
Method Article
ab initio 열화학에서 분자 클러스터의 대기 농도 계산
요약
약하게 결합된 분자 클러스터의 대기 농도는 유전 알고리즘과 반 경험적 및 ab initio 양자 화학을 활용하는 다단계 구성 샘플링 방법을 통해 발견되는 저에너지 구조의 열화학적 특성으로부터 계산할 수 있습니다.
초록
대기 에어로졸의 형성 과 성장에 대한 계산 연구는 가스 상 전자 구조 및 진동 주파수 계산에서 얻을 수있는 정확한 깁스 자유 에너지 표면을 필요로한다. 이러한 수량은 형상이 잠재적 에너지 표면의 최소값에 해당하는 대기 클러스터에 유효합니다. 최소 에너지 구조의 깁스 자유 에너지는 온도 및 압력과 같은 다양한 조건하에서 클러스터의 대기 농도를 예측하는 데 사용될 수 있다. 우리는 점점 더 정확한 스크리닝 계산의 시리즈 뒤에 유전 알고리즘 기반 구성 샘플링에 내장 된 계산 저렴한 절차를 제시한다. 이 절차는 반 경험적 모델을 사용하여 대규모 구성 세트의 형상을 생성하고 진화한 다음 일련의 높은 수준의 ab initio 수준의 이론에서 결과고유 구조를 구체화하는 것으로 시작합니다. 마지막으로, 열역학 보정은 최소 에너지 구조의 결과 집합에 대해 계산되고 깁스 의 형성, 평형 상수 및 대기 농도의 자유 에너지를 계산하는 데 사용됩니다. 우리는 주변 조건하에서 수화 글리신 클러스터의 연구에이 절차의 응용 프로그램을 제시한다.
서문
기후 변화의 대기 연구에서 가장 불확실한 매개 변수는 구름 입자가 들어오는 태양 복사를 반영하는 정확한 정도입니다. 가스에 부유하는 미립자 물질인 에어로졸은 들어오는 방사선을 산란시키는 구름 응축 핵(CCN)이라고 불리는 구름 입자를 형성하여대기의흡수와 후속 가열을 방지합니다 1. 이 순 냉각 효과에 대한 자세한 이해를 위해서는 에어로졸이 CCN으로 증가하는 것을 이해해야 하며, 이로 인해 작은 분자 클러스터가 에어로졸 입자로 증가하는 것을 이해해야 합니다. 최근 연구는 에어로졸 형성이 직경 3 nm 이하의 분자 클러스터에 의해 시작된다는 것을 제안했다2; 그러나,이 크기 정권은 실험 기술3,,4를사용하여 액세스하기 가 어렵습니다. 따라서 이러한 실험적 한계를 극복하기 위해서는 전산 모델링 접근법이 요구된다.
아래에 설명된 모델링 방법을 사용하여 수화 클러스터의 성장을 분석할 수 있습니다. 우리는 생물학적 전 환경에서 작은 성분에서 큰 생물학적 분자의 형성에 물의 역할에 관심이 있기 때문에, 우리는 글리신으로 우리의 접근 방식을 설명합니다. 이러한 연구 질문을 해결하는 데 필요한 과제와 도구는 대기 에어로졸 및 사전 핵 화 클러스터,,5,6,,7,,8,,9,,10,11,,12,13,,14,,15의연구에 관여하는 것과 매우 유사하다. 여기에서는, 우리는 격리된 글리신 분자에서 시작하는 수화한 글리신 클러스터를 검토하고 최대 5개의 물 분자의 일련의 단계별 추가에 선행됩니다. 최종 목표는 해수면에서 실온에서 대기 중의 글리(H2O)n=0-5 클러스터의 평형 농도와 100%의 상대 습도(RH)를 계산하는 것입니다.
이 sub 나노미터 분자 클러스터의 소수는 그밖 증기 분자를 추가하거나 기존 클러스터에 응고하여 메타안정 임계 클러스터 (직경 1-3 nm)로 증가합니다. 이러한 중요한 클러스터는 구름의 침전 효율과 입사광을 반사하는 능력에 직접적인 영향을 미치는 훨씬 더 큰 구름 응축 핵(CCN)의 형성으로 이어지는 유리한 성장 프로필을 가지고 있습니다. 따라서 분자 클러스터의 열역학과 평형 분포를 잘 이해하면 에어로졸이 지구 기후에 미치는 영향을 보다 정확하게 예측할 수 있습니다.
에어로졸 형성의 설명 모델은 분자 클러스터 형성의 정확한 열역학을 필요로합니다. 분자 클러스터 형성의 정확한 열역학의 계산은 클러스터의 잠재적 에너지 표면(PES)16에서글로벌 및 로컬 최소를 찾는 것을 포함하는 가장 안정적인 구성의 식별을 요구한다. 이러한 과정은 구성 샘플링이라고 하며 분자 역학(MD)17,18,,19,,20,몬테카를로(MC)21,,22,및 유전 알고리즘(GA)23,,24,,25를기초로 하는 것을 포함하는 다양한 기술을 통해 달성될 수 있다.,
이론의 높은 수준에서 대기 수화물의 구조와 열역학을 얻기 위해 수년에 걸쳐 다른 프로토콜이 개발되었습니다. 이러한 프로토콜은 (i) 구성 샘플링 방법, (ii) 구성 샘플링에 사용되는 로우 레벨 방법의 특성, 및 (iii) 후속 단계에서 결과를 구체화하는 데 사용되는 상위 레벨 방법의 계층구조의 선택에서 달랐다.
구성 샘플링 방법은 화학 직관26,무작위 샘플링27,,28,분자 역학 (MD)29,30,분지 호핑 (BH)31,및 유전 알고리즘 (GA)24,,25,,32를포함했다., 이러한 샘플링 방법에 사용되는 가장 일반적인 저수준 방법은 PM6, PM7 및 SCC-DFTB와 같은 힘 필드 또는 반 경험적 모델입니다. 이들은 종종 점점 더 큰 기초 세트와 야곱의 사다리(33)의높은 횡단에서 더 신뢰할 수있는 기능DFT 계산 뒤에 있습니다. 경우에 따라, 이들은 MP2, CCSD (T) 및 비용 효율적인 DLPNO-CCSD (T)34,,35와같은 더 높은 수준의 파형 방법에 선행된다.
Kildgaard 등36은 더 큰 클러스터에 대한 후보를 생성하기 위해 더 작은 수화 또는 비수화 클러스터 주위의 피보나치구체 37의 지점에서 물 분자가 추가되는 체계적인 방법을 개발했습니다. 비물리적 및 중복 후보는 가까운 접촉 임계값과 서로 다른 순응자 간의 근평균 제곱 거리에 따라 제거됩니다. PM6 반 경험적 방법과 DFT 및 웨이브 함수 방법의 계층 구조를 사용하여 후속 최적화는 높은 수준의 이론에서 낮은 에너지 적합성 세트를 얻는 데 사용됩니다.
인공 꿀벌 콜로니(ABC)알고리즘(38)은 최근 장 외가 ABCluster39라는프로그램에서 분자 클러스터를 연구하기 위해 구현한 새로운 구성 샘플링 접근법이다. Kubecka 등40은 구성 샘플링을 위해 ABCluster를 사용한 다음, 타이트결합 GFN-xTB 반경험적방법(41)을사용하여 낮은 수준의 재최적화를 수행하였다. 그들은 DFT 방법을 사용하여 구조와 에너지를 더욱 정제한 다음 DLPNO-CCSD(T)를 사용하여 최종 에너지를 추가했습니다.
방법에 관계없이 구성 샘플링은 PES에서 임의로 또는 임의로 생성된 포인트 분포로 시작합니다. 각 점은 해당 분자 클러스터의 특정 형상에 해당하며 샘플링 방법에 의해 생성됩니다. 그런 다음 PES의 "내리막" 방향을 따라 각 점에 대해 가장 가까운 로컬 최소를 찾습니다. 이렇게 발견된 최소의 집합은 적어도 얼마 동안 안정되어 있는 분자 클러스터의 그 기하학에 해당합니다. 여기서, PES의 형상과 표면의 각 지점에서의 에너지의 평가는 보다 정확한 물리적 설명이 더 많은 계산비용이 드는 에너지 계산을 초래하는 시스템의 물리적 설명에 민감할 것이다. 우리는 특히 OGOLEM25 프로그램에서 구현된 GA 방법을 사용할 것이며, 이는 다양한 글로벌 최적화 및 구성 샘플링 문제42,,43,,44,,45,초기 샘플링 포인트 세트를 생성하기 위해 성공적으로 적용되었다. PES는 MOPAC2016 프로그램 47에 구현된 PM7모델(46)에의해 설명될 것이다.46 이 조합은 MD 및 MC 방법에 비해 더 다양한 포인트를 생성하고 PES에 대한 보다 상세한 설명보다 더 빠르게 로컬 미니마를 발견하기 때문에 사용된다.
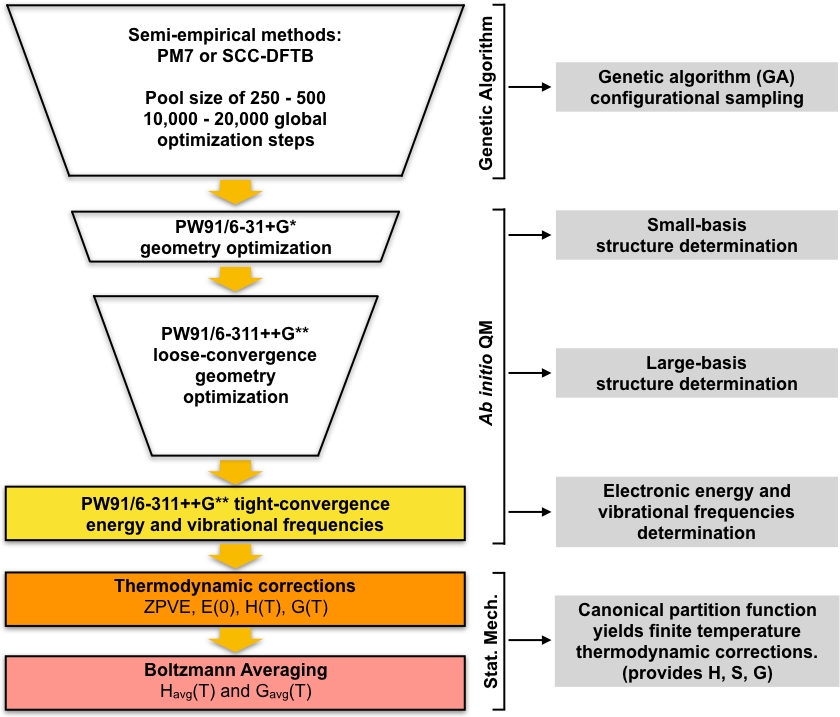
GA에 최적화된 로컬 미니마 세트는 일련의 스크리닝 단계의 시작 형상으로 사용되며, 이로 인해 최소 에너지가 낮아지지 않습니다. 프로토콜의 이 부분은 작은 기초 세트로 밀도 기능 이론(DFT)을 사용하여 고유한 GA 에 최적화된 구조 세트를 최적화하는 것으로 시작됩니다. 이 최적화 세트는 일반적으로 GA에 최적화된 반 경험적 구조에 비해 더 자세하게 모델링되는 더 작은 고유한 로컬 최소 구조 세트를 제공합니다. 그런 다음 더 큰 기초 집합을 사용하여 이 작은 구조 집합에서 또 다른 DFT 최적화가 수행됩니다. 다시 말하지만, 이 단계는 일반적으로 작은 기초 DFT 단계에 비해 더 자세하게 모델링되는 더 작은 구조세트를 제공할 것이다. 그런 다음 최종 고유 구조 세트가 더 엄격한 수렴에 최적화되고 고조파 진동 주파수가 계산됩니다. 이 단계 후 우리는 대기 중 클러스터의 평형 농도를 계산하는 데 필요한 모든 것을 갖습니다. 전체 적인 접근 방식은 그림 1에서다이어그램으로 요약됩니다. 우리는 PW9148 일반화 그라데이션 근사치 (GGA) 교환 상관 관계 기능 Pople49 50 기초 세트의 두 가지 변형과 함께 DFT의 구현 (작은 기초 단계에 대한 6-311 +G*와 큰 기초 단계에 대한 6-311 +G**)를 사용합니다. 이러한 교환 상관 기능 및 기초 세트의 이러한 특별한 조합은 대기클러스터(51,,52)에대한 정확한 깁스 프리 에너지 형성의 이전 성공으로 인해 선택되었다.
이 프로토콜은 사용자가 휴대용 배치시스템(53)을 사용하여 고성능 컴퓨팅(HPC) 클러스터에 액세스할 수 있다고 가정하고, MOPAC2016(http://openmopac.net/MOPAC2016.html)47,OGOLEM(https://www.ogolem.org)25,가우시안 09(https://gaussian.com)49,및 OpenBabel54(http://openbabel.org/wiki/Main_Page) 소프트웨어가 특정 설치 지시에 따라 설치된 소프트웨어이다. 이 프로토콜의 각 단계는 사용자의 $PATH 환경 변수에 포함된 디렉터리에 저장해야 하는 사내 셸 및 Python 2.7 스크립트 집합을 사용합니다. 위의 모든 프로그램을 실행하는 데 필요한 모든 환경 모듈 및 실행 권한도 사용자의 세션에 로드되어야 합니다. GA 코드(OGOLEM) 및 반경험적 코드(MOPAC)에 의한 디스크 및 메모리 사용량은 최신 컴퓨터 리소스 표준에 따라 매우 작습니다. OGOLEM/MOPAC의 전체 메모리 및 디스크 사용량은 사용하려는 스레드 수에 따라 달라지며, 그렇다 하더라도 대부분의 HPC 시스템의 기능에 비해 리소스 사용량이 작습니다. QM 메서드의 리소스 요구 는 클러스터의 크기와 사용된 이론 의 수준에 따라 달라집니다. 이 프로토콜을 사용하면 이론 의 수준이 달라져 서 에너지 구조의 최종 집합을 계산할 수 있으며, 일반적으로 빠른 계산으로 인해 결과의 정확도가 더 불확실해질 수 있다는 점입니다.
명확성을 위해 사용자의 로컬 컴퓨터는"로컬 컴퓨터"라고하며 액세스 권한이 있는 HPC 클러스터는"원격 클러스터"라고합니다.
프로토콜
1. 분리된 글리신과 물의 최소 에너지 구조 찾기
참고: 여기서 의 목표는 두 가지입니다: (i) 유전 알고리즘 구성 샘플링에 사용하기 위해 단리된 물과 글리신 분자의 최소 에너지 구조를 얻고, (ii) 대기 농도 계산에 사용하기 위해 이들 분자의 기체 상 에너지에 대한 열역학적 보정을 계산하는 것입니다.
- 로컬 컴퓨터에서 Avogadro의 새 세션을 엽니다.
- 빌드 > 삽입 > 펩티드를 클릭하고 인서트 펩타이드 창에서 글리신 단량체를 생성하려면 글리시를 선택합니다.
- 확장 > 가우시안을 클릭하고 텍스트 상자의 첫 번째 줄을 편집하여 '# pw91pw91/6-311++G** int(Acc2E=12,UltraFine) scf(수렴=12) 옵트(꽉, maxcyc=300) 프런시크'를 읽습니다. 입력 파일을 생성하고 저장을 glycine.com.
- 분자가 글리신이55를하는 것처럼 분자가 상당한 변형 유연성을 가지고 있다면, 글로벌 최소 구조 및 기타 저지대 적합자를 식별하기 위해 형태 분석을 수행하는 것이 중요합니다. OpenBabel54는 다양한 알고리즘과 빠른 힘 필드를 활용하는 강력한 형태 검색 도구를 제공합니다. conformers는 GA 및 후속 계산 중에 이완및 상호 변환할 수 있지만, 때로는 여러 GA 계산을 실행해야 하는 경우가 있습니다.
- 로컬 컴퓨터에서 Avogadro에서 새 세션을 엽니다.
- 빌드 > 삽입 > 조각 및 삽입 조각 창에서 "물"을 검색하여 물 좌표를 가져옵니다.
- 확장 > 가우시안을 클릭하고 텍스트 상자의 첫 번째 줄을 편집하여 '# pw91pw91/6-311++G** int(Acc2E=12,UltraFine) scf(수렴=12) 옵트(꽉, maxcyc=300) 프런시크'를 읽습니다. 입력 파일을 생성하고 저장을 water.com.
- 두 .com 파일을 원격 클러스터로 전송합니다. 원격 클러스터에 로그인하면 일괄 처리 제출 스크립트에서 Gaussian 09를 호출하여 계산을 시작합니다. 계산이 완료되면 OpenBabel을 호출하여 최소 에너지 구조의 카르테시안좌표(.xyz 파일)를 추출합니다. 글리신의 경우 실행할 명령은 다음과 같은 것입니다.
오바벨 -ig09 글리신.log -oxyz > 글리신.xyz
이 두 .xyz 파일은 다음 단계에서 GA 구성 샘플링에서 사용됩니다.
2. 글리 (H2O)n = 1-5 클러스터의 유전자 알고리즘 기반 구성 샘플링
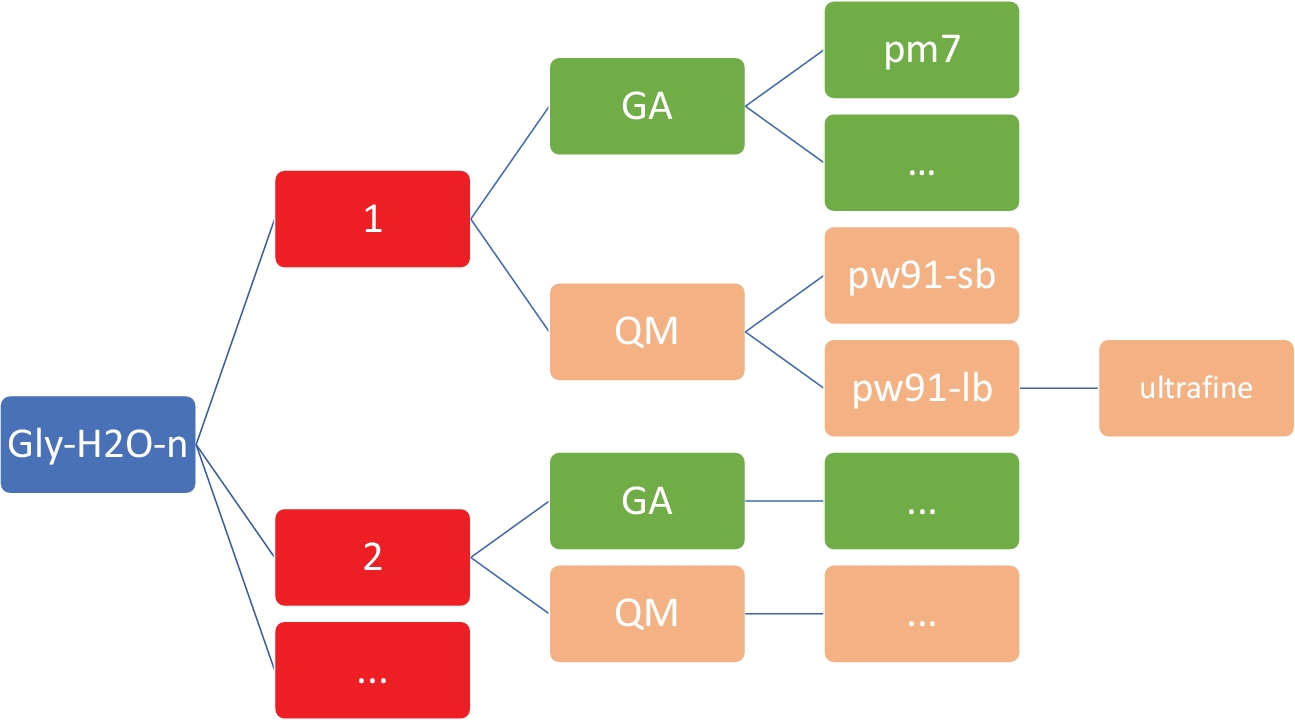
참고: 여기서 의 목표는 MOPAC47에서구현된 PM746 모델을 사용하여 저렴한 반 경험적 이론 수준에서 글리(H2 O)n=1-5에 대한 저에너지 구조 세트를 얻는 것입니다.2 그림 2와같이 작업 디렉토리에 정확한 조직과 구조가 있는 것이 필수적입니다. 이는 사용자 지정 셸 및 Python 스크립트가 오류 없이 작동하도록 하기 위한 것입니다.
- 필요한 모든 스크립트를 원격 클러스터에 복사하고 해당 위치를 추가하여 $PATH
- 모든 스크립트 및 템플릿 파일을 폴더(예: 스크립트)에 넣고 원격 클러스터에 복사합니다.
- 모든 스크립트가 실행 가능한지 확인
- 터미널에서 다음 명령을 입력하여 스크립트 디렉터리 위치를 $PATH 환경 변수에 추가합니다. 스크립트의 기본 위치는 $HOME/JoVE-demo/script로 설정되어 있지만 스크립트가 포함된 디렉터리를 가리키는 $SCRIPTS_HOME이라는 환경 변수를 정의하고 경로에 $SCRIPTS_HOME을 추가할 수 있습니다.
- 배쉬 쉘:
내보내기 SCRIPTS_HOME=/경로/에/스크립트
내보내기 PATH=${SCRIPTS_HOME}:${PATH} - Tcsh/Csh 쉘:
세텐프 SCRIPTS_HOME /경로 / 에 / 스크립트
setenv PATH ${SCRIPTS_HOME}:${PATH}
- 배쉬 쉘:
- 원격 클러스터에서 GA 계산을 설정하고 실행합니다.
- gly-h2o-n이라는 디렉토리를 만들었는데 여기서 n은 물 분자의 수입니다.
- gly-h2o-n 디렉터리 아래에 GA라는 하위 디렉터리를 만들어 유전자 알고리즘 계산을 실행합니다.
- OGOLEM 입력 파일(예: pm7.ogo), 단량체 카르테시안 좌표(예: 글리신.xyz, water.xyz) 및 PBS 일괄 제출 스크립트(예: run.pbs)를 GA 디렉토리에 복사합니다.
- OGOLEM 입력 파일 및 일괄 처리 제출 파일을 필요한 변경합니다.
- 계산을 제출합니다. 계산이 시작되면 OGOLEM은 GA 디렉터리에 OGOLEM 입력 파일(예: pm7)의 접두사로 명명된 새 디렉터리를 만들고 새로 생성된 좌표를 저장합니다.
- 계산이 완료되면 에너지 및 회전 상수를 컴파일하고 해당 정보를 사용하여 고유한 저에너지 구조가 결정됩니다.
- 글리-h2o-n/GA/pm7로 디렉토리 변경 및
- 에너지를 추출하고 명령을 통해 GA 에 최적화된 클러스터의 회전 상수를 계산합니다.
getRotConsts-GA.csh N 0 99
여기서 N은 분자 클러스터의 원자 수와 '0 99'는 GA 풀 크기가 100이고 인덱스는 0에서 99까지 실행된다는 것을 나타냅니다. 이렇게 하면 rotConstsData_C 라는 파일이 생성되며, 여기에는 GA에 최적화된 모든 클러스터 구성, 에너지 및 회전 상수의 정렬된 목록이 포함됩니다. - 명령을 실행합니다.
similarityAnalysis.py pm7 rotConstsData_C
여기서 pm7은 파일 이름 지정 레이블로 사용되어 고유한 GA 최적화 클러스터를 찾고 저장합니다. 이렇게 하면 고유한 GA 최적화 구성의 정렬된 목록이 포함된 고유구조스-pm7.data라는 파일이 생성됩니다. 이것은 이론의 PM7 수준에서 최적화된 Gly(H2O)n 클러스터에 대한 고유한 로컬 최소 구조 목록이며, 이러한 구조는 이제 DFT를 사용하여 구체화할 준비가 되었습니다.
- gly-h2o-n/GA 디렉토리로 이동하여 combine-GA.csh 스크립트를 사용하여 여러 유사한 GA 실행의 결과를 결합합니다. 구문은 다음과 입니다.
결합-GA.csh & 레이블> <와 GA 실행 및 >
이 특별한 경우 명령은 다음과 같은 것입니다.
결합 GA.csh pm7 pm7
글리-h2o-n/GA 디렉토리에서'고유 구조-pm7.data'라는이름의 새로운 고유 구조 목록을 생성합니다.
3. 작은 기초 세트와 QM 방법을 사용하여 정제
참고: 여기서 목표는 더 나은 양자-기계적 설명을 사용하여 Gly(H2O)n=1-5 클러스터 구조의 구성 샘플링을 구체화하여 더 작지만 더 정확한 Gly(H2 O)n=1-5 클러스터 구조 집합을 얻는 것입니다.2 이 단계의 시작 구조는 2단계의 출력입니다.
- 작은 기준 세트 DFT 계산을 준비하고 실행합니다.
- 글리 h2o-n 디렉토리 아래에 QM이라는 하위 디렉토리를 만듭니다. QM 디렉터리에서 pw91-sb라는다른 하위 디렉터리를 만듭니다.
- 글리-h2o-n/GA 디렉토리에서 QM/pw91-sb 디렉토리에 고유한 구조목록(고유구조-pm7.data)을복사합니다.
- 그 gly-h2o-n/QM/pw91-sb로디렉토리를 변경합니다.
- 명령을 사용하여 작은 기준 세트 DFT 구성 샘플링 스크립트를 실행합니다.
run-pw91-sb.csh 고유구조스-pm7.data sb QUEUE 10
여기서 sb는 이 계산 집합의 레이블이며 QUEUE는 컴퓨팅 클러스터에서 선호되는 큐이며 10개는 10개의 계산을 하나의 일괄 처리 작업으로 그룹화해야 한다는 것을 나타냅니다. 이 스크립트는 Gaussian 09에 대한 입력을 자동으로 생성하고 모든 계산을 제출합니다. 드라이 런을 수행하려면 'QUEUE'에 대한 '테스트'를 입력합니다.
- 제출된 계산이 완료되면 결과를 추출하고 분석합니다.
- 에너지를 추출하고 명령을 사용하여 소규모 로 최적화된 클러스터의 회전 상수를 계산합니다.
getRotConsts-dft-sb.csh pw91 N
여기서 pw91은 PW91 밀도 함수가 사용되었고 N은 클러스터의 원자 수임을 나타냅니다. 그렇게 하면 rotConstsData_C라는 파일이 생성됩니다. - 이제 명령을 통해 고유한 구조를 식별합니다.
similarityAnalysis.py sb rotConstsData_C
여기서 sb가 파일 이름 지정 레이블로 사용됩니다. 이제 파일 고유 구조-sb.data에저장된 이론의 PW91/6-31+G* 수준에서 최적화된 고유한 구성 목록이 표시됩니다.
- 에너지를 추출하고 명령을 사용하여 소규모 로 최적화된 클러스터의 회전 상수를 계산합니다.
- gly-h2o-n/QM 디렉토리로 이동하여 결합-QM.csh 스크립트를 사용하여 여러 유사한 QM 실행의 결과를 결합합니다. 구문은 다음과 입니다.
QM.csh&label>>
이 특별한 경우 명령은 다음과 같은 것입니다.
결합-QM.csh sb pw91-sb
글리-h2o-n/QM 디렉토리에서'고유 구조-sb.data'라는이름의 새로운 고유 구조 목록을 생성합니다.
4. 큰 기초 세트와 QM 방법을 사용하여 추가 정제
참고: 여기서 목표는 더 나은 양자-기계적 설명을 사용하여2Gly(H2 O)n=1-5 클러스터의 구성 샘플링을 더욱 구체화하는 것입니다. 이 단계의 시작 구조는 3단계의 출력입니다.
- 더 큰 기준 집합을 사용하여 보다 신뢰할 수 있는 계산을 제출합니다.
- QM 디렉토리 아래에 pw91-lb라는 하위 디렉토리를 만듭니다.
- 글리-h2o-n/QM 디렉토리에서 글리-h2o-n/QM/pw91-lb 디렉토리에 고유한 구조목록(고유Structures-sb.data)을복사하고 해당 디렉토리로 변경합니다.
- 다음과 같은 명령으로 대단위 DFT 구성 샘플링 스크립트를 실행합니다.
run-pw91-lb.csh 고유구조스-sb.data lb QUEUE 10
여기서 lb가 이 계산 집합의 레이블인 경우 QUEUE는 컴퓨팅 클러스터에서 선호되는 큐이며 10은 10개의 계산을 하나의 일괄 처리 작업으로 그룹화해야 한다는 것을 나타냅니다. 이 스크립트는 Gaussian 09에 대한 입력을 자동으로 생성하고 모든 계산을 제출합니다. 'QUEUE'에 대한 '테스트'를 입력하여 드라이 런 테스트를 수행합니다.
- 제출된 계산이 완료되면 데이터를 추출하고 분석합니다.
- 다음 명령을 통해 대용량 최적화 클러스터의 회전 상수를 계산합니다.
getRotConsts-dft-lb.csh pw91 N
여기서 pw91은 PW91 밀도 함수가 사용되었고 N은 클러스터의 원자 수임을 나타냅니다. - 이제 명령을 통해 고유한 구조를 식별합니다.
similarityAnalysis.py 파운드 rotConstsData_C
여기서 lb가 파일 이름 지정 레이블로 사용됩니다. 이제 파일 고유 구조-lb.data에저장된 이론의 PW91/6-311++G** 수준에서 최적화된 고유한 구성 목록이 있습니다.
- 다음 명령을 통해 대용량 최적화 클러스터의 회전 상수를 계산합니다.
5. 최종 에너지 및 열역학 보정 계산
참고: 여기서 목표는 원하는 열화학 적 보정을 계산하기 위해 큰 기초 세트와 초미세 통합 그리드를 사용하여 Gly(H2O)n = 1-5 클러스터의 진동 구조와 에너지를 얻는 것입니다.
- 이전 단계의 결과로 시작하여 보다 신뢰할 수 있는 계산을 제출합니다.
- QM/pw91-lb 디렉토리 아래에 초미세라는 하위 디렉토리를 만듭니다. 그런 다음 QM/pw91-lb 디렉토리에서 QM/pw91-lb/초미세 디렉토리에 고유한 구조목록(고유Structures-lb.data)을복사하고 해당 디렉토리로 변경합니다.
- 명령과 함께 초미세 대용량 DFT 스크립트를 제출합니다.
run-pw91-lb-ultrafine.csh 고유구조-lb.data uf QUEUE 10
uf가 이 계산 집합의 레이블인 경우 QUEUE는 컴퓨팅 클러스터에서 선호되는 큐이며 10개는 10개의 계산을 하나의 일괄 처리 작업으로 그룹화해야 한다는 것을 나타냅니다. 이 스크립트는 Gaussian 09에 대한 입력을 자동으로 생성하고 모든 계산을 제출합니다. 'QUEUE'에 대한 '테스트'를 입력하여 드라이 런 테스트를 수행합니다.
- 제출된 계산이 완료되면 데이터를 추출하고 분석합니다.
- 에너지를 추출하고 명령을 통해 대용량 최적화 클러스터의 회전 상수를 계산합니다.
getRotConsts-dft-lb-ultrafine.csh pw91 N
여기서 pw91은 PW91 밀도 함수가 사용되었고 N은 클러스터의 원자 수임을 나타냅니다. - 이제 명령을 통해 고유한 구조를 식별합니다.
similarityAnalysis.py uf rotConstsData_C
uf가 파일 이름 지정 레이블로 사용되는 경우 이제 파일 고유 구조-uf.data에저장된 이론의 PW91/6-311++G** 수준에서 최적화된 고유한 구성 목록이 있습니다.
- 에너지를 추출하고 명령을 통해 대용량 최적화 클러스터의 회전 상수를 계산합니다.
- 열역학 적 교정을 계산하는 데 필요한 정보의 최종 추출을 수행합니다. 이 정보를 사용하여 열역학 교정을 계산합니다.
- 최종 전자 에너지, 회전 상수 및 진동 주파수를 추출하고 이를 사용하여 명령을 사용하여 열역학 보정을 계산합니다.
런 열-pw91.csh 고유구조-uf.data - 'gly-h2o-n.xlsx'라는이름의 Excel 스프레드시트의'Raw_Energies'시트에 명령줄 출력을 복사/붙여넣습니다. 단량체 (글리신 및 물)뿐만 아니라 각 수화물의 가장 낮은 에너지 부재 (gly-h2o-n, n = 1,2, ...)에 대해이 작업을 수행해야합니다.
- 'gly-h2o-n.xlsx'스프레드시트의 첫 번째 시트에 원시 에너지가 추가되면 후속'Binding_Energies'및'Hydrate_Distribution'시트가 자동으로 업데이트됩니다. 특히,'Hydrate_Distribution'시트는 서로 다른 온도(예: 298.15K), 상대 습도(20%, 50%, 100%)에서 수화물의 평형 농도를 산출합니다. 및 물 ([H2O]) 및 글리신 ([글리신])의 초기 농도. 이러한 계산뒤에 있는 이론은 다음 단계에서 설명합니다.
- 최종 전자 에너지, 회전 상수 및 진동 주파수를 추출하고 이를 사용하여 명령을 사용하여 열역학 보정을 계산합니다.
6. 해수면에서 실온에서글리(H2O)n=0-5 클러스터의 대기 농도 계산
참고: 이전 단계에서 생성된 열역학 데이터를 스프레드시트에 먼저 복사하고 순차수화의 깁스 프리 에너지를 계산하여 이 작업을 수행합니다. 그런 다음 깁스 프리 에너지는 각 순차 수화에 대한 평형 상수를 계산하는 데 사용됩니다. 마지막으로, 선형 방정식 세트는 단량체, 온도 및 압력의 주어진 농도에 대한 수화물의 평형 농도를 얻기 위해 해결된다.
- 아래와 같이 글리신의 순차적 수화를 위한 화학적 평형 시스템을 설정하여 시작하십시오.

- K n =e-ΔG n/(kBT)를사용하여 평형 상수 K n을 계산하고, 여기서 n은 수화 수준이고, ΔGn은 nth 수화 반응의 깁스 자유 에너지 변화, kB는 Boltzmann의 상수이고, T는 온도이다. e

- 수화 및 무수화 글리신 클러스터의 평형 농도의 합이 단리글리신[Gly]0의 초기 농도와 같다는 가정을 이용하여 질량 보존을 위한0방정식을 설정한다. 평형 상수 식의 일부 대수 재배열을 사용하여 6 개의 동시 방정식의이 시스템을 다시 작성하십시오.

- 상기에 나타난 방정식의 시스템을 해결하여 글리(H2O)n=0-5의 평형2농도를 얻어 서 대기 중글리신의 농도에 대한 실험값56,57,,58을 이용하여, [Gly]0 = 2.9 x 106 cm-3,100% 상대 습도 및 298.15 K59의온도에서 대기 중의 물 농도 , [H2O] = 7.7 x 1017 cm-3.,
결과
이 프로토콜의 첫 번째 결과 집합은 구성 샘플링 절차를 통해2발견된 Gly(H2 O)n=1-5의 저에너지 구조 집합이어야 합니다. 이러한 구조는 PW91/6-311++G** 이론 수준에서 최적화되었으며 이 백서의 목적을 위해 정확하다고 가정합니다. PW91/6-311++G**가 이러한 클러스터의 결합 에너지를 지속적으로 과소 평가하거나 과대 평가한다는 증거는 없습니다. MP2/CBS32 및 [DLPNO-]CCSD(T)/CBS60,,61 추정 및실험(52)에 비해 결합 에너지를 예측하는 능력은 많은 변동을 나타낸다. 대부분의 다른 밀도 기능도 마찬가지입니다. 일반적으로 n = 1 - 5의 각 값은 가장 낮은 에너지 구조의 약 5 kcal mol-1 내에서 소수의 저에너지 구조를 산출해야 합니다. 여기서는 간결성을 위해 run-thermo-pw91.csh 스크립트에서 생성된 첫 번째 구조에 중점을 둡니다. 도 3은 글리(H2O)n=0-5 군집의 가장 낮은 전자 에너지 이소메를 나타낸다. 하나는 물 분자의 수가 증가함에 따라 수소 결합 네트워크가 복잡성에서 증가하는 것을 볼 수 있으며, 심지어 n = 5에서 3 차원 케이지와 같은 구조로 대부분 평면 네트워크에서 간다. 이 텍스트의 나머지 부분에서는 이 다섯 개의 특정 클러스터에 해당하는 에너지 및 열역학적 수량을 사용합니다.
표 1은 프로토콜을 수행하는 데 필요한 열역학적 수량을 포함합니다. 표 2는 전자 에너지, 진동 제로 포인트 보정 및 세 가지 다른 온도에서열역학적 보정이 인쇄되는 run-thermo-pw91.csh 스크립트의 출력의 예를 보여줍니다. 각 클러스터(행)에 대해 E[PW91/6-311++G**]는 Hartree 단위의 초미세 통합 그리드에서 계산된 이론의 PW91/6-311++G** 수준의 가스 상 전자 에너지와 kcalmol-1단위의 제로 포인트 진동 에너지(ZPVE)에 해당합니다.ZPVE 각 온도에서, 216.65 K, 273.15 K, 및 298.15 K, 열역학 보정이 열역학 적 보정이 나열되어, Kcal mol-1의단위로 형성의 엔트로피 , S cal mol-1의단위로 형성의 엔트로피 , 및 G는 kcal mol의 단위로 형성의 무료 에너지.1. 표 3은 순차적 수화뿐만 아니라 총 깁스 자유 에너지 변화의 계산을 나타낸다. 반응에 대한 수화의 총 깁스 자유 에너지 변화의 예

전자 에너지 EPW91의 계산으로 시작됩니다.

여기서 EPW91[Gly∙(H2O)]는 표 2 열 C에서 가져온 것이고, EPW91[글리] 및 EPW91[H2O]는 표 1 열 B에서 가져온다. 다음으로 반응의 제로포인트 진동E에너지의 변화를 포함시킴으로써 총 기위 에너지 변화 Δ E(0)를 계산한다.

을 사용하여 열 D를 구합니다. 여기서, ΔEPW91/6−311+G**는 표 3 열 C, EZPVE[글리 ∙(H2O)]로부터 표 2 열 D, 및 E ZPVE [글리] 및 EZPVE[H2O]표 1 열 C로부터 가져온다.ZPVE 간결성을 위해 실온 클러스터로 이동하므로 216.65 K 및 273.15 K 데이터를 건너뜁니다. 실온에서, 우리는 가스 상 에너지 변화를 수정하여 반응 ΔH의 엔탈피 변화를 계산합니다.

ΔEE(0)가 표 3 열 D에서 가져온 경우, Δ H[Gly∙(H2O)]는 표 2 열 K에서 가져온 것이고, ΔH[글리] 및 ΔH[H2O]는 표 1 열 J에서 가져온다.H 마지막으로, 우리는 반응 ΔG의 깁스 자유 에너지 변화를 계산합니다.

ΔH가 표 3 열 I에서 가져온 경우, S[Gly∙(H2 O)]는 표 2 열 L에서 가져온 것이고, S[Gly] 및2 S[H2O]는 표 1 열 K.에서 가져온 엔트로피 값을 이 단계에서 kcalmol-1 K-1의 단위로 변환해야 한다는 점에 유의하십시오.
이제 6단계에서와 같이 수화 글리신의 대기 농도를 계산하는 데 필요한 수량이 있습니다. 결과는 표 4에표시된 데이터와 유사해야 하지만 작은 수치 차이가 예상됩니다. 표 4는 6.2단계에서 6개의 방정식의 시스템의 제형으로부터 발견된 평형 수화물 농도를 하나의 매트릭스 방정식 및 그 후속 해로 나타낸다. 우리는 방정식 의 시스템이 다음과 같이 작성 될 수 있다는 사실을 인정하여 시작합니다

여기서Kn은 글리신의 순차적th 수화에 대한 평형 상수이며, w는 대기 중의 물의 농도이고, g는 대기 중의 고립된 글리신의 초기 농도이고, gn은 글리(H2 O)n의평형 농도이다.2 위의 방정식을 A= b로다시 작성하면 A-1이 행렬 A의 역인 x = A-1b를 얻습니다. 이 역은 표 4에 표시된 대로 기본 제공 스프레드시트 함수를 사용하여 쉽게 계산하여 최종 결과를 얻을 수 있습니다.
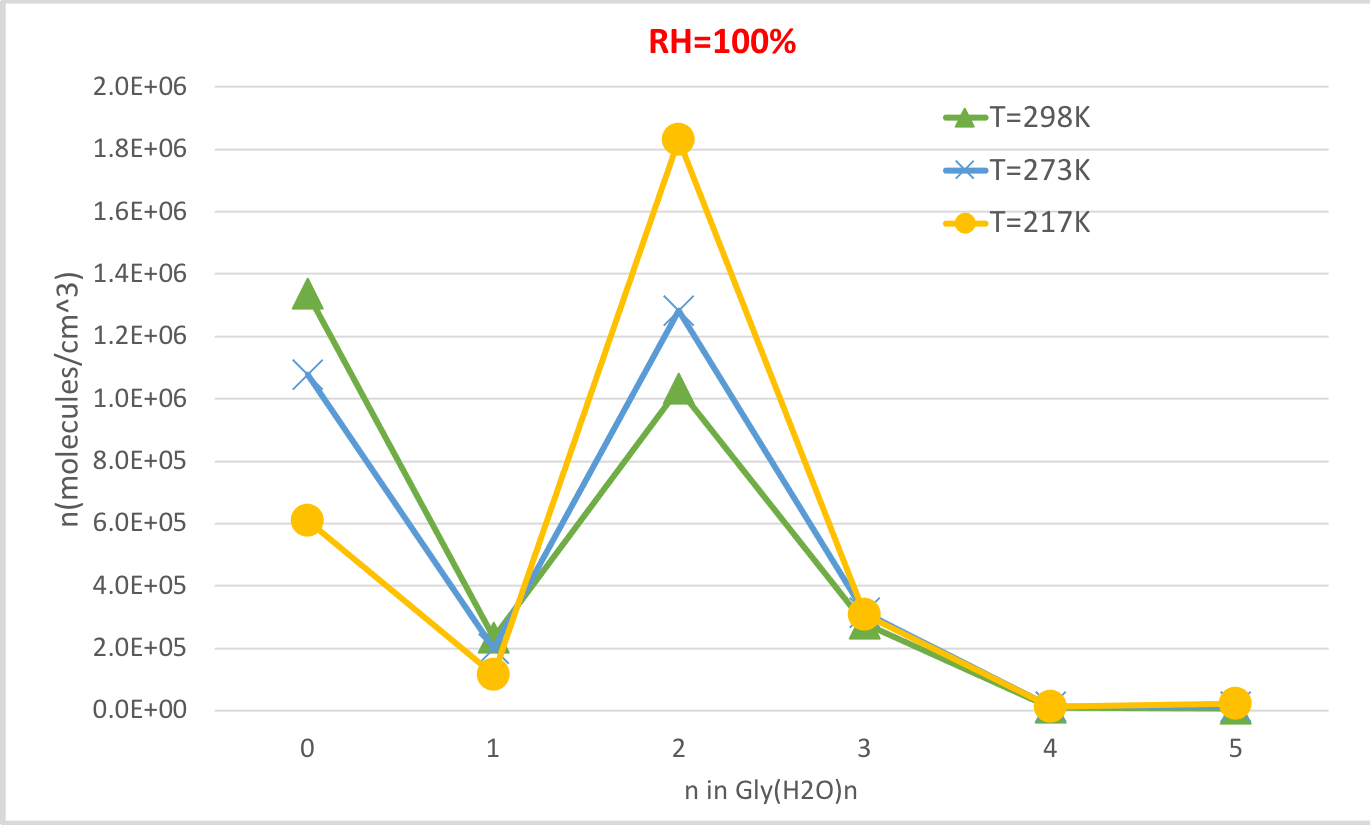
도 4는 100% 상대 습도 및 1 대기 압력에서 온도의 함수로서 표 4에서 계산된 수화 글리신의 평형 농도를 나타낸다. 온도가 298.15K에서 216.65K로 감소함에 따라 수화되지 않은 글리신 (n = 0)의 농도가 감소하고 수화 된 글리신의 농도가 증가한다는 것을 보여줍니다. 글리신 이수산염(n=2)은 특히 온도 감소와 함께 극적으로 증가하는 반면 다른 수화물의 농도 변화는 눈에 띄지 않습니다. 온도와 수화물 농도 사이의 이러한 역 상관 관계는 낮은 온도에서 수화물의 낮은 깁스 자유 에너지가 수화물의 형성을 선호한다는 기대와 일치합니다.
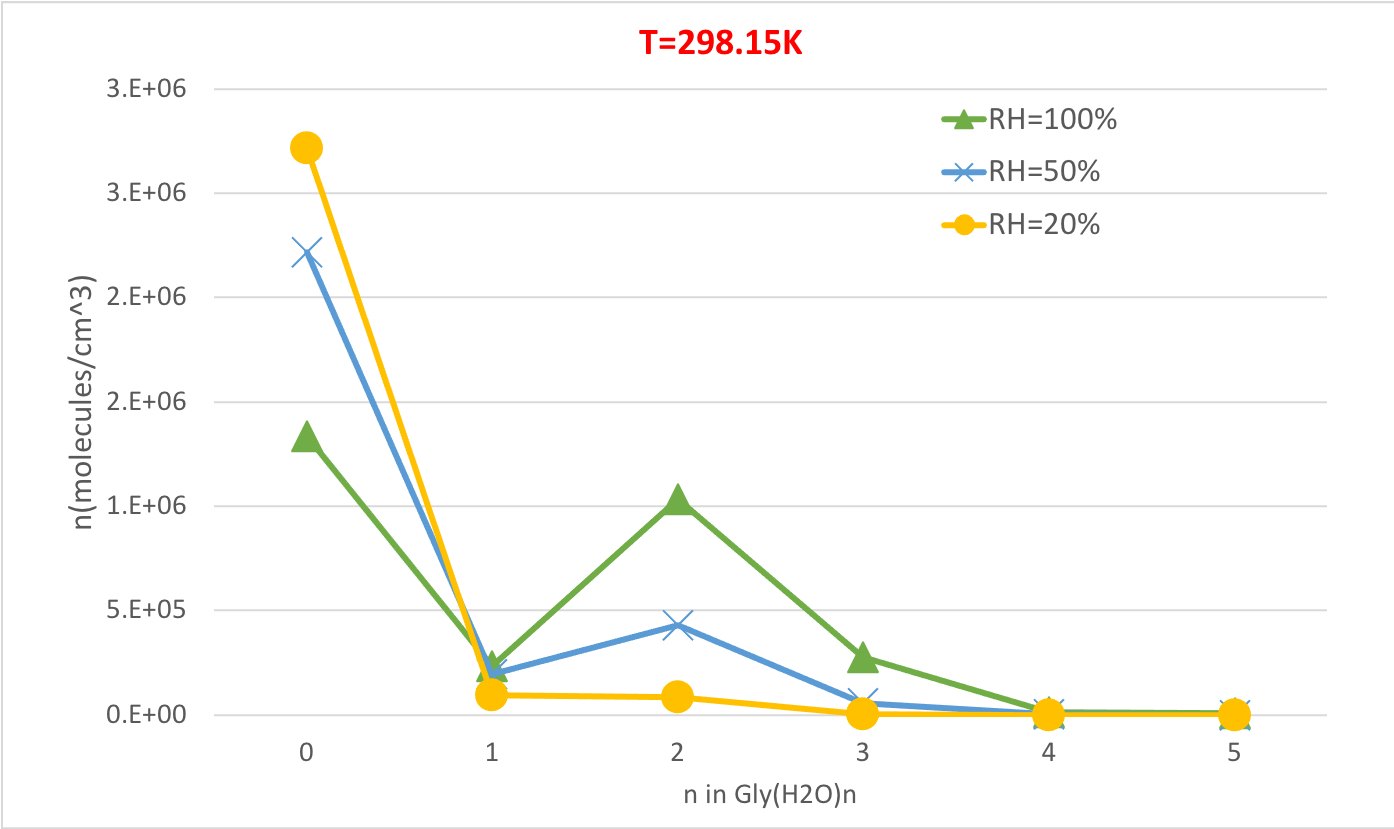
도 5는 298.15K 및 1 대기 압력에서 글리신 수화물의 평형 농도의 상대 습도 의존성을 나타낸 것이다. RH가 20%에서 100%로 증가함에 따라 수분 공급량(n>0)의 농도가 무수화 글리신(n=0)을 희생하여 증가한다는 것을 분명히 보여줍니다. 다시 한번 상대 습도와 수화물의 농도 사이의 직접적인 상관 관계는 더 높은 RH에서 더 많은 물 분자의 존재가 수화물의 형성을 촉진한다는 생각과 일치합니다.
제시된 바와 같이, 이 프로토콜은 대기 중의 수화 글리신 집단에 대한 질적 이해를 제공한다. 입방 센티미터당 290만 분자의 분리된 글리신의 초기 농도를 가정할 때, 우리는 수화되지 않은 글리신(n=0)이 T=216.65K 및 RH=100%를 제외한 대부분의 조건에서 가장 풍부한 종이라는 것을 알 수 있습니다. 세 가지 온도에서 가장 낮은 순차적 깁스 자유 에너지를 가진 이소화산염(n=2)은 여기서 고려된 조건에서 가장 풍부한 수화물입니다. 모노하이드레이트(n=1)와 더 큰 수화물(n≥3)은 무시할 수 있는 양으로 발견될 것으로 예측됩니다. 도 3을검사하면, n=1-4 클러스터의 풍부성은 클러스터의 수소 결합 네트워크의 안정성 및 변형과 관련될 수 있다. 이 클러스터는 다양한 수소 결합 링 구조의 것과 매우 유사한 기하학적 구조에서 글리신의 카르복실산 모티에 결합된 수소 분자를 가지고 있어 특히 안정적입니다.

그림 1: 현재 절차에 대한 회로도 설명입니다. 유전자 알고리즘(GA)에 의해 생성된 대규모 추측 구조 풀은 일련의 PW91 지오메트리 최적화에 의해 수렴된 구조 집합이 얻어질 때까지 정제됩니다. 이러한 구조의 진동 주파수는 계산되어 Gibbs 의 형성 자유 에너지를 계산하는 데 사용되며, 이는 주변 조건하에서 클러스터의 평형 농도를 계산하는 데 사용됩니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 2: 각 클러스터에 대한 대표 디렉터리 구조입니다. 이 프로토콜에 포함된 사내 스크립트에는 위에 표시된 디렉토리 구조가 필요하며, 여기서 n은 물 분자의 수입니다. 글리-h2o-n의각 n에는 다음과 같은 하위 디렉토리가 있습니다: GA/pm7 디렉토리가 있는 유전자 알고리즘용 GA, PW91/6-31+G*, PW91/6-311++G**의 QM/pw91-sb, QM/pw91+G**의 QM/pw91-lb 및 초미세 통합 그리드에 대한 최적화 및 최종 진동 계산을 위한 QM/pw91-lb/초미세 양자 역학용 QM. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 3: 글리(H2 O)2n=0-5의대표적인 저에너지 구조. 이 클러스터는 PW91/6-311++G** 이론 수준에서 최적화된 전자 에너지 글로벌 미니마였습니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 4: 글리(H22O)n=0-5의 온도 의존도는 100% 상대 습도 및 1 기압입니다. 수화물의 농도는 분자 cm-3의단위로 주어진다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 5: 글리(H2 O)2n=0-5의 상대 습도 의존도는 298.15 K 및 1 기압입니다. 수화물의 농도는 분자 cm-3의단위로 주어진다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
| E[PW91/6-311++G**] | 216.65 K | 273.15 K | 298.15 K | ||||||||
| LB-UF | ZPVE | □H | S | □G | □H | S | □G | □H | S | □G | |
| 물 | -76.430500 | 13.04 | 1.72 | 42.59 | 5.54 | 2.17 | 44.44 | 3.08 | 2.37 | 45.14 | 1.96 |
| 글리신 | -284.434838 | 48.55 | 2.65 | 69.53 | 36.14 | 3.70 | 73.81 | 32.09 | 4.22 | 75.61 | 30.22 |
표 1: 단량체 에너지. 전자 에너지는 Hartree 단위로, 다른 모든 수량은 kcal mol-1의 단위에 있습니다. 물과 글리신은 PW91/6-311++G** 이론 수준에서 최적화되었고 진동 주파수가 계산되었습니다. 1 atm의 압력과 298.15 K의 온도에 대한 열역학 적 보정은 thermo.pl 스크립트를 사용하여 계산되었습니다.
| E[PW91/6-311++G**] | 0 K | 216.65 K | 273.15 K | 298.15 K | ||||||||
| N | 이름 | LB-UF | ZPVE | □H | S | □G | □H | S | □G | □H | S | □G |
| 1 | 글리 h2o-1 | -360.88481 | 63.96 | 3.61 | 80.12 | 50.22 | 5.12 | 86.27 | 45.52 | 5.85 | 88.83 | 43.33 |
| 2 | 글리 h2o-2 | -437.33763 | 79.33 | 4.53 | 90.86 | 64.17 | 6.46 | 98.78 | 58.81 | 7.40 | 102.06 | 56.30 |
| 3 | 글리 h2o-3 | -513.78620 | 94.52 | 5.67 | 105.08 | 77.42 | 8.08 | 114.94 | 71.19 | 9.23 | 119.00 | 68.27 |
| 4 | 글리 h2o-4 | -590.23667 | 109.80 | 6.03 | 104.98 | 91.30 | 8.78 | 116.21 | 84.40 | 10.11 | 120.87 | 81.14 |
| 5 | 글리 h2o-5 | -666.68845 | 125.80 | 7.26 | 121.70 | 106.69 | 10.47 | 134.83 | 99.44 | 12.01 | 140.24 | 96.00 |
표 2: 클러스터 에너지. 그림 1에설명된 절차를 사용하여 발견된 최저 에너지 Gly(H2O)n=1-5 구조의 에너지. 전자 에너지는 Hartree 단위로, 다른 모든 수량은 kcal mol-1의 단위에 있습니다.
| 총 수분 공급: 글리 + nH2O & > 글리(H2O)n | 순차 적 수분 공급 : 글리 (H2O)n-1 + H2O & > 글리 (H2O)n | ||||||||||||||||
| E[PW91/6-311++G**] | 216.65 | 273.15 | 298.15 | 216.65 | 273.15 | 298.15 | |||||||||||
| N | 시스템 이름 | LB-UF | □E(0) | □H(T) | □G(T) | □H(T) | □G(T) | □H(T) | □G(T) | LB-UF | □E(0) | □H(T) | □G(T) | H(T) | □G(T) | □H(T) | □G(T) |
| 1 | 글리 h2o-1 | -12.22 | -9.85 | -10.61 | -3.68 | -10.61 | -1.87 | -10.59 | -1.07 | -12.22 | -9.85 | -10.61 | -3.68 | -10.61 | -1.87 | -10.59 | -1.07 |
| 2 | 글리 h2o-2 | -26.22 | -21.53 | -23.10 | -9.27 | -23.11 | -5.66 | -23.09 | -4.06 | -14.00 | -11.68 | -12.49 | -5.59 | -12.50 | -3.79 | -12.50 | -2.99 |
| 3 | 글리 h2o-3 | -37.56 | -30.72 | -32.88 | -12.90 | -32.87 | -7.69 | -32.82 | -5.38 | -11.34 | -9.19 | -9.78 | -3.63 | -9.76 | -2.03 | -9.73 | -1.32 |
| 4 | 글리 h2o-4 | -50.10 | -40.34 | -43.48 | -15.87 | -43.54 | -8.71 | -43.51 | -5.55 | -12.54 | -9.62 | -10.60 | -2.97 | -10.67 | -1.02 | -10.69 | -0.17 |
| 5 | 글리 h2o-5 | -63.45 | -51.41 | -55.42 | -20.58 | -55.51 | -11.48 | -55.48 | -7.45 | -13.35 | -11.07 | -11.94 | -4.71 | -11.97 | -2.77 | -11.97 | -1.90 |
표 3: 수화 에너지. Gcal mol-1단위로 글리(H2O)n=1-5에 대한 순차적 수화의 수화 및 에너지의 총 에너지. 여기서, E[PW91/6-311++G**]는 전자 에너지의 변화이며, □E(0)는 제로포인트 진동 에너지(ZPVE)가 에너지의 변화를 보정하고, □H(T)는 온도 T에서 엔탈피 변화이고, □G(T)는 각 글리(Gly)의 수화의 깁스 자유 에너지 변화(H2)의 에너지 변화이다.2n=1-5
| 온도 및 상대 습도의 함수로 평형 수화물 분포 | |||||||||
| T = 298.15K | T = 273.15K | T = 216.65K | |||||||
| 글리(H2O)n | RH = 100 % | RH=50% | RH =20% | RH = 100 % | RH=50% | RH =20% | RH = 100 % | RH=50% | RH =20% |
| 0 | 1.3E+06 | 2.2E+06 | 2.7E+06 | 1.1E+06 | 2.0E+06 | 2.7E+06 | 6.1E+05 | 1.5E+06 | 2.5E+06 |
| 1 | 2.3E+05 | 1.9E+05 | 9.5E+04 | 2.0E+05 | 1.9E+05 | 9.9E+04 | 1.2E+05 | 1.5E+05 | 9.5E+04 |
| 2 | 1.0E+06 | 4.3E+05 | 8.4E+04 | 1.3E+06 | 6.1E+05 | 1.3E+05 | 1.8E+06 | 1.1E+06 | 3.0E+05 |
| 3 | 2.8E+05 | 5.8E+04 | 4.5E+03 | 3.2E+05 | 7.4E+04 | 6.3E+03 | 3.1E+05 | 9.6E+04 | 1.0E+04 |
| 4 | 1.1E+04 | 1.1E+03 | 3.4E+01 | 1.3E+04 | 1.5E+03 | 5.0E+01 | 1.1E+04 | 1.8E+03 | 7.5E+01 |
| 5 | 7.5E+03 | 3.9E+02 | 4.9E+00 | 1.2E+04 | 7.2E+02 | 9.7E+00 | 2.4E+04 | 1.9E+03 | 3.1E+01 |
표 4: 글리(H2O)n=0-5의 평형 수화물 농도를 함수 온도(T=298.15K, 273.15K, 216.65K) 및 상대 습도(RH=100%, 50%, 20%). 수화물의 농도는 분자의 단위로 주어진다cm -3 실험 값56,,57,,58, [글리]0 = 2.9 x 106 cm-3 및 [H2O] = 7.7 x 1017 cm-3,1.6 x10 17 cm-3 및 9.9 x 1014 cm-3 100% T00% T00. 273.15 K, 및 216.65 K, 각각59.
추가 파일. 이 파일을 다운로드하려면 여기를 클릭하십시오.
토론
이 프로토콜에 의해 생성된 데이터의 정확도는 주로 세 가지에 따라 달라집니다: (i) 단계 2에 의해 샘플링 된 구성의 다양성, (ii) 시스템의 전자 구조의 정확성, (iii) 열역학 적 교정의 정확성. 포함된 스크립트를 편집하여 메서드를 수정하여 이러한 각 요소를 해결할 수 있습니다. 첫 번째 요소는 임의로 생성된 구조의 더 큰 초기 풀, GA의 더 많은 반복 및 GA에 관련된 기준의 느슨한 정의를 사용하여 쉽게 극복할 수 있습니다. 또한, 상이한 물리적 설명의 효과를 탐구하기 위해 자기 일관된 전하 밀도-기능적 타이트 바인딩(SCC-DFTB)모델 및 유효 단편 전위(EFP)63 모델과 같은 상이한 반경험적 방법을 사용할 수 있다. 여기서 주요 제한 사항은 공유 결합을 형성하거나 끊을 수있는 방법의 무능력이며, 이는 단량체가 동결된다는 것을 의미합니다. GA 절차는 반 경험적 설명에 따라 이러한 냉동 단량체의 가장 안정적인 상대 적 위치를 찾습니다.
시스템의 전자 구조의 정확도는 다양한 방법으로 향상 될 수있다, 그 계산 비용 각각. 하나는 M06-2X64 및 wB97X-V65,또는 이러한 Møller-Plesset66,,67,,68(MPn) 섭동 이론 및 결합 클러스터69(CC) 방법과 같은 더 나은 밀도 기능을 선택할 수 있다.68 기능 계층구조에서는 PW91과 같은 일반화된 그라데이션 근사치(GGA) 기능에서 wB97X-D 및 M06-2X와 같은 메타-GGA 하이브리드 기능과 같은 범위 분리하이브리드 기능으로 전환시 성능이 향상됩니다.
DFT 방법의 단점은 정확한 값을 향한 체계적인 수렴이 불가능하다는 것입니다. 그러나 DFT 방법은 계산 비용이 저렴하며 다양한 응용 프로그램에 대한 다양한 기능이 있습니다.
MP2 및 CCSD(T)와 같은 파형 함수 방법을 사용하여 계산된 에너지는 증가하는 추기경 수의 상관 관계 일관된 기준 집합과 함께([8-]cc-pV[D,T,Q,...] Z) 전체 기준 설정 한도를 체계적으로 수렴하지만 시스템 크기가 증가함에 따라 각 계산의 계산 비용이 엄중해집니다. 전자 구조의 추가 개선은 명시적으로 상관된 기준세트(70)를 사용하고 전체 기초 세트(CBS)한계로 추정함으로써 달성될 수 있다. 우리의 최근 연구는 밀도 장착 명시적으로 상관 2 차 Møller-Plesset (DF-MP2-F12) 교란 접근 MP2 / CBS계산32에접근 에너지를 산출 제안한다. 다른 전자 구조 방법을 사용하는 현재 프로토콜의 수정에는 두 단계가 포함됩니다: (i) 소프트웨어에 의해 주어진 구문에 따라 템플릿 입력 파일을 준비하고, (ii) run-pw91-sb.csh, run-pw91-lb.csh,및 run-pw91-lb-ultrafine.csh 스크립트를 편집하여 올바른 입력 파일 구문뿐만 아니라 올바른 스크립트를 생성합니다.
마지막으로, 열역학 적 교정의 정확도는 전자 구조 방법뿐만 아니라 전 세계적으로 최소 한도에 대한 PES의 설명에 따라 달라집니다. PES에 대한 정확한 설명은 매우 비용이 많이 드는 작업인 분력 필드72,,73(QFF)과 같은 핵 자유도의 변위와 관련하여 PES의 3차 및 고차 유도체의 계산을 요구합니다. 현재 프로토콜은 진동 주파수에 대한 고조파 발진기 근사치를 사용하므로 PES의 최대 두 번째 유도체만 계산해야 합니다. 이 접근법은 실제 PES및 고조파 PES의 큰 차이로 인해 매우 플로피 분자 및 대칭 이중 웰 전위와 같은 높은 조화성을 가진 시스템에서 문제가됩니다. 더욱이, 계산적으로 까다로운 전자 구조 방법으로부터 고품질 PES를 갖는 비용은 진동 주파수 계산에 대한 비용 의 문제를 복합화할 뿐이다. 이를 극복하기 위한 한 가지 방법은 고품질 의 전자 구조 계산과 낮은 품질의 PES에서 계산된 진동 주파수를 사용하여 비용과 정확도 간의 균형을 맞추는 것입니다. 현재 프로토콜은 이전 단락에 설명된 바와 같이 상이한 PES 설명을 사용하도록 수정될 수 있다; 그러나 스크립트 및 템플릿의 진동 주파수 키워드를 편집하여 조화진동 주파수를 계산할 수도 있습니다.
모든 구성 샘플링 프로토콜에 대한 두 가지 중요한 문제는 잠재적인 에너지 표면을 샘플링하는 초기 방법과 각 클러스터를 식별하는 데 사용되는 기준입니다. 우리는 전작에서 다양한 방법을 광범위하게 사용했습니다. 첫 번째 문제, 잠재적 인 에너지 표면을 샘플링하기위한 초기 방법, 우리는 이러한 요인에 따라 반 경험적 방법으로 GA를 사용하는 선택을했다. 화학적 직관26,무작위 샘플링 및 분자 역학(MD)29,,30을이용한 구성 샘플링은 물 클러스터18에대한 우리의 연구에서 관찰한 바와 같이 10단량 이상의 클러스터에 대해 정기적으로 가시적 글로벌 미니마를 찾지 못한다. 우리는 성공적으로(H2O)1174의복잡한 PES를 연구하기 위해 분지 호핑 (BH)을 사용했지만, BH 알고리즘이 찾지 못한 잠재적 인 저에너지 이섬의 수동 포함이 필요했습니다. 물 클러스터의 글로벌 최소 를 찾는 BH 및 GA의 성능의 비교,(H2O)n=10-20 GA가 지속적으로 BH75보다더 빠른 글로벌 최소를 발견것을 입증했다. OGOLEM 및 CLUSTER에서 구현된 GA는 모든 분자 클러스터에 적용할 수 있으며 클래식포스 필드, 반경험적, 밀도 기능성 및 ab initio 기능을 갖춘 방대한 수의 패키지와 인터페이스할 수 있기 때문에 매우 다재다능합니다. PM7의 선택은 속도와 합리적인 정확도에 의해 구동됩니다. 사실상 다른 모든 반 경험적 방법은 계산 비용이 상당히 높을 것입니다.
두 번째 문제에 관해서는, 우리는 전자 에너지, 다이폴 모멘트, 중첩 RMSD 및 회전 상수에 이르기까지 독특한 구조를 식별하기 위해 다른 기준을 사용하여 탐구했다. 다이폴 모멘트 를 사용하는 것은 두 다이폴 모멘트 구성 요소가 분자의 배향에 의존하고 총 다이폴 모멘트는 구조가 동일하거나 고유하다고 결정하는 임계값을 설정하기 어려웠던 방식으로 기하학적 차이에 매우 민감했기 때문에 어려웠습니다. 전자 에너지와 회전 상수의 조합이 가장 유용합니다.
두 구조물을 고유하게 간주하기 위한 현재 기준은 0.10 kcal mol-1의 에너지 차이 임계값과 1%의 회전 상수 차이를 기반으로 합니다. 따라서 에너지가 0.10 kcal mol-1 (~0.00015 a.u.) 이상 차이가 있는 경우 두 구조가 다른 것으로 간주됩니다. 그리고 그들의 세 회전 상수 (A, B, C)는 1 % 이상 다릅니다. 수년에 걸쳐 상당한 내부 벤치 마크는 이러한 임계 값이 합리적인 선택으로 나타났습니다. 당사의 구성 샘플링 접근법 및 스크리닝 방법론은 암모니아 및아민(32)을함유하는 강하게 결합된 삼차 황산염 수화물뿐만 아니라 물76,,77로 복잡한 다방향족 탄화수소와 같은 매우 약하게 결합된 클러스터에 적용되었다. 고려해야 할 다른 프로토네이션 상태가 있는 클러스터의 경우, 가장 좋은 방법은 다양한 PROTONATION 상태의 단량체로 시작하여 다양한 GA 계산을 실행하는 것입니다. 이렇게 하면 다른 프로토네이션 상태를 가진 구조물이 신중하게 고려됩니다. 그러나 낮은 수준의 DFT 계산을 통해 형상 최적화 과정에서 프로토네이션 상태가 변경될 수 있으며, 따라서 시작 형상에 관계없이 가장 안정적인 프로토네이션 상태를 생성할 수 있습니다.
GA 구성 샘플링 방법은 GA 코드가 GA 실행 과정에서 단량체가 다른 구성을 채택할 수 있도록 하는 일반적인 비매개 변수화 방법과 인터페이스되어 있는 한 플로피 분자에서도 잘 작동해야 합니다. 예를 들어, PM7과 GA를 인터페이싱하면 단량체의 구조가 변경될 수 있지만, 프로토네이션 상태가 변경될 때 와 같이 채권이 파손되면 구조물이 허용할 수 없는 후보로 버려질 수 있습니다.
우리는 고조파 근사치의 단점을 수정하는 다른 방법을 고려했다, 특히 낮은 진동 주파수에서 발생하는 사람들. 준 고조파 근사치를 현재 방법론에 통합하는 것은 어렵지 않습니다. 그러나, 준 고조파 방법에 대한 질문이 여전히있다, 그것은 적용 될 아래의 컷오프 주파수에 관해서 특히. 또한, 기존의 지혜가 RRHO 근사치에 비해 개선되어야한다고 제안하더라도 준 RRHO 근사치의 신뢰성을 검토하는 엄격한 벤치마킹 작업은 없습니다.
이렇게 제시된 프로토콜은 비협조적으로 결합된 가스 상 분자 클러스터의 임의의 시스템에 일반화될 수 있다. 또한 스크립트 및 템플릿을 편집함으로써 임의의 반경험적 방법, 전자 구조 방법 및 소프트웨어, 진동 분석 방법 및 소프트웨어를 사용하는 것이 일반화될 수 있다. 이는 사용자가 Linux 명령줄 인터페이스, Python 스크립팅 및 고성능 컴퓨팅에 익숙하다고 가정합니다. 익숙하지 않은 구문과 Linux 운영 체제의 모양과 스크립팅 경험 부족은 이 프로토콜의 가장 큰 함정이며 새로운 학생들이 가장 어려움을 겪는 곳입니다. 이 프로토콜은 주로 에어로졸 형성에 황산과 암모니아의 효과에 초점을 맞추고, 우리 그룹에서 년 동안 구현의 다양한 성공적으로 사용되어왔다. 이 프로토콜의 추가 개선은 더 많은 전자 구조 소프트웨어, 유전 알고리즘의 대체 구현 및 전자 및 진동 에너지의 빠른 계산을 위한 최신 방법의 사용에 대한 보다 강력한 인터페이스를 포함합니다. 이 프로토콜의 현재 응용 프로그램은 현재 대기에서 에어로졸 형성의 초기 단계와 프리바이오틱 환경에서 더 큰 생물학적 분자의 형성에서 아미노산의 중요성을 탐구하고 있습니다.
공개
없음.
감사의 말
이 프로젝트는 보조금 CHE-1229354, CHE-1662030, CHE-1721511, 그리고 CHE-1903871 국립 과학 재단에서 지원되었다 (GCS), 아놀드와 메이블 베크만 재단 베크만 장학생 상 (AGG), 배리 M. 골드 워터 장학금 (AGG). 머큐리 컨소시엄(http://www.mercuryconsortium.org)의 고성능 컴퓨팅 리소스가 사용되었습니다.
자료
| Name | Company | Catalog Number | Comments |
| Avogadro | https://avogadro.cc | Open-source molecular visualization program | |
| Gaussian [09/16] Software | http://www.gaussian.com/ | Commercial ab initio electronic structure program | |
| MOPAC 2016 | http://openmopac.net/MOPAC2016.html | Open-source semi-empirical program | |
| OGOLEM Software | https://www.ogolem.org | Genetic algorithm-based global optimization program | |
| OpenBabel | http://openbabel.org/wiki/Main_Page | Open-source cheminformatics library | |
| calcRotConsts.py | Shields Group, Department of Chemistry, Furman University | Python script to compute rotational constants | |
| calcSymmetry.csh | Shields Group, Department of Chemistry, Furman University | Shell script to calculate symmetry number of a molecule given Cartesian coordinates | |
| combine-GA.csh | Shields Group, Department of Chemistry, Furman University | Shell script to combine energy and rotational constants from different GA directories | |
| combine-QM.csh | Shields Group, Department of Chemistry, Furman University | Shell script to combine energy and rotational constants from different QM directories | |
| gaussianE.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract Gaussian 09 energies | |
| gaussianFreqs.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract Gaussian 09 vibrational frequencies | |
| getrotconsts | Shields Group, Department of Chemistry, Furman University | Executable to calculate rotational constants given a molecule's Cartesian coordinates | |
| getRotConsts-dft-lb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of large basis DFT optimized structures | |
| getRotConsts-dft-lb-ultrafine.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of ultrafine DFT optimized structures | |
| getRotConsts-dft-sb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of small basis DFT optimized structures | |
| getRotConsts-GA.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of genetic algorithm optimized structures | |
| global-minimum-coords.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of global minimum structures of gly-(h2o)n, where n=0-5 | |
| make-thermo-gaussian.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract data from Gaussian output files and make input files for the thermo.pl script | |
| ogolem-input-file.ogo | Shields Group, Department of Chemistry, Furman University | Ogolem sample input file | |
| ogolem-submit-script.pbs | Shields Group, Department of Chemistry, Furman University | PBS batch submission file for Ogolem calculations | |
| README.docx | Shields Group, Department of Chemistry, Furman University | Clarifications to help readers use the scripts effectively | |
| runogolem.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run OGOLEM | |
| run-pw91-lb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of large basis DFT optimization calculations | |
| run-pw91-lb-ultrafine.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of ultrafine DFT optimization calculations | |
| run-pw91-sb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of small basis DFT optimization calculations | |
| run-thermo-pw91.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute the thermodynamic corrections for a batch of DFT optimized structures | |
| similarityAnalysis.py | Shields Group, Department of Chemistry, Furman University | Python script to determine unique structures based on rotational constants and energies | |
| symmetry | Shields Group, Department of Chemistry, Furman University | Executable to calculate molecular symmetry given Cartesian coordinates | |
| symmetry.c | (C) 1996, 2003 S. Patchkovskii, Serguei.Patchkovskii@sympatico.ca | C code to determine the molecular symmstry of a molecule given Cartesian coordinates | |
| template-marcy.pbs | Shields Group, Department of Chemistry, Furman University | Template for a PBS submit script which uses OGOLEM | |
| template-pw91.com | Shields Group, Department of Chemistry, Furman University | Template Gaussian 09 input | |
| template-pw91-HL.com | Shields Group, Department of Chemistry, Furman University | Template Gaussian 09 input for ultrafine DFT optimization | |
| thermo.pl | https://www.nist.gov/mml/csd/chemical-informatics-research-group/products-and-services/program-computing-ideal-gas | Perl open-source script to compute ideal gas thermodynamic corrections | |
| gly-h2o-n.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet for the complete protocol | |
| table-1.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-2.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-3.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-4.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| water.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of water | |
| glycine.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of glycine |
참고문헌
- Foster, P., Ramaswamy, V., Solomon, S., Qin, D., Manning, M., Chen, Z., Marquis, M., Averyt, K. B., Tignor, M., Miller, H. L. . Climate Change 2007 The Scientific Basis. , (2007).
- Kulmala, M., et al. Toward direct measurement of atmospheric nucleation. Science. 318 (5847), 89-92 (2007).
- Sipila, M., et al. The role of sulfuric acid in atmospheric nucleation. Science. 327 (5970), 1243-1246 (2010).
- Jiang, J., et al. First measurement of neutral atmospheric cluster and 1 - 2 nm particle number size distributions during nucleation events. Aerosol Science and Technology. 45 (4), (2011).
- Dunn, M. E., Pokon, E. K., Shields, G. C. Thermodynamics of forming water clusters at various Temperatures and Pressures by Gaussian-2, Gaussian-3, Complete Basis Set-QB3, and Complete Basis Set-APNO model chemistries; implications for atmospheric chemistry. Journal of the American Chemical Society. 126 (8), 2647-2653 (2004).
- Pickard, F. C., Pokon, E. K., Liptak, M. D., Shields, G. C. Comparison of CBSQB3, CBSAPNO, G2, and G3 thermochemical predictions with experiment for formation of ionic clusters of hydronium and hydroxide ions complexed with water. Journal of Chemical Physics. 122, 024302 (2005).
- Pickard, F. C., Dunn, M. E., Shields, G. C. Comparison of Model Chemistry and Density Functional Theory Thermochemical Predictions with Experiment for Formation of Ionic Clusters of the Ammonium Cation Complexed with Water and Ammonia; Atmospheric Implications. Journal of Physical Chemistry A. 109 (22), 4905-4910 (2005).
- Alongi, K. S., Dibble, T. S., Shields, G. C., Kirschner, K. N. Exploration of the Potential Energy Surfaces, Prediction of Atmospheric Concentrations, and Vibrational Spectra of the HO2•••(H2O)n (n=1-2) Hydrogen Bonded Complexes. Journal of Physical Chemistry A. 110 (10), 3686-3691 (2006).
- Allodi, M. A., Dunn, M. E., Livada, J., Kirschner, K. N. Do Hydroxyl Radical-Water Clusters, OH(H2O)n, n=1-5, Exist in the Atmosphere. Journal of Physical Chemistry A. 110 (49), 13283-13289 (2006).
- Kirschner, K. N., Hartt, G. M., Evans, T. M., Shields, G. C. In Search of CS2(H2O)n=1-4 Clusters. Journal of Chemical Physics. 126, 154320 (2007).
- Hartt, G. M., Kirschner, K. N., Shields, G. C. Hydration of OCS with One to Four Water Molecules in Atmospheric and Laboratory Conditions. Journal of Physical Chemistry A. 112 (19), 4490-4495 (2008).
- Morrell, T. E., Shields, G. C. Atmospheric Implications for Formation of Clusters of Ammonium and 110 Water Molecules. Journal of Physical Chemistry A. 114 (12), 4266-4271 (2010).
- Temelso, B., et al. Quantum Mechanical Study of Sulfuric Acid Hydration: Atmospheric Implications. Journal of Physical Chemistry A. 116 (9), 2209 (2012).
- Husar, D. E., Temelso, B., Ashworth, A. L., Shields, G. C. Hydration of the Bisulfate Ion: Atmospheric Implications. Journal of Physical Chemistry A. 116 (21), 5151-5163 (2012).
- Bustos, D. J., Temelso, B., Shields, G. C. Hydration of the Sulfuric Acid – Methylamine Complex and Implications for Aerosol Formation. Journal of Physical Chemistry A. 118 (35), 7430-7441 (2014).
- Wales, D. J., Scheraga, H. A. Global optimization of clusters, crystals, and biomolecules. Science. 27 (5432), 1368-1372 (1999).
- Day, M. B., Kirschner, K. N., Shields, G. C. Global search for minimum energy (H2O)n clusters, n = 3 - 5. The Journal of Physical Chemistry A. 109 (30), 6773-6778 (2005).
- Shields, R. M., Temelso, B., Archer, K. A., Morrell, T. E., Shields, G. C. Accurate predictions of water cluster formation, (H2O)n=2-10. The Journal of Physical Chemistry A. 114 (43), 11725-11737 (2010).
- Temelso, B., Archer, K. A., Shields, G. C. Benchmark structures and binding energies of small water clusters with anharmonicity corrections. The Journal of Physical Chemistry A. 115 (43), 12034-12046 (2011).
- Temelso, B., Shields, G. C. The role of anharmonicity in hydrogen-bonded systems: The case of water clusters. The Journal of Chemical Theory and Computation. 7 (9), 2804-2817 (2011).
- Von Freyberg, B., Braun, W. Efficient search for all low energy conformations of polypeptides by Monte Carlo methods. The Journal of Computational Chemistry. 12 (9), 1065-1076 (1991).
- Rakshit, A., Yamaguchi, T., Asada, T., Bandyopadhyay, P. Understanding the structure and hydrogen bonding network of (H2O)32 and (H2O)33: An improved Monte Carlo temperature basin paving (MCTBP) method of quantum theory of atoms in molecules (QTAIM) analysis. RSC Advances. 7 (30), 18401-18417 (2017).
- Deaven, D. M., Ho, K. M. Molecular geometry optimization with a genetic algorithm. Physical Review Letters. 75, 288-291 (1995).
- Hartke, B. Application of evolutionary algorithms to global cluster geometry optimization. Applications of Evolutionary Computation in Chemistry. , (2004).
- Dieterich, J. M., Hartke, B. OGOLEM: Global cluster structure optimization for arbitrary mixtures of flexible molecules. A multiscaling, object-oriented approach. Molecular Physics. 108 (3-4), 279-291 (2010).
- Herb, J., Nadykto, A. B., Yu, F. Large ternary hydrogen-bonded pre-nucleation clusters in the Earth's atmosphere. Chemical Physics Letters. 518, 7-14 (2011).
- Ortega, I. K., et al. From quantum chemical formation free energies to evaporation rates. Atmospheric Chemistry and Physics. 12 (1), 225-235 (2012).
- Elm, J., Bilde, M., Mikkelsen, K. V. Influence of Nucleation Precursors on the Reaction Kinetics of Methanol with the OH Radical. Journal of Physical Chemistry A. 117 (30), 6695-6701 (2013).
- Loukonen, V., et al. Enhancing effect of dimethylamine in sulfuric acid nucleation in the presence of water - a computational study. Atmospheric Chemistry and Physics. 10 (10), 4961-4974 (2010).
- Temelso, B., Phan, T. N., Shields, G. C. Computational study of the hydration of sulfuric acid dimers: implications for acid dissociation and aerosol formation. Journal of Physical Chemistry A. 116 (39), 9745-9758 (2012).
- Jiang, S., et al. Study of Cl-(H2O)n (n = 1-4) using basin-hopping method coupled with density functional theory. Journal of Computational Chemistry. 35 (2), 159-165 (2014).
- Temelso, B., et al. Effect of mixing ammonia and alkylamines on sulfate aerosol formation. Journal of Physical Chemistry A. 122 (6), 1612-1622 (2018).
- Perdew, J. P., Ruzsinszky, A., Tao, J. Prescription for the design and selection of density functional approximations: More constraint satisfaction with fewer fits. Journal of Chemical Physics. 123, 062201 (2005).
- Riplinger, C., Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. Journal of Chemical Physics. 138, 034106 (2013).
- Riplinger, C., Pinski, P., Becker, U., Valeev, E. F., Neese, F. Sparse maps--A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory. Journal of Chemical Physics. 144 (2), 024109 (2016).
- Kildgaard, J. V., Mikkelsen, K. V., Bilde, M., Elm, J. Hydration of atmospheric molecular clusters: a new method for systematic configurational sampling. Journal of Physical Chemistry A. 122 (22), 5026-5036 (2018).
- González, &. #. 1. 9. 3. ;. Measurement of areas on a sphere Using Fibonacci and latitude-longitude lattices. Mathematical Geosciences. 42, 49-64 (2010).
- Karaboga, D., Basturk, B. On the performance of artificial bee colony (ABC) algorithm. Applied Soft Computing. 8 (1), 687-697 (2008).
- Zhang, J., Doig, M. Global optimization of rigid molecules using the artificial bee colony algorithm. Physical Chemistry Chemical Physics. 18 (4), 3003-3010 (2016).
- Kubecka, J., Besel, V., Kurten, T., Myllys, N., Vehkamaki, H. Configurational sampling of noncovalent (atmospheric) molecular clusters: sulfuric acid and guanidine. Journal of Physical Chemistry A. 123 (28), 6022-6033 (2019).
- Grimme, S., Bannwarth, C., Shushkov, P. A Robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent Interactions of large molecular systems parametrized for all spd-block elements (Z = 1-86). Journal of Chemical Theory and Computation. 13 (5), 1989-2009 (2017).
- Buck, U., Pradzynski, C. C., Zeuch, T., Dieterich, J. M., Hartke, B. A size resolved investigation of large water clusters. Physical Chemistry Chemical Physics. 16 (15), 6859 (2014).
- Forck, R. M., et al. Structural diversity in sodium doped water trimers. Physical Chemistry Chemical Physics. 14 (25), 9054-9057 (2012).
- Witt, C., Dieterich, J. M., Hartke, B. Cluster structures influenced by interaction with a surface. Physical Chemistry Chemical Physics. 20 (23), 15661-15670 (2018).
- Freitbert, A., Dieterich, J. M., Hartke, B. Exploring self-organization of molecular tether molecules on a gold surface by global structure optimization. The Journal of Computational Chemistry. 40 (22), 1978-1989 (2019).
- Stewart, J. J. P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. The Journal of Molecular Modeling. 19 (1), 1-32 (2013).
- Burke, K., Perdew, J. P., Wang, Y. Derivation of a generalized gradient approximation: The PW91 density functional. Electronic Density Functional Theory. , 81-111 (1998).
- Frisch, M. J., et al. . Gaussian 09, Revision A.02. , (2016).
- Ditchfield, R., Hehre, W. J., Pople, J. A. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. The Journal of Chemical Physics. 54 (2), 724 (1971).
- Elm, J., Bilde, M., Mikkelsen, K. V. Assessment of density functional theory in predicting structures and free energies of reaction of atmospheric prenucleation clusters. The Journal of Chemical Theory and Computation. 8 (6), 2071-2077 (2012).
- Elm, J., Mikkelsen, K. V. Computational approaches for efficiently modelling of small atmospheric clusters. Chemical Physics Letters. 615, 26-29 (2014).
- Bayucan, A., et al. . PBS Portable Batch System. , (1999).
- O'Boyle, N. M., et al. Open Babel: An open chemical toolbox. Journal of Cheminformatics. 3, 33 (2011).
- Csaszar, A. G. Conformers of gaseous glycine. Journal of the American Chemical Society. 114 (24), 9568-9575 (1992).
- Zhang, Q., Anastasio, C. Free and combined amino compounds in atmospheric fine particles (PM2.5) and fog waters from Northern California. Atmospheric Environment. 37 (16), 2247-2258 (2003).
- Matsumoto, K., Uematsu, M. Free amino acids in marine aerosols over the western North Pacific Ocean. Atmospheric Environment. 39 (11), 2163-2170 (2005).
- Mandalakis, M., Apostolaki, M., Stephanou, E. G. Trace analysis of free and combined amino acids in atmospheric aerosols by gas chromatography-mass spectrometry. Journal of Chromatography A. 1217 (1), 143-150 (2010).
- Seinfeld, J. H., Pandis, S. N. . Atmospheric Chemistry and Physics, 3rd Ed. , (2016).
- Myllys, N., Elm, J., Halonen, R., Kurten, T., Vehkamaki, H. Coupled cluster evaluation of atmospheric acid-base clusters with up to 10 molecules. The Journal of Physical Chemistry A. 120 (4), 621-630 (2016).
- Elm, J., Bilde, M., Mikkelsen, K. V. Assessment of binding energies of atmospherically relevant clusters. Physical Chemistry Chemical Physics. 15 (39), (2013).
- Elstner, M. The SCC-DFTB method and its application to biological systems. Theoretical Chemistry Accounts. 116 (1-3), 316-325 (2006).
- Kaliman, I. A., Slipchenko, L. V. LIBEFP: A new parallel implementation of the effective fragment potential method as a portable software library. The Journal of Computational Chemistry. 34 (26), 2284-2292 (2013).
- Zhao, Y., Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and trasition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theoretical Chemistry Accounts. 120 (1-3), 215-241 (2008).
- Mardirossian, N., Head-Gordon, M. wB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy. Physical Chemistry Chemical Physics. 16 (21), 9904-9924 (2014).
- Head-Gordon, M., Pople, J. A., Frisch, M. J. MP2 energy evaluation by direct methods. Chemical Physics Letters. 153 (6), 503-506 (1988).
- Pople, J. A., Seeger, R., Krishnan, R. Variational configuration interaction methods and comparison with perturbation theory. The International Journal of Quantum Chemistry. 12, 149-163 (1977).
- Pople, J. A., Binkley, J. S., Seeger, R. Theoretical models incorporating electron correlation. The International Journal of Quantum Chemistry. 10 (10), 1-19 (1976).
- Monkhorst, H. J. Calculation of properties with the coupled-cluster method. The International Journal of Quantum Chemistry. 12 (11), 421-432 (1977).
- Klopper, W., Manby, F. R., Ten-No, S., Valeev, E. F. R12 methods in explicitly correlated molecular electronic structure theory. International Reviews in Physical Chemistry. 25, 427-468 (2006).
- Hattig, C. Optimization of auxiliary basis sets for RI-MP2 and RI-CC2 calculations: Core-valence and quintuple-z basis sets for H to Ar and QZVPP basis sets for Li to Kr. Physical Chemistry Chemical Physics. 7 (1), 59-66 (2005).
- Barone, V. Anharmonic vibrational properties by a fully automated second-order perturbative approach. The Journal of Chemical Physics. 122, 014108 (2005).
- Barone, V. Vibrational zero-point energies and thermodynamic functions beyond the harmonic approximation. The Journal of Chemical Physics. 120 (7), 3059-3065 (2004).
- Temelso, B., et al. Exploring the Rich Potential Energy Surface of (H2O)11 and Its Physical Implications. Journal of Chemical Theory and Computation. 14 (2), 1141-1153 (2018).
- Kabrede, H., Hentschke, R. Global minima of water clusters (H2O)N, N≤25, described by three empirical potentials. Journal of Physical Chemistry B. 107 (16), (2003).
- Steber, A. L., et al. Capturing the Elusive Water Trimer from the Stepwise Growth of Water on the Surface of a Polycyclic Aromatic Hydrocarbon Acenaphthene. Journal of Physical Chemistry Letters. 8 (23), 5744-5750 (2017).
- Perez, C., et al. Corrannulene and its complex with water: A tiny cup of water. Physical Chemistry Chemical Physics. 19 (22), 14214-14223 (2017).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유