
Method Article
Calcolo delle concentrazioni atmosferiche degli ammassi molecolari dalla termochimica dell'initio ab
In questo articolo
Riepilogo
Le concentrazioni atmosferiche di ammassi molecolari debolmente legati possono essere calcolate dalle proprietà termochimiche delle strutture a bassa energia trovate attraverso una metodologia di campionamento configurazionele a più fasi utilizzando un algoritmo genetico e chimica quantistica semi-empirica e ab.
Abstract
Lo studio computazionale della formazione e della crescita degli aerosol atmosferici richiede un'accurata superficie di energia libera di Gibbs, che può essere ottenuta dalla struttura elettronica della fase gassosa e dai calcoli delle frequenze vibrazionali. Queste quantità sono valide per quei cluster atmosferici le cui geometrie corrispondono a un minimo sulle loro potenziali superfici energetiche. L'energia libera di Gibbs della struttura di energia minima può essere utilizzata per prevedere le concentrazioni atmosferiche dell'ammasso in una varietà di condizioni come temperatura e pressione. Vi presentiamo una procedura computazionalmente poco costosa basata su un campionamento di configurazione basato su algoritmi genetici seguito da una serie di calcoli di screening sempre più accurati. La procedura inizia generando ed evolvendo le geometrie di un ampio set di configurazioni utilizzando modelli semi-empirici, quindi affina le strutture uniche risultanti a una serie di livelli di teoria ab inizialiti. Infine, le correzioni termodinamiche vengono calcolate per il set risultante di strutture di energia minima e utilizzate per calcolare le energie libere di Gibbs di formazione, costanti di equilibrio e concentrazioni atmosferiche. Presentiamo l'applicazione di questa procedura allo studio dei cluster di glicina idratati in condizioni ambientali.
Introduzione
Il parametro più incerto negli studi atmosferici sul cambiamento climatico è la misura esatta in cui le particelle di nube riflettono la radiazione solare in arrivo. Gli aerosol, che sono particolati sospesi in un gas, formano particelle di nube chiamate nuclei di condensazione nuvolosa (CCN) che disperdono le radiazioni in entrata, impedendone così l'assorbimento e il successivo riscaldamento dell'atmosfera1. Una comprensione dettagliata di questo effetto di raffreddamento netto richiede una comprensione della crescita degli aerosol nelle CCN, che a sua volta richiede la comprensione della crescita di piccoli ammassi molecolari in particelle di aerosol. Recenti lavori hanno suggerito che la formazione di aerosol è avviata da ammassi molecolari di 3 nm di diametro o meno2; tuttavia, questo regime di dimensioni è difficile da accedere utilizzando tecniche sperimentali3,4. Pertanto, si desidera un approccio di modellazione computazionale per superare questa limitazione sperimentale.
Utilizzando il nostro approccio di modellazione descritto di seguito, possiamo analizzare la crescita di qualsiasi ammasso idratato. Poiché siamo interessati al ruolo dell'acqua nella formazione di grandi molecole biologiche da piccoli costituenti in ambienti pre-biotici, illustriamo il nostro approccio con la glicina. Le sfide incontrate e gli strumenti necessari per affrontare queste domande di ricerca sono molto simili a quelle coinvolte nello studio degli aerosol atmosferici e degli ammassi di prenupollazione5,6,7,8,9,10,11,12,13,14,15. Qui esaminiamo gli ammassi di glicina idratati a partire da una molecola isolata di glicina seguita da una serie di aggiunte graduali fino a cinque molecole d'acqua. L'obiettivo finale è quello di calcolare le concentrazioni di equilibrio dei cluster Di Gly(H2O)n-0-5 nell'atmosfera a temperatura ambiente a livello del mare e di un'umidità relativa (RH) del 100 %.
Un piccolo numero di questi ammassi molecolari sub-nanometrici cresce in un ammasso critico metastabile (1-3 nm di diametro) aggiungendo altre molecole di vapore o coagulando su ammassi esistenti. Questi cluster critici hanno un profilo di crescita favorevole che porta alla formazione di nuclei di condensazione delle nuvole (CCN) molto più grandi (fino a 50-100 nm), che influenzano direttamente l'efficienza delle precipitazioni delle nuvole e la loro capacità di riflettere la luce incidente. Pertanto, avere una buona comprensione della termodinamica dei cluster molecolari e delle loro distribuzioni di equilibrio dovrebbe portare a previsioni più accurate dell'impatto degli aerosol sul clima globale.
Un modello descrittivo di formazione di aerosol richiede una termodinamica accurata della formazione di ammassi molecolari. Il calcolo della termodinamica accurata della formazione molecolare dei cluster richiede l'identificazione delle configurazioni più stabili, che comporta la ricerca del minimo minimo globale e locale sulla superficie energetica potenziale del cluster (PES)16. Questo processo è chiamato campionamento di configurazione e può essere realizzato attraverso una varietà di tecniche, tra cui quelle basate sulla dinamica molecolare (MD)17,18,19,20, Monte Carlo (MC)21,22,e algoritmi genetici (GA)23,24,25.
Nel corso degli anni sono stati sviluppati diversi protocolli per ottenere la struttura e la termodinamica degli idrati atmosferici ad un alto livello di teoria. Questi protocolli differivano nella scelta del metodo di campionamento configurazionele (i) , della natura (ii) del metodo di basso livello utilizzato nel campionamento di configurazione e (iii) la gerarchia dei metodi di livello superiore utilizzati per perfezionare i risultati nei passaggi successivi.
I metodi di campionamento configurazionele includevano l'intuizione chimica26, il campionamento casuale27,28, la dinamica molecolare (MD)29,30, il basin hopping (BH)31e l'algoritmo genetico (GA)24,25,32. I metodi di basso livello più comuni impiegati con questi metodi di campionamento sono i campi di forza o i modelli semi-empirici come PM6, PM7 e SCC-DFTB. Questi sono spesso seguiti da calcoli DFT con set di base sempre più grandi e funzioni più affidabili dai gradini più alti della scala di Jacob33. In alcuni casi, questi sono seguiti da metodi di funzione d'onda di livello superiore come MP2, CCSD(T) e il rapporto costi-economico DLPNO-CCSD(T)34,35.
Kildgaard et al.36 ha sviluppato un metodo sistematico in cui le molecole d'acqua vengono aggiunte in punti sulle sfere di Fibonacci37 intorno a piccoli cluster idratati o non idratati per generare candidati per ammassi più grandi. I candidati non fisici e ridondanti vengono rimossi in base alle soglie di contatto ravvicinate e alla distanza root-mean-square tra diversi conformatori. Le successive ottimizzazioni utilizzando il metodo semi-empirico PM6 e una gerarchia di Metodi DFT e di funzione d'onda vengono utilizzati per ottenere un insieme di conformatori a bassa energia ad un alto livello di teoria.
L'algoritmo a forma di colonia artificiale delle api (ABC)38 è un nuovo approccio di campionamento configurazionele che è stato recentemente implementato da .hang et al. per studiare gli ammassi molecolari in un programma chiamato ABCluster39. Kubecka et al.40 utilizzato ABCluster per il campionamento di configurazione seguito da rioptimization di basso livello utilizzando il metodo semi-empirico GFN-xTBstretto-associazione 41. Hanno ulteriormente perfezionato le strutture e le energie utilizzando i metodi DFT seguiti dalle energie finali usando DLPNO-CCSD(T).
Indipendentemente dal metodo, il campionamento di configurazione inizia con una distribuzione dei punti generata casualmente o non casualmente sul PES. Ogni punto corrisponde a una geometria specifica del cluster molecolare in questione e viene generato dal metodo di campionamento. Quindi il minimo locale più vicino si trova per ogni punto seguendo la direzione "in discesa" sul PES. L'insieme dei minimi così trovati corrisponde a quelle geometrie del cluster molecolare che sono stabili, almeno per qualche tempo. Qui, la forma del PES e la valutazione dell'energia in ogni punto della superficie saranno sensibili alla descrizione fisica del sistema in cui una descrizione fisica più accurata si traduce in un calcolo dell'energia più costoso dal punto di vista computazionale. Utilizzeremo specificamente il metodo GA implementato nel programma OGOLEM25, che è stato applicato con successo a una serie di problemi di ottimizzazione globale e di campionamento di configurazione42,43,44,45, per generare il set iniziale di punti di campionamento.45 Il PES sarà descritto dal modello PM746 implementato nel programma MOPAC201647. Questa combinazione è impiegata perché genera una maggiore varietà di punti rispetto ai metodi MD e MC e trova il minimo locale più veloce di descrizioni più dettagliate del PES.
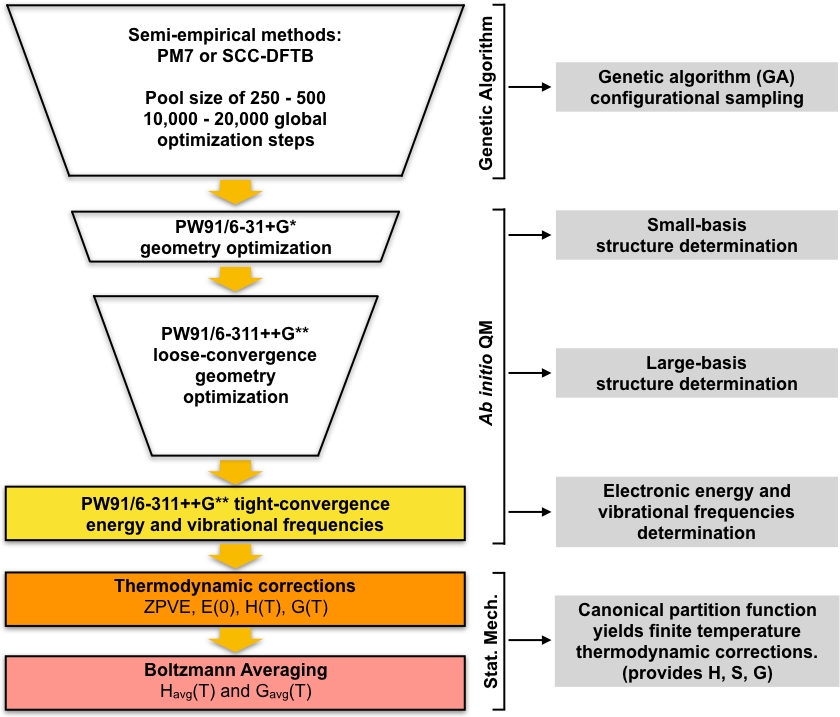
L'insieme dei minimi locali ottimizzati per GA sono considerati come geometrie di partenza per una serie di passaggi di screening, che portano a una serie di energia minima bassa. Questa parte del protocollo inizia ottimizzando l'insieme di strutture uniche ottimizzate per GA utilizzando la teoria funzionale della densità (DFT) con un piccolo set di base. Questo insieme di ottimizzazioni darà generalmente un set più piccolo di strutture minime locali uniche che sono modellate in modo più dettagliato rispetto alle strutture semi-empiriche ottimizzate GA. Quindi un altro ciclo di ottimizzazioni DFT viene eseguito su questo set più piccolo di strutture utilizzando un set di base più grande. Anche in questo caso, questo passaggio darà generalmente un insieme più piccolo di strutture uniche che sono modellate in modo più dettagliato rispetto al piccolo passo base DFT. L'insieme finale di strutture uniche viene quindi ottimizzato per una convergenza più stretta e vengono calcolate le frequenze vibrazionali armoniche. Dopo questo passo abbiamo tutto ciò di cui abbiamo bisogno per calcolare le concentrazioni di equilibrio dei cluster nell'atmosfera. L'approccio generale è riassunto diagrammaticamente figura 1. Useremo la correlazione di correlazione di correlazione di scambio PW9148 generalized-gradient (GGA) funzionale nell'implementazione Gaussian0949 di DFT insieme a due varianti del set di base Pople50 (6-31 , G per la piccola fase di base e 6-311 , G , per il passo di base grande). Questa particolare combinazione di scambio-correlazione funzionale e set di base è stata scelta a causa del suo precedente successo nel calcolo accurate Gibbs energie libere di formazione per gli ammassi atmosferici51,52.
Questo protocollo presuppone che l'utente abbia accesso a un cluster HPC (High Performance Computing) con il sistema batch portatile53 (PBS), MOPAC2016 (http://openmopac.net/MOPAC2016.html)47, OGOLEM (https://www.ogolem.org)25, Gaussian 09 (https://gaussian.com)49e il software OpenBabel54 (http://openbabel.org/wiki/Main_Page) installato seguendo le istruzioni di installazione specifiche. Ogni passaggio di questo protocollo usa anche un set di script di shell in-house e Python 2.7 che devono essere salvati in una directory inclusa nella variabile di ambiente $PATH dell'utente. Tutti i moduli ambientali necessari e le autorizzazioni di esecuzione per eseguire tutti i programmi di cui sopra devono anche essere caricati nella sessione dell'utente. L'utilizzo del disco e della memoria da parte del codice GA (OGOLEM) e dei codici semi-empirici (MOPAC) sono molto piccoli per i moderni standard di risorse del computer. L'utilizzo complessivo della memoria e del disco per OGOLEM/MOPAC dipende dal numero di thread che si desidera utilizzare e, anche in questo momento, l'utilizzo delle risorse sarà ridotto rispetto alle funzionalità della maggior parte dei sistemi HPC. Le esigenze di risorse dei metodi QM dipendono dalle dimensioni dei cluster e dal livello di teoria utilizzato. Il vantaggio di utilizzare questo protocollo è che si può variare il livello di teoria per essere in grado di calcolare l'insieme finale di strutture a bassa energia, tenendo presente che di solito calcoli più veloci portano a una maggiore incertezza nella precisione dei risultati.
Per motivi di chiarezza, il computer locale dell'utente sarà indicato come "computer locale" mentre il cluster HPC a cui hanno accesso sarà indicato come "cluster remoto".
Protocollo
1. Trovare la struttura energetica minima di glicina e acqua isolati
NOTA: L'obiettivo qui è duplice: i) ottenere strutture energetiche minime di molecole di acqua e glicina isolate da utilizzare nel campionamento configurazionele dell'algoritmo genetico (ii) e calcolare le correzioni termodinamiche alle energie della fase gassosa di queste molecole per l'uso nel calcolo delle concentrazioni atmosferiche.
- Sul computer locale, aprire una nuova sessione di Avogadro.
- Fate clic su Compila (Build) > Inserisci (Insert) > Peptide (Gly) dalla finestra Inserisci Peptide (Insert Peptide) per generare un monomero glicinano nella finestra di visualizzazione.
- Fare clic su Estensioni > Gaussiano e modificare la prima riga nella casella di testo in modo da leggere '''pw91pw91/6-311'''int(Acc2E'12,UltraFine) scf(conver'12) opt(tight,maxcyc'300) freq'. Fare clic su Genera e salvare il file di input come glycine.com.
- Si prega di notare che se la molecola ha una significativa flessibilità conformazionale, come la glicina fa55, è fondamentale eseguire analisi conformazionali per identificare la struttura minima globale e altri conformatori bassi. OpenBabel54 fornisce robusti strumenti di ricerca conformazionale che utilizzano diversi algoritmi e campi di forza rapida. Mentre i conformatori sono autorizzati a rilassarsi e interconvertirsi durante GA e calcoli successivi, a volte è necessario eseguire più calcoli GA, ognuno a partire da un conformatore diverso.
- Sul computer locale, aprire una nuova sessione in Avogadro.
- Fare clic su Build > Inserisci > Frammento e cercare "acqua" dalla finestra Inserisci frammento per ottenere le coordinate dell'acqua.
- Fare clic su Estensioni > Gaussiano e modificare la prima riga nella casella di testo in modo da leggere '''pw91pw91/6-311'''int(Acc2E'12,UltraFine) scf(conver'12) opt(tight,maxcyc'300) freq'. Fare clic su Genera e salvare il file di input come water.com.
- Trasferire i due file .com nel cluster remoto. Una volta effettuato l'accesso al cluster remoto, chiamare Gaussian 09 in uno script di invio batch per avviare il calcolo. Al termine dei calcoli, estrarre le coordinate cartesiane (file .xyz) delle strutture energetiche minime chiamando OpenBabel. Per la glicina, il comando da eseguire è:
obabel -ig09 glycine.log -oxyz > glicine.xyz
Questi due file .xyz verranno utilizzati dal campionamento di configurazione GA nel passaggio successivo.
2. Campionamento di configurazione basato su algoritmi genetici di Gly(H2O)n-1-5 cluster
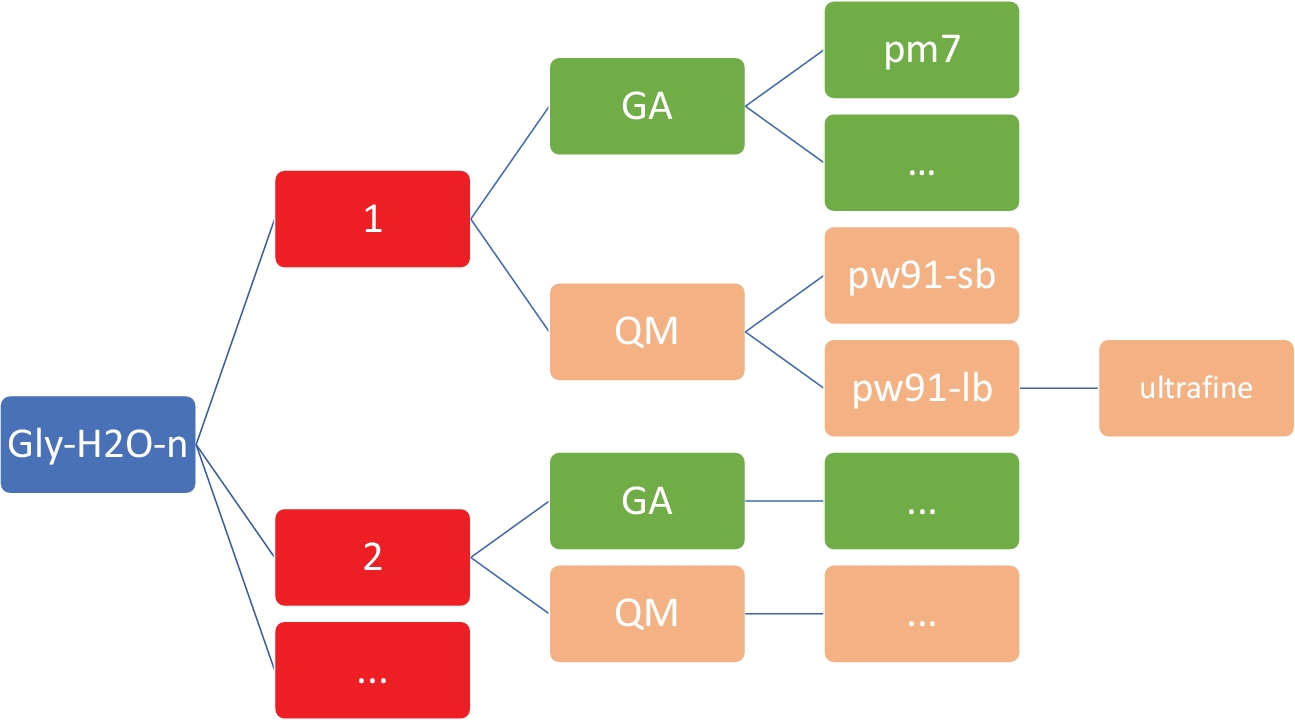
NOTA: L'obiettivo qui è quello di ottenere una serie di strutture a bassa energia per Gly (H2O)n-1-5 al livello semi-empirico poco costoso della teoria, utilizzando il modello PM746 implementato in MOPAC47. È fondamentale che la directory di lavoro abbia l'organizzazione e la struttura esatte, come illustrato nella Figura 2. Questo per garantire che la shell personalizzata e gli script Python funzionino senza errori.
- Copiare tutti gli script necessari nel cluster remoto e aggiungerne la posizione in $PATH
- Inserire tutti gli script e i file modello in una cartella (script ad esempio) e copiarli nel cluster remoto
- Assicurarsi che tutti gli script siano eseguibili
- Aggiungere il percorso della directory degli script alla variabile di ambiente $PATH immettendo i seguenti comandi in un terminale. Il percorso predefinito degli script è impostato su $HOME/JoVE-demo/scripts, tuttavia, è possibile definire una variabile ambientale denominata $SCRIPTS_HOME che punta alla directory contenente gli script e aggiungere $SCRIPTS_HOME al proprio percorso
- Bash shell:
esportare SCRIPTS_HOME/percorso/a/script
percorso di esportazione : SCRIPTS_HOME " : PERCORSO " PATH - Shell Tcsh/Csh:
setenv SCRIPTS_HOME /percorso/a/script
PERCORSO setenv SCRIPTS_HOME
- Bash shell:
- Nel cluster remoto, impostare ed eseguire un calcolo GA:
- Creare una directory chiamata gly-h2o-n dove n è il numero di molecole d'acqua.
- Creare una sottodirectory denominata GA sotto la directory gly-h2o-n per eseguire calcoli dell'algoritmo genetico.
- Copiare i file di input OGOLEM (Ad esempio pm7.ogo), monomere coordinate cartesiane (ad esempio glycine.xyz, water.xyz) e lo script di invio batch PBS (Ad esempio run.pbs) nella directory GA.
- Apportare le modifiche necessarie al file di input OGOLEM e al file di invio batch.
- Inviare il calcolo. All'inizio del calcolo, OGOLEM creerà una nuova directory denominata come prefisso del file di input OGOLEM (Eg. pm7) nella directory GA e memorizzerà le coordinate appena generate.
- Una volta completato il calcolo, compilare le energie e le costanti rotazionali e utilizzare tali informazioni per determinare quali sono le strutture uniche a bassa energia:
- Passare alla directory gly-h2o-n/GA/pm7 e
- Estrarre le energie e calcolare le costanti di rotazione dei cluster ottimizzati per GA con il comando:
getRotConsts-GA.csh N 0 99
dove N è il numero di atomi nel cluster molecolare e '0 99' indica che la dimensione del pool GA è 100, con indici che passano da 0 a 99. Questo genererà un file chiamato rotConstsData_C che contiene un elenco ordinato di tutte le configurazioni cluster ottimizzate per GA, le loro energie e le loro costanti di rotazione. - Eseguire il comando:
rotConstsData_C similarityAnalysis.py 7
dove pm7 verrà utilizzato come etichetta di denominazione dei file, per trovare e salvare i cluster unici ottimizzati per GA. Verrà generato un file denominato uniqueStructures-pm7.data che contiene un elenco ordinato delle configurazioni uniche ottimizzate per GA. Questo è un elenco di strutture minime locali uniche per il cluster Gly(H2O)n ottimizzato al livello di teoria PM7, e queste strutture sono ora pronte per essere perfezionate utilizzando DFT.
- Vai fino alla directory gly-h2o-n/GA e combina i risultati di più serie GA comparabili utilizzando lo script combine-GA.csh. La sintassi è la seguente:
combine-GA.csh
In questo caso particolare, il comando:
combinare-GA.csh pm7 pm7
verrà generato un nuovo elenco di strutture univoche denominato 'uniqueStructures-pm7.data' nella directory gly-h2o-n/GA.
3. Perfezionamento con il metodo QM con una base ridotta impostata
NOTA: L'obiettivo qui è quello di perfezionare il campionamento di configurazione dei cluster Gly(H2O)n-1-5 utilizzando una migliore descrizione quantistica-meccanica per ottenere un set più piccolo ma più accurato di strutture a grillo Di Gly(H2O)n-1-5. Le strutture iniziali per questo passaggio sono le uscite del passaggio 2.The starting structures for this step are the outputs of Step 2.
- Preparare ed eseguire il calcolo DFT di piccola base:
- Creare una sottodirectory denominata QM nella directory gly-h2o-n. Nella directory QM creare un'altra sottodirectory denominata pw91-sb.
- Copiare l'elenco di strutture univoche (uniqueStructures-pm7.data) dalla directory gly-h2o-n/GA nella directory QM/pw91-sb.
- Passare alla directory gly-h2o-n/QM/pw91-sb.
- Eseguire lo script di campionamento di configurazione DFT di base ridotto utilizzando il comando:
run-pw91-sb.csh uniqueStructures-pm7.data sb QUEUE 10
dove sb è un'etichetta per questo set di calcoli, QUEUE è la coda preferita nel cluster di calcolo e 10 indica che 10 calcoli devono essere raggruppati in un unico processo batch. Questo script genererà automaticamente gli input per Gaussian 09 e invierà tutti i calcoli. Immettere 'test' per la 'QUEUE' per eseguire una corsa a secco.
- Una volta completati i calcoli inviati, estrarre e analizzare i risultati.
- Estrarre le energie e calcolare le costanti di rotazione dei cluster ottimizzati per piccola base utilizzando il comando:
getRotConsts-dft-sb.csh pw91 N
dove pw91 indica che è stata utilizzata la densità PW91 funzionale, e N è il numero di atomi nel cluster. Verrà creato un file denominato rotConstsData_C. - Ora identificare le strutture uniche con il comando:
rotConstsData_C sb similarityAnalysis.py
dove sb viene utilizzato come etichetta di denominazione dei file. Ci sarà ora un elenco di configurazioni uniche ottimizzate al livello di teoria PW91/6-31-G , salvato nel file uniqueStructures-sb.data.
- Estrarre le energie e calcolare le costanti di rotazione dei cluster ottimizzati per piccola base utilizzando il comando:
- Vai fino alla directory gly-h2o-n/QM e combina i risultati di più esecuzioni QM comparabili utilizzando lo script combine-QM.csh. La sintassi è la seguente:
combine-QM.csh
In questo caso particolare, il comando:
combinare-QM.csh sb pw91-sb
verrà generato un nuovo elenco di strutture univoche denominato 'uniqueStructures-sb.data' nella directory gly-h2o-n/QM.
4. Ulteriore perfezionamento con il metodo QM con un set di base di grandi dimensioni
NOTA: L'obiettivo qui è quello di perfezionare ulteriormente il campionamento di configurazione dei cluster Gly(H2O)n-1-5 utilizzando una migliore descrizione quantistica-meccanica. Le strutture iniziali per questo passaggio sono le uscite del passaggio 3.The starting structures for this step are the outputs of Step 3.
- Inviare calcoli più affidabili utilizzando un set di base più ampio.
- Creare una sottodirectory denominata pw91-lb nella directory QM.
- Copiare l'elenco di strutture univoche (uniqueStructures-sb.data) dalla directory gly-h2o-n/QM nella directory gly-h2o-n/QM/pw91-lb e passare a tale directory.
- Eseguire lo script di campionamento di configurazione DFT su larga base con il comando:
run-pw91-lb.csh uniqueStructures-sb.data lb QUEUE 10
dove lb è un'etichetta per questo set di calcoli, QUEUE è la coda preferita nel cluster di calcolo e 10 indica che 10 calcoli devono essere raggruppati in un unico processo batch. Questo script genererà automaticamente gli input per Gaussian 09 e invierà tutti i calcoli. Immettere 'test' per il 'QUEUE' per eseguire un test di esecuzione a secco.
- Una volta completati i calcoli inviati, estrarre e analizzare i dati
- Calcolare le costanti di rotazione dei cluster ottimizzati per la base di grandi dimensioni con il comando:
getRotConsts-dft-lb.csh pw91 N
dove pw91 indica che è stata utilizzata la densità PW91 funzionale, e N è il numero di atomi nel cluster. - Ora identificare le strutture uniche con il comando:
similarityAnalysis.py lb rotConstsData_C
dove lb viene utilizzato come etichetta di denominazione dei file. È ora che si dispone di un elenco di configurazioni univoche ottimizzate al livello di teoria PW91/6-311, G, salvato nel file uniqueStructures-lb.data.
- Calcolare le costanti di rotazione dei cluster ottimizzati per la base di grandi dimensioni con il comando:
5. Calcoli finali di correzione dell'energia e della termodinamica
NOTA: L'obiettivo qui è quello di ottenere la struttura vibrazionale e le energie dei cluster Gly(H2O)n-1-5 utilizzando un ampio set di base e una griglia di integrazione ultrafine per calcolare le correzioni termochimiche desiderate.
- A partire dai risultati del passaggio precedente, inviare calcoli più affidabili.
- Creare una sottodirectory denominata ultrafine nella directory QM/pw91-lb. Copiare quindi l'elenco di strutture univoche (uniqueStructures-lb.data) dalla directory QM/pw91-lb alla directory QM/pw91-lb/ultrafine e passare a tale directory.
- Inviare lo script DFT ultrafine base di grandi dimensioni con il comando:
run-pw91-lb-ultrafine.csh uniqueStructures-lb.data uf QUEUE 10
dove uf è un'etichetta per questo set di calcoli, QUEUE è la coda preferita nel cluster di calcolo e 10 indica che 10 calcoli devono essere raggruppati in un unico processo batch. Questo script genererà automaticamente gli input per Gaussian 09 e invierà tutti i calcoli. Immettere 'test' per il 'QUEUE' per eseguire un test di esecuzione a secco.
- Una volta completati i calcoli inviati, estrarre e analizzare i dati
- Estrarre le energie e calcolare le costanti di rotazione dei cluster ottimizzati per la base larga con il comando:
getRotConsts-dft-lb-ultrafine.csh pw91 N
dove pw91 indica che è stata utilizzata la densità PW91 funzionale, e N è il numero di atomi nel cluster. - Ora identificare le strutture uniche con il comando:
rotConstsData_C similarityAnalysis.py uf
dove uf viene utilizzato come etichetta di denominazione dei file. È ora presente un elenco di configurazioni uniche ottimizzate al livello di teoria PW91/6-311, g, salvato nel file uniqueStructures-uf.data.
- Estrarre le energie e calcolare le costanti di rotazione dei cluster ottimizzati per la base larga con il comando:
- Eseguire un'estrazione finale delle informazioni necessarie per calcolare le correzioni termodinamiche. Utilizzare queste informazioni per calcolare le correzioni termodinamiche.
- Estrarre le energie elettroniche finali, le costanti di rotazione e le frequenze vibrazionali e utilizzarle per calcolare le correzioni termodinamiche utilizzando il comando:
run-thermo-pw91.csh uniqueStructures-uf.data - Copiare/incollare l'output della riga di comando nel foglio 'Raw_Energies' del foglio di calcolo di Excel denominato 'gly-h2o-n.xlsx'. Avresti bisogno di farlo per i monomeri (glicina e acqua) così come il membro più basso consumo di energia di ogni idrato (glily-h2o-n, dove n. 1,2, ...).
- Man mano che le energie grezze vengono aggiunte al primo foglio del foglio di calcolo 'gly-h2o-n.xlsx', i successivi fogli 'Binding_Energies' e 'Hydrate_Distribution' vengono aggiornati automaticamente. In particolare, il foglio 'Hydrate_Distribution' produce la concentrazione di equilibrio degli idrati a temperature diverse (Ad es. 298,15K), umidità relativa (20%, 50%, 100%) e le concentrazioni iniziali di acqua ([H2O]) e glicina ([Glicina]). La teoria alla base di questi calcoli è descritta nel passaggio successivo.
- Estrarre le energie elettroniche finali, le costanti di rotazione e le frequenze vibrazionali e utilizzarle per calcolare le correzioni termodinamiche utilizzando il comando:
6. Calcolo delle concentrazioni atmosferiche di gruppi di Gly(H2O)n-0-5 a temperatura ambiente a livello del mare
NOTA: Questa operazione viene eseguita copiando prima i dati termodinamici generati nel passaggio precedente in un foglio di calcolo e calcolando le energie libere di Gibbs di idratazione sequenziale. Quindi, le energie libere di Gibbs vengono utilizzate per calcolare le costanti di equilibrio per ogni idratazione sequenziale. Infine, una serie di equazioni lineari sono risolte per ottenere la concentrazione di equilibrio degli idrati per una data concentrazione di monomeri, temperatura e pressione.
- Iniziare con l'impostazione di un sistema di equilibri chimici per l'idratazione sequenziale della glicina, come illustrato di seguito:

- Calcolare le costanti di equilibrio Kn utilizzando Kn - e-z -Gn/(kBT), dove n è il livello di idratazione, nn è il cambiamento di energia libera di Gibbs della reazione di idratazione nth, kB è la costante di Boltzmann e T è la temperatura.

- Impostare l'equazione per la conservazione della massa, utilizzando l'assunto che la somma delle concentrazioni di equilibrio dei cluster di glicina idratati e non idratati sia uguale alla concentrazione iniziale di glicina isolata [Gly]0. Riscrivere questo sistema di sei equazioni simultanee, utilizzando qualche riarrangiamento algebrico delle espressioni costanti di equilibrio, come

- Risolvere il sistema di equazioni mostrato sopra per ottenere le concentrazioni di equilibrio di Gly(H2O)n - 0-5 utilizzando un valore sperimentale56,57,58 per la concentrazione di glicina nell'atmosfera, [Gly]0 - 2,9 x 106 cm-3, e la concentrazione di acqua nell'atmosfera al 100% di umidità relativa e una temperatura di 298.15 K59, [H2O] - 7,7 x10 17 cm-3.
Risultati
La prima serie di risultati di questo protocollo dovrebbe essere un insieme di strutture a bassa energia di Gly(H2O)n-1-5 trovate attraverso la procedura di campionamento configurazionele. Queste strutture sono state ottimizzate ai fini della teoria PW91/6-311, e si presume che siano accurate ai fini di questo documento. Non vi è alcuna prova che suggerisca che il PW91/6-311, la svalutazione o la sovrastima costantemente dell'energia vincolante di questi cluster. La sua capacità di prevedere le energie di legame rispetto a MP2/CBS32 e [DLPNO-]CCSD(T)/CBS60,61 stime e l'esperimento52 mostra un sacco di fluttuazioni. Lo stesso vale per la maggior parte delle altre funzioni di densità. In genere, ogni valore di n - 1 – 5 dovrebbe produrre una manciata di strutture a bassa energia entro circa 5 kcal mol-1 della struttura a energia più bassa. Qui, ci concentriamo sulla prima struttura prodotta dallo script run-thermo-pw91.csh per brevità. La figura 3 mostra i più bassi isomeri di energia elettronica dei cluster Gly(H2O)n-0-5. Si può vedere che la rete di legame dell'idrogeno cresce in complessità con l'aumentare del numero di molecole d'acqua, e passa anche da una rete per lo più planare a una struttura tridimensionale simile a una gabbia a n . 5. Il resto di questo testo utilizza le energie e le quantità termodinamiche corrispondenti a questi cinque cluster specifici.
La tabella 1 contiene le quantità termodinamiche necessarie per eseguire il protocollo. La tabella 2 mostra un esempio dell'output dello script run-thermo-pw91.csh in cui vengono stampate le energie elettroniche, le correzioni vibrazionali a zero punti e le correzioni termodinamiche a tre diverse temperature. Per ogni ammasso (riga), E[PW91/6-311'G'] corrisponde alla fase gassosa delle energie elettroniche al livello di teoria PW91/6-311ZPVE-1 Ad ogni temperatura, 216.65 K, 273.15 K, e 298.15 K, sono elencate le correzioni termodinamiche, -H l'enthalpy di formazione in unità di kcal mol-1, S entropia di formazione in unità di cal mol-1,e SG il Gibbs energia libera di formazione in unità di kcal mol-1. La tabella 3 mostra un esempio di calcolo del cambiamento totale di idratazione dell'energia libera di Gibbs, nonché dell'idratazione sequenziale. Un esempio di calcolo del cambiamento totale di energia libera di Gibbs di idratazione per la reazione

inizia con il calcolo dell'energia elettronica EPW91 come

dove EPW91[Gly(H2O)] è tratto dalla tabella 2 colonna C, e EPW91[Gly] e EPW91[H2O] sono tratti dalla tabella 1 colonna B. Successivamente calcoliamo il cambio di energia totale della fase gassosa, E (0)includendo il cambiamento nell'energia vibrazionale a punto zero della reazione come

per ottenere la colonna D. In questoPW91/6−311++G** caso, la colonna D è ricavata dalla tabella 3 c// di seguito dallaH2tabellaZPVE 3, dallacolonna C della tabella 1 [G.ZPVE Per brevità, passeremo ai cluster di temperatura ambiente, quindi salteremo i dati 216.65 K e 273.15 K. A temperatura ambiente, calcoliamo quindi il cambio implacabile della reazioneH

dove la modificadi E(0) è tratta dalla tabella 3 colonna D, l'operazioneH[Gly[H2O)] è tratta dalla tabella 2 colonna K, e l'operazioneh[Gly] e la colonna[H2O] sono tratte dalla tabella 1 colonna J. Infine, calcoliamo il cambio di energia liberaG di Gibbs della reazione

dovela colonna 3 n. 3 è tratta dalla tabella 3, s[Gly[H2O)] è presa dalla tabella 2 colonna L, mentre S[Gly] e S[H2O] sono tratti dalla tabella 1 colonna K. Notate qui che i valori di entropia devono essere convertiti in unità di kcal mol-1 K-1 durante questo passaggio.
Ora abbiamo le quantità necessarie per calcolare le concentrazioni atmosferiche della glicina idratata, come mostrato nel passaggio 6. I risultati dovrebbero essere simili ai dati riportati nella tabella 4, ma sono previste piccole differenze numeriche. La tabella 4 mostra le concentrazioni idratanti di equilibrio trovate dalla formulazione del sistema di sei equazioni nel passo 6.2 in un'equazione di matrice e la sua soluzione successiva. Iniziamo riconoscendo il fatto che il sistema di equazioni può essere scritto come

dove Kn è la costante di equilibrio per l'idratazionesequenziale della glicina, w è la concentrazione di acqua nell'atmosfera, g è la concentrazione iniziale di glicina isolata nell'atmosfera, e gn è la concentrazione di equilibrio di Gly(H2O)n. Se riscriviamo l'equazione di cui sopra come Ax x , b, otteniamo x , A, 1b dove Aè l'inverso della matrice A. Questo inverso può essere facilmente calcolato utilizzando le funzioni predefinite del foglio di calcolo, come mostrato nella Tabella 4 per ottenere i risultati finali.
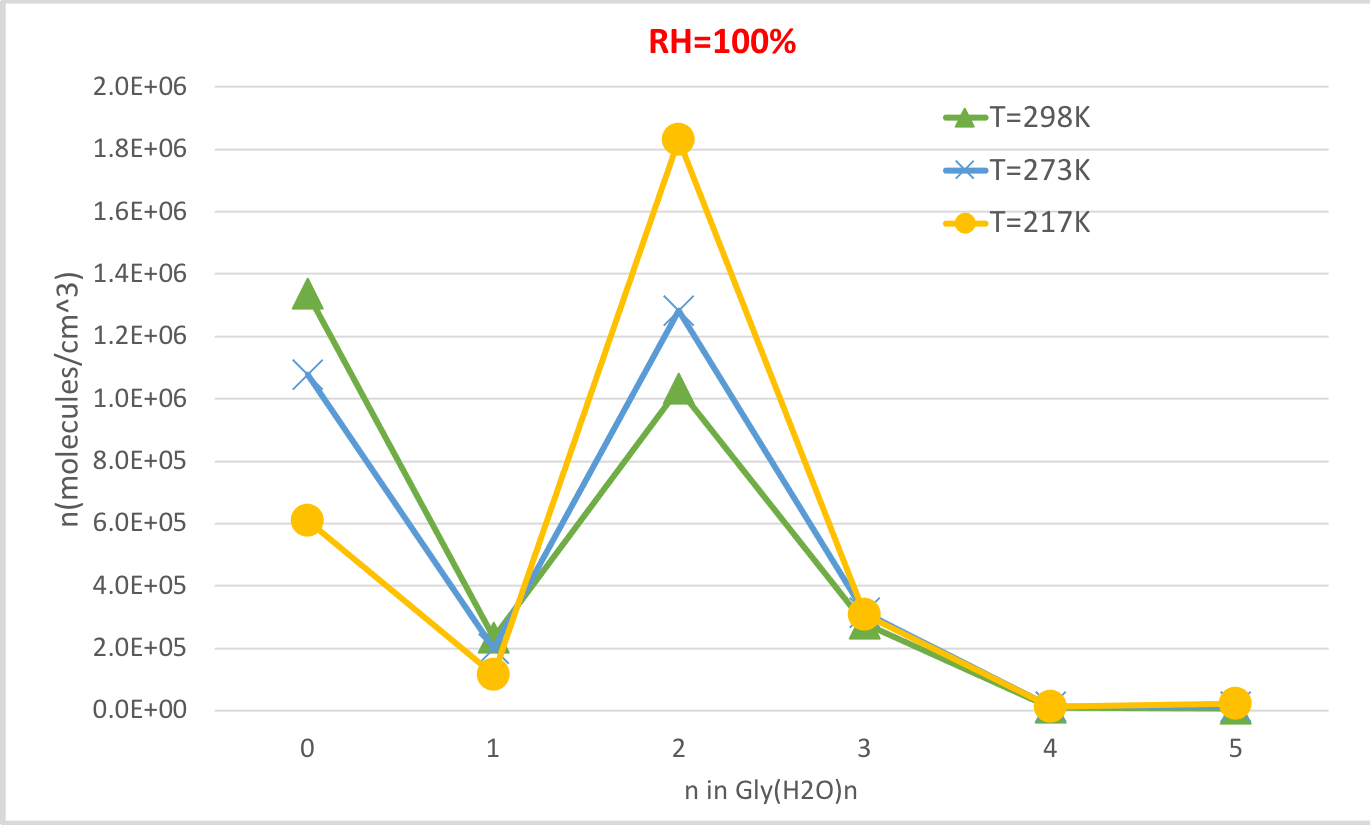
La figura 4 mostra la concentrazione di equilibrio della glicina idratata calcolata nella tabella 4 in funzione della temperatura al 100% di umidità relativa e della pressione dell'atmosfera di 1%. Essa mostra che, con la diminuzione della temperatura da 298.15K a 216.65K, diminuisce la concentrazione di glicina non idratata (n. 0) e aumenta quella della glicina idratata. La disidratazione della glicina (n. 2) aumenta in particolare con la diminuzione della temperatura, mentre il cambiamento nella concentrazione di altri idrati è meno evidente. Queste correlazioni inverse tra temperatura e concentrazione idratata sono coerenti con l'aspettativa che basse energie libere di Gibbs di idratazioni a temperature più basse favoriscano la formazione di idrati.
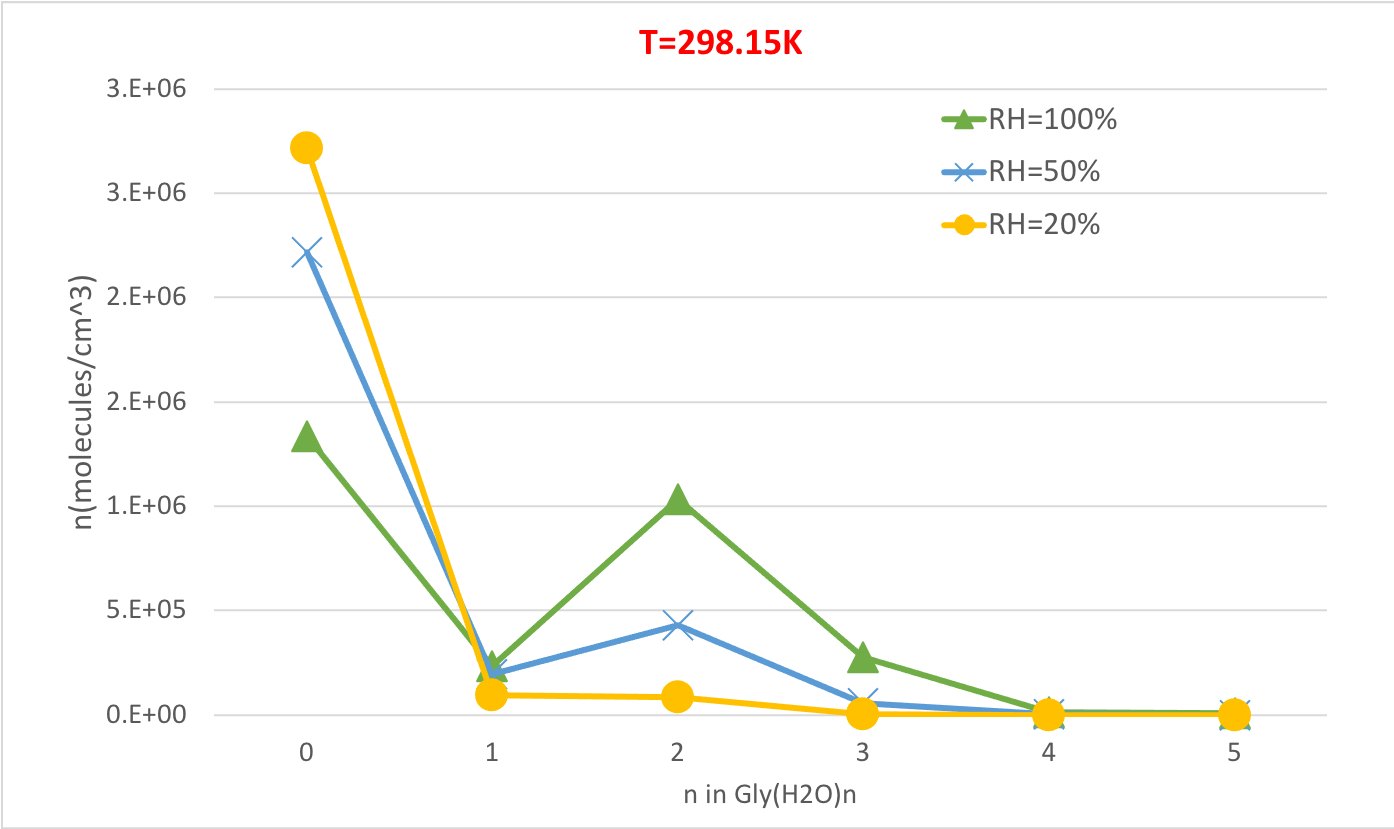
La figura 5 illustra la dipendenza relativa dall'umidità della concentrazione di equilibrio degli idrati di glicina a 298,15K e 1 pressione dell'atmosfera. Dimostra chiaramente che con l'aumento della RH dal 20% al 100%, la concentrazione di idrati (n>0) aumenta a scapito della glicina non idratata (n-0). Ancora una volta la correlazione diretta tra l'umidità relativa e la concentrazione degli idrati è coerente con l'idea che la presenza di più molecole d'acqua a più RMe superiore favorisca la formazione di idrati.
Come presentato, questo protocollo fornisce una comprensione qualitativa delle popolazioni di glicina idratate nell'atmosfera. Supponendo una concentrazione iniziale di glicina isolata di 2,9 milioni di molecole per centimetro cubo, vediamo che la glicina disidratata (n.0) è la specie più abbondante nella maggior parte delle condizioni, ad eccezione di T.216.65K e RH-100%. Il disidrato,che ha la più bassa energia sequenziale Gibbs libera di idratazione a tutte e tre le temperature, è l'idrato più abbondante alle condizioni considerate qui. Si prevede che il monoidrato (n. 1) e gli idrati più grandi (n-3) si trovino in quantità trascurabili. Al momento dell'ispezione della Figura 3, l'abbondanza dei cluster n - 1-4 può essere correlata alla stabilità e alla tensione nella rete di legame dell'idrogeno dei cluster. Questi ammassi hanno le molecole d'acqua legate all'idrogeno dell'acido carboxylico della glicina in una geometria molto simile a quelle di varie strutture ad anello legate all'idrogeno, rendendole particolarmente stabili.

Figura 1: Descrizione schematica della procedura corrente. Un ampio pool di strutture di congettura generate dall'algoritmo genetico (GA) è perfezionato da una serie di ottimizzazioni della geometria PW91 fino a quando non si ottiene una serie di strutture convergenti. Le frequenze vibrazionali di queste strutture vengono calcolate e utilizzate per calcolare l'energia libera di Gibbs di formazione, che a sua volta viene utilizzata per calcolare le concentrazioni di equilibrio dei cluster in condizioni ambientali. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: struttura di directory rappresentativa per ogni cluster. Gli script interni inclusi in questo protocollo richiedono la struttura di directory mostrata sopra, dove n è il numero di molecole d'acqua. Per ogni n in gly-h2o-n, ci sono le seguenti sottodirectory: GA per algoritmo genetico con una directory GA/pm7, QM per la meccanica quantistica con QM/pw91-sb per PW91/6-31-G, QM/pw91-lb per PW91/6-311, e QM/pw91-lb/ultrafine per ottimizzazioni e calcoli vibrazionali finali su reti di integrazione ultrafini. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: Strutture rappresentative a bassa energia di Gly(H2O)n-0-5. Questi cluster sono stati il minimo globale di energia elettronica ottimizzato al livello di teoria PW91/6-311. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: Dipendenza dalla temperatura di Gly(H2O)n-0-5 come umidità relativa al 100% e pressione di 1 atm. La concentrazione degli idrati è data in unità di molecole cm-3. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5: Dipendenza relativa dall'umidità di Gly(H2O)n-0-5 come 298,15 K e 1 atm di pressione. La concentrazione degli idrati è data in unità di molecole cm-3. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
| E[PW91/6-311 | 216,65 K | 273.15 K | 298,15 K | ||||||||
| LB-UF | VANE (VACOM) | H | S (in vi | G (G) | H | S (in vi | G (G) | H | S (in vi | G (G) | |
| Acqua | -76.430500 | 13.04 | 1.72 | 42.59 | 5.54 | 2.17 | 44.44 | 3.08 | 2.37 | 45.14 | 1.96 |
| Glicina | -284.434838 | 48.55 | 2.65 | 69.53 | 36.14 | 3.70 | 73.81 | 32.09 | 4.22 | 75.61 | 30.22 |
Tabella 1: Energie dei monomeri. Le energie elettroniche sono in unità di Hartree, mentre tutte le altre quantità sono in unità di kcal mol-1. L'acqua e la glicina sono state ottimizzate al livello teorico e le frequenze vibrazionali PW91/6-311. Le correzioni termodinamiche per una pressione di 1 atm e temperatura di 298.15 K sono state calcolate utilizzando lo script thermo.pl.
| E[PW91/6-311 | 0 K | 216,65 K | 273.15 K | 298,15 K | ||||||||
| N | Nome | LB-UF | VANE (VACOM) | H | S (in vi | G (G) | H | S (in vi | G (G) | H | S (in vi | G (G) |
| 1 | glily-h2o-1 | -360.88481 | 63.96 | 3.61 | 80.12 | 50.22 | 5.12 | 86.27 | 45.52 | 5.85 | 88.83 | 43.33 |
| 2 | glily-h2o-2 | -437.33763 | 79.33 | 4.53 | 90.86 | 64.17 | 6.46 | 98.78 | 58.81 | 7.40 | 102.06 | 56.30 |
| 3 | glily-h2o-3 | -513.78620 | 94.52 | 5.67 | 105.08 | 77.42 | 8.08 | 114.94 | 71.19 | 9.23 | 119.00 | 68.27 |
| 4 | glily-h2o-4 | -590.23667 | 109.80 | 6.03 | 104.98 | 91.30 | 8.78 | 116.21 | 84.40 | 10.11 | 120.87 | 81.14 |
| 5 | glily-h2o-5 | -666.68845 | 125.80 | 7.26 | 121.70 | 106.69 | 10.47 | 134.83 | 99.44 | 12.01 | 140.24 | 96.00 |
Tabella 2: Energie a grappolo. Le energie dellestrutture Gly(H2O) a energia più bassa si trovano utilizzando la procedura descritta nella Figura 1. Le energie elettroniche sono in unità di Hartree, mentre tutte le altre quantità sono in unità di kcal mol-1.
| Idratazione totale: Gly & nH2O <-> Gly(H2O)n | Idratazione sequenziale: Gly(H2O)n-1 - H2O <-> Gly(H2O)n | ||||||||||||||||
| E[PW91/6-311 | 216.65 | 273.15 | 298.15 | 216.65 | 273.15 | 298.15 | |||||||||||
| N | nome del sistema | LB-UF | E(0) (0) | H(T) | G(T) | H(T) | G(T) | H(T) | G(T) | LB-UF | E(0) (0) | H(T) | G(T) | H(T) | G(T) | H(T) | G(T) |
| 1 | glily-h2o-1 | -12.22 | -9.85 | -10.61 | -3.68 | -10.61 | -1.87 | -10.59 | -1.07 | -12.22 | -9.85 | -10.61 | -3.68 | -10.61 | -1.87 | -10.59 | -1.07 |
| 2 | glily-h2o-2 | -26.22 | -21.53 | -23.10 | -9.27 | -23.11 | -5.66 | -23.09 | -4.06 | -14.00 | -11.68 | -12.49 | -5.59 | -12.50 | -3.79 | -12.50 | -2.99 |
| 3 | glily-h2o-3 | -37.56 | -30.72 | -32.88 | -12.90 | -32.87 | -7.69 | -32.82 | -5.38 | -11.34 | -9.19 | -9.78 | -3.63 | -9.76 | -2.03 | -9.73 | -1.32 |
| 4 | glily-h2o-4 | -50.10 | -40.34 | -43.48 | -15.87 | -43.54 | -8.71 | -43.51 | -5.55 | -12.54 | -9.62 | -10.60 | -2.97 | -10.67 | -1.02 | -10.69 | -0.17 |
| 5 | glily-h2o-5 | -63.45 | -51.41 | -55.42 | -20.58 | -55.51 | -11.48 | -55.48 | -7.45 | -13.35 | -11.07 | -11.94 | -4.71 | -11.97 | -2.77 | -11.97 | -1.90 |
Tabella 3: Energie di idratazione. L'energia totale di idratazione e l'energia dell'idratazione sequenziale per Gly(H2O)n-1-5 in unità di kcal mol-1. In questo caso, E[PW91/6-311,G'] è il cambiamento nell'energia elettronica, e è l'energia vibrazionale a punto zero (PVE) corretto cambiamento di energia, .H(T) è il cambiamento enthalpy alla temperatura T, e G (G(T) è il cambiamento di energia libera di Gibbs di idratazione di ogni Gly(H2O)n.
| Equilibrium Hydrate Distribution in funzione della temperatura e dell'umidità relativa | |||||||||
| 298,15K | 273.15K | 216,65K | |||||||
| Gly(H2O)n | RH-100% | RH-50% | RH-20% | RH-100% | RH-50% | RH-20% | RH-100% | RH-50% | RH-20% |
| 0 | 1.3E-06 | 2.2E-06 | 2.7E-06 | 1.1E-06 | 2,0E-06 | 2.7E-06 | 6.1E-05 | 1.5E-06 (in inglese) | 2.5E-06 |
| 1 | 2.3E-05 | 1.9E-05 | Ore 9.5E-04 | 2,0E-05 | 1.9E-05 | 9.9E-04 | 1.2E-05 | 1.5E-05 | Ore 9.5E-04 |
| 2 | 1,0E-06 | 4.3E-05 | 8.4E-04 | 1.3E-06 | 6.1E-05 | 1.3E-05 | 1.8E-06 | 1.1E-06 | 3,0E,05 |
| 3 | 2.8E-05 | 5,8E-04 | 4,5E-03 | 3.2E-05 | Ore su 7.4E-04 | 6.3E-03 | 3.1E-05 | Ore 9.6E-04 | 1,0E-04 |
| 4 | 1.1E-04 | 1.1E-03 | 3.4E-01 | 1.3E-04 | 1.5E-03 | Al numero 5.0E-01 | 1.1E-04 | 1.8E-03 | Ore su 7.5E-01 |
| 5 | Ore su 7.5E-03 | Ore 3.9E-02 | 4.9E-00 | 1.2E-04 | Ore 7.2E-02 | Ore 9.7E-00 | 2.4E-04 | 1.9E-03 | 3.1E-01 |
Tabella 4: Equilibrium idrata le concentrazioni di Gly (H2O)n -0-5 come temperatura di funzione (T 298.15K, 273.15K, 216.65K) e l'umidità relativa (RH-100%, 50%, 20%). La concentrazione degli idrati è data in unità di molecole cm-3 assumendo valori sperimentali56,57,58, di [Gly]0 - 2,9 x 106 cm-3 e [H2O] - 7,7 x 1 017 cm-3, 1,6 x 1017 cm-3 e 9,9 x10 14 cm-3 a 100% di umidità relativa e T 298,15 K, 273,15 K, e 216,65 K, rispettivamente59.
File supplementari. Fare clic qui per scaricare questi file.
Discussione
L'accuratezza dei dati generati da questo protocollo dipende principalmente da tre cose: (i) la varietà di configurazioni campionate dal punto 2, (ii) l'accuratezza della struttura elettronica del sistema, (iii) e l'accuratezza delle correzioni termodinamiche. Ognuno di questi fattori può essere affrontato modificando il metodo modificando gli script inclusi. Il primo fattore è facilmente superata con l'uso di un pool iniziale più ampio di strutture generate casualmente, più numerose iterazioni del GA e una definizione più allentata dei criteri coinvolti nella GA. Inoltre, si può utilizzare un metodo semi-empirico diverso, come il modello di adassociazione stretta-vincolo a tenuta stretta a densità di carica auto-consistente (SCC-DFTB)62 e il potenziale di frammento efficace (EFP)63 modello al fine di esplorare gli effetti delle diverse descrizioni fisiche. La limitazione principale qui è l'incapacità del metodo di formare o rompere legami covalenti, il che significa che i monomeri sono congelati. La procedura GA trova solo le posizioni relative più stabili di questi monomeri congelati secondo la descrizione semi-empirica.
L'accuratezza della struttura elettronica del sistema può essere migliorata in diversi modi, ognuno con il suo costo computazionale. Si può scegliere una migliore densità funzionale, come M06-2X64 e wB97X-V65, o metodo meccanico quantistico (QM) come il M 'ller-Plesset66,67,68 (MPn) teorie di perturbazione e accoppiato-cluster69 (CC) metodi al fine di migliorare la descrizione fisica del sistema. Nella gerarchia delle funzioni, le prestazioni in genere migliorano passando da funzioni di approssimazione generalizzata(GGA) come PW91 a funzioni ibride separate da intervalli come funzioni ibride wB97X-D e meta-GGA come M06-2X.
Lo svantaggio dei metodi DFT è che non è possibile una convergenza sistematica verso un valore accurato; tuttavia, i metodi DFT sono computazionalmente economici e c'è una vasta gamma di funzioni per una vasta gamma di applicazioni.
Energie calcolate utilizzando metodi di funzione d'onda come MP2 e CCSD(T) in combinazione con set di base coerenti di correlazione di numero cardinale crescente ([aug-]cc-pV[D,T,Q,...] Convergere sistematicamente verso il limite completo del loro set di basi completo, ma il costo computazionale di ogni calcolo diventa proibitivo man mano che la dimensione del sistema cresce. Un ulteriore perfezionamento della struttura elettronica può essere effettuato utilizzando set di base di cui al massimo correlazioneesplicitamente ed estrapolando il limite completo del base set (CBS)71. Il nostro recente lavoro suggerisce che un approccio perturbativo di seconda densità ad fitted a densità ha correlato esplicitamente il m'ller-Plesset (DF-MP2-F12) produce energie che si avvicinano a quella dei calcoli MP2/CBS32. La modifica del protocollo corrente per l'utilizzo di diversi metodi di struttura elettronica prevede due passaggi: (i) preparare un file di input del modello seguendo la sintassi fornita dal software (ii) e modificare il run-pw91-sb.csh, run-pw91-lb.cshe gli script run-pw91-lb ultrafine.csh per generare la sintassi corretta del file di input, nonché lo script di invio corretto per il software.
Infine, l'accuratezza delle correzioni termodinamiche dipende dal metodo della struttura elettronica e dalla descrizione del PES intorno al minimo globale. Una descrizione accurata del PES richiede il calcolo di derivati di terzo e più ordine del PES rispetto agli spostamenti nei gradi di libertà nucleari, come il campo di forza quartica72,73 (QFF), che è un compito eccezionalmente costoso. Il protocollo attuale utilizza l'approssimazione armonica dell'oscillatore alle frequenze vibrazionali, con conseguente necessità di calcolare solo fino a seconde derivate del PES. Questo approccio diventa problematico nei sistemi ad alta armonicità, come molecole molto floppy e potenziali simmetrici a doppio pozzo a causa della grande differenza nel reale PES e nell'armonica PES. Inoltre, il costo di avere un PES di alta qualità da un metodo di struttura elettronica computazionalmente esigente non fa che aggravare il problema del costo per i calcoli della frequenza vibrazionale. Un approccio per superare questo è quello di utilizzare le energie elettroniche da un calcolo della struttura elettronica di alta qualità insieme a frequenze vibrazionali calcolate su un PES di qualità inferiore, con conseguente equilibrio tra costo e precisione. Il protocollo attuale può essere modificato per utilizzare diverse descrizioni PES come descritto nel paragrafo precedente; tuttavia, si possono anche modificare le parole chiave di frequenza vibrazionale negli script e nei modelli per calcolare le frequenze vibrazionali anarmoniche.
Due questioni cruciali per qualsiasi protocollo di campionamento configurazionele sono il metodo iniziale per il campionamento della potenziale superficie energetica e i criteri utilizzati per identificare ogni cluster. Abbiamo fatto ampio uso di una varietà di metodi nel nostro lavoro precedente. Per il primo problema, il metodo iniziale per il campionamento della potenziale superficie energetica, abbiamo scelto di utilizzare GA con metodi semi-empirici basati su questi fattori. Il campionamento configurazionele utilizzando l'intuizione chimica26, il campionamento casuale e la dinamica molecolare (MD)29,30, non riescono a trovare regolarmente minime globali putativo per cluster più grandi di 10 monomeri, come abbiamo osservato nei nostri studi sui cluster d'acqua18. Abbiamo utilizzato con successo il bacino hopping (BH) per studiare il complesso PES di (H2O)1174, ma ha richiesto l'inclusione manuale di alcuni potenziali isomeri a bassa energia che l'algoritmo BH non ha trovato. Un confronto tra le prestazioni di BH e GA nel trovare il minimo globale di cluster d'acqua, (H2O)n-10-20 ha dimostrato che GA ha trovato costantemente il minimo globale più veloce di BH75. GA come implementato in OGOLEM e CLUSTER è molto versatile perché può essere applicato a qualsiasi cluster molecolare e può interfacciarsi con un vasto numero di pacchetti con campo di forza classico, semi-empirico, densità funzionale, e ab initio capacità. La scelta del PM7 è guidata dalla sua velocità e dalla sua ragionevole precisione. Praticamente qualsiasi altro metodo semi-empirico avrebbe costi computazionali significativamente più elevati.
Per quanto riguarda il secondo numero, abbiamo esplorato utilizzando diversi criteri per identificare strutture uniche che vanno dalle energie elettroniche, ai momenti di dipolo, agli RMSD sovrapposti e alle costanti rotazionali. L'uso dei momenti di dipolo si è rivelato difficile perché entrambi i componenti del momento del dipolo dipendevano dall'orientamento della molecola e il momento totale del dipolo era molto sensibile alle differenze di geometria in modo tale che era difficile impostare soglie determinando che le strutture fossero uguali o uniche. Una combinazione di energie elettroniche e costanti rotazionali si è rivelata più utile.
Gli attuali criteri per rimettere due strutture univoche si basano su una soglia di differenza di energia di 0,10 kcal mol-1 e su una differenza costante rotazionale dell'1%. Pertanto, due strutture sono considerate diverse se le loro energie differiscono di più di 0,10 kcal mol-1 (0,00015 a.u.) E una qualsiasi delle loro tre costanti rotazionali (A, B, C) differiscono di oltre l'1%. Nel corso degli anni, tali soglie sono stati ritenuti disincono di riferimento. Il nostro approccio di campionamento di configurazione e la metodologia di screening sono stati applicati a cluster molto debolmente legati come gli idrocarburi poliaromatici complessi con acqua76,77 e idrati di solfato ternario fortemente legati contenenti ammoniaca e ammine32. Per i cluster in cui ci sono diversi stati di protonazione da considerare, l'approccio migliore è quello di eseguire vari calcoli GA, ognuno a partire da monomeri in diversi stati di protonazione. Ciò garantisce che le strutture con diversi stati di protonazione siano attentamente considerate. Tuttavia, i calcoli DFT di basso livello spesso consentono agli stati di protonazione di cambiare nel corso dell'ottimizzazione della geometria, producendo così lo stato di protonazione più stabile indipendentemente dalla geometria iniziale.
I nostri metodi di campionamento di configurazione GA dovrebbero funzionare bene anche per le molecole floppy, purché i codici GA siano interfacciati con metodi generali non parametrizzati che consentono ai monomeri di adottare configurazioni diverse nel corso dell'esecuzione GA. Ad esempio, interfacciarsi con GA con PM7 consentirebbe alle strutture dei monomeri di cambiare, ma se i loro legami si romperebbero come accadrebbe quando cambiano gli stati di protonazione, le strutture possono essere scartate come candidati inaccettabili.
Abbiamo considerato diversi modi per correggere le carenze dell'approssimazione armonica, in particolare quelle derivanti da basse frequenze vibrazionali. Incorporare l'approssimazione quasi-armonica nella metodologia attuale non è difficile. Tuttavia, ci sono ancora domande sul metodo quasi-armonico, soprattutto quando si tratta della frequenza di taglio al di sotto della quale verrà applicato. Inoltre, non ci sono lavori rigorosi di benchmarking che esaminano l'affidabilità dell'approssimazione quasi-RRHO, anche se la saggezza convenzionale suggerisce che dovrebbe essere un miglioramento rispetto all'approssimazione RRHO.
Il protocollo così presentato può essere generalizzato a qualsiasi sistema di ammassi molecolari di fase gassosa non covalentmente legati. Può anche essere generalizzato utilizzare qualsiasi metodo semi-empirico, metodo di struttura elettronica e software, e metodo di analisi vibrazionale e software modificando gli script e modelli. Si presuppone che l'utente abbia familiarità con l'interfaccia della riga di comando Linux, lo scripting Python e l'elaborazione ad alte prestazioni. La sintassi e l'aspetto sconosciuti del sistema operativo Linux e la mancanza di esperienza di scripting è la più grande trappola in questo protocollo ed è dove i nuovi studenti lottano di più. Questo protocollo è stato utilizzato con successo in una varietà di implementazioni per anni nel nostro gruppo, concentrandosi principalmente sugli effetti dell'acido solforico e dell'ammoniaca sulla formazione di aerosol. Ulteriori miglioramenti a questo protocollo comporteranno un'interfaccia più robusta a un software più efficace per la struttura elettronica, implementazioni alternative dell'algoritmo genetico e, eventualmente, l'uso di metodi più recenti per calcoli più veloci delle energie elettroniche e vibrazionali. Le nostre attuali applicazioni di questo protocollo stanno esplorando l'importanza degli amminoacidi nelle prime fasi della formazione di aerosol nell'atmosfera attuale e nella formazione di molecole biologiche più grandi in ambienti prebiotici.
Divulgazioni
Nessuno.
Riconoscimenti
Questo progetto è stato sostenuto dalle sovvenzioni CHE-1229354, CHE-1662030, CHE-1721511 e CHE-1903871 dalla National Science Foundation (GCS), dall'Arnold and Mabel Beckman Foundation Beckman Scholar Award (AGG) e dalla borsa di studio Barry M. Goldwater Scholarship (AGG). Sono state utilizzate risorse di calcolo ad alte prestazioni del Consorzio MERCURY (http://www.mercuryconsortium.org).
Materiali
| Name | Company | Catalog Number | Comments |
| Avogadro | https://avogadro.cc | Open-source molecular visualization program | |
| Gaussian [09/16] Software | http://www.gaussian.com/ | Commercial ab initio electronic structure program | |
| MOPAC 2016 | http://openmopac.net/MOPAC2016.html | Open-source semi-empirical program | |
| OGOLEM Software | https://www.ogolem.org | Genetic algorithm-based global optimization program | |
| OpenBabel | http://openbabel.org/wiki/Main_Page | Open-source cheminformatics library | |
| calcRotConsts.py | Shields Group, Department of Chemistry, Furman University | Python script to compute rotational constants | |
| calcSymmetry.csh | Shields Group, Department of Chemistry, Furman University | Shell script to calculate symmetry number of a molecule given Cartesian coordinates | |
| combine-GA.csh | Shields Group, Department of Chemistry, Furman University | Shell script to combine energy and rotational constants from different GA directories | |
| combine-QM.csh | Shields Group, Department of Chemistry, Furman University | Shell script to combine energy and rotational constants from different QM directories | |
| gaussianE.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract Gaussian 09 energies | |
| gaussianFreqs.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract Gaussian 09 vibrational frequencies | |
| getrotconsts | Shields Group, Department of Chemistry, Furman University | Executable to calculate rotational constants given a molecule's Cartesian coordinates | |
| getRotConsts-dft-lb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of large basis DFT optimized structures | |
| getRotConsts-dft-lb-ultrafine.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of ultrafine DFT optimized structures | |
| getRotConsts-dft-sb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of small basis DFT optimized structures | |
| getRotConsts-GA.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of genetic algorithm optimized structures | |
| global-minimum-coords.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of global minimum structures of gly-(h2o)n, where n=0-5 | |
| make-thermo-gaussian.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract data from Gaussian output files and make input files for the thermo.pl script | |
| ogolem-input-file.ogo | Shields Group, Department of Chemistry, Furman University | Ogolem sample input file | |
| ogolem-submit-script.pbs | Shields Group, Department of Chemistry, Furman University | PBS batch submission file for Ogolem calculations | |
| README.docx | Shields Group, Department of Chemistry, Furman University | Clarifications to help readers use the scripts effectively | |
| runogolem.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run OGOLEM | |
| run-pw91-lb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of large basis DFT optimization calculations | |
| run-pw91-lb-ultrafine.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of ultrafine DFT optimization calculations | |
| run-pw91-sb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of small basis DFT optimization calculations | |
| run-thermo-pw91.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute the thermodynamic corrections for a batch of DFT optimized structures | |
| similarityAnalysis.py | Shields Group, Department of Chemistry, Furman University | Python script to determine unique structures based on rotational constants and energies | |
| symmetry | Shields Group, Department of Chemistry, Furman University | Executable to calculate molecular symmetry given Cartesian coordinates | |
| symmetry.c | (C) 1996, 2003 S. Patchkovskii, Serguei.Patchkovskii@sympatico.ca | C code to determine the molecular symmstry of a molecule given Cartesian coordinates | |
| template-marcy.pbs | Shields Group, Department of Chemistry, Furman University | Template for a PBS submit script which uses OGOLEM | |
| template-pw91.com | Shields Group, Department of Chemistry, Furman University | Template Gaussian 09 input | |
| template-pw91-HL.com | Shields Group, Department of Chemistry, Furman University | Template Gaussian 09 input for ultrafine DFT optimization | |
| thermo.pl | https://www.nist.gov/mml/csd/chemical-informatics-research-group/products-and-services/program-computing-ideal-gas | Perl open-source script to compute ideal gas thermodynamic corrections | |
| gly-h2o-n.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet for the complete protocol | |
| table-1.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-2.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-3.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-4.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| water.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of water | |
| glycine.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of glycine |
Riferimenti
- Foster, P., Ramaswamy, V., Solomon, S., Qin, D., Manning, M., Chen, Z., Marquis, M., Averyt, K. B., Tignor, M., Miller, H. L. . Climate Change 2007 The Scientific Basis. , (2007).
- Kulmala, M., et al. Toward direct measurement of atmospheric nucleation. Science. 318 (5847), 89-92 (2007).
- Sipila, M., et al. The role of sulfuric acid in atmospheric nucleation. Science. 327 (5970), 1243-1246 (2010).
- Jiang, J., et al. First measurement of neutral atmospheric cluster and 1 - 2 nm particle number size distributions during nucleation events. Aerosol Science and Technology. 45 (4), (2011).
- Dunn, M. E., Pokon, E. K., Shields, G. C. Thermodynamics of forming water clusters at various Temperatures and Pressures by Gaussian-2, Gaussian-3, Complete Basis Set-QB3, and Complete Basis Set-APNO model chemistries; implications for atmospheric chemistry. Journal of the American Chemical Society. 126 (8), 2647-2653 (2004).
- Pickard, F. C., Pokon, E. K., Liptak, M. D., Shields, G. C. Comparison of CBSQB3, CBSAPNO, G2, and G3 thermochemical predictions with experiment for formation of ionic clusters of hydronium and hydroxide ions complexed with water. Journal of Chemical Physics. 122, 024302 (2005).
- Pickard, F. C., Dunn, M. E., Shields, G. C. Comparison of Model Chemistry and Density Functional Theory Thermochemical Predictions with Experiment for Formation of Ionic Clusters of the Ammonium Cation Complexed with Water and Ammonia; Atmospheric Implications. Journal of Physical Chemistry A. 109 (22), 4905-4910 (2005).
- Alongi, K. S., Dibble, T. S., Shields, G. C., Kirschner, K. N. Exploration of the Potential Energy Surfaces, Prediction of Atmospheric Concentrations, and Vibrational Spectra of the HO2•••(H2O)n (n=1-2) Hydrogen Bonded Complexes. Journal of Physical Chemistry A. 110 (10), 3686-3691 (2006).
- Allodi, M. A., Dunn, M. E., Livada, J., Kirschner, K. N. Do Hydroxyl Radical-Water Clusters, OH(H2O)n, n=1-5, Exist in the Atmosphere. Journal of Physical Chemistry A. 110 (49), 13283-13289 (2006).
- Kirschner, K. N., Hartt, G. M., Evans, T. M., Shields, G. C. In Search of CS2(H2O)n=1-4 Clusters. Journal of Chemical Physics. 126, 154320 (2007).
- Hartt, G. M., Kirschner, K. N., Shields, G. C. Hydration of OCS with One to Four Water Molecules in Atmospheric and Laboratory Conditions. Journal of Physical Chemistry A. 112 (19), 4490-4495 (2008).
- Morrell, T. E., Shields, G. C. Atmospheric Implications for Formation of Clusters of Ammonium and 110 Water Molecules. Journal of Physical Chemistry A. 114 (12), 4266-4271 (2010).
- Temelso, B., et al. Quantum Mechanical Study of Sulfuric Acid Hydration: Atmospheric Implications. Journal of Physical Chemistry A. 116 (9), 2209 (2012).
- Husar, D. E., Temelso, B., Ashworth, A. L., Shields, G. C. Hydration of the Bisulfate Ion: Atmospheric Implications. Journal of Physical Chemistry A. 116 (21), 5151-5163 (2012).
- Bustos, D. J., Temelso, B., Shields, G. C. Hydration of the Sulfuric Acid – Methylamine Complex and Implications for Aerosol Formation. Journal of Physical Chemistry A. 118 (35), 7430-7441 (2014).
- Wales, D. J., Scheraga, H. A. Global optimization of clusters, crystals, and biomolecules. Science. 27 (5432), 1368-1372 (1999).
- Day, M. B., Kirschner, K. N., Shields, G. C. Global search for minimum energy (H2O)n clusters, n = 3 - 5. The Journal of Physical Chemistry A. 109 (30), 6773-6778 (2005).
- Shields, R. M., Temelso, B., Archer, K. A., Morrell, T. E., Shields, G. C. Accurate predictions of water cluster formation, (H2O)n=2-10. The Journal of Physical Chemistry A. 114 (43), 11725-11737 (2010).
- Temelso, B., Archer, K. A., Shields, G. C. Benchmark structures and binding energies of small water clusters with anharmonicity corrections. The Journal of Physical Chemistry A. 115 (43), 12034-12046 (2011).
- Temelso, B., Shields, G. C. The role of anharmonicity in hydrogen-bonded systems: The case of water clusters. The Journal of Chemical Theory and Computation. 7 (9), 2804-2817 (2011).
- Von Freyberg, B., Braun, W. Efficient search for all low energy conformations of polypeptides by Monte Carlo methods. The Journal of Computational Chemistry. 12 (9), 1065-1076 (1991).
- Rakshit, A., Yamaguchi, T., Asada, T., Bandyopadhyay, P. Understanding the structure and hydrogen bonding network of (H2O)32 and (H2O)33: An improved Monte Carlo temperature basin paving (MCTBP) method of quantum theory of atoms in molecules (QTAIM) analysis. RSC Advances. 7 (30), 18401-18417 (2017).
- Deaven, D. M., Ho, K. M. Molecular geometry optimization with a genetic algorithm. Physical Review Letters. 75, 288-291 (1995).
- Hartke, B. Application of evolutionary algorithms to global cluster geometry optimization. Applications of Evolutionary Computation in Chemistry. , (2004).
- Dieterich, J. M., Hartke, B. OGOLEM: Global cluster structure optimization for arbitrary mixtures of flexible molecules. A multiscaling, object-oriented approach. Molecular Physics. 108 (3-4), 279-291 (2010).
- Herb, J., Nadykto, A. B., Yu, F. Large ternary hydrogen-bonded pre-nucleation clusters in the Earth's atmosphere. Chemical Physics Letters. 518, 7-14 (2011).
- Ortega, I. K., et al. From quantum chemical formation free energies to evaporation rates. Atmospheric Chemistry and Physics. 12 (1), 225-235 (2012).
- Elm, J., Bilde, M., Mikkelsen, K. V. Influence of Nucleation Precursors on the Reaction Kinetics of Methanol with the OH Radical. Journal of Physical Chemistry A. 117 (30), 6695-6701 (2013).
- Loukonen, V., et al. Enhancing effect of dimethylamine in sulfuric acid nucleation in the presence of water - a computational study. Atmospheric Chemistry and Physics. 10 (10), 4961-4974 (2010).
- Temelso, B., Phan, T. N., Shields, G. C. Computational study of the hydration of sulfuric acid dimers: implications for acid dissociation and aerosol formation. Journal of Physical Chemistry A. 116 (39), 9745-9758 (2012).
- Jiang, S., et al. Study of Cl-(H2O)n (n = 1-4) using basin-hopping method coupled with density functional theory. Journal of Computational Chemistry. 35 (2), 159-165 (2014).
- Temelso, B., et al. Effect of mixing ammonia and alkylamines on sulfate aerosol formation. Journal of Physical Chemistry A. 122 (6), 1612-1622 (2018).
- Perdew, J. P., Ruzsinszky, A., Tao, J. Prescription for the design and selection of density functional approximations: More constraint satisfaction with fewer fits. Journal of Chemical Physics. 123, 062201 (2005).
- Riplinger, C., Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. Journal of Chemical Physics. 138, 034106 (2013).
- Riplinger, C., Pinski, P., Becker, U., Valeev, E. F., Neese, F. Sparse maps--A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory. Journal of Chemical Physics. 144 (2), 024109 (2016).
- Kildgaard, J. V., Mikkelsen, K. V., Bilde, M., Elm, J. Hydration of atmospheric molecular clusters: a new method for systematic configurational sampling. Journal of Physical Chemistry A. 122 (22), 5026-5036 (2018).
- González, &. #. 1. 9. 3. ;. Measurement of areas on a sphere Using Fibonacci and latitude-longitude lattices. Mathematical Geosciences. 42, 49-64 (2010).
- Karaboga, D., Basturk, B. On the performance of artificial bee colony (ABC) algorithm. Applied Soft Computing. 8 (1), 687-697 (2008).
- Zhang, J., Doig, M. Global optimization of rigid molecules using the artificial bee colony algorithm. Physical Chemistry Chemical Physics. 18 (4), 3003-3010 (2016).
- Kubecka, J., Besel, V., Kurten, T., Myllys, N., Vehkamaki, H. Configurational sampling of noncovalent (atmospheric) molecular clusters: sulfuric acid and guanidine. Journal of Physical Chemistry A. 123 (28), 6022-6033 (2019).
- Grimme, S., Bannwarth, C., Shushkov, P. A Robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent Interactions of large molecular systems parametrized for all spd-block elements (Z = 1-86). Journal of Chemical Theory and Computation. 13 (5), 1989-2009 (2017).
- Buck, U., Pradzynski, C. C., Zeuch, T., Dieterich, J. M., Hartke, B. A size resolved investigation of large water clusters. Physical Chemistry Chemical Physics. 16 (15), 6859 (2014).
- Forck, R. M., et al. Structural diversity in sodium doped water trimers. Physical Chemistry Chemical Physics. 14 (25), 9054-9057 (2012).
- Witt, C., Dieterich, J. M., Hartke, B. Cluster structures influenced by interaction with a surface. Physical Chemistry Chemical Physics. 20 (23), 15661-15670 (2018).
- Freitbert, A., Dieterich, J. M., Hartke, B. Exploring self-organization of molecular tether molecules on a gold surface by global structure optimization. The Journal of Computational Chemistry. 40 (22), 1978-1989 (2019).
- Stewart, J. J. P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. The Journal of Molecular Modeling. 19 (1), 1-32 (2013).
- Burke, K., Perdew, J. P., Wang, Y. Derivation of a generalized gradient approximation: The PW91 density functional. Electronic Density Functional Theory. , 81-111 (1998).
- Frisch, M. J., et al. . Gaussian 09, Revision A.02. , (2016).
- Ditchfield, R., Hehre, W. J., Pople, J. A. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. The Journal of Chemical Physics. 54 (2), 724 (1971).
- Elm, J., Bilde, M., Mikkelsen, K. V. Assessment of density functional theory in predicting structures and free energies of reaction of atmospheric prenucleation clusters. The Journal of Chemical Theory and Computation. 8 (6), 2071-2077 (2012).
- Elm, J., Mikkelsen, K. V. Computational approaches for efficiently modelling of small atmospheric clusters. Chemical Physics Letters. 615, 26-29 (2014).
- Bayucan, A., et al. . PBS Portable Batch System. , (1999).
- O'Boyle, N. M., et al. Open Babel: An open chemical toolbox. Journal of Cheminformatics. 3, 33 (2011).
- Csaszar, A. G. Conformers of gaseous glycine. Journal of the American Chemical Society. 114 (24), 9568-9575 (1992).
- Zhang, Q., Anastasio, C. Free and combined amino compounds in atmospheric fine particles (PM2.5) and fog waters from Northern California. Atmospheric Environment. 37 (16), 2247-2258 (2003).
- Matsumoto, K., Uematsu, M. Free amino acids in marine aerosols over the western North Pacific Ocean. Atmospheric Environment. 39 (11), 2163-2170 (2005).
- Mandalakis, M., Apostolaki, M., Stephanou, E. G. Trace analysis of free and combined amino acids in atmospheric aerosols by gas chromatography-mass spectrometry. Journal of Chromatography A. 1217 (1), 143-150 (2010).
- Seinfeld, J. H., Pandis, S. N. . Atmospheric Chemistry and Physics, 3rd Ed. , (2016).
- Myllys, N., Elm, J., Halonen, R., Kurten, T., Vehkamaki, H. Coupled cluster evaluation of atmospheric acid-base clusters with up to 10 molecules. The Journal of Physical Chemistry A. 120 (4), 621-630 (2016).
- Elm, J., Bilde, M., Mikkelsen, K. V. Assessment of binding energies of atmospherically relevant clusters. Physical Chemistry Chemical Physics. 15 (39), (2013).
- Elstner, M. The SCC-DFTB method and its application to biological systems. Theoretical Chemistry Accounts. 116 (1-3), 316-325 (2006).
- Kaliman, I. A., Slipchenko, L. V. LIBEFP: A new parallel implementation of the effective fragment potential method as a portable software library. The Journal of Computational Chemistry. 34 (26), 2284-2292 (2013).
- Zhao, Y., Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and trasition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theoretical Chemistry Accounts. 120 (1-3), 215-241 (2008).
- Mardirossian, N., Head-Gordon, M. wB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy. Physical Chemistry Chemical Physics. 16 (21), 9904-9924 (2014).
- Head-Gordon, M., Pople, J. A., Frisch, M. J. MP2 energy evaluation by direct methods. Chemical Physics Letters. 153 (6), 503-506 (1988).
- Pople, J. A., Seeger, R., Krishnan, R. Variational configuration interaction methods and comparison with perturbation theory. The International Journal of Quantum Chemistry. 12, 149-163 (1977).
- Pople, J. A., Binkley, J. S., Seeger, R. Theoretical models incorporating electron correlation. The International Journal of Quantum Chemistry. 10 (10), 1-19 (1976).
- Monkhorst, H. J. Calculation of properties with the coupled-cluster method. The International Journal of Quantum Chemistry. 12 (11), 421-432 (1977).
- Klopper, W., Manby, F. R., Ten-No, S., Valeev, E. F. R12 methods in explicitly correlated molecular electronic structure theory. International Reviews in Physical Chemistry. 25, 427-468 (2006).
- Hattig, C. Optimization of auxiliary basis sets for RI-MP2 and RI-CC2 calculations: Core-valence and quintuple-z basis sets for H to Ar and QZVPP basis sets for Li to Kr. Physical Chemistry Chemical Physics. 7 (1), 59-66 (2005).
- Barone, V. Anharmonic vibrational properties by a fully automated second-order perturbative approach. The Journal of Chemical Physics. 122, 014108 (2005).
- Barone, V. Vibrational zero-point energies and thermodynamic functions beyond the harmonic approximation. The Journal of Chemical Physics. 120 (7), 3059-3065 (2004).
- Temelso, B., et al. Exploring the Rich Potential Energy Surface of (H2O)11 and Its Physical Implications. Journal of Chemical Theory and Computation. 14 (2), 1141-1153 (2018).
- Kabrede, H., Hentschke, R. Global minima of water clusters (H2O)N, N≤25, described by three empirical potentials. Journal of Physical Chemistry B. 107 (16), (2003).
- Steber, A. L., et al. Capturing the Elusive Water Trimer from the Stepwise Growth of Water on the Surface of a Polycyclic Aromatic Hydrocarbon Acenaphthene. Journal of Physical Chemistry Letters. 8 (23), 5744-5750 (2017).
- Perez, C., et al. Corrannulene and its complex with water: A tiny cup of water. Physical Chemistry Chemical Physics. 19 (22), 14214-14223 (2017).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati