
Method Article
Computação de Concentrações Atmosféricas de Aglomerados Moleculares a partir de ab initio Termoquímica
Neste Artigo
Resumo
As concentrações atmosféricas de aglomerados moleculares fracamente ligados podem ser computadas a partir das propriedades termoquímicas de estruturas de baixa energia encontradas através de uma metodologia de amostragem configuracional de várias etapas utilizando um algoritmo genético e química quântica semi-empírica e ab initio.
Resumo
O estudo computacional da formação e crescimento de aerossóis atmosféricos requer uma superfície de energia livre gibbs precisa, que pode ser obtida a partir da estrutura eletrônica da fase do gás e cálculos de frequência vibracional. Essas quantidades são válidas para esses aglomerados atmosféricos cujas geometrias correspondem a um mínimo em suas superfícies energéticas potenciais. A energia livre gibbs da estrutura de energia mínima pode ser usada para prever concentrações atmosféricas do aglomerado sob uma variedade de condições como temperatura e pressão. Apresentamos um procedimento computacionalmente barato construído em uma amostragem de configuração baseada em algoritmo genético seguida por uma série de cálculos de triagem cada vez mais precisos. O procedimento começa gerando e evoluindo as geometrias de um grande conjunto de configurações usando modelos semi-empíricos e, em seguida, refina as estruturas únicas resultantes em uma série de níveis de teoria ab initio de alto nível. Finalmente, as correções termodinâmicas são calculadas para o conjunto resultante de estruturas de energia mínima e usadas para calcular as energias livres de formação, constantes de equilíbrio e concentrações atmosféricas. Apresentamos a aplicação deste procedimento ao estudo de aglomerados de glicina hidratado em condições ambientais.
Introdução
O parâmetro mais incerto nos estudos atmosféricos da mudança climática é a extensão exata em que as partículas de nuvens refletem a radiação solar recebida. Os aerossóis, que são material particulado suspenso em um gás, formam partículas de nuvens chamadas núcleos de condensação de nuvens (CCN) que espalham radiação recebida, impedindo assim sua absorção e o subsequente aquecimento da atmosfera1. Uma compreensão detalhada desse efeito de resfriamento líquido requer uma compreensão do crescimento dos aerossóis em CCNs, o que, por sua vez, requer uma compreensão do crescimento de pequenos aglomerados moleculares em partículas de aerossol. Trabalhos recentes sugerem que a formação de aerossol é iniciada por aglomerados moleculares de 3 nm de diâmetro ou menos2; no entanto, este regime de tamanho é de difícil acesso usando técnicas experimentais3,4. Portanto, deseja-se uma abordagem computacional de modelagem para superar essa limitação experimental.
Usando nossa abordagem de modelagem descrita abaixo, podemos analisar o crescimento de qualquer cluster hidratado. Como estamos interessados no papel da água na formação de grandes moléculas biológicas de constituintes menores em ambientes pré-bióticos, ilustramos nossa abordagem com glicina. Os desafios encontrados e as ferramentas necessárias para abordar essas questões de pesquisa são muito semelhantes aos envolvidos no estudo de aerossóis atmosféricos e aglomerados de prenucleação55,6,,77,88,9,,10,,11,,12,,13,,14,,15. Aqui, examinamos aglomerados de glicina hidratados a partir de uma molécula de glicina isolada, seguida por uma série de adições stepwise de até cinco moléculas de água. O objetivo final é calcular as concentrações de equilíbrio dos aglomerados Gly(H2O)n=0-5 na atmosfera a temperatura ambiente ao nível do mar e uma umidade relativa (RH) de 100 %.
Um pequeno número desses aglomerados moleculares subnanômetros cresce em um aglomerado crítico metastável (1-3 nm de diâmetro) adicionando outras moléculas de vapor ou coagulando em aglomerados existentes. Esses clusters críticos têm um perfil de crescimento favorável que leva à formação de núcleos de condensação de nuvens muito maiores (até 50-100 nm) (CCN), que afetam diretamente a eficiência de precipitação das nuvens, bem como sua capacidade de refletir a luz incidente. Portanto, ter uma boa compreensão da termodinâmica dos aglomerados moleculares e suas distribuições de equilíbrio deve levar a previsões mais precisas do impacto dos aerossóis no clima global.
Um modelo descritivo de formação de aerossóis requer termodinâmica precisa da formação de aglomerados moleculares. A computação de termodinâmica precisa da formação de clustermolecular requer a identificação das configurações mais estáveis, o que envolve encontrar a minima global e local na superfície potencial de energia do cluster (PES)16. Este processo é chamado de amostragem configuracional e pode ser alcançado através de uma variedade de técnicas, incluindo as baseadas na dinâmica molecular (DM)17,18,19,20, Monte Carlo (MC)21,22, e algoritmos genéticos (GA)23,24,25.
Diferentes protocolos foram desenvolvidos ao longo dos anos para obter a estrutura e a termodinâmica dos hidratos atmosféricos em um alto nível de teoria. Esses protocolos diferem na escolha do (i) método de amostragem configuracional, (ii) natureza do método de baixo nível utilizado na amostragem configuracional e (iii) a hierarquia dos métodos de nível superior utilizados para refinar os resultados nas etapas subsequentes.
Os métodos de amostragem configuracional incluíram intuição química26, amostragem aleatória27,28, dinâmica molecular (DM)29,30, salto de bacia (BH)31, e algoritmo genético (GA)24,25,32. Os métodos de baixo nível mais comuns empregados com esses métodos de amostragem são campos de força ou modelos semi-empíricos como PM6, PM7 e SCC-DFTB. Estes são frequentemente seguidos por cálculos DFT com conjuntos de base cada vez maiores e funcionais mais confiáveis dos degraus mais altos da escada de Jacó33. Em alguns casos, estes são seguidos por métodos de função de onda de nível mais alto, como MP2, CCSD(T) e o custo eficiente DLPNO-CCSD(T)34,35.
Kildgaard et al.36 desenvolveram um método sistemático onde moléculas de água são adicionadas em pontos nas esferas de Fibonacci37 em torno de pequenos aglomerados hidratados ou não hidratados para gerar candidatos para aglomerados maiores. Candidatos não físicos e redundantes são removidos com base em limiares de contato próximos e distância quadrada entre diferentes conformers. Otimizações subseqüentes usando o método semi-empírico PM6 e uma hierarquia de métodos de DFT e função de onda são usadas para obter um conjunto de conformers de baixa energia em um alto nível de teoria.
O algoritmo da colônia de abelhas artificiais (ABC)38 é uma nova abordagem de amostragem de configuração que foi recentemente implementada por Zhang et al. para estudar clusters moleculares em um programa chamado ABCluster39. Kubecka et al.40 utilizaram o ABCluster para amostragem de configuração seguido de reotimizações de baixo nível usando o método semi-empírico GFN-xTB de ligação apertada41. Eles refinaram ainda mais as estruturas e energias usando métodos DFT seguidos de energias finais usando DLPNO-CCSD(T).
Independentemente do método, a amostragem configuracional começa com uma distribuição de pontos gerada aleatoriamente ou não no PES. Cada ponto corresponde a uma geometria específica do aglomerado molecular em questão e é gerada pelo método de amostragem. Em seguida, o mínimo local mais próximo é encontrado para cada ponto seguindo a direção "downhill" no PES. O conjunto de minima assim encontrado corresponde às geometrias do aglomerado molecular que são estáveis, pelo menos por algum tempo. Aqui, a forma do PES e a avaliação da energia em cada ponto da superfície serão sensíveis à descrição física do sistema onde uma descrição física mais precisa resulta em um cálculo de energia mais computacionalmente caro. Utilizaremos especificamente o método GA implementado no programa OGOLEM25, que foi aplicado com sucesso a uma variedade de problemas globais de otimização e amostragem de configuração42,,43,,44,45,para gerar o conjunto inicial de pontos de amostragem. O PES será descrito pelo modelo PM746 implementado no programa MOPAC201647. Essa combinação é empregada porque gera uma variedade maior de pontos em comparação com os métodos MD e MC e encontra o minima local mais rápido do que descrições mais detalhadas do PES.
O conjunto de minima locais otimizados por GA são tomados como geometrias iniciais para uma série de etapas de triagem, que levam a um conjunto de energia mínima baixa. Esta parte do protocolo começa otimizando o conjunto de estruturas otimizadas por GA exclusivas usando a teoria de densidade funcional (DFT) com um pequeno conjunto de bases. Este conjunto de otimizações geralmente dará um conjunto menor de estruturas mínimas locais únicas que são modeladas com mais detalhes em comparação com as estruturas semi-empíricas otimizadas pelo GA. Em seguida, outra rodada de otimizações de DFT são realizadas neste conjunto menor de estruturas usando um conjunto de basemaior. Mais uma vez, esta etapa geralmente dará um conjunto menor de estruturas únicas que são modeladas com mais detalhes em comparação com o pequeno passo dft base. O conjunto final de estruturas únicas são então otimizados para uma convergência mais apertada e as frequências vibracionais harmônicas são calculadas. Após esta etapa temos tudo o que precisamos para calcular as concentrações de equilíbrio dos aglomerados na atmosfera. A abordagem geral é resumida diagramaticamente na Figura 1. Utilizaremos a aproximação de gradiente generalizadaPW91 48 (GGA) na implementação gaussiana49 do DFT, juntamente com duas variações do conjunto base Pople50 (6-31+G* para a pequena etapa base e 6-311++G** para a grande etapa base). Esta combinação particular de conjunto funcional e de base de correlação de troca foi escolhida devido ao seu sucesso anterior na computação precisa de energias livres de formação de Gibbs para aglomerados atmosféricos51,52.
Este protocolo pressupõe que o usuário tenha acesso a um cluster de computação de alto desempenho (HPC) com o sistema de lote portátil53 (PBS), MOPAC2016 (http://openmopac.net/MOPAC2016.html)47, OGOLEM (https://www.ogolem.org)25, Gaussian 09 (https://gaussian.com)49e OpenBabel54 (http://openbabel.org/wiki/Main_Page) software instalado seguindo suas instruções específicas de instalação. Cada etapa deste protocolo também usa um conjunto de scripts shell interno e Python 2.7 que devem ser salvos em um diretório que está incluído na variável $PATH ambiental do usuário. Todos os módulos ambientais necessários e permissões de execução para executar todos os programas acima também devem ser carregados na sessão do usuário. O uso de disco e memória pelo código GA (OGOLEM) e códigos semi-empíricos (MOPAC) são muito pequenos para os padrões modernos de recursos do computador. O uso geral de memória e disco para OGOLEM/MOPAC depende de quantos threads se quer usar e, mesmo assim, o uso de recursos será pequeno em comparação com as capacidades da maioria dos sistemas HPC. As necessidades de recursos dos métodos QM dependem do tamanho dos clusters e do nível de teoria utilizado. A vantagem de usar este protocolo é que se pode variar o nível da teoria para poder calcular o conjunto final de estruturas de baixa energia, tendo em mente que geralmente cálculos mais rápidos levam a mais incerteza na precisão dos resultados.
Para uma questão de clareza, o computador local do usuário será referido como "computador local", enquanto o cluster HPC a que eles têm acesso será referido como "cluster remoto".
Protocolo
1. Encontrar a estrutura de energia mínima de glicina isolada e água
NOTA: O objetivo aqui é duas vezes: (i) obter estruturas energéticas mínimas de moléculas isoladas de água e glicina para uso na amostragem de configuração do algoritmo genético, (ii) e calcular as correções termodinâmicas das energias da fase gasosa dessas moléculas para uso no cálculo das concentrações atmosféricas.
- No seu computador local, abra uma nova sessão de Avogadro.
- Clique em Build > Insira > Peptídeo e selecione Gly na janela Inserir peptídeo para gerar um monômero de glicina na janela de visualização.
- Clique em Extensões > Gaussian e edite a primeira linha na caixa de texto para ler '# pw91pw91/6-311++G** int (Acc2E=12,UltraFine) scf (conver=12) opt (tight,maxcyc=300) freq'. Clique em Gerar e salvar o arquivo de entrada como glycine.com.
- Observe que se a molécula tem uma flexibilidade conformacional significativa, como a glicina faz55, é fundamental realizar análises conformacionais para identificar a estrutura mínima global e outros conformers de baixa altitude. OpenBabel54 fornece ferramentas de busca conformacionais robustas utilizando diferentes algoritmos e campos de força rápida. Enquanto os conformers são autorizados a relaxar e interconverter durante os cálculos ga e subsequentes, às vezes é necessário executar vários cálculos de GA, cada um começando com um conformer diferente.
- No seu computador local, abra uma nova sessão em Avogadro.
- Clique em Build > Insert > Fragmente e procure por "água" na janela Insert Fragment para obter as coordenadas de água.
- Clique em Extensões > Gaussian e edite a primeira linha na caixa de texto para ler '# pw91pw91/6-311++G** int (Acc2E=12,UltraFine) scf (conver=12) opt (tight,maxcyc=300) freq'. Clique em Gerar e salvar o arquivo de entrada como water.com.
- Transfira os dois arquivos .com para o cluster remoto. Depois de entrar no cluster remoto, ligue para Gaussian 09 em um script de envio em lote para iniciar o cálculo. Quando os cálculos terminarem, extraia as coordenadas cartesianas (arquivos.xyz) das estruturas de energia mínimas, chamando OpenBabel. Para glicina, o comando a ser executado é:
obabel -ig09 glicina.log -oxyz > glicina.xyz
Estes dois arquivos .xyz serão usados pela amostragem de configuração GA na próxima etapa.
2. Amostragem de configuração baseada em algoritmo genético de aglomerados Gly(H2O)n=1-5
NOTA: O objetivo aqui é obter um conjunto de estruturas de baixa energia para Gly(H2O)n=1-5 no nível semi-empírico barato da teoria, utilizando o modelo PM746 implementado no MOPAC47. É imprescindível que o diretório de trabalho tenha a organização e estrutura exatas como mostra a Figura 2. Isso é para garantir que os scripts personalizados shell e Python funcionem sem falhas.
- Copie todos os scripts necessários para o cluster remoto e adicione sua localização ao $PATH
- Coloque todos os scripts e arquivos de modelo em uma pasta (scripts por exemplo) e copie-os para o cluster remoto
- Certifique-se de que todos os scripts são executáveis
- Adicione a localização do diretório de scripts à variável ambiental $PATH inserindo os seguintes comandos em um terminal. A localização padrão dos scripts está definida como $HOME/JoVE-demo/scripts, no entanto, pode-se definir uma variável ambiental chamada $SCRIPTS_HOME apontando para o diretório contendo os scripts e adicionar $SCRIPTS_HOME ao seu caminho
- Bash shell:
SCRIPTS_HOME de exportação=/path/to/scripts
exportar PATH=${SCRIPTS_HOME}:${PATH} - Concha Tcsh/Csh:
setenv SCRIPTS_HOME /path/to/scripts
setenv PATH ${SCRIPTS_HOME}:${PATH}
- Bash shell:
- No cluster remoto, configure e execute um cálculo GA:
- Crie um diretório chamado gly-h2o-n onde n é o número de moléculas de água.
- Crie um subdiretório chamado GA sob o diretório gly-h2o-n para executar cálculos de algoritmogenético.
- Copie os arquivos de entrada OGOLEM (Por exemplo, pm7.ogo), as coordenadas cartesianas monômeras (Por exemplo, glycine.xyz, water.xyz) e o script de envio em lote PBS (Eg. run.pbs) no diretório GA.
- Faça as alterações necessárias ao arquivo de entrada OGOLEM e ao arquivo de envio em lote.
- Envie o cálculo. Quando o cálculo começar, o OGOLEM criará um novo diretório nomeado como o prefixo do arquivo de entrada OGOLEM (Por exemplo, pm7) no diretório GA e armazenará coordenadas recém-geradas lá.
- Uma vez que o cálculo esteja concluído, compile as energias e as constantes rotacionais e use essas informações para determinar quais são as estruturas únicas de baixa energia:
- Alterar diretório para gly-h2o-n/GA/pm7 e
- Extrair as energias e calcular as constantes rotacionais dos clusters otimizados por GA com o comando:
getRotConsts-GA.csh N 0 99
onde N é o número de átomos no aglomerado molecular e '0 99' indica que o tamanho da piscina ga é de 100, com índices que vão de 0 a 99. Isso gerará um arquivo chamado rotConstsData_C que contém uma lista classificada de todas as configurações de cluster otimizadas pelo GA, suas energias e suas constantes de rotação. - Execute o comando:
similarityAnalysis.py rotConstsData_C pm7
onde o pm7 será usado como um rótulo de nomeação de arquivo, para encontrar e salvar os clusters otimizados pelo GA exclusivos. Isso gerará um arquivo chamado uniqueStructures-pm7.data que contém uma lista classificada das configurações otimizadas pelo GA exclusivos. Esta é uma lista de estruturas mínimas locais únicas para o cluster Gly(H2O)n otimizado no nível de teoria PM7, e essas estruturas estão agora prontas para serem refinadas usando DFT.
- Suba para o diretório gly-h2o-n/GA e combine os resultados de várias corridas de GA comparáveis usando o script combine-GA.csh. A sintaxe é:
combine-GA.csh
3. Refinamento usando o método QM com um pequeno conjunto de base
NOTA: O objetivo aqui é refinar a amostragem de configuração dos clusters Gly(H2O)n=1-5 usando uma melhor descrição quântica-mecânica para obter um conjunto menor, mas mais preciso, de estruturas de cluster Gly(H2O)n=1-5. As estruturas de partida para esta etapa são as saídas do Passo 2.
- Preparar e executar o pequeno cálculo DFT definido com base:
- Crie um subdiretório chamado QM sob o diretório gly-h2o-n. Sob o diretório QM, crie outro subdiretório chamado pw91-sb.
- Copie a lista de estruturasexclusivas (uniqueStructures-pm7.data) do diretório gly-h2o-n/GA para o diretório QM/pw91-sb.
- Alterar diretório para aquele gly-h2o-n/QM/pw91-sb.
- Execute o pequeno script de amostragem configuracional DFT definido com base pequena usando o comando:
run-pw91-sb.csh uniqueStructures-pm7.data sb QUEUE 10
onde sb é um rótulo para este conjunto de cálculos, queue é a fila preferida no cluster de computação, e 10 indica que 10 cálculos devem ser agrupados em um trabalho em lote. Este script gerará automaticamente as entradas para Gaussian 09 e enviará todos os cálculos. Digite 'teste' para 'Queue' para fazer uma corrida a seco.
- Uma vez que os cálculos apresentados estejam concluídos, extraia e analise os resultados.
- Extrair as energias e calcular as constantes rotacionais dos clusters otimizados de base pequena usando o comando:
getRotConsts-dft-sb.csh pw91 N
onde pw91 indica que a densidade PW91 funcional foi usada, e N é o número de átomos no cluster. Isso criará um arquivo chamado rotConstsData_C. - Agora identifique as estruturas únicas com o comando:
similarityAnalysis.py rotConstsData_C
onde sb é usado como um rótulo de nomeação de arquivo. Agora haverá uma lista de configurações exclusivas otimizadas no nível de teoria PW91/6-31+G* salvo no arquivo exclusivoStructures-sb.data.
- Extrair as energias e calcular as constantes rotacionais dos clusters otimizados de base pequena usando o comando:
- Suba para o diretório gly-h2o-n/QM e combine os resultados de várias corridas de QM comparáveis usando o script combine-QM.csh. A sintaxe é:
combine-QM.csh
4. Refinamento adicional usando o método QM com um conjunto de bases grande
NOTA: O objetivo aqui é refinar ainda mais a amostragem de configuração dos clusters Gly(H2O)n=1-5 usando uma melhor descrição quântica-mecânica. As estruturas de partida para esta etapa são as saídas do Passo 3.
- Envie cálculos mais confiáveis usando um conjunto de bases maior.
- Crie um subdiretório chamado pw91-lb sob o diretório QM.
- Copie a lista de estruturasexclusivas (uniqueStructures-sb.data)do diretório gly-h2o-n/QM para o diretório gly-h2o-n/QM/pw91-lb e mude para esse diretório.
- Execute o script de amostragem configuracional DFT de grande base com o comando:
run-pw91-lb.csh uniqueStructures-sb.data lb QUEUE 10
onde lb é um rótulo para este conjunto de cálculos, queue é a fila preferida no cluster de computação, e 10 indica que 10 cálculos devem ser agrupados em um trabalho em lote. Este script gerará automaticamente as entradas para Gaussian 09 e enviará todos os cálculos. Digite 'teste' para 'Queue' para fazer um teste de execução a seco.
- Uma vez que os cálculos enviados estejam concluídos, extraia e analise os dados
- Calcule as constantes rotacionais dos clusters otimizados de grande base com o comando:
getRotConsts-dft-lb.csh pw91 N
onde pw91 indica que a densidade PW91 funcional foi usada, e N é o número de átomos no cluster. - Agora identifique as estruturas únicas com o comando:
similarityAnalysis.py lb rotConstsData_C
onde lb é usado como um rótulo de nomeação de arquivo. Agora você tem uma lista de configurações exclusivas otimizadas no nível de teoria PW91/6-311++G** salvo no arquivo exclusivoStructures-lb.data.
- Calcule as constantes rotacionais dos clusters otimizados de grande base com o comando:
5. Cálculos finais de energia e correção termodinâmica
NOTA: O objetivo aqui é obter a estrutura vibracional e as energias dos aglomerados Gly(H2O)n=1-5 usando um conjunto de base grande e uma grade de integração ultrafina, a fim de calcular as correções termoquímicas desejadas.
- Começando com os resultados da etapa anterior, envie cálculos mais confiáveis.
- Crie um subdiretório chamado ultrafino sob o diretório QM/pw91-lb. Em seguida, copie a lista de estruturasexclusivas (uniqueStructures-lb.data) do diretório QM/pw91-lb para o diretório QM/pw91-lb/ultrafine e mude para esse diretório.
- Envie o script DFT de base ultrafina com o comando:
run-pw91-lb-ultrafine.csh uniqueStructures-lb.data uf Queue 10
onde uf é um rótulo para este conjunto de cálculos, queue é a fila preferida no cluster de computação, e 10 indica que 10 cálculos devem ser agrupados em um trabalho em lote. Este script gerará automaticamente as entradas para Gaussian 09 e enviará todos os cálculos. Digite 'teste' para 'Queue' para fazer um teste de execução a seco.
- Uma vez que os cálculos enviados estejam concluídos, extraia e analise os dados
- Extrair as energias e calcular as constantes rotacionais dos clusters otimizados de grande base com o comando:
getRotConsts-dft-lb-ultrafine.csh pw91 N
onde pw91 indica que a densidade PW91 funcional foi usada, e N é o número de átomos no cluster. - Agora identifique as estruturas únicas com o comando:
similarityAnalysis.py rotConstsData_C uf
onde uf é usado como um rótulo de nomeação de arquivo. Agora você tem uma lista de configurações exclusivas otimizadas no nível de teoria PW91/6-311++G** salvo no arquivo exclusivoStructures-uf.data.
- Extrair as energias e calcular as constantes rotacionais dos clusters otimizados de grande base com o comando:
- Realizar uma extração final das informações necessárias para calcular correções termodinâmicas. Use essas informações para calcular as correções termodinâmicas.
- Extrair as energias eletrônicas finais, constantes rotacionais e frequências vibracionais, e usá-las para calcular correções termodinâmicas usando o comando:
run-thermo-pw91.csh uniqueStructures-uf.data - Copiar/colar a saída de linha de comando para a folha 'Raw_Energies' da planilha do Excel chamada 'gly-h2o-n.xlsx'. Você precisaria fazer isso para os monômeros (glicina e água) bem como para o membro de menor energia de cada hidrato (gly-h2o-n, onde n=1,2, ...).
- À medida que as energias brutas são adicionadas à primeira folha da planilha 'gly-h2o-n.xlsx', as folhas subsequentes 'Binding_Energies' e 'Hydrate_Distribution' são automaticamente atualizadas. Em particular, a folha 'Hydrate_Distribution' produz a concentração de equilíbrio de hidratos em diferentes temperaturas (Por exemplo, 298,15K), umidade relativa (20%, 50%, 100%) e concentrações iniciais de água ([H2O]) e glicina ([Glicina]). A teoria por trás desses cálculos é descrita no próximo passo.
- Extrair as energias eletrônicas finais, constantes rotacionais e frequências vibracionais, e usá-las para calcular correções termodinâmicas usando o comando:
6. Computação de concentrações atmosféricas de Gly(H2O)n=0-5 aglomerados à temperatura ambiente ao nível do mar
NOTA: Isso é feito primeiro copiando os dados termodinâmicos gerados na etapa anterior em uma planilha e calculando as energias livres de Gibbs de hidratação sequencial. Em seguida, as energias livres gibbs são usadas para calcular constantes de equilíbrio para cada hidratação seqüencial. Finalmente, um conjunto de equações lineares são resolvidos para obter a concentração de equilíbrio dos hidratos para uma dada concentração de monômeros, temperatura e pressão.
- Comece por criar um sistema de equilíbrio químico para a hidratação sequencial da glicina, conforme mostrado abaixo:

- Calcule as constantes de equilíbrio Kn usando Kn = e-Δ Gn/(kBT),onde n é o nível de hidratação, ΔGn é a mudança de energia livre gibbs da nth reação de hidratação, kB é constante de Boltzmann, e T é a temperatura.

- Configurar a equação para a conservação da massa, utilizando-se a suposição de que a soma das concentrações de equilíbrio dos aglomerados de glicina hidratado e não hidratado é igual à concentração inicial de glicina isolada [Gly]0. Reescrever este sistema de seis equações simultâneas, usando algum rearranjo algébrico das expressões constantes de equilíbrio, como

- Resolver o sistema de equações mostrado acima para obter as concentrações de equilíbrio de Gly(H2O)n = 0-5 usando um valor experimental56,57,58 para a concentração de glicina na atmosfera, [Gly]0 = 2,9 x 106 cm-3, e a concentração de água na atmosfera a 100% umidade relativa e uma temperatura de 298,15 K59, [H2O] = 7,7 x 1017 cm-3.
Resultados
O primeiro conjunto de resultados deste protocolo deve ser um conjunto de estruturas de baixa energia de Gly(H2O)n=1-5 encontradas através do procedimento de amostragem configuracional. Essas estruturas foram otimizadas no nível de teoria PW91/6-311++G** e são consideradas precisas para o propósito deste artigo. Não há evidências que sugiram que o PW91/6-311++G** subestime ou superestime consistentemente a energia vinculante desses clusters. Sua capacidade de prever energias vinculantes em relação a MP2/CBS32 e [DLPNO-]CCSD(T)/CBS60,61 estimativas e experimento52 mostra muitas flutuações. O mesmo acontece com a maioria das outras funcionais de densidade. Geralmente, cada valor de n = 1 – 5 deve produzir um punhado de estruturas de baixa energia dentro de cerca de 5 kcal mol-1 da estrutura de menor energia. Aqui, focamos na primeira estrutura produzida pelo roteiro run-thermo-pw91.csh para brevidade. A Figura 3 mostra os menores isômeros de energia eletrônica dos aglomerados Gly(H2O)n=0-5. Pode-se ver que a rede de ligação de hidrogênio cresce em complexidade à medida que o número de moléculas de água aumenta, e até passa de uma rede planar principalmente para uma estrutura tridimensional semelhante a uma gaiola em n = 5. O restante deste texto utiliza as energias e quantidades termodinâmicas correspondentes a esses cinco clusters específicos.
A Tabela 1 contém as quantidades termodinâmicas necessárias para a realização do protocolo. A Tabela 2 mostra um exemplo da saída do script run-thermo-pw91.csh onde as energias eletrônicas, correções vibracionais de ponto zero e as correções termodinâmicas a três temperaturas diferentes são impressas. Para cada cluster (linha), E[PW91/6-311++G**] corresponde às energias eletrônicas de fase de gás no nível de teoria PW91/6-311++G** calculado em grades de integração ultrafinas em unidades de Hartree, bem como a energia vibracional de ponto zero(ZPVE)em unidades de kcal mol-1. A cada temperatura, 216,65 K, 273,15 K e 298,15 K, as correções termodinâmicas são listadas, ¥H a entalpia de formação em unidades de kcal mol-1, S a entropia de formação em unidades de cal mol-1, e ¥G a gibbs livre energia de formação em unidades de kcal mol-1. A Tabela 3 mostra um exemplo de computação da mudança de energia total de Gibbs livre de hidratação, bem como para hidratação seqüencial. Um exemplo de computação da mudança de energia livre de Gibbs total para a reação

começa com o cálculo da energia eletrônica EPW91 como

onde EPW91[Gly⁄(H2O)] é retirado da tabela 2 coluna C, e EPW91[Gly] e EPW91[H2O] são retirados da coluna B da Tabela 1. Em seguida, calculamos a mudança de energia total da fase do gás ΔE(0) incluindo a mudança na energia vibracional de ponto zero da reação como

para obter a coluna D. Aqui, ΔEPW91/6-311++G** é tirada da coluna C da Tabela 3, EZPVE[Gly ◗ (H2O)] da Tabela 2 coluna D, e EZPVE[Gly] e EZPVE[H2O] da Coluna 1 coluna C. Por uma questão de brevidade, passaremos para clusters de temperatura ambiente, então pulamos os dados de 216,65 K e 273,15 K. À temperatura ambiente, então calculamos a mudança entalpia da reação ΔH corrigindo a mudança de energia da fase do gás como

onde ΔE(0) é retirado da tabela 3 coluna D, ΔH[Gly⁄(H2O)] é retirado da tabela 2 coluna K, e ΔH[Gly] e ΔH[H2O] são retirados da tabela 1 coluna J. Finalmente, calculamos a mudança de energia livre Gibbs da reação ΔG como

onde ΔH é retirado da coluna I da Tabela 3, S[Gly⁄(H2O)] é retirado da tabela 2 coluna L, e S[Gly] e S[H2O] são retirados da tabela 1 coluna K. Observe aqui que os valores de entropia devem ser convertidos em unidades de kcal mol-1 K-1 durante esta etapa.
Temos agora as quantidades necessárias para calcular as concentrações atmosféricas de glicina hidratada, como mostrado no Passo 6. Os resultados devem se assemelhar aos dados apresentados na Tabela 4,mas pequenas diferenças numéricas são esperadas. A Tabela 4 mostra as concentrações de hidrato de equilíbrio encontradas a partir da formulação do sistema de seis equações na Etapa 6.2 em uma equação matricial e sua solução subsequente. Começamos reconhecendo o fato de que o sistema de equações pode ser escrito como

onde Kn é a constante de equilíbrio para a hidratação sequencial nth de glicina, w é a concentração de água na atmosfera, g é a concentração inicial de glicina isolada na atmosfera, e gn é a concentração de equilíbrio de Gly(H2O)n. Se reescrevermos a equação acima como Ax = b,obtemos x = A−1b onde A−1 é o inverso da matriz A. Este inverso pode ser facilmente computado usando funções de planilha incorporadas, como mostrado na Tabela 4 para obter os resultados finais.
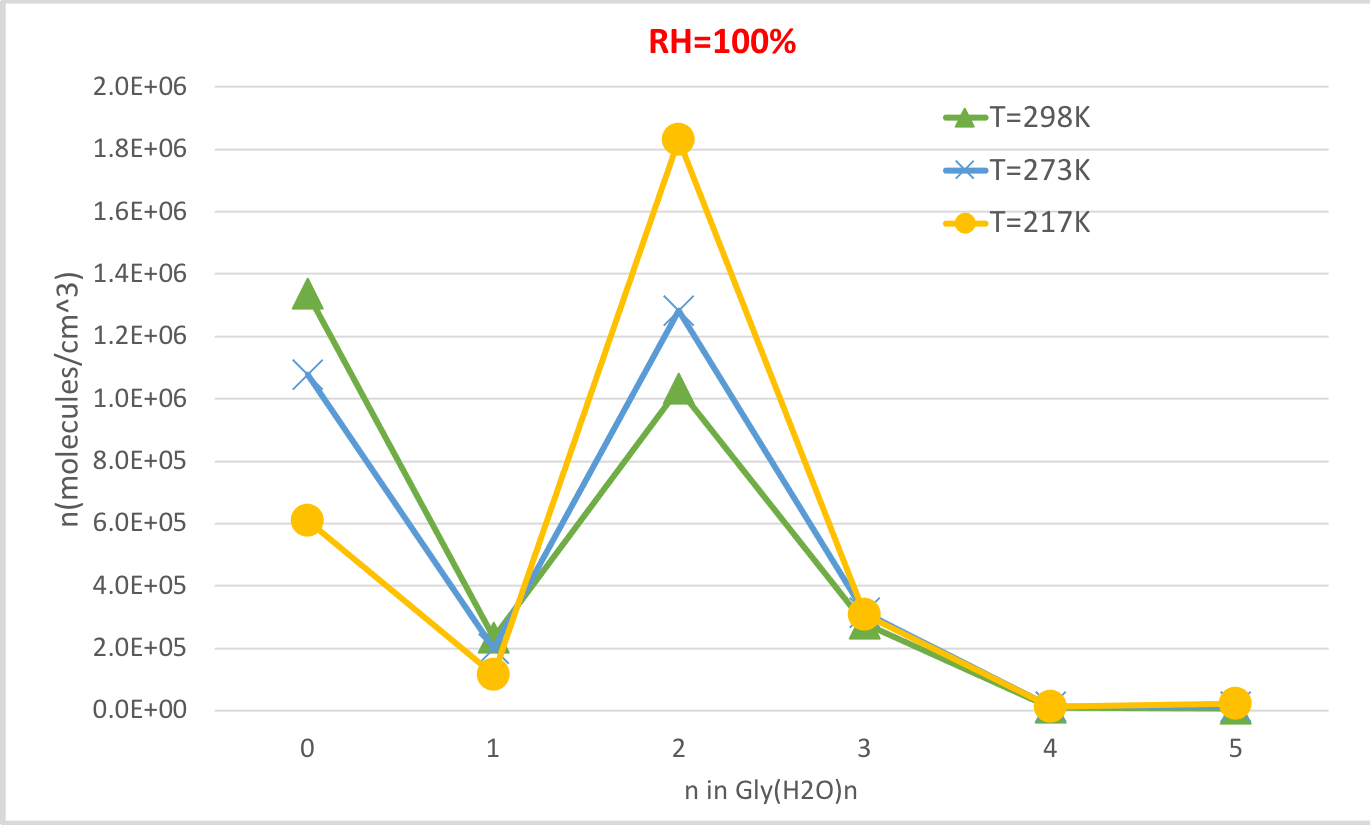
A Figura 4 mostra a concentração de equilíbrio da glicina hidratada calculada na Tabela 4 em função da temperatura a 100% de umidade relativa e 1 pressão atmosférica. Mostra que, à medida que a temperatura diminui de 298,15K para 216,65K, a concentração de glicina não hidratada (n=0) diminui e as de glicina hidratada aumentam. O dihidrato de glicina (n=2) em particular aumenta drasticamente com a temperatura decrescente, enquanto a mudança na concentração de outros hidratos é menos perceptível. Esta correlação inversa entre temperatura e concentração hidratada é consistente com a expectativa de que as energias livres de Gibbs de hidratação a temperaturas mais baixas favoreçam a formação de hidratos.
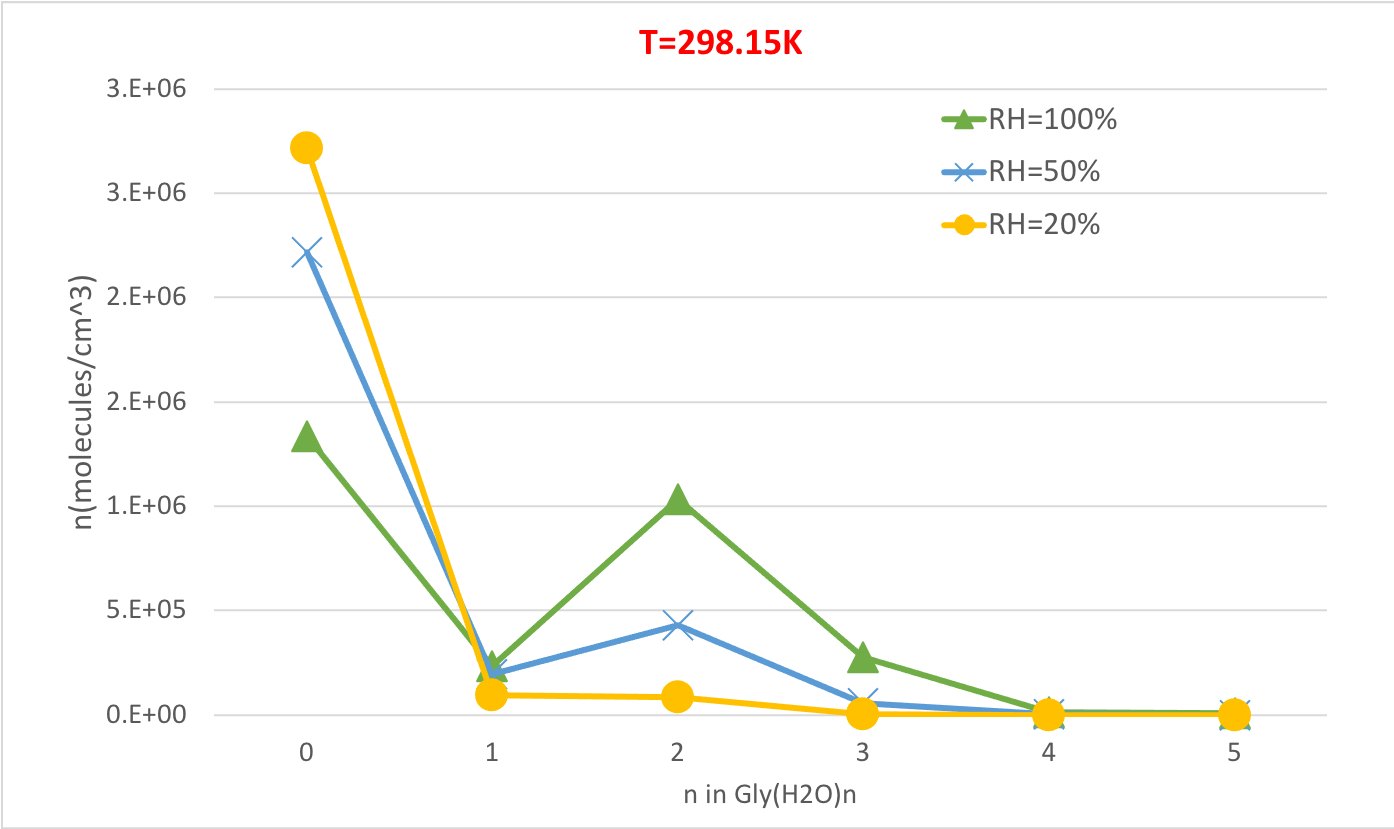
A Figura 5 ilustra a dependência da umidade relativa da concentração de equilíbrio de hidratos de glicina a 298,15K e 1 pressão atmosférica. Demonstra claramente que, à medida que o RH aumenta de 20% para 100%, a concentração de hidratados (n>0) aumenta em detrimento da glicina não hidratada (n=0). Mais uma vez, a correlação direta entre a umidade relativa e a concentração de hidratos é consistente com a ideia de que a presença de mais moléculas de água no RH mais alto promove a formação de hidratos.
Como apresentado, este protocolo dá uma compreensão qualitativa das populações de glicina hidratada na atmosfera. Assumindo uma concentração inicial de glicina isolada de 2,9 milhões de moléculas por centímetro cúbico, vemos que a glicina não hidratada (n=0) é a espécie mais abundante na maioria das condições, exceto T=216,65K e RH=100%. O dihidrato (n=2), que tem a menor energia livre sequencial de Gibbs de hidratação em todas as três temperaturas, é o hidratado mais abundante nas condições aqui consideradas. Prevê-se que o monohidrato (n=1) e os hidratos maiores (n≥3) sejam encontrados em quantidades insignificantes. Após a inspeção da Figura 3,a abundância dos aglomerados n = 1-4 pode estar relacionada à estabilidade e tensão na rede de ligação de hidrogênio dos aglomerados. Esses aglomerados têm as moléculas de água de hidrogênio ligadas ao ácido carboxílico moiety de glicina em uma geometria muito semelhante à de várias estruturas de anel ligados a hidrogênio, tornando-as especialmente estáveis.

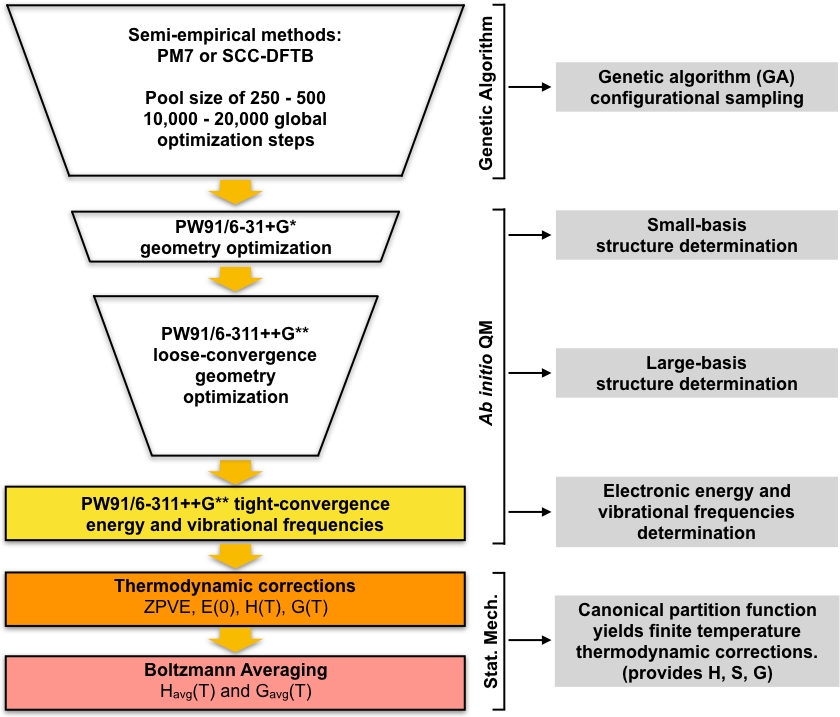
Figura 1: Descrição esquemmática do procedimento atual. Um grande conjunto de estruturas de adivinhação geradas pelo algoritmo genético (GA) é refinado por uma série de otimizações de geometria PW91 até que um conjunto de estruturas convergentes sejam obtidos. As freqüências vibracionais dessas estruturas são computadas e usadas para calcular a energia livre de formação de Gibbs, que por sua vez é usada para calcular as concentrações de equilíbrio dos aglomerados em condições ambientais. Clique aqui para ver uma versão maior desta figura.
{kind=link}

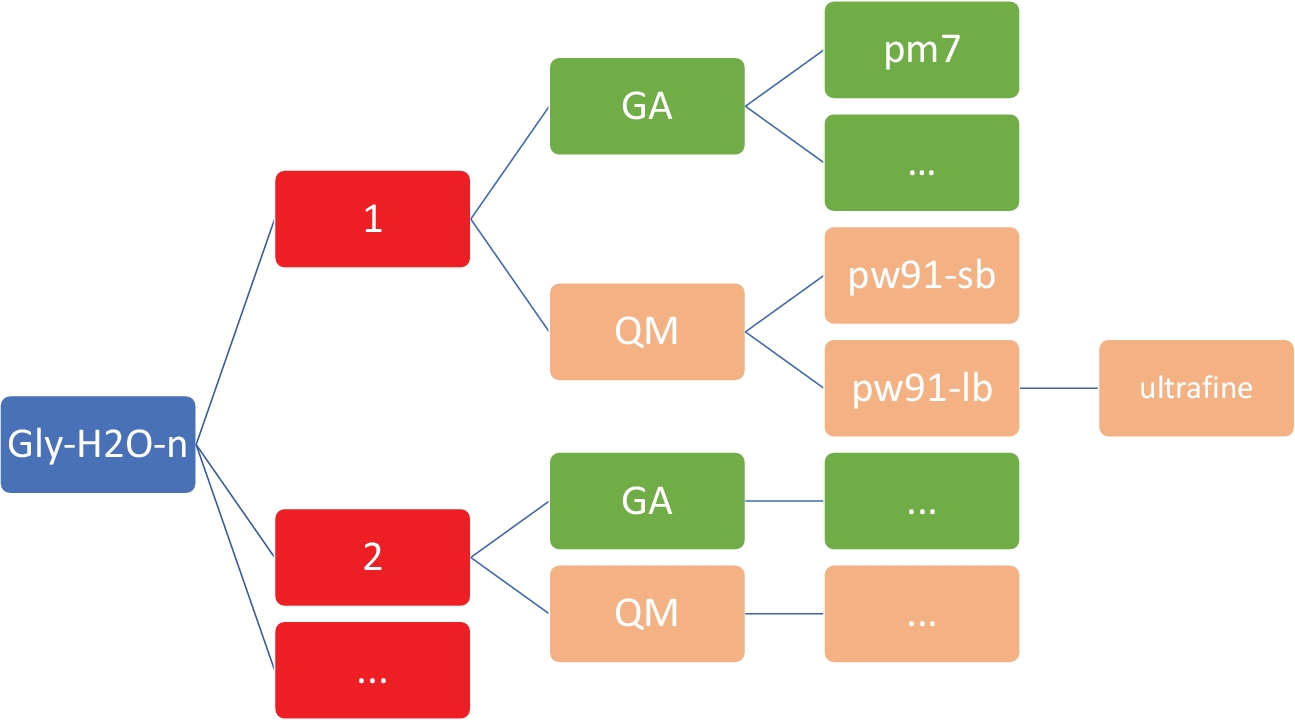
Figura 2: Estrutura representativa do diretório para cada cluster. Os scripts interno incluídos neste protocolo requerem a estrutura do diretório mostrada acima, onde n é o número de moléculas de água. Para cada n em gly-h2o-n,existem os seguintes subdiretórios: GA para algoritmo genético com um diretório GA/pm7, QM para mecânica quântica com QM/pw91-sb para PW91/6-31+G*, QM/pw91-lb para PW91/6-311++G**, e QM/pw91-lb/ultrafine para otimizações e cálculos vibracionais finais em grades de integração ultrafina. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Estruturas representativas de baixa energia de Gly(H2O)n=0-5. Esses clusters foram o minima global de energia eletrônica otimizado no nível de teoria PW91/6-311++G***. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: Dependência de temperatura de Gly(H2O)n=0-5 como 100% umidade relativa e 1 pressão de atm. A concentração dos hidratos é dada em unidades de moléculas cm-3. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: Dependência de umidade relativa de Gly(H2O)n=0-5 como 298,15 K e 1 pressão de atm. A concentração dos hidratos é dada em unidades de moléculas cm-3. Clique aqui para ver uma versão maior desta figura.
{kind=link}
| E[PW91/6-311++G**] | 216,65 Mil | 273,15 Mil | 298,15 Mil | ||||||||
| LB-UF | ZPVE | ♦H | S | ♦G | ♦H | S | ♦G | ♦H | S | ♦G | |
| Água | -76.430500 | 13.04 | 1.72 | 42.59 | 5.54 | 2.17 | 44.44 | 3.08 | 2.37 | 45.14 | 1.96 |
| Glicina | -284.434838 | 48.55 | 2.65 | 69.53 | 36.14 | 3.70 | 73.81 | 32.09 | 4.22 | 75.61 | 30.22 |
Tabela 1: Energias monômeras. As energias eletrônicas estão em unidades de Hartree, enquanto todas as outras quantidades estão em unidades de kcal mol-1. A água e a glicina foram otimizadas no nível de teoria e frequências vibracionais PW91/6-311++G**. As correções termodinâmicas para uma pressão de 1 atm e temperatura de 298,15 K foram calculadas utilizando-se o script thermo.pl.
| E[PW91/6-311++G**] | 0 K | 216,65 Mil | 273,15 Mil | 298,15 Mil | ||||||||
| N | Nome | LB-UF | ZPVE | ♦H | S | ♦G | ♦H | S | ♦G | ♦H | S | ♦G |
| 1 | gly-h2o-1 | -360.88481 | 63.96 | 3.61 | 80.12 | 50.22 | 5.12 | 86.27 | 45.52 | 5.85 | 88.83 | 43.33 |
| 2 | gly-h2o-2 | -437.33763 | 79.33 | 4.53 | 90.86 | 64.17 | 6.46 | 98.78 | 58.81 | 7.40 | 102.06 | 56.30 |
| 3 | gly-h2o-3 | -513.78620 | 94.52 | 5.67 | 105.08 | 77.42 | 8.08 | 114.94 | 71.19 | 9.23 | 119.00 | 68.27 |
| 4 | gly-h2o-4 | -590.23667 | 109.80 | 6.03 | 104.98 | 91.30 | 8.78 | 116.21 | 84.40 | 10.11 | 120.87 | 81.14 |
| 5 | gly-h2o-5 | -666.68845 | 125.80 | 7.26 | 121.70 | 106.69 | 10.47 | 134.83 | 99.44 | 12.01 | 140.24 | 96.00 |
Tabela 2: Energias de cluster. As energias das estruturas gly(H2O)de menor energia encontradas usando nosso procedimento descrito na Figura 1. As energias eletrônicas estão em unidades de Hartree, enquanto todas as outras quantidades estão em unidades de kcal mol-1.
| Hidratação Total: Gly + nH2O <-> Gly(H2O)n | Hidratação Sequencial: Gly(H2O)n-1 + H2O <-> Gly(H2O)n | ||||||||||||||||
| E[PW91/6-311++G**] | 216.65 | 273.15 | 298.15 | 216.65 | 273.15 | 298.15 | |||||||||||
| N | nome do sistema | LB-UF | ♦E(0) | ♦H(T) | ♦G(T) | ♦H(T) | ♦G(T) | ♦H(T) | ♦G(T) | LB-UF | ♦E(0) | ♦H(T) | ♦G(T) | H(T) | ♦G(T) | ♦H(T) | ♦G(T) |
| 1 | gly-h2o-1 | -12.22 | -9.85 | -10.61 | -3.68 | -10.61 | -1.87 | -10.59 | -1.07 | -12.22 | -9.85 | -10.61 | -3.68 | -10.61 | -1.87 | -10.59 | -1.07 |
| 2 | gly-h2o-2 | -26.22 | -21.53 | -23.10 | -9.27 | -23.11 | -5.66 | -23.09 | -4.06 | -14.00 | -11.68 | -12.49 | -5.59 | -12.50 | -3.79 | -12.50 | -2.99 |
| 3 | gly-h2o-3 | -37.56 | -30.72 | -32.88 | -12.90 | -32.87 | -7.69 | -32.82 | -5.38 | -11.34 | -9.19 | -9.78 | -3.63 | -9.76 | -2.03 | -9.73 | -1.32 |
| 4 | gly-h2o-4 | -50.10 | -40.34 | -43.48 | -15.87 | -43.54 | -8.71 | -43.51 | -5.55 | -12.54 | -9.62 | -10.60 | -2.97 | -10.67 | -1.02 | -10.69 | -0.17 |
| 5 | gly-h2o-5 | -63.45 | -51.41 | -55.42 | -20.58 | -55.51 | -11.48 | -55.48 | -7.45 | -13.35 | -11.07 | -11.94 | -4.71 | -11.97 | -2.77 | -11.97 | -1.90 |
Tabela 3: Energias de hidratação. A energia total de hidratação e energia de hidratação seqüencial para Gly(H2O)n=1-5 em unidades de kcal mol-1. Aqui, E[PW91/6-311++G**] é a mudança na energia eletrônica, ¥E(0) é a energia vibracional de ponto zero (ZPVE) corrigida mudança de energia, ¥H(T) é a mudança de entalpia na temperatura T, e ¥G(T) é a mudança de energia livre de Gibbs de hidratação de cada aglomerado Gly(H2O)n=1-5.
| Distribuição de Hidratação do Equilíbrio em função da temperatura e umidade relativa | |||||||||
| T=298,15K | T=273.15K | T=216,65K | |||||||
| Gly(H2O)n | RH=100% | RH=50% | RH=20% | RH=100% | RH=50% | RH=20% | RH=100% | RH=50% | RH=20% |
| 0 | 1.3E+06 | 2.2E+06 | 2.7E+06 | 1.1E+06 | 2.0E+06 | 2.7E+06 | 6.1E+05 | 1.5E+06 | 2.5E+06 |
| 1 | 2.3E+05 | 1.9E+05 | 9.5E+04 | 2.0E+05 | 1.9E+05 | 9.9E+04 | 1.2E+05 | 1.5E+05 | 9.5E+04 |
| 2 | 1.0E+06 | 4.3E+05 | 8.4E+04 | 1.3E+06 | 6.1E+05 | 1.3E+05 | 1.8E+06 | 1.1E+06 | 3.0E+05 |
| 3 | 2.8E+05 | 5.8E+04 | 4.5E+03 | 3.2E+05 | 7.4E+04 | 6.3E+03 | 3.1E+05 | 9.6E+04 | 1.0E+04 |
| 4 | 1.1E+04 | 1.1E+03 | 3.4E+01 | 1.3E+04 | 1.5E+03 | 5.0E+01 | 1.1E+04 | 1.8E+03 | 7.5E+01 |
| 5 | 7.5E+03 | 3.9E+02 | 4.9E+00 | 1.2E+04 | 7.2E+02 | 9.7E+00 | 2.4E+04 | 1.9E+03 | 3.1E+01 |
Tabela 4: Concentrações de hidrato de equilíbrio de Gly(H2O)n=0-5 como temperatura de função (T=298,15K, 273,15K, 216,65K) e umidade relativa (RH=100%, 50%, 20%). A concentração dos hidratos é dada em unidades de moléculas cm-3 assumindo valores experimentais56,57,58, de [Gly]0 = 2,9 x 106 cm-3 e [H2O] = 7,7 x 1017 cm-3, 1,6 x 1017 cm-3 e 9,9 x 1014 cm-3 a 100% umidade relativa e T = 298,15 K, 273,15 K, e 216,65 K, respectivamente59.
Arquivos Suplementares. Clique aqui para baixar esses arquivos.
Discussão
A precisão dos dados gerados por este protocolo depende principalmente de três coisas: (i) a variedade de configurações amostrados pela Etapa 2, (ii) a precisão da estrutura eletrônica do sistema, (iii) e a precisão das correções termodinâmicas. Cada um desses fatores pode ser abordado modificando o método editando os scripts incluídos. O primeiro fator é facilmente superado com o uso de um maior pool inicial de estruturas geradas aleatoriamente, mais numerosas iterações do Ga, e uma definição mais frouxa dos critérios envolvidos no Ga. Além disso, pode-se usar um método semi-empírico diferente, como o modelo de tight-binding tight-binding (SCC-DFTB)62 e o modelo potencial de fragmento efetivo (EFP)63, a fim de explorar os efeitos de diferentes descrições físicas. A principal limitação aqui é a incapacidade do método de formar ou quebrar ligações covalentes, o que significa que os monômeros estão congelados. O procedimento de AG encontra apenas as posições relativas mais estáveis desses monômeros congelados de acordo com a descrição semi-empírica.
A precisão da estrutura eletrônica do sistema pode ser melhorada de várias maneiras, cada uma com seu custo computacional. Pode-se escolher uma melhor densidade funcional, como m06-2X64 e wB97X-V65, ou métodos mecânicos quânticos (QM) como as teorias de perturbação Møller-Plesset66,67,68 (MPn) e métodos de cluster acoplado69 (CC) a fim de melhorar a descrição física do sistema. Na hierarquia das funcionais, o desempenho geralmente melhora ao passar de funis de aproximação de gradiente generalizado (GGA) como PW91 para funções híbridas separadas em faixa, como funções híbridas wB97X-D e meta-GGA como M06-2X.
A desvantagem dos métodos DFT é que não é possível uma convergência sistemática para um valor preciso; no entanto, os métodos DFT são computacionalmente baratos e há uma grande variedade de funcionais para uma grande variedade de aplicações.
Energias calculadas usando métodos de função de onda como MP2 e CCSD(T) em conjunto com conjuntos de base consistentes de correlação de aumento do número cardeal ([aug-]cc-pV[D,T,Q,...] Z) convergem para seu limite completo de fixação de base sistematicamente, mas o custo computacional de cada cálculo torna-se proibitivo à medida que o tamanho do sistema cresce. Um refinamento adicional da estrutura eletrônica pode ser realizado usando conjuntos de base explicitamente correlacionados70 e extrapolando para o limite completo do conjunto de base (CBS)71. Nosso trabalho recente sugere que uma abordagem perturbativa de segunda ordem explicitamente correlacionada de segunda ordem Møller-Plesset (DF-MP2-F12) produz energias que se aproximam das computações MP2/CBS32. A modificação do protocolo atual para usar diferentes métodos de estrutura eletrônica envolve duas etapas: (i) preparar um arquivo de entrada de modelo seguindo a sintaxe dada pelo software, (ii) e editar a sintaxe de arquivo de entrada run-pw91-sb.csh, run-pw91-lb.csh,e scripts run-pw91-lb-ultrafine.csh para gerar a sintaxe de arquivo de entrada correta, bem como o script de envio correto para o software.
Por fim, a precisão das correções termodinâmicas depende do método de estrutura eletrônica, bem como da descrição do PES em torno do mínimo global. Uma descrição precisa do PES requer a computação de derivados de terceira e alta ordem do PES em relação aos deslocamentos nos graus nucleares de liberdade, como o campo de força quartic72,73 (QFF), que é uma tarefa excepcionalmente cara. O protocolo atual utiliza a aproximação harmônica do oscilador às frequências vibracionais, resultando na necessidade de calcular apenas até os segundos derivados do PES. Essa abordagem torna-se problemática em sistemas com alta anharmonicidade, como moléculas muito disquetes e potenciais simétricos de dois poços devido à grande diferença no verdadeiro PES e no PES harmônico. Além disso, o custo de ter um PES de alta qualidade a partir de um método de estrutura eletrônica computacionalmente exigente só compõe o problema do custo para os cálculos de frequência vibracional. Uma abordagem para superar isso é usar as energias eletrônicas a partir de um cálculo de estrutura eletrônica de alta qualidade, juntamente com frequências vibracionais computadas em um PES de menor qualidade, resultando em um equilíbrio entre custo e precisão. O protocolo atual pode ser modificado para usar diferentes descrições de PES como descrito no parágrafo anterior; no entanto, pode-se também editar as palavras-chave de freqüência vibracional nos scripts e modelos para calcular frequências vibracionais anharmônicas.
Duas questões cruciais para qualquer protocolo de amostragem configuracional são o método inicial para a amostragem da superfície de energia potencial e os critérios utilizados para identificar cada cluster. Fizemos uso extensivo de uma variedade de métodos em nosso trabalho anterior. Para a primeira questão, o método inicial de amostragem da superfície potencial de energia, fizemos a escolha de usar a AI com métodos semi-empíricos com base nesses fatores. A amostragem configuracional utilizando intuição química26,amostragem aleatória e dinâmica molecular (DM)29,30,não encontra ma global putativo regularmente para aglomerados maiores que 10 monômeros, como observamos em nossos estudos de aglomerados de água18. Temos usado com sucesso o salto de bacia (BH) para estudar o complexo PES de (H2O)1174,mas exigiu a inclusão manual de alguns potenciais isôsimos de baixa energia que o algoritmo de BH não encontrou. Uma comparação do desempenho de BH e GA em encontrar o mínimo global de aglomerados de água, (H2O)n=10-20 demonstrou que a Ga encontrou consistentemente o mínimo global mais rápido que BH75. Ga como implementado no OGOLEM e CLUSTER é muito versátil porque pode ser aplicado a qualquer cluster molecular e pode interagir com um grande número de pacotes com capacidades clássicas de campo de força, semi-empírica, funcional de densidade e ab initio. A escolha do PM7 é impulsionada por sua velocidade e precisão razoável. Praticamente qualquer outro método semi-empírico teria um custo computacional significativamente maior.
Quanto à segunda edição, exploramos utilizando diferentes critérios para identificar estruturas únicas que vão desde energias eletrônicas, momentos dipolo, sobreposição de RMSDs e constantes rotacionais. O uso de momentos de dipolo mostrou-se difícil porque tanto os componentes do momento do dipolo dependiam da orientação da molécula quanto o momento total do dipolo era muito sensível às diferenças de geometria de tal forma que era difícil definir limiares determinando se as estruturas são iguais ou únicas. Uma combinação de energias eletrônicas e constantes rotacionais provou ser mais útil.
O critério atual para a análise de duas estruturas únicas baseia-se em um limiar de diferença de energia de 0,10 kcal mol-1 e diferença constante rotacional de 1%. Portanto, duas estruturas são consideradas diferentes se suas energias diferem em mais de 0,10 kcal mol-1 (~0,00015 a.u.) E qualquer uma de suas três constantes rotacionais (A, B, C) diferem em mais de 1%. Referências internas substanciais ao longo dos anos consideraram esses limiares escolhas razoáveis. Nossa metodologia de abordagem e triagem de configuração tem sido aplicada a aglomerados muito fracos, como hidrocarbonetos poliaromáticos complexos com água76,77, bem como hidratos de sulfato ternário fortemente ligados contendo amônia e aminas32. Para clusters onde há diferentes estados de protonação a serem considerados, a melhor abordagem é executar vários cálculos de A, cada um começando com monômeros em diferentes estados de protonação. Isso garante que estruturas com diferentes estados de protonação sejam cuidadosamente consideradas. No entanto, os cálculos dft de baixo nível muitas vezes permitem que os estados de protonação mudem durante o curso da otimização da geometria, produzindo assim o estado de protonação mais estável, independentemente da geometria inicial.
Nossos métodos de amostragem de configuração ga devem funcionar bem mesmo para moléculas disquetes, desde que os códigos GA sejam interfaceados com métodos gerais e não parametrizados que permitem que os monômeros adotem diferentes configurações durante o curso da execução de GA. Por exemplo, a interligação de Ga com o PM7 permitiria que as estruturas dos monômeros mudassem, mas se seus vínculos se rompessem como aconteceria quando os estados de protonação mudassem, as estruturas podem ser descartadas como candidatos inaceitáveis.
Consideramos diferentes formas de corrigir as deficiências da aproximação harmônica, especialmente aquelas decorrentes de baixas frequências vibracionais. Incorporar a aproximação quase harmônica na metodologia atual não é difícil. No entanto, ainda há dúvidas sobre o método quase harmônico, especialmente quando se trata da frequência de corte abaixo da qual será aplicado. Além disso, não há trabalhos rigorosos de benchmarking examinando a confiabilidade da aproximação quase-RRHO, embora a sabedoria convencional sugira que deve ser uma melhoria em relação à aproximação da RRHO.
O protocolo assim apresentado pode ser generalizado para qualquer sistema de aglomerados moleculares de fase de gás não covalentemente ligados. Também pode ser generalizado para usar qualquer método semi-empírico, método de estrutura eletrônica e software, e método de análise vibracional e software editando os scripts e modelos. Isso pressupõe que o usuário esteja confortável com a interface de linha de comando Linux, scripting Python e computação de alto desempenho. A sintaxe e o visual desconhecidos do sistema operacional Linux e a falta de experiência em scripts é a maior armadilha neste protocolo e é onde os novos alunos mais lutam. Este protocolo tem sido usado com sucesso em uma variedade de implementações durante anos em nosso grupo, focando principalmente nos efeitos do ácido sulfúrico e amônia na formação de aerossóis. Outras melhorias neste protocolo envolverão uma interface mais robusta para software de estrutura mais eletrônica, implementações alternativas do algoritmo genético e, possivelmente, o uso de métodos mais novos para cálculos mais rápidos de energias eletrônicas e vibracionais. Nossas aplicações atuais deste protocolo estão explorando a importância dos aminoácidos nos estágios iniciais da formação de aerossóis na atmosfera atual e na formação de moléculas biológicas maiores em ambientes prebióticos.
Divulgações
Nenhum.
Agradecimentos
Este projeto foi apoiado pelas subvenções CHE-1229354, CHE-1662030, CHE-1721511 e CHE-1903871 da National Science Foundation (GCS), do Arnold and Mabel Beckman Foundation Beckman Scholar Award (AGG) e da Barry M. Goldwater Scholarship (AGG). Foram utilizados recursos de computação de alto desempenho do Consórcio MERCURY (http://www.mercuryconsortium.org).
Materiais
| Name | Company | Catalog Number | Comments |
| Avogadro | https://avogadro.cc | Open-source molecular visualization program | |
| Gaussian [09/16] Software | http://www.gaussian.com/ | Commercial ab initio electronic structure program | |
| MOPAC 2016 | http://openmopac.net/MOPAC2016.html | Open-source semi-empirical program | |
| OGOLEM Software | https://www.ogolem.org | Genetic algorithm-based global optimization program | |
| OpenBabel | http://openbabel.org/wiki/Main_Page | Open-source cheminformatics library | |
| calcRotConsts.py | Shields Group, Department of Chemistry, Furman University | Python script to compute rotational constants | |
| calcSymmetry.csh | Shields Group, Department of Chemistry, Furman University | Shell script to calculate symmetry number of a molecule given Cartesian coordinates | |
| combine-GA.csh | Shields Group, Department of Chemistry, Furman University | Shell script to combine energy and rotational constants from different GA directories | |
| combine-QM.csh | Shields Group, Department of Chemistry, Furman University | Shell script to combine energy and rotational constants from different QM directories | |
| gaussianE.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract Gaussian 09 energies | |
| gaussianFreqs.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract Gaussian 09 vibrational frequencies | |
| getrotconsts | Shields Group, Department of Chemistry, Furman University | Executable to calculate rotational constants given a molecule's Cartesian coordinates | |
| getRotConsts-dft-lb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of large basis DFT optimized structures | |
| getRotConsts-dft-lb-ultrafine.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of ultrafine DFT optimized structures | |
| getRotConsts-dft-sb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of small basis DFT optimized structures | |
| getRotConsts-GA.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of genetic algorithm optimized structures | |
| global-minimum-coords.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of global minimum structures of gly-(h2o)n, where n=0-5 | |
| make-thermo-gaussian.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract data from Gaussian output files and make input files for the thermo.pl script | |
| ogolem-input-file.ogo | Shields Group, Department of Chemistry, Furman University | Ogolem sample input file | |
| ogolem-submit-script.pbs | Shields Group, Department of Chemistry, Furman University | PBS batch submission file for Ogolem calculations | |
| README.docx | Shields Group, Department of Chemistry, Furman University | Clarifications to help readers use the scripts effectively | |
| runogolem.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run OGOLEM | |
| run-pw91-lb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of large basis DFT optimization calculations | |
| run-pw91-lb-ultrafine.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of ultrafine DFT optimization calculations | |
| run-pw91-sb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of small basis DFT optimization calculations | |
| run-thermo-pw91.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute the thermodynamic corrections for a batch of DFT optimized structures | |
| similarityAnalysis.py | Shields Group, Department of Chemistry, Furman University | Python script to determine unique structures based on rotational constants and energies | |
| symmetry | Shields Group, Department of Chemistry, Furman University | Executable to calculate molecular symmetry given Cartesian coordinates | |
| symmetry.c | (C) 1996, 2003 S. Patchkovskii, Serguei.Patchkovskii@sympatico.ca | C code to determine the molecular symmstry of a molecule given Cartesian coordinates | |
| template-marcy.pbs | Shields Group, Department of Chemistry, Furman University | Template for a PBS submit script which uses OGOLEM | |
| template-pw91.com | Shields Group, Department of Chemistry, Furman University | Template Gaussian 09 input | |
| template-pw91-HL.com | Shields Group, Department of Chemistry, Furman University | Template Gaussian 09 input for ultrafine DFT optimization | |
| thermo.pl | https://www.nist.gov/mml/csd/chemical-informatics-research-group/products-and-services/program-computing-ideal-gas | Perl open-source script to compute ideal gas thermodynamic corrections | |
| gly-h2o-n.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet for the complete protocol | |
| table-1.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-2.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-3.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-4.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| water.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of water | |
| glycine.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of glycine |
Referências
- Foster, P., Ramaswamy, V., Solomon, S., Qin, D., Manning, M., Chen, Z., Marquis, M., Averyt, K. B., Tignor, M., Miller, H. L. . Climate Change 2007 The Scientific Basis. , (2007).
- Kulmala, M., et al. Toward direct measurement of atmospheric nucleation. Science. 318 (5847), 89-92 (2007).
- Sipila, M., et al. The role of sulfuric acid in atmospheric nucleation. Science. 327 (5970), 1243-1246 (2010).
- Jiang, J., et al. First measurement of neutral atmospheric cluster and 1 - 2 nm particle number size distributions during nucleation events. Aerosol Science and Technology. 45 (4), (2011).
- Dunn, M. E., Pokon, E. K., Shields, G. C. Thermodynamics of forming water clusters at various Temperatures and Pressures by Gaussian-2, Gaussian-3, Complete Basis Set-QB3, and Complete Basis Set-APNO model chemistries; implications for atmospheric chemistry. Journal of the American Chemical Society. 126 (8), 2647-2653 (2004).
- Pickard, F. C., Pokon, E. K., Liptak, M. D., Shields, G. C. Comparison of CBSQB3, CBSAPNO, G2, and G3 thermochemical predictions with experiment for formation of ionic clusters of hydronium and hydroxide ions complexed with water. Journal of Chemical Physics. 122, 024302 (2005).
- Pickard, F. C., Dunn, M. E., Shields, G. C. Comparison of Model Chemistry and Density Functional Theory Thermochemical Predictions with Experiment for Formation of Ionic Clusters of the Ammonium Cation Complexed with Water and Ammonia; Atmospheric Implications. Journal of Physical Chemistry A. 109 (22), 4905-4910 (2005).
- Alongi, K. S., Dibble, T. S., Shields, G. C., Kirschner, K. N. Exploration of the Potential Energy Surfaces, Prediction of Atmospheric Concentrations, and Vibrational Spectra of the HO2•••(H2O)n (n=1-2) Hydrogen Bonded Complexes. Journal of Physical Chemistry A. 110 (10), 3686-3691 (2006).
- Allodi, M. A., Dunn, M. E., Livada, J., Kirschner, K. N. Do Hydroxyl Radical-Water Clusters, OH(H2O)n, n=1-5, Exist in the Atmosphere. Journal of Physical Chemistry A. 110 (49), 13283-13289 (2006).
- Kirschner, K. N., Hartt, G. M., Evans, T. M., Shields, G. C. In Search of CS2(H2O)n=1-4 Clusters. Journal of Chemical Physics. 126, 154320 (2007).
- Hartt, G. M., Kirschner, K. N., Shields, G. C. Hydration of OCS with One to Four Water Molecules in Atmospheric and Laboratory Conditions. Journal of Physical Chemistry A. 112 (19), 4490-4495 (2008).
- Morrell, T. E., Shields, G. C. Atmospheric Implications for Formation of Clusters of Ammonium and 110 Water Molecules. Journal of Physical Chemistry A. 114 (12), 4266-4271 (2010).
- Temelso, B., et al. Quantum Mechanical Study of Sulfuric Acid Hydration: Atmospheric Implications. Journal of Physical Chemistry A. 116 (9), 2209 (2012).
- Husar, D. E., Temelso, B., Ashworth, A. L., Shields, G. C. Hydration of the Bisulfate Ion: Atmospheric Implications. Journal of Physical Chemistry A. 116 (21), 5151-5163 (2012).
- Bustos, D. J., Temelso, B., Shields, G. C. Hydration of the Sulfuric Acid – Methylamine Complex and Implications for Aerosol Formation. Journal of Physical Chemistry A. 118 (35), 7430-7441 (2014).
- Wales, D. J., Scheraga, H. A. Global optimization of clusters, crystals, and biomolecules. Science. 27 (5432), 1368-1372 (1999).
- Day, M. B., Kirschner, K. N., Shields, G. C. Global search for minimum energy (H2O)n clusters, n = 3 - 5. The Journal of Physical Chemistry A. 109 (30), 6773-6778 (2005).
- Shields, R. M., Temelso, B., Archer, K. A., Morrell, T. E., Shields, G. C. Accurate predictions of water cluster formation, (H2O)n=2-10. The Journal of Physical Chemistry A. 114 (43), 11725-11737 (2010).
- Temelso, B., Archer, K. A., Shields, G. C. Benchmark structures and binding energies of small water clusters with anharmonicity corrections. The Journal of Physical Chemistry A. 115 (43), 12034-12046 (2011).
- Temelso, B., Shields, G. C. The role of anharmonicity in hydrogen-bonded systems: The case of water clusters. The Journal of Chemical Theory and Computation. 7 (9), 2804-2817 (2011).
- Von Freyberg, B., Braun, W. Efficient search for all low energy conformations of polypeptides by Monte Carlo methods. The Journal of Computational Chemistry. 12 (9), 1065-1076 (1991).
- Rakshit, A., Yamaguchi, T., Asada, T., Bandyopadhyay, P. Understanding the structure and hydrogen bonding network of (H2O)32 and (H2O)33: An improved Monte Carlo temperature basin paving (MCTBP) method of quantum theory of atoms in molecules (QTAIM) analysis. RSC Advances. 7 (30), 18401-18417 (2017).
- Deaven, D. M., Ho, K. M. Molecular geometry optimization with a genetic algorithm. Physical Review Letters. 75, 288-291 (1995).
- Hartke, B. Application of evolutionary algorithms to global cluster geometry optimization. Applications of Evolutionary Computation in Chemistry. , (2004).
- Dieterich, J. M., Hartke, B. OGOLEM: Global cluster structure optimization for arbitrary mixtures of flexible molecules. A multiscaling, object-oriented approach. Molecular Physics. 108 (3-4), 279-291 (2010).
- Herb, J., Nadykto, A. B., Yu, F. Large ternary hydrogen-bonded pre-nucleation clusters in the Earth's atmosphere. Chemical Physics Letters. 518, 7-14 (2011).
- Ortega, I. K., et al. From quantum chemical formation free energies to evaporation rates. Atmospheric Chemistry and Physics. 12 (1), 225-235 (2012).
- Elm, J., Bilde, M., Mikkelsen, K. V. Influence of Nucleation Precursors on the Reaction Kinetics of Methanol with the OH Radical. Journal of Physical Chemistry A. 117 (30), 6695-6701 (2013).
- Loukonen, V., et al. Enhancing effect of dimethylamine in sulfuric acid nucleation in the presence of water - a computational study. Atmospheric Chemistry and Physics. 10 (10), 4961-4974 (2010).
- Temelso, B., Phan, T. N., Shields, G. C. Computational study of the hydration of sulfuric acid dimers: implications for acid dissociation and aerosol formation. Journal of Physical Chemistry A. 116 (39), 9745-9758 (2012).
- Jiang, S., et al. Study of Cl-(H2O)n (n = 1-4) using basin-hopping method coupled with density functional theory. Journal of Computational Chemistry. 35 (2), 159-165 (2014).
- Temelso, B., et al. Effect of mixing ammonia and alkylamines on sulfate aerosol formation. Journal of Physical Chemistry A. 122 (6), 1612-1622 (2018).
- Perdew, J. P., Ruzsinszky, A., Tao, J. Prescription for the design and selection of density functional approximations: More constraint satisfaction with fewer fits. Journal of Chemical Physics. 123, 062201 (2005).
- Riplinger, C., Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. Journal of Chemical Physics. 138, 034106 (2013).
- Riplinger, C., Pinski, P., Becker, U., Valeev, E. F., Neese, F. Sparse maps--A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory. Journal of Chemical Physics. 144 (2), 024109 (2016).
- Kildgaard, J. V., Mikkelsen, K. V., Bilde, M., Elm, J. Hydration of atmospheric molecular clusters: a new method for systematic configurational sampling. Journal of Physical Chemistry A. 122 (22), 5026-5036 (2018).
- González, &. #. 1. 9. 3. ;. Measurement of areas on a sphere Using Fibonacci and latitude-longitude lattices. Mathematical Geosciences. 42, 49-64 (2010).
- Karaboga, D., Basturk, B. On the performance of artificial bee colony (ABC) algorithm. Applied Soft Computing. 8 (1), 687-697 (2008).
- Zhang, J., Doig, M. Global optimization of rigid molecules using the artificial bee colony algorithm. Physical Chemistry Chemical Physics. 18 (4), 3003-3010 (2016).
- Kubecka, J., Besel, V., Kurten, T., Myllys, N., Vehkamaki, H. Configurational sampling of noncovalent (atmospheric) molecular clusters: sulfuric acid and guanidine. Journal of Physical Chemistry A. 123 (28), 6022-6033 (2019).
- Grimme, S., Bannwarth, C., Shushkov, P. A Robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent Interactions of large molecular systems parametrized for all spd-block elements (Z = 1-86). Journal of Chemical Theory and Computation. 13 (5), 1989-2009 (2017).
- Buck, U., Pradzynski, C. C., Zeuch, T., Dieterich, J. M., Hartke, B. A size resolved investigation of large water clusters. Physical Chemistry Chemical Physics. 16 (15), 6859 (2014).
- Forck, R. M., et al. Structural diversity in sodium doped water trimers. Physical Chemistry Chemical Physics. 14 (25), 9054-9057 (2012).
- Witt, C., Dieterich, J. M., Hartke, B. Cluster structures influenced by interaction with a surface. Physical Chemistry Chemical Physics. 20 (23), 15661-15670 (2018).
- Freitbert, A., Dieterich, J. M., Hartke, B. Exploring self-organization of molecular tether molecules on a gold surface by global structure optimization. The Journal of Computational Chemistry. 40 (22), 1978-1989 (2019).
- Stewart, J. J. P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. The Journal of Molecular Modeling. 19 (1), 1-32 (2013).
- Burke, K., Perdew, J. P., Wang, Y. Derivation of a generalized gradient approximation: The PW91 density functional. Electronic Density Functional Theory. , 81-111 (1998).
- Frisch, M. J., et al. . Gaussian 09, Revision A.02. , (2016).
- Ditchfield, R., Hehre, W. J., Pople, J. A. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. The Journal of Chemical Physics. 54 (2), 724 (1971).
- Elm, J., Bilde, M., Mikkelsen, K. V. Assessment of density functional theory in predicting structures and free energies of reaction of atmospheric prenucleation clusters. The Journal of Chemical Theory and Computation. 8 (6), 2071-2077 (2012).
- Elm, J., Mikkelsen, K. V. Computational approaches for efficiently modelling of small atmospheric clusters. Chemical Physics Letters. 615, 26-29 (2014).
- Bayucan, A., et al. . PBS Portable Batch System. , (1999).
- O'Boyle, N. M., et al. Open Babel: An open chemical toolbox. Journal of Cheminformatics. 3, 33 (2011).
- Csaszar, A. G. Conformers of gaseous glycine. Journal of the American Chemical Society. 114 (24), 9568-9575 (1992).
- Zhang, Q., Anastasio, C. Free and combined amino compounds in atmospheric fine particles (PM2.5) and fog waters from Northern California. Atmospheric Environment. 37 (16), 2247-2258 (2003).
- Matsumoto, K., Uematsu, M. Free amino acids in marine aerosols over the western North Pacific Ocean. Atmospheric Environment. 39 (11), 2163-2170 (2005).
- Mandalakis, M., Apostolaki, M., Stephanou, E. G. Trace analysis of free and combined amino acids in atmospheric aerosols by gas chromatography-mass spectrometry. Journal of Chromatography A. 1217 (1), 143-150 (2010).
- Seinfeld, J. H., Pandis, S. N. . Atmospheric Chemistry and Physics, 3rd Ed. , (2016).
- Myllys, N., Elm, J., Halonen, R., Kurten, T., Vehkamaki, H. Coupled cluster evaluation of atmospheric acid-base clusters with up to 10 molecules. The Journal of Physical Chemistry A. 120 (4), 621-630 (2016).
- Elm, J., Bilde, M., Mikkelsen, K. V. Assessment of binding energies of atmospherically relevant clusters. Physical Chemistry Chemical Physics. 15 (39), (2013).
- Elstner, M. The SCC-DFTB method and its application to biological systems. Theoretical Chemistry Accounts. 116 (1-3), 316-325 (2006).
- Kaliman, I. A., Slipchenko, L. V. LIBEFP: A new parallel implementation of the effective fragment potential method as a portable software library. The Journal of Computational Chemistry. 34 (26), 2284-2292 (2013).
- Zhao, Y., Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and trasition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theoretical Chemistry Accounts. 120 (1-3), 215-241 (2008).
- Mardirossian, N., Head-Gordon, M. wB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy. Physical Chemistry Chemical Physics. 16 (21), 9904-9924 (2014).
- Head-Gordon, M., Pople, J. A., Frisch, M. J. MP2 energy evaluation by direct methods. Chemical Physics Letters. 153 (6), 503-506 (1988).
- Pople, J. A., Seeger, R., Krishnan, R. Variational configuration interaction methods and comparison with perturbation theory. The International Journal of Quantum Chemistry. 12, 149-163 (1977).
- Pople, J. A., Binkley, J. S., Seeger, R. Theoretical models incorporating electron correlation. The International Journal of Quantum Chemistry. 10 (10), 1-19 (1976).
- Monkhorst, H. J. Calculation of properties with the coupled-cluster method. The International Journal of Quantum Chemistry. 12 (11), 421-432 (1977).
- Klopper, W., Manby, F. R., Ten-No, S., Valeev, E. F. R12 methods in explicitly correlated molecular electronic structure theory. International Reviews in Physical Chemistry. 25, 427-468 (2006).
- Hattig, C. Optimization of auxiliary basis sets for RI-MP2 and RI-CC2 calculations: Core-valence and quintuple-z basis sets for H to Ar and QZVPP basis sets for Li to Kr. Physical Chemistry Chemical Physics. 7 (1), 59-66 (2005).
- Barone, V. Anharmonic vibrational properties by a fully automated second-order perturbative approach. The Journal of Chemical Physics. 122, 014108 (2005).
- Barone, V. Vibrational zero-point energies and thermodynamic functions beyond the harmonic approximation. The Journal of Chemical Physics. 120 (7), 3059-3065 (2004).
- Temelso, B., et al. Exploring the Rich Potential Energy Surface of (H2O)11 and Its Physical Implications. Journal of Chemical Theory and Computation. 14 (2), 1141-1153 (2018).
- Kabrede, H., Hentschke, R. Global minima of water clusters (H2O)N, N≤25, described by three empirical potentials. Journal of Physical Chemistry B. 107 (16), (2003).
- Steber, A. L., et al. Capturing the Elusive Water Trimer from the Stepwise Growth of Water on the Surface of a Polycyclic Aromatic Hydrocarbon Acenaphthene. Journal of Physical Chemistry Letters. 8 (23), 5744-5750 (2017).
- Perez, C., et al. Corrannulene and its complex with water: A tiny cup of water. Physical Chemistry Chemical Physics. 19 (22), 14214-14223 (2017).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados