
Method Article
Tinción e imágenes de alta resolución de modelos organoides y esferoides tridimensionales
* Estos autores han contribuido por igual
En este artículo
Resumen
Aquí, proporcionamos protocolos detallados, robustos y complementarios para realizar imágenes de tinción y resolución subcelular de modelos de cultivo celular tridimensionales fijos que van desde 100 μm hasta varios milímetros, lo que permite la visualización de su morfología, composición de tipo celular e interacciones.
Resumen
Los modelos de cultivo celular tridimensional (3D) in vitro, como organoides y esferoides, son herramientas valiosas para muchas aplicaciones, incluido el desarrollo y el modelado de enfermedades, el descubrimiento de fármacos y la medicina regenerativa. Para explotar plenamente estos modelos, es crucial estudiarlos a nivel celular y subcelular. Sin embargo, caracterizar tales modelos de cultivo celular 3D in vitro puede ser técnicamente desafiante y requiere experiencia específica para realizar análisis efectivos. Aquí, este documento proporciona protocolos detallados, robustos y complementarios para realizar imágenes de tinción y resolución subcelular de modelos de cultivo celular 3D in vitro fijos que van desde 100 μm hasta varios milímetros. Estos protocolos son aplicables a una amplia variedad de organoides y esferoides que difieren en su célula de origen, morfología y condiciones de cultivo. Desde la recolección de estructuras 3D hasta el análisis de imágenes, estos protocolos se pueden completar en 4-5 días. Brevemente, las estructuras 3D se recolectan, fijan y luego pueden procesarse mediante incrustación de parafina y tinción histológica / inmunohistoquímica, o directamente inmunomarcadas y preparadas para la limpieza óptica y la reconstrucción 3D (200 μm de profundidad) mediante microscopía confocal.
Introducción
En las últimas décadas, los avances en la biología de células madre y las tecnologías de cultivo 3D in vitro han anunciado una revolución en biología y medicina. Los modelos celulares de mayor complejidad en 3D se han vuelto muy populares, ya que permiten que las células crezcan e interactúen con un marco extracelular circundante, recapitulando estrechamente aspectos de los tejidos vivos, incluida su arquitectura, organización celular e interacciones, o incluso características de difusión. Como tal, los modelos de cultivo celular 3D pueden proporcionar información única sobre el comportamiento de las células en tejidos en desarrollo o enfermos in vitro. Los organoides y los esferoides son estructuras 3D multicelulares, que van desde varios micrómetros hasta milímetros, y son las estructuras 3D in vitro más prominentes. Ambos pueden cultivarse dentro de un andamio de soporte que incluye (i) hidrogeles derivados de animales (extracto de membrana basal, colágeno), plantas (alginato / agarosa) o sintetizados a partir de productos químicos, o (ii) matrices inertes que contienen poros para promover la proliferación y el crecimiento celular.
Los organoides y esferoides también pueden desarrollarse sin la presencia de un andamio de soporte al depender de las células para autoensamblarse en grupos. Esto se basa en diferentes técnicas, como el uso de materiales no adhesivos para inhibir la unión celular, la tensión superficial y la fuerza gravitacional (por ejemplo, técnicas de caída colgante) o la rotación circular constante de los vasos (por ejemplo, cultivo de hilanderas). En todos los casos, estas técnicas facilitan las interacciones célula-célula y célula-matriz para superar las limitaciones del cultivo celular monocapa tradicional1. Los términos "organoides" y "esferoides" se han utilizado indistintamente en el pasado, pero existen diferencias clave entre estos dos modelos de cultivo celular 3D. Los organoides son grupos celulares 3D in vitro derivados de células madre pluripotentes o células madre específicas de tejido, en los que las células se autoorganizan espontáneamente en progenitoras y tipos de células diferenciadas y que recapitulan al menos algunas funciones del órgano de interés2. Los esferoides comprenden una gama más amplia de estructuras 3D multicelulares formadas bajo condiciones no adherentes y pueden surgir de una gran diversidad de tipos de células, como líneas celulares inmortalizadas o células primarias3. Por lo tanto, inherentes a sus orígenes intrínsecos de células madre, los organoides tienen una mayor propensión al autoensamblaje, la viabilidad y la estabilidad que los esferoides.
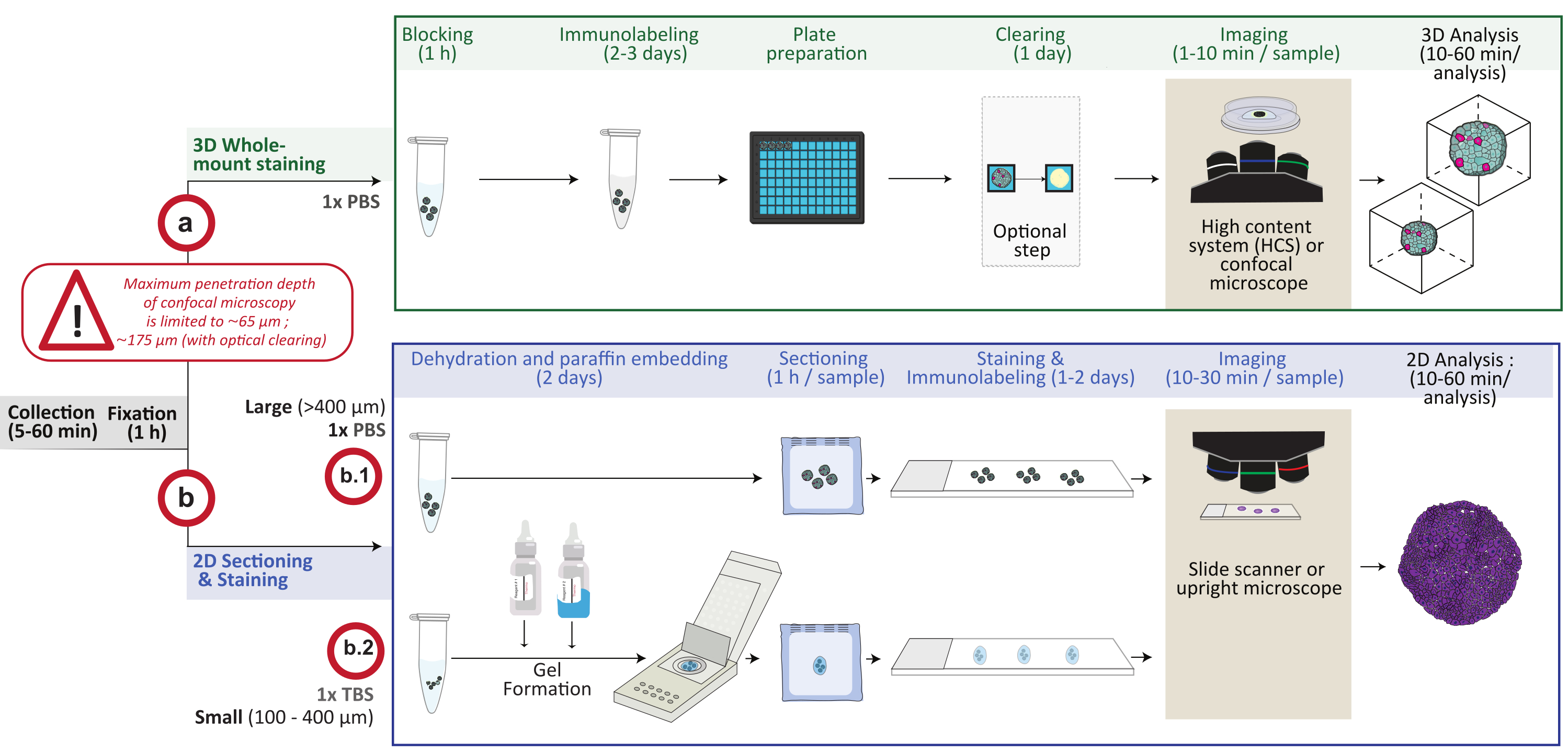
Sin embargo, en esencia, estos dos modelos son estructuras 3D compuestas por múltiples células, y las técnicas desarrolladas para estudiarlas son, por lo tanto, muy similares. Por ejemplo, se necesitan potentes enfoques de imagen a nivel de resolución de una sola célula para sondear la complejidad celular tanto de organoides como de esferoides. Aquí, al resumir la experiencia de este grupo y la de los líderes en el campo de los organoides4, este documento describe procedimientos detallados para realizar tinción bidimensional (2D) y 3D de montaje completo, imágenes y análisis de la composición celular y subcelular y la organización espacial de organoides y esferoides que van desde 100 μm hasta varios milímetros. De hecho, este procedimiento presenta dos tipos diferentes y complementarios de tinción y adquisición de imágenes para analizar una gran variedad de tamaños y tipos de modelos de cultivo celular 3D in vitro. El uso de uno (análisis 3D de montaje completo) u otro (análisis de sección 2D) dependerá del modelo estudiado y de las respuestas buscadas. El análisis 3D de montaje completo por microscopía confocal puede, por ejemplo, aplicarse para visualizar células en cultivo 3D de hasta 200 μm de profundidad, independientemente del tamaño total de la estructura 3D, mientras que el análisis de secciones 2D proporciona información sobre muestras de cualquier tamaño, aunque a nivel 2D. Este procedimiento se ha aplicado con éxito a través de una variedad de organoides4,5 y esferoides derivados de células humanas y murinas, procedentes de diferentes capas germinales embrionarias. La descripción general del procedimiento se muestra en la figura 1. Se indican las etapas principales, las relaciones entre ellas, los pasos decisivos y el momento esperado.

Figura 1: Descripción general esquemática del procedimiento. Los modelos de cultivo celular 3D in vitro se recogen y fijan, luego se preparan para la tinción 3D de montaje completo (opción a) o se incrustan en parafina para seccionamiento y tinción 2D (opción b). Para experimentos de tinción 3D de montaje completo, las estructuras 3D fijas se inmunomarcan siguiendo el paso de fijación. Se puede realizar un paso opcional de limpieza óptica para mejorar la calidad de imagen y la profundidad de la microscopía óptica al reducir la dispersión de la luz durante el procesamiento de imágenes. Las imágenes se capturan en un microscopio confocal invertido o en un sistema confocal de alto contenido y se analizan utilizando el software apropiado. Para la incrustación de parafina, las estructuras 3D se procesan directamente (opción b.1 para estructuras grandes ≥ 400 μm) o se incluyen en un gel (b.2; estructuras pequeñas ≤ 400 μm) para deshidratación e incrustación de parafina. Luego se cortan y tiñen los bloques de parafina (tinción histológica o inmunoquímica). Las imágenes de las secciones 2D se obtienen en un escáner de diapositivas digital o un microscopio vertical y se analizan en una plataforma de análisis de imágenes utilizando un análisis cuantitativo digital rápido. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Protocolo
NOTA: Se debe esperar una pérdida del ≤25% del número inicial de estructuras 3D durante los pasos que implican cambios de reactivos y lavado en el siguiente procedimiento. Planifique utilizar un número final de al menos diez estructuras 3D, con un tamaño que oscile entre 100 y 500 μm, por condición probada para realizar análisis de imágenes cualitativos y cuantitativos. Si es necesario, para estructuras más grandes, corte los extremos de las puntas de pipeta de 1 ml para evitar romper las estructuras. Para todos los pasos, si la sedimentación de la estructura 3D es demasiado larga, las células se pueden girar suavemente a 50 × g durante 5 minutos a temperatura ambiente (RT). Dependiendo del problema investigado, se deben considerar las ventajas / desventajas de tal paso de giro, ya que la centrifugación puede comprometer la forma de las estructuras 3D. Evite girar a >100 × g.
1. Recopilación y fijación de modelos de cultivo celular 3D
NOTA: Tenga cuidado de no aspirar las estructuras 3D, que solo estarán unidas libremente a la pared del tubo.
- Recolección de modelos de cultivo celular 3D incrustados en una matriz
NOTA: Esta sección describe la recuperación de estructuras 3D cultivadas en gotas de un extracto de membrana basal del sarcoma murino de Engelbreth-Holm-Swarm (BME), pero puede adaptarse a otras matrices. Consulte la discusión para conocer los puntos cruciales relacionados con ECM.- Retire el medio de cultivo de los pozos sin interrumpir la matriz 3D. Cubra previamente el interior y el exterior de una punta de pipeta de 1 ml con proteína (llamada punta prerecubierta de 1 ml en adelante) sumergiendo toda la longitud de la punta en albúmina sérica bovina (BSA) al 0,1% en solución salina tamponada con fosfato (PBS) (llamada solución PBS-BSA al 0,1% en lo sucesivo) y pipeteando 1 ml de esta solución hacia arriba y hacia abajo dos veces.
NOTA: Este recubrimiento previo evitará que las células se peguen a la punta y minimizará cualquier pérdida. - Recubra previamente el interior de un tubo de centrífuga (15 ml) con proteína (llamado tubo de centrífuga prerecubierto en adelante) llenando repetidamente con solución PBS-BSA al 0,1% y vaciando el tubo.
NOTA: Esto evitará que las células se peguen al tubo y minimizará cualquier pérdida. - Usando la punta prerecubierta de 1 ml, resuspenda cuidadosamente las estructuras 3D del pozo usando 1 ml de 1x PBS helado y transfiera suavemente la suspensión que contiene las estructuras 3D al tubo de centrífuga prerecubierto.
- Agregue suavemente 13 ml de PBS 1x helado y permita que las estructuras 3D sedimenten en el hielo durante al menos 10 minutos.
NOTA: Si es necesario, centrifugar durante 5 min a 50 × g a 4 °C. Evite girar >100 × g, ya que esto comprometerá la forma de las estructuras 3D. - Retire el sobrenadante. Usando una punta prerecubierta de 1 ml, resuspenda suavemente las estructuras 3D en 1 ml de PBS 1x helado. Repita los pasos 1.1.4 a 1.1.5 para obtener un pellet homogéneo sin ningún residuo de matriz 3D.
NOTA: La eliminación eficiente de matrices está influenciada por el tipo de matriz, el número y el tamaño de las estructuras 3D y requiere optimización para diferentes condiciones de cultivo. Para estructuras 3D cultivadas en BME, la recuperación de la eliminación de la matriz suele tardar entre 45 y 60 minutos. - Con una punta prerecubierta de 1 ml, transfiera la suspensión PBS de 1 ml 1x que contiene las estructuras 3D a un tubo centrífugo prerecubierto de 1,5 ml y continúe con la sección 1.3.
- Retire el medio de cultivo de los pozos sin interrumpir la matriz 3D. Cubra previamente el interior y el exterior de una punta de pipeta de 1 ml con proteína (llamada punta prerecubierta de 1 ml en adelante) sumergiendo toda la longitud de la punta en albúmina sérica bovina (BSA) al 0,1% en solución salina tamponada con fosfato (PBS) (llamada solución PBS-BSA al 0,1% en lo sucesivo) y pipeteando 1 ml de esta solución hacia arriba y hacia abajo dos veces.
- Recolección de modelos de cultivo celular 3D flotantes
- Usando una punta prerecubierta de 1 ml, recoja y transfiera cuidadosamente las estructuras 3D a un tubo centrífugo prerecubierto de 1,5 ml. Permita que las estructuras 3D sedimenten o giren durante 5 minutos a 50 × g en RT.
- Retire el sobrenadante. Usando una punta prerecubierta de 1 ml, resuspenda las estructuras 3D en 1 ml de 1x PBS. Continúe con la sección 1.3.
- Fijación de modelos de cultivo celular 3D
- Permita que las estructuras 3D sedimenten. Retire con cuidado el sobrenadante; bajo una campana extractora, resuspenda suavemente las estructuras 3D en 1 ml de formalina utilizando una punta prerecubierta de 1 ml.
NOTA: La formalina contiene formaldehído, que es peligroso. Manipule el producto químico en una campana química. Use guantes de goma y gafas protectoras para los ojos. - Incubar las estructuras 3D durante 30 minutos en RT.
NOTA: Se requiere un paso de fijación de 30 minutos con formalina para la inmunotinción de una amplia gama de estructuras 3D (que varían en tamaño, forma y origen). Sin embargo, en general, los tiempos de fijación más largos (>3 h) son más adecuados para preservar la fluorescencia de las proteínas reporteras. - Permita que las estructuras 3D sedimenten o giren durante 5 minutos a 50 × g en RT. Retire suavemente la formalina y reemplácela con 1 ml de 1x PBS. Repita este paso de lavado en 1x PBS dos veces. Conservar las muestras a 4 °C y proceder con la sección 2 o la sección 3.
NOTA: El protocolo se puede pausar aquí, y las celdas se pueden mantener a 4 ° C para el almacenamiento a largo plazo (> 1 año).
- Permita que las estructuras 3D sedimenten. Retire con cuidado el sobrenadante; bajo una campana extractora, resuspenda suavemente las estructuras 3D en 1 ml de formalina utilizando una punta prerecubierta de 1 ml.
2.3D tinción de montaje completo, imágenes y análisis de modelos de cultivo celular 3D
NOTA: Como los organoides están unidos libremente a la pared del tubo, manéjelos suavemente ya que todos los cambios de reactivos siguientes pueden causar la pérdida de muestra. Antes de comenzar, asegúrese de la disponibilidad de los controles correctos para la tinción. Los controles positivos y negativos pueden ser células, en las que se sabe que la proteína de interés está sobreexpresada o ausente, respectivamente. Incubar muestras sin el anticuerpo primario para determinar si la señal observada se debe a la unión no específica del anticuerpo secundario. Como algunas células tienden a mostrar altos niveles de autofluorescencia, use controles desprovistos de anticuerpos secundarios para determinar si la fluorescencia observada proviene de la autofluorescencia de fondo. El inmunoetiquetado y la visualización fluorescente del reportero se pueden combinar.
- Tinción 3D de montaje completo
- Prepare la solución de bloqueo de permeabilización (PB) complementando 1x PBS con 0.1% -1% de un surfactante no iónico (ver la Tabla de materiales), 1% de dimetilsulfóxido, 1% de BSA y 1% de suero de burro (o del animal en el que se criaron los anticuerpos secundarios).
NOTA: Optimice cuidadosamente la concentración del surfactante no iónico dependiendo de la localización del objetivo: membrana (0-0.5%), citoplasma (0.5-1%) y núcleo (1%). Esta solución se puede almacenar a 4°C durante un máximo de 1 mes. La BSA generalmente funciona bien para el paso de bloqueo, pero en caso de alto ruido de fondo, realice una prueba empírica para obtener los mejores resultados posibles para una combinación dada de anticuerpos. - Transfiera los organoides del tubo de centrífuga de 1,5 ml a un tubo de 0,5 ml utilizando una punta prerecubierta de 1 ml. Deje que los organoides sedimenten, retire suavemente el 1x PBS y reemplácelo con 0.5 ml de solución de PB. Incubar los organoides con una suave agitación horizontal (30-50 rpm) durante 1 h a RT.
- Deje que los organoides sedimenten, retire suavemente la solución de PB y lave dos veces en 1 mL PBS-BSA 0.1% durante 3 min.
NOTA: Esperar 3 minutos permite que las estructuras sedimenten en el fondo del tubo. - Retire suavemente el PBS-BSA al 0,1% y agregue 250 μL de anticuerpo primario diluido a la concentración adecuada en la solución PB:1x PBS (1:10). Para preparar 10 ml de solución de PB:1x PBS (1:10), diluir 1 ml de solución de PB en 9 ml de 1x PBS. Incubar durante 2-3 días con una suave agitación horizontal (30-50 rpm) a 4 °C.
NOTA: Un tiempo de incubación de anticuerpos apropiado es crucial para una penetración de anticuerpos adecuada, ya que las estructuras 3D a veces pueden alcanzar tamaños grandes. - Deje que los organoides sedimenten y retire suavemente la solución de anticuerpos primarios. Lavar 5x en PBS-BSA 0.1% durante 3 min por lavado y luego 2x en 1 mL PBS-BSA 0.1% durante 15 min por lavado con agitación horizontal suave.
- Añadir 250 μL de anticuerpo secundario diluido a 1:250 en PB:1x PBS (1:10) solución. Incubar durante 24 h a 4°C con una suave agitación horizontal (30-50 rpm). Para este paso, proteja las muestras de la luz.
- Añadir 250 μL de solución madre de Hoechst 33342 (20 μM) diluida a 1:1000 en PB:1x PBS (1:10) y incubar durante otras 2 h a 4 °C con una agitación horizontal suave (30-50 rpm).
- Deje sedimentar los organoides y retire suavemente la solución que contiene el anticuerpo secundario + Hoechst 33342. Lave los organoides 5 veces en 1 ml de 1x PBS durante 3 minutos por lavado y luego 2 veces en 1 ml de 1x PBS durante 15 minutos por lavado con agitación horizontal suave (30-50 rpm).
NOTA: Es crucial lavar extensamente las muestras para evitar el ruido de fondo o la pérdida de señal. - Almacenar las muestras en PBS a 4 °C hasta la adquisición de la imagen. Continúe con la sección 2.2.
NOTA: El protocolo se puede pausar aquí, y las muestras se pueden almacenar a 4 °C durante varios meses, protegidas de la luz.
- Prepare la solución de bloqueo de permeabilización (PB) complementando 1x PBS con 0.1% -1% de un surfactante no iónico (ver la Tabla de materiales), 1% de dimetilsulfóxido, 1% de BSA y 1% de suero de burro (o del animal en el que se criaron los anticuerpos secundarios).
- Preparación de muestras para imágenes confocales
- Usando una punta prerecubierta de 1 ml, transfiera cuidadosamente los organoides a 50 μL del 1x PBS por pocillo en una microplaca de poliestireno negro de 96 pocillos. Continúe con el paso 2.2.3 o la sección 2.3.
NOTA: En esta etapa, la muestra puede protegerse de la luz y almacenarse a 4 °C durante muchas semanas. - Claro
NOTA: El paso de limpieza es opcional y se puede utilizar para inmunomarcar organoides o para detectar fluorescencia endógena. El aclaramiento puede causar la contracción de la estructura 3D, pero no cambia la morfología general, excepto para los organoides esféricos de una sola capa con grandes lúmenes4. Para estos organoides quísticos, omita el paso de limpieza y realice imágenes de tejido profundo6.- Prepare una solución de limpieza de glicerol-fructosa 2,5 M que contenga 50% v/v de glicerol, 11% v/v de agua destilada y 45% p/v de fructosa mezclando en un agitador magnético al menos durante la noche hasta que la solución esté completamente solubilizada y homogénea. Conservar a 4 °C en la oscuridad hasta 1 mes.
- Retire la mayor cantidad posible de PBS 1x sin tocar los organoides. Añadir 200 μL de la solución de aclaramiento con una punta de pipeta de 1 ml después de retirar el extremo y volver a suspender suavemente para evitar la formación de burbujas. Incubar a RT durante al menos 12 h y continuar con la sección 3.
NOTA: Como la solución de limpieza es viscosa, los volúmenes pequeños son difíciles de manejar. Para facilitar la manipulación, asegúrese de que la solución esté en RT y pipetear lentamente. Para una limpieza óptima, deje que la muestra sedimente en la solución de limpieza durante al menos 24 horas antes de obtener la imagen. Si las estructuras 3D están flotando en el momento de la adquisición, realice un giro opcional durante 10 minutos a <100 × g en RT, o permita más tiempo (uno o varios días) para dejar que sedimenten. El protocolo se puede pausar en este paso antes de proceder a la obtención de imágenes si está protegido de la luz y almacenado a 4 ° C (durante semanas) o -20 ° C (durante meses).
- Usando una punta prerecubierta de 1 ml, transfiera cuidadosamente los organoides a 50 μL del 1x PBS por pocillo en una microplaca de poliestireno negro de 96 pocillos. Continúe con el paso 2.2.3 o la sección 2.3.
- Adquisición y análisis de imágenes
NOTA: Se requerirá tecnología de seccionamiento de imágenes para obtener imágenes de estructuras 3D.- Utilice microscopios confocales, y favorezca los objetivos de inmersión con mayor apertura numérica (NA) en comparación con el aire. Elija objetivos de ampliación (10x, 20x, 40x) de acuerdo con el tamaño de las estructuras 3D, la reconstrucción de imágenes (costura) y las soluciones utilizadas para el análisis.
- Al seleccionar el modo de adquisición, tenga en cuenta la profundidad de enfoque del objetivo utilizado para definir el paso para el apilamiento Z; permiten una representación 3D óptima.
NOTA: Las soluciones de análisis de imágenes varían y el análisis deberá ajustarse al software utilizado. Por ejemplo, este protocolo de análisis se estableció en un software de análisis de alto contenido (consulte la Tabla de materiales y la Figura suplementaria 1 para obtener más detalles) y proporciona datos sobre la segmentación de objetos, el cálculo de propiedades y la selección de poblaciones celulares dentro de un objeto reconstruido en 3D.
3. Sección 2D, tinción, imágenes y análisis de modelos de cultivo celular 3D
NOTA: Los modelos de cultivo celular 3D varían en tamaño. Continúe con la sección 3.1 o 3.2 para una incrustación eficiente de parafina (Figura 2). Deje suficiente tiempo para la sedimentación de la estructura 3D antes de cualquier lavado y cambio de reactivo. Tenga cuidado de no aspirar los organoides que flotarán en el fondo del tubo. Para la incrustación de parafina, consulte la Figura 2 para obtener instrucciones.
- Incrustación de parafina de grandes modelos de cultivo celular 3D (Ø ≥ 400 μm)
- El día antes de la incrustación, precaliente dos matraces de 150 ml llenos de parafina (baños de parafina), un pequeño molde metálico por muestra y pinzas finas a 65 °C.
- Usando una punta prerecubierta de 1 ml, transfiera cuidadosamente los organoides en 1x PBS a un tubo de vidrio de fondo plano con una tapa de botella forrada de politetrafluoroetileno. Deje que los organoides sedimenten, retire con cuidado el 1x PBS y reemplácelo con etanol al 70%. Incubar durante al menos 30 min.
- Deje que los organoides sedimenten y retire con cuidado el etanol al 70%. Reemplácelo con 1 ml de solución Y de eosina lista para usar. Mueva el tubo y manténgalo durante al menos 30 minutos. Retire cuidadosamente la solución de eosina y deshidrate los organoides en tres lavados sucesivos con 1 ml de etanol al 100% durante ~ 30 minutos cada uno.
NOTA: El etanol, un líquido inflamable y volátil, causa irritación severa de los ojos y las vías respiratorias. Manipularlo en una campana extractora y usar gafas protectoras para los ojos. - Retire cuidadosamente el etanol al 100% y, bajo una campana química, limpie los organoides en 3 lavados sucesivos con 1 ml de xileno durante ~ 30 minutos cada uno.
NOTA: El xileno es un líquido tóxico inflamable cuyos vapores pueden causar irritación. Manipularlo en una campana extractora. Evite el contacto directo con la piel y use guantes de goma y gafas protectoras para los ojos. - Bajo una campana química, prepare un casete de tejido microgemelo blanco colocando un trozo de almohadilla de biopsia (previamente empapada en xileno) dentro de uno de los compartimentos del casete. Transfiera cuidadosamente las estructuras 3D utilizando una pipeta Pasteur de plástico prerecubierta de 2 ml a la almohadilla de biopsia. Cúbralos con otra almohadilla de biopsia empapada en xileno para evitar que los organoides se muevan y cierre el casete.
- Si se procesan varias muestras, coloque el casete en un baño de xileno para esperar un procesamiento posterior. Una vez que todas las muestras se transfieran a casetes, coloque los casetes en un baño de parafina precalentado durante 30 minutos a 65 ° C. Transfiera los casetes a un baño de parafina precalentado fresco durante la noche.
- Después de la impregnación de parafina, tome un molde de incrustación precalentado y agréguele la parafina calentada. Coloque la almohadilla de biopsia que contiene las estructuras 3D en el molde y agite suavemente hasta que todos los organoides caigan al fondo del molde. Coloque con mucho cuidado las estructuras 3D en el centro del molde utilizando pinzas finas precalentadas. Continúe con la sección 3.3.
NOTA: Tenga cuidado de no interrumpir las estructuras 3D con los fórceps; Empuje, pero no los pellizque.
- Incrustación de parafina de modelos de cultivo celular 3D pequeños (Ø ≤ 400 μm)
- El día antes de la incrustación, precaliente dos matraces de 150 ml llenos de parafina (baños de parafina), un pequeño molde metálico por muestra y pinzas finas a 65 °C.
- Retire con cuidado el 1x PBS de la suspensión organoide. Realice suavemente 3 lavados en 1 ml de 1x solución salina tamponada con Tris (TBS). Retire la mayor cantidad posible de 1x TBS sin tocar los organoides.
NOTA: Tenga cuidado de no aspirar la muestra. Si es necesario, realice un giro de 5 minutos a 50 x g a RT. Las trazas restantes de fosfato interferirán con los siguientes pasos, especialmente evitando la polimerización en gel. Por lo tanto, no utilice soluciones PBS durante ningún paso de procesamiento. Para este paso, se utilizó un kit comercial, que contenía casetes, Reactivo # 1 (fluido claro) y Reactivo # 2 (fluido coloreado), para facilitar el procedimiento incrustado en parafina sin perder potencialmente pequeños fragmentos (ver Tabla de materiales). Siga las instrucciones del kit. Los casetes están premontados con papeles de respaldo e inserciones de cartón ya en su lugar. - Agregue 2 gotas de Reactivo #2 en el tubo y mezcle suavemente golpeando el tubo. Agregue 2 gotas de Reactivo # 1 y mezcle nuevamente golpeando para que el gel se solidifique. Usando las pinzas finas, retire el gel del tubo y colóquelo en el pozo del casete.
- Bajo la campana extractora, deshidratar la muestra colocando el casete en baños sucesivos de la siguiente manera (use los matraces de 150 ml y use etanol fresco o xileno para cada baño): etanol 70%, 30 min; etanol 96%, 30 min; etanol 100%, tres lavados, 30 min cada uno; xileno, tres lavados, 30 min cada uno.
- Coloque los casetes en un baño de parafina precalentado durante 30 minutos a 65 ° C y transfiéralos a un baño de parafina precalentado fresco durante la noche. Después de la impregnación de parafina, tome un molde de incrustación precalentado y agregue parafina calentada en él. Abra el casete, desaloje cuidadosamente el gel con pinzas finas y coloque el gel que contiene las estructuras 3D en el centro del molde de incrustación. Continúe con la sección 3.3.
- Pasos comunes para la incorporación de parafina
- Transfiera suavemente el molde a un área fría para permitir que la parafina se solidifique en una capa delgada, lo que mantendrá las estructuras 3D en la posición adecuada. Agregue un cassette de pañuelo de papel encima del molde y agregue parafina caliente para cubrir este cassette de plástico. Retire el molde una vez que esté completamente solidificado y continúe con la sección 3.4.
NOTA: Los bloques de parafina se pueden almacenar a temperatura ambiente durante años.
- Transfiera suavemente el molde a un área fría para permitir que la parafina se solidifique en una capa delgada, lo que mantendrá las estructuras 3D en la posición adecuada. Agregue un cassette de pañuelo de papel encima del molde y agregue parafina caliente para cubrir este cassette de plástico. Retire el molde una vez que esté completamente solidificado y continúe con la sección 3.4.

Figura 2: Descripción general del procedimiento para la incorporación de parafina de modelos de cultivo celular 3D in vitro grandes y pequeños.
A) Procedimiento normalizado para la incorporación de parafina. Después de la fijación y deshidratación, las estructuras 3D se tiñen con eosina para facilitar su visualización (arriba y abajo a la izquierda). Las estructuras 3D se colocan cuidadosamente en la almohadilla de biopsia (azul) en el casete utilizando una pipeta Pasteur de 2 ml (centro). Después de la impregnación de parafina, las estructuras 3D se dejan caer suavemente en la parafina líquida con fórceps y se agitan suavemente en la almohadilla de biopsia. Las pequeñas estructuras 3D se pierden durante este paso, ya que no se pueden liberar de la almohadilla (abajo a la derecha: incrustación fallida). Solo se incrustarán grandes estructuras 3D (arriba a la derecha: incrustación exitosa). Las puntas de flecha apuntan a culturas 3D. (B) Alternativa al protocolo estándar de incrustación de parafina. Después de haber fijado pequeñas estructuras 3D, se utiliza un kit comercial para mantener las células en un gel y facilitar su transferencia al molde después de la impregnación de parafina (derecha: incrustación exitosa). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Sección y tinción de bloques
- Cortar secciones de 4 μm utilizando un micrótomo estándar y realizar técnicas histológicas e inmunohistoquímicas estándar. Continúe con la sección 3.5.
NOTA: Se utilizaron diapositivas específicas (ver Tabla de materiales) para una mejor adhesión de las secciones. Los portaobjetos se pueden almacenar a temperatura ambiente o a 4 °C durante años.
- Cortar secciones de 4 μm utilizando un micrótomo estándar y realizar técnicas histológicas e inmunohistoquímicas estándar. Continúe con la sección 3.5.
- Adquisición y análisis de imágenes
- Realice imágenes utilizando un escáner digital de diapositivas o un microscopio vertical, y analice los datos utilizando una plataforma para un análisis cuantitativo digital rápido que informa datos de expresión morfológica y multiplexada célula por celda en secciones completas de la estructura 3D (consulte la Figura complementaria 2 para obtener más detalles).
NOTA: El objetivo 20x es utilizado rutinariamente por este grupo.
- Realice imágenes utilizando un escáner digital de diapositivas o un microscopio vertical, y analice los datos utilizando una plataforma para un análisis cuantitativo digital rápido que informa datos de expresión morfológica y multiplexada célula por celda en secciones completas de la estructura 3D (consulte la Figura complementaria 2 para obtener más detalles).
Resultados
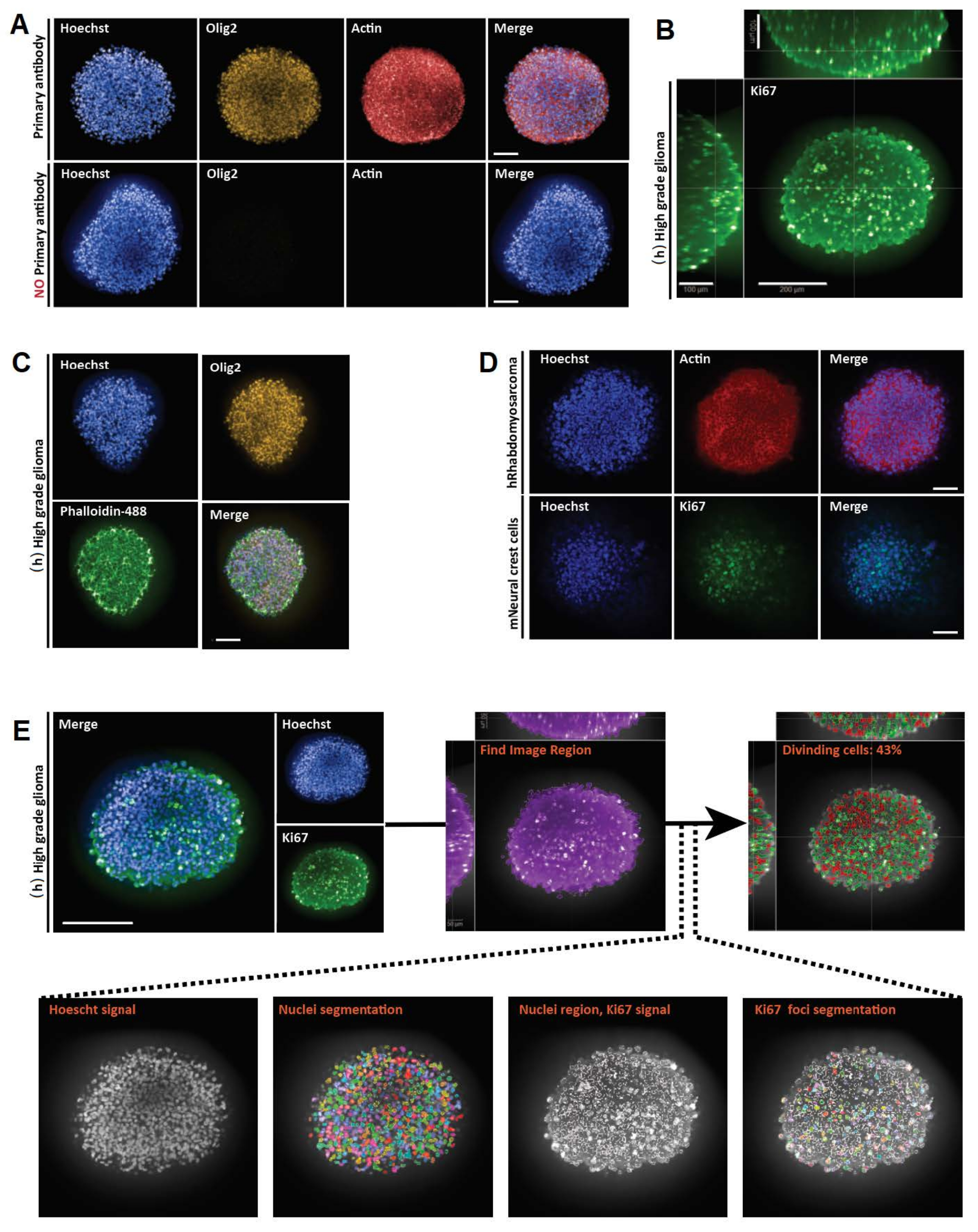
Este protocolo proporciona una visión general de los pasos críticos para la tinción de montaje completo 2D y 3D, así como imágenes y análisis cuantitativos de modelos de cultivo celular 3D (Figura 3 y Figura 4). Es aplicable a una amplia gama de modelos de cultivo celular 3D, desde esferoides hasta organoides de diferentes especies o tejidos huéspedes, y permite la adquisición de información precisa y cuantitativa sobre arquitectura, organización celular e interacciones a nivel celular y subcelular (Figura 3 y Figura 4). Los laboratorios pueden necesitar optimizar las técnicas histológicas e inmunohistoquímicas 2D y las concentraciones de anticuerpos de acuerdo con sus propias necesidades.
Ambos métodos proporcionan información biológica valiosa. La tinción 3D de montaje completo y la microscopía confocal proporcionan información visual sobre la composición celular y la posición espacial con un campo de profundidad de hasta 200 μm (Figura 3B). Sin embargo, la sección 2D es conveniente para que las estructuras 3D más grandes revelen rasgos morfológicos celulares detallados en toda la sección de estructuras 3D que de otro modo pueden ser difíciles de observar in situ debido a la dispersión de la luz que compromete la resolución en muestras más grandes. Además, ambas técnicas pueden proporcionar datos cuantitativos. De hecho, la resolución obtenida permite la aplicación de algoritmos de segmentación celular y subcelular para la cuantificación del número de células y la detección de la presencia de varios marcadores celulares en diferentes subtipos celulares (Figura 3F y Figura 4). En resumen, las técnicas de imagen descritas aquí son reproducibles, simples y complementarias y representan herramientas valiosas para estudiar la heterogeneidad celular.

Figura 3: Resultados representativos para montaje completo 3D, imágenes y análisis de secciones ópticas 3D y 2D. (A) Imágenes confocales de esferoide de glioma humano (h) de alto grado cultivado durante una semana y etiquetado con Hoechst (azul), Olig2 (amarillo) y Actina (rojo) (objetivo de agua 20x). Para todas las imágenes adquiridas, los ajustes del microscopio se establecieron utilizando un control positivo (arriba), y luego el control negativo se visualizó utilizando ajustes idénticos para controlar la falta de fluorescencia en ausencia de anticuerpos primarios (abajo). (B) Representación ortogonal 3D de montaje completo de la tinción de Ki67 realizada en (h) esferoide de glioma de alto grado cultivado durante una semana (aclaramiento de glicerol-fructosa; objetivo de agua 20x, confocal). (C) Imágenes confocales de (h) esferoide de glioma de alto grado cultivado durante una semana y marcado con Hoechst (azul), Olig2 (amarillo) y faloidina-488 (verde) (aclaramiento de glicerol-fructosa; objetivo de agua 20x). (D) Imágenes confocales de esferoides humanos (h) de rabdomiosarcoma (arriba) y ratón (m) de células de la cresta neural (abajo) cultivados durante una semana y marcados con Hoechst (azul), Actina (rojo) y Ki67 (verde), respectivamente (eliminación de glicerol-fructosa; objetivo seco 20x). (E) Imágenes confocales de (h) esferoide de glioma de alto grado cultivado durante una semana y etiquetado con Hoechst (azul) y Ki67 (verde) (limpieza de glicerol-fructosa; objetivo de agua 40x) (arriba a la izquierda). Las imágenes segmentadas en el canal Hoechst y las regiones nucleares Ki67-positivas (+) en el canal verde se generaron utilizando software de análisis de alto contenido (ver Figura suplementaria 1 y Tabla de materiales) (abajo). La salida dada es el porcentaje de núcleos Ki67+ por estructura 3D segmentada (arriba a la derecha). Barra de escala = 100 μm. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

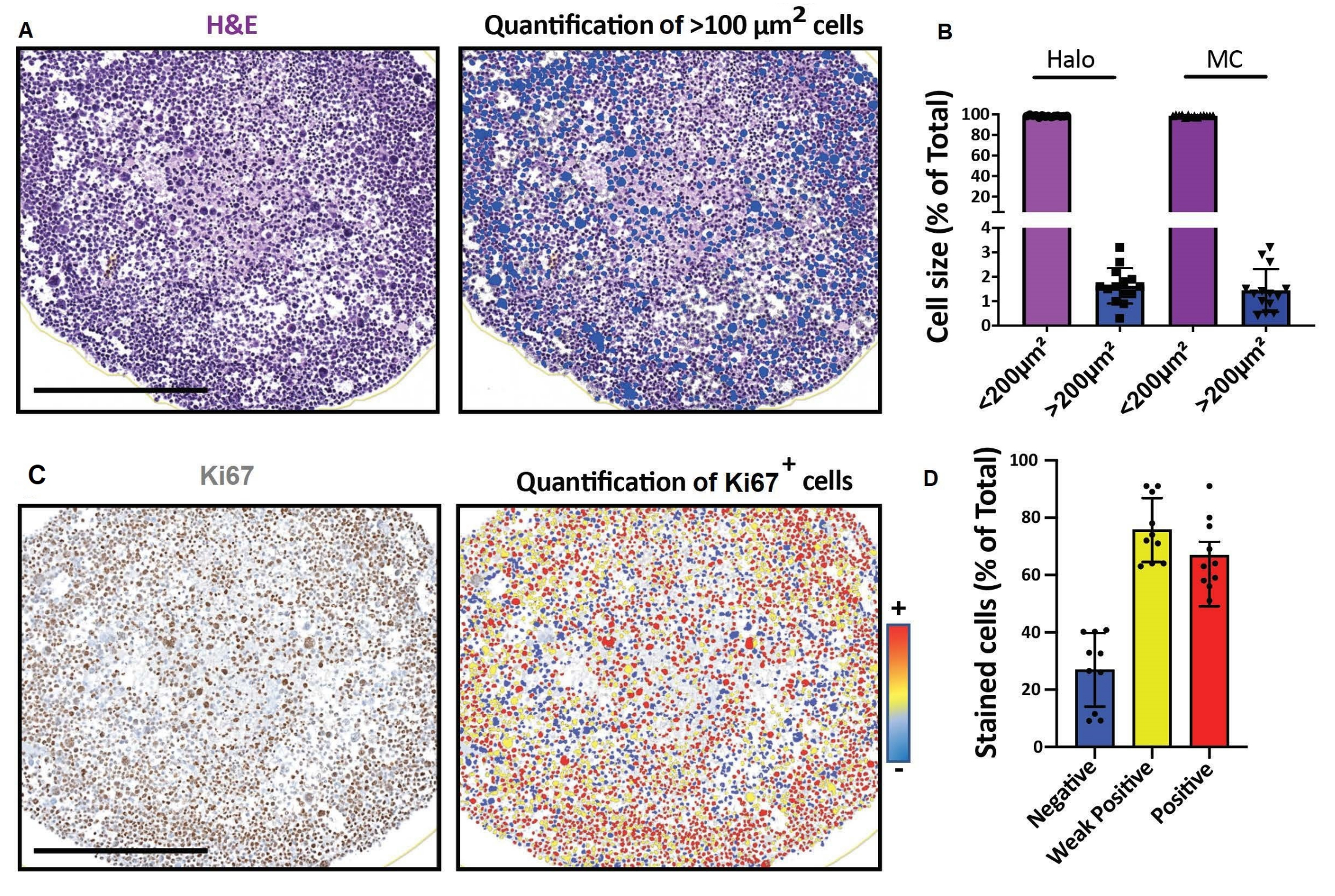
Figura 4: Resultados representativos para imágenes y análisis de secciones ópticas 2D. (A, D) Imágenes de sección 2D de un modelo celular 3D (esferoides de rabdomiosarcoma humano cultivados durante un mes) obtenidas con un escáner digital de diapositivas y analizadas en una plataforma para un análisis cuantitativo digital rápido. (A) Tinción H&E y detección de células según su tamaño. Barra de escala = 500 μm. (B) El histograma muestra el porcentaje de células < 100 μm2 y > 100μm2 detectadas utilizando software para un análisis cuantitativo digital rápido (izquierda: Halo) o conteo manual (derecha: MC). (C) Tinción de Ki67 y detección de células según la intensidad de su señal de 3,3'-diaminobencidina (DAB). Negativo (azul), débilmente positivo (amarillo), positivo (rojo). Barra de escala = 500 μm. (D) El histograma muestra el porcentaje de células Ki67 negativas, débilmente positivas y positivas. Abreviaturas: H&E = hematoxilina y eosina; MC = conteo manual. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Figura complementaria 1: Descripción general de los pasos en el software de análisis de imágenes. Los análisis se basan en la asociación de bloques de construcción. Cada bloque de construcción corresponde a una función (segmentación, cálculo, asociación, definición de salida) y ofrece múltiples algoritmos y selecciones de variables para que coincidan con la muestra biológica que se está fotografiando. El software proporciona múltiples protocolos de análisis RMS (Ready Made Solution) que se pueden usar y modificar fácilmente. Los protocolos de análisis de imágenes integrados se pueden guardar, aplicar a diferentes conjuntos de datos y compartir entre usuarios. Brevemente, el protocolo de análisis implica segmentación secuencial de objetos: esferoide, núcleos y, finalmente, bolsas Ki67 (A488). Luego, la intensidad media de los bolsillos Ki67 se calcula para discriminar aún más los eventos positivos. Finalmente, los núcleos que abarcan bolsas positivas de Ki67 se seleccionan positivamente. Haga clic aquí para descargar este archivo.
Figura complementaria 2: Descripción general de los pasos del procedimiento del software de análisis cuantitativo. Paso 1. Cargue los archivos utilizando la pestaña Estudios . Los archivos se abrirán en la sección Acciones de imagen . Paso 2. Abra la pestaña Anotaciones y, a continuación, haga clic en Acciones de capa para diseñar una nueva capa alrededor de la estructura con la herramienta circular de la barra de herramientas. Para estructuras no circulares, se puede utilizar la herramienta de lápiz en su lugar. Paso 3. La barra de herramientas se puede utilizar para diseñar anotaciones y visualizar la cuantificación con la  herramienta. Paso 4. Abra la pestaña Análisis y seleccione las mejores condiciones para el análisis de la muestra (pueden ser necesarios varios ensayos aquí). Paso 4.1. Utilice la sección Selección de manchas para configurar la condición de tinción. En el caso de varias manchas, estas se pueden agregar y renombrar, y el color virtual se puede modificar. La detección de localización puede ser específica nuclear o tinción citoplasmática. Paso 4.2. Utilice la sección Detección de celdas para configurar la detección de celdas. Esta sección será la más importante para el análisis. La sección Umbral de contraste nuclear permitirá la detección de todos los núcleos. Se debe prestar atención en caso de que haya múltiples tamaños de población, el software puede detectar varias células en lugar de una grande única. Las secciones de agresividad de tamaño nuclear y segmentación nuclear se pueden usar para cuantificar los rangos de población de tamaño celular. Paso 5. Descripción sobre cómo ejecutar el análisis de muestras. Siga los pasos que se muestran en la figura. La sección Capa de anotación ejecutará la configuración solo en esta diapositiva. La cuantificación se puede visualizar utilizando la herramienta. Repita los pasos 4.1-5 hasta que se logre la cuantificación adecuada. Pasos 6-6.1. Estos pasos le permiten dibujar una figura utilizando el software. Paso 7. Los gráficos de cuantificación obtenidos a través del software se pueden guardar. Paso 8. Los datos se pueden exportar. Haga clic aquí para descargar este archivo.
herramienta. Paso 4. Abra la pestaña Análisis y seleccione las mejores condiciones para el análisis de la muestra (pueden ser necesarios varios ensayos aquí). Paso 4.1. Utilice la sección Selección de manchas para configurar la condición de tinción. En el caso de varias manchas, estas se pueden agregar y renombrar, y el color virtual se puede modificar. La detección de localización puede ser específica nuclear o tinción citoplasmática. Paso 4.2. Utilice la sección Detección de celdas para configurar la detección de celdas. Esta sección será la más importante para el análisis. La sección Umbral de contraste nuclear permitirá la detección de todos los núcleos. Se debe prestar atención en caso de que haya múltiples tamaños de población, el software puede detectar varias células en lugar de una grande única. Las secciones de agresividad de tamaño nuclear y segmentación nuclear se pueden usar para cuantificar los rangos de población de tamaño celular. Paso 5. Descripción sobre cómo ejecutar el análisis de muestras. Siga los pasos que se muestran en la figura. La sección Capa de anotación ejecutará la configuración solo en esta diapositiva. La cuantificación se puede visualizar utilizando la herramienta. Repita los pasos 4.1-5 hasta que se logre la cuantificación adecuada. Pasos 6-6.1. Estos pasos le permiten dibujar una figura utilizando el software. Paso 7. Los gráficos de cuantificación obtenidos a través del software se pueden guardar. Paso 8. Los datos se pueden exportar. Haga clic aquí para descargar este archivo.
Discusión
El cultivo celular es una herramienta indispensable para descubrir mecanismos biológicos fundamentales involucrados en el desarrollo, función, regeneración y alteración de tejidos y órganos, y enfermedades. Aunque el cultivo celular 2D monocapa ha predominado, investigaciones recientes se han desplazado hacia cultivos que generan estructuras 3D que reflejan más las respuestas celulares in vivo, debido en particular a la organización espacial adicional y los contactos célula-célula que influyen en la expresión génica y el comportamiento celular y, por lo tanto, podrían proporcionar datos más predictivos7. Sin embargo, quedan muchos desafíos, incluida la necesidad de técnicas de tinción e imágenes fáciles de usar para la visualización microscópica detallada y la evaluación de estructuras 3D complejas a nivel celular y subcelular. En ese contexto, se han proporcionado protocolos detallados, robustos y complementarios para realizar tinción e imágenes de resolución celular y subcelular de modelos de cultivo celular 3D in vitro fijos que van desde 100 μm hasta varios milímetros de tamaño.
Este procedimiento presenta dos estrategias diferentes para tratar una gran variedad de tamaños y tipos de modelos de cultivo celular 3D in vitro. La elección de uno (análisis 3D de montaje completo) u otro (análisis de sección 2D) dependerá del modelo utilizado y del problema investigado. El análisis 3D de montaje completo por microscopía confocal permite la visualización de células con un campo de profundidad de hasta 200 μm, independientemente del tamaño total de la estructura 3D, mientras que la sección 2D es aplicable a muestras de cualquier tamaño, pero la visualización sigue siendo 2D dimensional. A continuación se presentan algunas sugerencias para la solución de problemas y consideraciones técnicas.
La pérdida de estructuras 3D durante el flujo de trabajo es el inconveniente más común. Pueden permanecer adheridos a las puntas y tubos, por lo que las puntas y tubos de recubrimiento previo con solución PBS-BSA al 0,1% son clave. Además, es crucial dejar que las estructuras 3D sedimenten entre los cambios de reactivo y realizar todo el pipeteo con mucho cuidado. Como se mencionó en el procedimiento, para todos los pasos, si la sedimentación de la estructura 3D es demasiado larga, las células se pueden girar suavemente a 50 × g durante 5 minutos a RT. Dependiendo del objetivo del estudio, se deben considerar las ventajas / desventajas de tal paso de hilado, ya que la centrifugación puede comprometer la forma de las estructuras 3D. Además, se debe tener cuidado para preservar esta morfología durante la etapa de fijación porque los organoides quísticos tienden a colapsar. La fijación de estructuras de menos de 400 μm de tamaño debe evitar cambios estructurales.
Para un inmunomarcaje óptimo, la recuperación de organoides de sus matrices 3D es un paso crucial. La matriz 3D puede impedir la penetración adecuada de anticuerpos o conducir a una alta tinción de fondo debido a la unión no específica a la matriz. La eliminación de la MCE puede alterar la morfología de los segmentos externos de los organoides (especialmente en el caso de pequeñas protuberancias celulares que se extienden desde estructuras 3D estudiadas) y dificultar parcialmente los análisis. Para tales estructuras 3D, la matriz se puede conservar durante todo el procedimiento; Sin embargo, las condiciones de cultivo deben adaptarse cuidadosamente para cultivar células en una cantidad mínima de matriz para evitar una penetración insuficiente de soluciones y anticuerpos y para evitar pasos sucesivos de lavado destinados a reducir el ruido de fondo excesivo 6,8.
El paso de limpieza óptica descrito en este protocolo en la sección de tinción de montaje completo 3D es pertinente para la obtención de imágenes de estructuras 3D de hasta 150-200 μm de profundidad en lugar de 50-80 μm sin aclaración. En comparación con otras metodologías de limpieza que a menudo requieren varias semanas y utilizan agentes de limpieza tóxicos, eneste protocolo se utilizó un paso de limpieza rápido y seguro previamente publicado 4,9. Además, este paso de limpieza es reversible, y se pueden agregar nuevos anticuerpos a la tinción inicial sin pérdida de resolución o brillo4. Sin embargo, dependiendo del modelo de cultivo celular 3D estudiado, una profundidad de 150-200 μm podría no ser suficiente para obtener imágenes de la estructura 3D de manera informativa, y este protocolo de limpieza puede causar cambios en la morfología general de organoides esféricos monocapa con grandes lúmenes4. Los usuarios deben diseñar cuidadosamente su experimento y, si es necesario, optimizar el tiempo de la etapa de permeabilización / bloqueo (para permitir la penetración de anticuerpos y solución), la etapa de limpieza (para penetrar más profundo que 200 μm, las muestras deben estar totalmente despejadas) y la adquisición de imágenes. Las dos tecnologías más frecuentes disponibles en las instalaciones básicas serían la microscopía de lámina de luz y la microscopía confocal. Los usuarios deberán elegir cuidadosamente una tecnología basada en el tamaño de sus estructuras 3D y su pregunta biológica10. Sin embargo, en comparación con la microscopía confocal, la resolución de microscopía de lámina de luz obtenida para tales estructuras profundas sigue siendo subóptima para obtener la resolución subcelular.
Aquí, se ha informado de un proceso detallado y robusto que se dedica a la incorporación de parafina de muestras individuales. Curiosamente, Gabriel et al. desarrollaron recientemente un protocolo que incorpora cultivos celulares 3D en parafina con un mayor rendimiento. Utilizaron un molde de polidimetilsiloxano (PDMS) para confinar 96 estructuras 3D en un patrón de micromatriz en un bloque, proporcionando nuevas perspectivas para estudios sobre modelos tumorales 3D que abarcan más grupos, puntos de tiempo, condiciones de tratamiento y réplicas11. Sin embargo, este método requiere amplias habilidades y maquinaria, especialmente para la fabricación del premolde utilizado para crear moldes PDMS.
En resumen, este artículo describe dos enfoques diferentes, complementarios y adaptables que permiten la adquisición de información precisa y cuantitativa sobre la composición arquitectónica y celular de los modelos celulares 3D. Ambos parámetros son cruciales para estudiar procesos biológicos como la heterogeneidad celular intratumoral y su papel en la resistencia a los tratamientos.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Este trabajo fue apoyado por el Premio a la Innovación Robert J. Arceci # 604303 de St Baldrick.
Materiales
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| Biopsy pad Q Path blue | VWR | 720-2254 | |
| Cassettes macrostar III Blc couv. Char. x1500 | VWR | 720-2233 | |
| Cassette microtwin white | VWR | 720-2183 | |
| Chemical hood | Erlab | FI82 5585-06 | |
| Filter tips 1000 µL | Star lab Tip-One | S1122-1730 | |
| Fine forceps | Pyramid innovation | R35002-E | |
| Flat-bottom glass tubes with PTFE lined 2 mL | Fisher Scientific | 11784259 | Excellent for environmental samples, pharmaceuticals and diagnostic reagents. PTFE is designed for the ultimate in product safety. PTFE provides totally inert inner seal and surface facing the sample or product. |
| Glass bottom dish plate 35 mm | Ibedi | 2018003 | |
| Horizontal agitation | N-BIOTEK | NB-205 | |
| Incubator prewarmed to 65 °C | Memmert Incubator | LAB129 | |
| Inox molds 15x15 | VWR | 720-1918 | |
| Microscope Slides Matsunami TOMO-11/90 | Roche diagnostics | 8082286001 | these slides are used for a better adhesion of sections |
| Microtome | Microm Microtech France | HM340E | |
| Panoramic scan II | 3dhistech | 2397612 | |
| Paraffin embedding equipment | Leica | EG1150C | |
| Plastic pipette Pasteur 2 mL | VWR | 612-1681 | |
| Q Path flacon 150mL cape blanc x250 | VWR | 216-1308 | Good for environmental samples, pharmaceuticals and diagnostic reagents. Polypropylene (PP) are rigid, solid, provide excellent stress crack and impact resistance and have a good oil and alcohol barrier and chemical resistance. PE-lined cap is stress crack resistant and offers excellent sealing characteristics. |
| Set of micropipettors (p200, p1000) | Thermo Scientific | 11877351 (20-200) 11887351(p1000) | |
| OPERA PHENIX | PerkinElmer | HH14000000 | |
| SP5 inverted confocal microscope | Leica | LSM780 | |
| Tissue cassette | VWR | 720-0228 | |
| Zeiss Axiomager microscope | Leica | SIP 60549 | |
| Reagent | |||
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | A7030-100G | |
| Cytoblock (kit) | Thermofisher Scientific | 10066588 | |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | 57648266 | CAUTION: toxic and flammable. Vapors may cause irritation. Manipulate in a fume hood. Avoid direct contact with skin. Wear rubber gloves, protective eye goggles. |
| Eosin aqueous 1% | Sigma-Aldrich | HT110316 | |
| Ethanol 96% | VWR | 83804.360 | CAUTION: Causes severe eye irritation. Flammable liquid and vapor. Causes respiratory tract irritation. Manipulate in a fume hood. Wear protective eye goggles. |
| Ethanol 100% | VWR | 20821.365 | CAUTION: Causes severe eye irritation. Flammable liquid and vapor. Causes respiratory tract irritation. Manipulate in a fume hood. Wear protective eye goggles. |
| Formalin 4% | Microm Microtech France | F/40877-36 | CAUTION: Formalin contains formaldehyde which is hazardous. Manipulate in a fume hood. Avoid direct contact with skin. Wear rubber gloves and protective eye goggles. |
| Fructose | Sigma-Aldrich | F0127 | |
| Gill hematoxylin type II | Microm Microtech France | F/CP813 | |
| Glycerol | Sigma-Aldrich | G5516 | 500 mL |
| Hoechst 33342 | Life Technologies | H3570 | CAUTION: Suspected of causing genetic defects. Avoid direct contact with skin. Wear rubber gloves and protective eye goggles. |
| Normal donkey serum | Sigma-Aldrich | D9663 | 10 mL |
| Paraffin Wax tek III | Sakura | 4511 | |
| Phosphate Buffer Saline (PBS) 1 X | Gibco | 14190-094 | |
| Tris-Buffered Saline (TBS) 10X | Microm Microtech France | F/00801 | 100 mL |
| Triton X-100 | Sigma-Aldrich | T8532 | CAUTION: Triton X100 is hazardous. Avoid contact with skin and eyes. |
| Xylene | Sigma-Aldrich | 534056 | CAUTION: Xylene is toxic and flammable. Vapors may cause irritation. Manipulate in a fume hood. Avoid direct contact with skin. Wear rubber gloves, protective eye goggles. |
| Solutions | |||
| Clearing solution | Glycerol-Fructose clearing solution is 60% (vol/w) glycerol and 2.5 M fructose. To prepare 10 mL of this solution, mix 6 mL of glycerol and 4.5 g of fructose. Complete to 10 mL with dH2O. Use a magnetic stirrer overnight. Refractive index = 1.4688 at room temperature (RT: 19–23 °C). Store at 4 °C in dark for up to 1 month. | ||
| PBS-BSA 0,1% solution | To prepare 0,1% (vol/wt) PBS-BSA 0,1% solution, dissolve 500 mg of BSA in 50 mL of PBS-1X (store at 4°C for up to 2 weeks). And dilute 1mL of this solution into 9mL of PBS-1X. This solution can be used to precoat the tip and centrifugation tube. | ||
| Permeabilisation-blocking solution (PB solution) | The PBSDT blocking solution is PBS-1X supplemented with 0.1% – 1% Tritonx-100 (depending on the protein localization membrane/nucleus), 1% DMSO, 1% BSA and 1% donkey serum (or from the animal in which the secondary antibodies were raised). This solution can be stored at 4°C for up to 1 month. | ||
| PB:PBS-1X (1:10) solution | PB:PBS-1X (1:10) solution is a 10 time diluted PB solution. To prepare 10 mL of this solution dilute 1 mL of PB solution in 9 mL of PBS-1X. | ||
| Software | |||
| Halo software | Indicalabs | NM 87114 | |
| Harmony software | PerkinElmer | HH17000010 |
Referencias
- Ryu, N. E., Lee, S. H., Park, H. Spheroid culture system methods and applications for mesenchymal stem cells. Cells. 8 (12), 1-13 (2019).
- Bartfeld, S., Clevers, H. Stem cell-derived organoids and their application for medical research and patient treatment. Journal of Molecular Medicine. 95 (7), 729-738 (2017).
- Cui, X., Hartanto, Y., Zhang, H. Advances in multicellular spheroids formation. Journal of the Royal Society, Interface. 14 (127), (2017).
- Dekkers, J. F., et al. High-resolution 3D imaging of fixed and cleared organoids. Nature Protocols. 14, 1756-1771 (2019).
- Broutier, L., et al. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nature Protocols. 11 (9), 1724-1743 (2016).
- Rezanejad, H., Lock, J. H., Sullivan, B. A., Bonner-Weir, S. Generation of pancreatic ductal organoids and whole-mount immunostaining of intact organoids. Current Protocols in Cell Biology. 83 (1), 82 (2019).
- Edmondson, R., Broglie, J. J., Adcock, A. F., Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay and Drug Development Technologies. 12 (4), 207-218 (2014).
- McCray, T., Richards, Z., Marsili, J., Prins, G. S., Nonn, L. Handling and assessment of human primary prostate organoid culture. Journal of Visualized Experiments: JoVE. (143), e59051 (2019).
- Ueda, H. R., et al. Tissue clearing and its applications in neuroscience. Nature Reviews: Neuroscience. 21 (2), 61-79 (2020).
- Lazzari, G., et al. Light sheet fluorescence microscopy versus confocal microscopy: in quest of a suitable tool to assess drug and nanomedicine penetration into multicellular tumor spheroids. European Journal of Pharmaceutics and Biopharmaceutics. 142, 195-203 (2019).
- Gabriel, J., Brennan, D., Elisseeff, J. H., Beachley, V. Microarray embedding/sectioning for parallel analysis of 3D cell spheroids. Scientific Reports. 9, 16287 (2019).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados