepare a template input file following the syntax given by the software, (ii) and edit the run-pw91-sb.csh, run-pw91-lb.csh, and run-pw91-lb-ultrafine.csh scripts to generate the correct input file syntax as well as the correct submit script for the software.
Lastly, the accuracy of the thermodynamic corrections depends on the electronic structure method as well as the description of the PES around the global minimum. An accurate description of the PES requires the computation of third- and higher-order derivatives of the PES with respect to displacements in the nuclear degrees of freedom, such as the quartic force field72,73 (QFF), which is an exceptionally costly task. The current protocol uses the harmonic oscillator approximation to the vibrational frequencies, resulting in the need to compute only up to second derivatives of the PES. This approach becomes problematic in systems with high anharmonicity, such as very floppy molecules and symmetric double-well potentials due to the large difference in the true PES and the harmonic PES. Furthermore, the cost of having a high-quality PES from a computationally demanding electronic structure method only compounds the problem of cost for vibrational frequency calculations. One approach to overcome this is to use the electronic energies from a high-quality electronic structure calculation along with vibrational frequencies computed on a lower quality PES, resulting in a balance between cost and accuracy. The current protocol can be modified to use different PES descriptions as described in the previous paragraph; however, one may also edit the vibrational frequency keywords in the scripts and templates to compute anharmonic vibrational frequencies.
Two crucial issues for any configurational sampling protocol are the initial method for sampling the potential energy surface and the criteria used to identify each cluster. We have made extensive use of a variety of methods in our previous work. For the first issue, the initial method for sampling the potential energy surface, we have made the choice of using GA with semi-empirical methods based on these factors. Configurational sampling using chemical intuition26, random sampling, and molecular dynamics (MD)29,30, fail to find putative global minima regularly for clusters larger than 10 monomers, as we observed in our studies of water clusters18. We have successfully used basin hopping (BH) to study the complex PES of (H2O)1174, but it required the manual inclusion of some potential low energy isomers the BH algorithm did not find. A comparison of the performance of BH and GA in finding the global minimum of water clusters, (H2O)n=10-20 demonstrated that GA consistently found the global minimum faster than BH75. GA as implemented in OGOLEM and CLUSTER is very versatile because it can be applied to any molecular cluster and it can interface with a vast number of packages with classical force field, semi-empirical, density functional, and ab initio capabilities. The choice of PM7 is driven by its speed and reasonable accuracy. Virtually any other semi-empirical method would have significantly higher computational cost.
As for the second issue, we have explored using different criteria to identify unique structures ranging from electronic energies, dipole moments, overlap RMSDs and rotational constants. Using dipole moments proved difficult because both the dipole moment components were dependent on the molecule's orientation and the total dipole moment was very sensitive to geometry differences in such a way that it was difficult to set thresholds determining is structures are the same or unique. A combination of electronic energies and rotational constants proved to be most useful.
The current criteria for deeming two structures unique is based on an energy difference threshold of 0.10 kcal mol-1 and rotational constant difference of 1%. Therefore, two structures are considered different if their energies differ by more than 0.10 kcal mol-1 (~0.00015 a.u.) AND any of their three rotational constants (A, B, C) differ by more than 1%. Substantial internal benchmarks over the years found these thresholds to be reasonable choices. Our configurational sampling approach and screening methodology has been applied to very weakly bound clusters such as polyaromatic hydrocarbons complexed with water76,77 as well as strongly bound ternary sulfate hydrates containing ammonia and amines32. For clusters where there are different protonation states to be considered, the best approach is to run various GA calculations, each starting with monomers in different protonation states. This ensures that structures with different protonation states are carefully considered. However, the low-level DFT calculations often allow protonation states to change during the course of the geometry optimization, thereby yielding the most stable protonation state regardless of the starting geometry.
Our GA configurational sampling methods should work well even for floppy molecules as long as the GA codes are interfaced with general, non-parameterized methods that allow the monomers to adopt different configurations during the course of the GA run. For example, interfacing GA with PM7 would allow monomers' structures to change, but if their bonds break as would happen when protonation states change, the structures may get discarded as unacceptable candidates.
We have considered different ways of correcting the shortcomings of the harmonic approximation, especially those arising from low vibrational frequencies. Incorporating the quasi-harmonic approximation into the current methodology is not difficult. However, there are still questions about the quasi-harmonic method, especially when it comes to the cutoff frequency below which it will be applied. Also, there are no rigorous benchmarking works examining the reliability of the quasi-RRHO approximation even though conventional wisdom suggests it should be an improvement over RRHO approximation.
The protocol thus presented may be generalized to any system of noncovalently-bound gas phase molecular clusters. It may also be generalized to use any semi-empirical method, electronic structure method and software, and vibrational analysis method and software by editing the scripts and templates. This assumes that the user is comfortable with the Linux command-line interface, Python scripting, and high-performance computing. The unfamiliar syntax and look of the Linux operating system and lack of scripting experience is the largest pitfall in this protocol and is where new students struggle the most. This protocol has been used successfully in a variety of implementations for years in our group, mostly focusing on the effects of sulfuric acid and ammonia on aerosol formation. Further improvements to this protocol will involve a more robust interface to more electronic structure software, alternative implementations of the genetic algorithm, and possibly the use of newer methods for faster computations of electronic and vibrational energies. Our current applications of this protocol are exploring the importance of amino acids in the early stages of aerosol formation in the current atmosphere and in the formation of larger biological molecules in prebiotic environments.
Disclosures
None.
Acknowledgements
This project was supported by grants CHE-1229354, CHE-1662030, CHE-1721511, and CHE-1903871 from the National Science Foundation (GCS), the Arnold and Mabel Beckman Foundation Beckman Scholar Award (AGG), and the Barry M. Goldwater Scholarship (AGG). High-performance computing resources of the MERCURY Consortium (http://www.mercuryconsortium.org) were used.
Materials
| Name | Company | Catalog Number | Comments |
| Avogadro | https://avogadro.cc | Open-source molecular visualization program | |
| Gaussian [09/16] Software | http://www.gaussian.com/ | Commercial ab initio electronic structure program | |
| MOPAC 2016 | http://openmopac.net/MOPAC2016.html | Open-source semi-empirical program | |
| OGOLEM Software | https://www.ogolem.org | Genetic algorithm-based global optimization program | |
| OpenBabel | http://openbabel.org/wiki/Main_Page | Open-source cheminformatics library | |
| calcRotConsts.py | Shields Group, Department of Chemistry, Furman University | Python script to compute rotational constants | |
| calcSymmetry.csh | Shields Group, Department of Chemistry, Furman University | Shell script to calculate symmetry number of a molecule given Cartesian coordinates | |
| combine-GA.csh | Shields Group, Department of Chemistry, Furman University | Shell script to combine energy and rotational constants from different GA directories | |
| combine-QM.csh | Shields Group, Department of Chemistry, Furman University | Shell script to combine energy and rotational constants from different QM directories | |
| gaussianE.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract Gaussian 09 energies | |
| gaussianFreqs.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract Gaussian 09 vibrational frequencies | |
| getrotconsts | Shields Group, Department of Chemistry, Furman University | Executable to calculate rotational constants given a molecule's Cartesian coordinates | |
| getRotConsts-dft-lb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of large basis DFT optimized structures | |
| getRotConsts-dft-lb-ultrafine.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of ultrafine DFT optimized structures | |
| getRotConsts-dft-sb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of small basis DFT optimized structures | |
| getRotConsts-GA.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of genetic algorithm optimized structures | |
| global-minimum-coords.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of global minimum structures of gly-(h2o)n, where n=0-5 | |
| make-thermo-gaussian.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract data from Gaussian output files and make input files for the thermo.pl script | |
| ogolem-input-file.ogo | Shields Group, Department of Chemistry, Furman University | Ogolem sample input file | |
| ogolem-submit-script.pbs | Shields Group, Department of Chemistry, Furman University | PBS batch submission file for Ogolem calculations | |
| README.docx | Shields Group, Department of Chemistry, Furman University | Clarifications to help readers use the scripts effectively | |
| runogolem.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run OGOLEM | |
| run-pw91-lb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of large basis DFT optimization calculations | |
| run-pw91-lb-ultrafine.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of ultrafine DFT optimization calculations | |
| run-pw91-sb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of small basis DFT optimization calculations | |
| run-thermo-pw91.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute the thermodynamic corrections for a batch of DFT optimized structures | |
| similarityAnalysis.py | Shields Group, Department of Chemistry, Furman University | Python script to determine unique structures based on rotational constants and energies | |
| symmetry | Shields Group, Department of Chemistry, Furman University | Executable to calculate molecular symmetry given Cartesian coordinates | |
| symmetry.c | (C) 1996, 2003 S. Patchkovskii, Serguei.Patchkovskii@sympatico.ca | C code to determine the molecular symmstry of a molecule given Cartesian coordinates | |
| template-marcy.pbs | Shields Group, Department of Chemistry, Furman University | Template for a PBS submit script which uses OGOLEM | |
| template-pw91.com | Shields Group, Department of Chemistry, Furman University | Template Gaussian 09 input | |
| template-pw91-HL.com | Shields Group, Department of Chemistry, Furman University | Template Gaussian 09 input for ultrafine DFT optimization | |
| thermo.pl | https://www.nist.gov/mml/csd/chemical-informatics-research-group/products-and-services/program-computing-ideal-gas | Perl open-source script to compute ideal gas thermodynamic corrections | |
| gly-h2o-n.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet for the complete protocol | |
| table-1.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-2.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-3.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-4.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| water.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of water | |
| glycine.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of glycine |
References
- Foster, P., Ramaswamy, V. Climate Change 2007 The Scientific Basis. Solomon, S., Qin, D., Manning, M., Chen, Z., Marquis, M., Averyt, K. B., Tignor, M., Miller, H. L. , Cambridge University Press. Cambridge, U.K. (2007).
- Kulmala, M., et al. Toward direct measurement of atmospheric nucleation. Science. 318 (5847), 89-92 (2007).
- Sipila, M., et al. The role of sulfuric acid in atmospheric nucleation. Science. 327 (5970), 1243-1246 (2010).
- Jiang, J., et al. First measurement of neutral atmospheric cluster and 1 - 2 nm particle number size distributions during nucleation events. Aerosol Science and Technology. 45 (4), (2011).
- Dunn, M. E., Pokon, E. K., Shields, G. C. Thermodynamics of forming water clusters at various Temperatures and Pressures by Gaussian-2, Gaussian-3, Complete Basis Set-QB3, and Complete Basis Set-APNO model chemistries; implications for atmospheric chemistry. Journal of the American Chemical Society. 126 (8), 2647-2653 (2004).
- Pickard, F. C., Pokon, E. K., Liptak, M. D., Shields, G. C. Comparison of CBSQB3, CBSAPNO, G2, and G3 thermochemical predictions with experiment for formation of ionic clusters of hydronium and hydroxide ions complexed with water. Journal of Chemical Physics. 122, 024302(2005).
- Pickard, F. C., Dunn, M. E., Shields, G. C. Comparison of Model Chemistry and Density Functional Theory Thermochemical Predictions with Experiment for Formation of Ionic Clusters of the Ammonium Cation Complexed with Water and Ammonia; Atmospheric Implications. Journal of Physical Chemistry A. 109 (22), 4905-4910 (2005).
- Alongi, K. S., Dibble, T. S., Shields, G. C., Kirschner, K. N. Exploration of the Potential Energy Surfaces, Prediction of Atmospheric Concentrations, and Vibrational Spectra of the HO2•••(H2O)n (n=1-2) Hydrogen Bonded Complexes. Journal of Physical Chemistry A. 110 (10), 3686-3691 (2006).
- Allodi, M. A., Dunn, M. E., Livada, J., Kirschner, K. N. Do Hydroxyl Radical-Water Clusters, OH(H2O)n, n=1-5, Exist in the Atmosphere. Journal of Physical Chemistry A. 110 (49), 13283-13289 (2006).
- Kirschner, K. N., Hartt, G. M., Evans, T. M., Shields, G. C. In Search of CS2(H2O)n=1-4 Clusters. Journal of Chemical Physics. 126, 154320(2007).
- Hartt, G. M., Kirschner, K. N., Shields, G. C. Hydration of OCS with One to Four Water Molecules in Atmospheric and Laboratory Conditions. Journal of Physical Chemistry A. 112 (19), 4490-4495 (2008).
- Morrell, T. E., Shields, G. C. Atmospheric Implications for Formation of Clusters of Ammonium and 110 Water Molecules. Journal of Physical Chemistry A. 114 (12), 4266-4271 (2010).
- Temelso, B., et al. Quantum Mechanical Study of Sulfuric Acid Hydration: Atmospheric Implications. Journal of Physical Chemistry A. 116 (9), 2209(2012).
- Husar, D. E., Temelso, B., Ashworth, A. L., Shields, G. C. Hydration of the Bisulfate Ion: Atmospheric Implications. Journal of Physical Chemistry A. 116 (21), 5151-5163 (2012).
- Bustos, D. J., Temelso, B., Shields, G. C. Hydration of the Sulfuric Acid – Methylamine Complex and Implications for Aerosol Formation. Journal of Physical Chemistry A. 118 (35), 7430-7441 (2014).
- Wales, D. J., Scheraga, H. A. Global optimization of clusters, crystals, and biomolecules. Science. 27 (5432), 1368-1372 (1999).
- Day, M. B., Kirschner, K. N., Shields, G. C. Global search for minimum energy (H2O)n clusters, n = 3 - 5. The Journal of Physical Chemistry A. 109 (30), 6773-6778 (2005).
- Shields, R. M., Temelso, B., Archer, K. A., Morrell, T. E., Shields, G. C. Accurate predictions of water cluster formation, (H2O)n=2-10. The Journal of Physical Chemistry A. 114 (43), 11725-11737 (2010).
- Temelso, B., Archer, K. A., Shields, G. C. Benchmark structures and binding energies of small water clusters with anharmonicity corrections. The Journal of Physical Chemistry A. 115 (43), 12034-12046 (2011).
- Temelso, B., Shields, G. C. The role of anharmonicity in hydrogen-bonded systems: The case of water clusters. The Journal of Chemical Theory and Computation. 7 (9), 2804-2817 (2011).
- Von Freyberg, B., Braun, W. Efficient search for all low energy conformations of polypeptides by Monte Carlo methods. The Journal of Computational Chemistry. 12 (9), 1065-1076 (1991).
- Rakshit, A., Yamaguchi, T., Asada, T., Bandyopadhyay, P. Understanding the structure and hydrogen bonding network of (H2O)32 and (H2O)33: An improved Monte Carlo temperature basin paving (MCTBP) method of quantum theory of atoms in molecules (QTAIM) analysis. RSC Advances. 7 (30), 18401-18417 (2017).
- Deaven, D. M., Ho, K. M. Molecular geometry optimization with a genetic algorithm. Physical Review Letters. 75, 288-291 (1995).
- Hartke, B. Application of evolutionary algorithms to global cluster geometry optimization. Applications of Evolutionary Computation in Chemistry. , Springer. Berlin. (2004).
- Dieterich, J. M., Hartke, B. OGOLEM: Global cluster structure optimization for arbitrary mixtures of flexible molecules. A multiscaling, object-oriented approach. Molecular Physics. 108 (3-4), 279-291 (2010).
- Herb, J., Nadykto, A. B., Yu, F. Large ternary hydrogen-bonded pre-nucleation clusters in the Earth's atmosphere. Chemical Physics Letters. 518, 7-14 (2011).
- Ortega, I. K., et al. From quantum chemical formation free energies to evaporation rates. Atmospheric Chemistry and Physics. 12 (1), 225-235 (2012).
- Elm, J., Bilde, M., Mikkelsen, K. V. Influence of Nucleation Precursors on the Reaction Kinetics of Methanol with the OH Radical. Journal of Physical Chemistry A. 117 (30), 6695-6701 (2013).
- Loukonen, V., et al. Enhancing effect of dimethylamine in sulfuric acid nucleation in the presence of water - a computational study. Atmospheric Chemistry and Physics. 10 (10), 4961-4974 (2010).
- Temelso, B., Phan, T. N., Shields, G. C. Computational study of the hydration of sulfuric acid dimers: implications for acid dissociation and aerosol formation. Journal of Physical Chemistry A. 116 (39), 9745-9758 (2012).
- Jiang, S., et al. Study of Cl-(H2O)n (n = 1-4) using basin-hopping method coupled with density functional theory. Journal of Computational Chemistry. 35 (2), 159-165 (2014).
- Temelso, B., et al. Effect of mixing ammonia and alkylamines on sulfate aerosol formation. Journal of Physical Chemistry A. 122 (6), 1612-1622 (2018).
- Perdew, J. P., Ruzsinszky, A., Tao, J. Prescription for the design and selection of density functional approximations: More constraint satisfaction with fewer fits. Journal of Chemical Physics. 123, 062201(2005).
- Riplinger, C., Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. Journal of Chemical Physics. 138, 034106(2013).
- Riplinger, C., Pinski, P., Becker, U., Valeev, E. F., Neese, F. Sparse maps--A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory. Journal of Chemical Physics. 144 (2), 024109(2016).
- Kildgaard, J. V., Mikkelsen, K. V., Bilde, M., Elm, J. Hydration of atmospheric molecular clusters: a new method for systematic configurational sampling. Journal of Physical Chemistry A. 122 (22), 5026-5036 (2018).
- González, Á Measurement of areas on a sphere Using Fibonacci and latitude-longitude lattices. Mathematical Geosciences. 42, 49-64 (2010).
- Karaboga, D., Basturk, B. On the performance of artificial bee colony (ABC) algorithm. Applied Soft Computing. 8 (1), 687-697 (2008).
- Zhang, J., Doig, M. Global optimization of rigid molecules using the artificial bee colony algorithm. Physical Chemistry Chemical Physics. 18 (4), 3003-3010 (2016).
- Kubecka, J., Besel, V., Kurten, T., Myllys, N., Vehkamaki, H. Configurational sampling of noncovalent (atmospheric) molecular clusters: sulfuric acid and guanidine. Journal of Physical Chemistry A. 123 (28), 6022-6033 (2019).
- Grimme, S., Bannwarth, C., Shushkov, P. A Robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent Interactions of large molecular systems parametrized for all spd-block elements (Z = 1-86). Journal of Chemical Theory and Computation. 13 (5), 1989-2009 (2017).
- Buck, U., Pradzynski, C. C., Zeuch, T., Dieterich, J. M., Hartke, B. A size resolved investigation of large water clusters. Physical Chemistry Chemical Physics. 16 (15), 6859(2014).
- Forck, R. M., et al. Structural diversity in sodium doped water trimers. Physical Chemistry Chemical Physics. 14 (25), 9054-9057 (2012).
- Witt, C., Dieterich, J. M., Hartke, B. Cluster structures influenced by interaction with a surface. Physical Chemistry Chemical Physics. 20 (23), 15661-15670 (2018).
- Freitbert, A., Dieterich, J. M., Hartke, B. Exploring self-organization of molecular tether molecules on a gold surface by global structure optimization. The Journal of Computational Chemistry. 40 (22), 1978-1989 (2019).
- Stewart, J. J. P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. The Journal of Molecular Modeling. 19 (1), 1-32 (2013).
- Stewart, J. J. P. MOPAC2012 Computational Chemistry. , Available from: http://openmopac.net (2012).
- Burke, K., Perdew, J. P., Wang, Y. Derivation of a generalized gradient approximation: The PW91 density functional. Electronic Density Functional Theory. , Springer. Boston, MA. 81-111 (1998).
- Frisch, M. J., et al. Gaussian 09, Revision A.02. , Gaussian, Inc. Wallingford, CT. (2016).
- Ditchfield, R., Hehre, W. J., Pople, J. A. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. The Journal of Chemical Physics. 54 (2), 724(1971).
- Elm, J., Bilde, M., Mikkelsen, K. V. Assessment of density functional theory in predicting structures and free energies of reaction of atmospheric prenucleation clusters. The Journal of Chemical Theory and Computation. 8 (6), 2071-2077 (2012).
- Elm, J., Mikkelsen, K. V. Computational approaches for efficiently modelling of small atmospheric clusters. Chemical Physics Letters. 615, 26-29 (2014).
- Bayucan, A., et al. PBS Portable Batch System. , MRJ Technology Solutions. Mountain View, CA. (1999).
- O'Boyle, N. M., et al. Open Babel: An open chemical toolbox. Journal of Cheminformatics. 3, 33(2011).
- Csaszar, A. G. Conformers of gaseous glycine. Journal of the American Chemical Society. 114 (24), 9568-9575 (1992).
- Zhang, Q., Anastasio, C. Free and combined amino compounds in atmospheric fine particles (PM2.5) and fog waters from Northern California. Atmospheric Environment. 37 (16), 2247-2258 (2003).
- Matsumoto, K., Uematsu, M. Free amino acids in marine aerosols over the western North Pacific Ocean. Atmospheric Environment. 39 (11), 2163-2170 (2005).
- Mandalakis, M., Apostolaki, M., Stephanou, E. G. Trace analysis of free and combined amino acids in atmospheric aerosols by gas chromatography-mass spectrometry. Journal of Chromatography A. 1217 (1), 143-150 (2010).
- Seinfeld, J. H., Pandis, S. N. Atmospheric Chemistry and Physics, 3rd Ed. , John Wiley & Sons. Hoboken, N.J. (2016).
- Myllys, N., Elm, J., Halonen, R., Kurten, T., Vehkamaki, H. Coupled cluster evaluation of atmospheric acid-base clusters with up to 10 molecules. The Journal of Physical Chemistry A. 120 (4), 621-630 (2016).
- Elm, J., Bilde, M., Mikkelsen, K. V. Assessment of binding energies of atmospherically relevant clusters. Physical Chemistry Chemical Physics. 15 (39), (2013).
- Elstner, M. The SCC-DFTB method and its application to biological systems. Theoretical Chemistry Accounts. 116 (1-3), 316-325 (2006).
- Kaliman, I. A., Slipchenko, L. V. LIBEFP: A new parallel implementation of the effective fragment potential method as a portable software library. The Journal of Computational Chemistry. 34 (26), 2284-2292 (2013).
- Zhao, Y., Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and trasition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theoretical Chemistry Accounts. 120 (1-3), 215-241 (2008).
- Mardirossian, N., Head-Gordon, M. wB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy. Physical Chemistry Chemical Physics. 16 (21), 9904-9924 (2014).
- Head-Gordon, M., Pople, J. A., Frisch, M. J. MP2 energy evaluation by direct methods. Chemical Physics Letters. 153 (6), 503-506 (1988).
- Pople, J. A., Seeger, R., Krishnan, R. Variational configuration interaction methods and comparison with perturbation theory. The International Journal of Quantum Chemistry. 12, 149-163 (1977).
- Pople, J. A., Binkley, J. S., Seeger, R. Theoretical models incorporating electron correlation. The International Journal of Quantum Chemistry. 10 (10), 1-19 (1976).
- Monkhorst, H. J. Calculation of properties with the coupled-cluster method. The International Journal of Quantum Chemistry. 12 (11), 421-432 (1977).
- Klopper, W., Manby, F. R., Ten-No, S., Valeev, E. F. R12 methods in explicitly correlated molecular electronic structure theory. International Reviews in Physical Chemistry. 25, 427-468 (2006).
- Hattig, C. Optimization of auxiliary basis sets for RI-MP2 and RI-CC2 calculations: Core-valence and quintuple-z basis sets for H to Ar and QZVPP basis sets for Li to Kr. Physical Chemistry Chemical Physics. 7 (1), 59-66 (2005).
- Barone, V. Anharmonic vibrational properties by a fully automated second-order perturbative approach. The Journal of Chemical Physics. 122, 014108(2005).
- Barone, V. Vibrational zero-point energies and thermodynamic functions beyond the harmonic approximation. The Journal of Chemical Physics. 120 (7), 3059-3065 (2004).
- Temelso, B., et al. Exploring the Rich Potential Energy Surface of (H2O)11 and Its Physical Implications. Journal of Chemical Theory and Computation. 14 (2), 1141-1153 (2018).
- Kabrede, H., Hentschke, R. Global minima of water clusters (H2O)N, N≤25, described by three empirical potentials. Journal of Physical Chemistry B. 107 (16), (2003).
- Steber, A. L., et al. Capturing the Elusive Water Trimer from the Stepwise Growth of Water on the Surface of a Polycyclic Aromatic Hydrocarbon Acenaphthene. Journal of Physical Chemistry Letters. 8 (23), 5744-5750 (2017).
- Perez, C., et al. Corrannulene and its complex with water: A tiny cup of water. Physical Chemistry Chemical Physics. 19 (22), 14214-14223 (2017).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved